")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Folate receptor-targeted liposomes enhanced the antitumor potency of imatinib through the combination of active targeting and molecular targeting
Authors Ye P, Zhang W, Yang T, Lu Y, Lu M, Gai Y, Ma X, Xiang G
Received 6 January 2014
Accepted for publication 11 March 2014
Published 7 May 2014 Volume 2014:9(1) Pages 2167—2178
DOI https://doi.org/10.2147/IJN.S60178
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Peng Ye,1,2 Wendian Zhang,1 Tan Yang,1 Yao Lu,1 Miao Lu,1 Yongkang Gai,1 Xiang Ma,1 Guangya Xiang1
1School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Pharmacy, Wuhan University, Renmin Hospital, Wuhan, People’s Republic of China
Purpose: Imatinib inhibits platelet-derived growth factor receptor (PDGFR), and evidence shows that PDGFR participates in the development and progression of cervical cancer. Although imatinib has exhibited preclinical activity against cervical cancer, only minimal clinical therapeutic efficacy was observed. This poor therapeutic efficacy may be due to insufficient drug delivery to the tumor cells and plasma protein binding. Therefore, the purpose of this study was to explore a novel folate receptor (FR)-targeted delivery system via imatinib-loaded liposomes to enhance drug delivery to tumor cells and to reduce plasma protein binding.
Methods: Imatinib was remote-loaded into FR-targeted liposomes which were prepared by thin film hydration followed by polycarbonate membrane extrusion. Encapsulation efficiency, mean size diameter, and drug retention were characterized and cellular uptake, cell cytotoxicity, and cell apoptosis on cervical cancer HeLa cells were evaluated. Comparative pharmacokinetic studies were also carried out with FR-targeted imatinib liposomes, simple imatinib liposomes, and free imatinib.
Results: High encapsulation efficiency (>90%), appropriate mean particle size (143.5 nm), and zeta potential (–15.97 mV) were obtained for FR-targeted imatinib liposomes. The drug release profile showed minimal imatinib leakage (<5%) in phosphate-buffered saline (PBS) at pH =7.4 within 72 hours of incubation, while more leakage (>25%) was observed in PBS at pH =5.5. This indicates that these liposomes possess a certain degree of pH sensitivity. Cytotoxicity assays demonstrated that the FR-targeted imatinib liposomes promoted a six-fold IC50 reduction on the non-targeted imatinib liposomes from 910 to 150 µM. In addition, FR-targeted imatinib liposomes enhanced HeLa cell apoptosis in vitro compared to the non-targeted imatinib liposomes. Pharmacokinetic parameters indicated that both targeted and non-targeted liposomes exhibited long circulation properties in Kunming mice.
Conclusion: These findings indicate that the nano-sized FR-targeted PDGFR antagonist imatinib liposomes may constitute a promising strategy in cervical cancer therapy through the combination of active targeting and molecular targeting.
Keywords: liposomes, imatinib, folate receptor, tumor targeting, PDGFR
Introduction
Cervical cancer is the fourth leading cause of cancer death in females worldwide.1 Standard treatment for more advanced cervical cancer is radiation combined with chemotherapy or concurrent chemoradiation.2,3 The suboptimal clinical outcome arising from chemoradiation emphasizes the need for new drugs and/or drug delivery platforms.
Imatinib mesylate (Gleevec®; Novartis, Basel, Switzerland) is the standard treatment for chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GIST).4,5 Additionally, imatinib prevents platelet-derived growth factor receptor (PDGFR) activation and the subsequent signaling pathway, which is responsible for cell proliferation, migration, and angiogenesis.6–8 PDGFR is an attractive therapeutic target for cervical cancer, since PDGFR and its ligands are frequently expressed in cervical cancer and play an important role in tumor pathogenesis. Moreover, cervical cancer cell line growth could be inhibited by the PDGFR antagonist imatinib.9–11 However, imatinib has shown minimal therapeutic activity as second-line treatment of recurrent or metastatic cervical cancer expressing PDGFR in clinical trials.12 Similar minimal therapeutic results were also obtained with other solid tumors like prostate and ovarian cancer in the clinical study.13,14 The reason for the treatment failure of imatinib is most likely that the drug concentration in the body cannot reach the effective anti-tumor concentration. In vitro studies showed that the IC50 of imatinib on PDGFR highly expressed tumor cell lines, such as prostate, ovarian, and cervical cancer, was in the 10–15 μM range, which is far higher than the plasma concentration arising from the usual imatinib dose of 400 mg/day.9,15–17 Furthermore, nonspecific drug binding in the plasma, particularly alpha-1-acid glycoprotein (AGP) binding can decrease the free, and therefore active, concentration of the drug, and hence reduce the tumor cellular concentration of imatinib.18 Therefore, considering the referred nonspecific drug binding with AGP and the fact that the allowed imatinib dose cannot reach the effective cancer cellular concentration, we hypothesized that loading of imatinib in a drug delivery system would be a promising therapeutic approach in cervical cancer.
Liposomes are small synthetic lipid vesicles that exhibit biocompatible and biodegradable features for their application as drug delivery systems to protect the drugs from premature inactivation and to modulate pharmacokinetics, biodistribution, and drug solubility.19 Liposomes have been designed as a delivery system to improve the overall therapeutic index of encapsulated anticancer drugs, due to preferential delivery and accumulation at tumor tissues as compared to free drugs.20,21 Through controlling the particle size at ~100–200 nm and surface modification with biocompatible polymers (such as polyethylene glycol [PEG]), liposomes can exhibit prolonged systemic circulation time, escape their uptake by macrophages, and increase tumor localization by up to ten-fold or more as a result of the enhanced permeability and retention (EPR) effect.22,23 Meanwhile, encapsulated imatinib can avoid nonspecific binding such as AGP and decrease systemic toxicity.
On the basis of the EPR effect, liposomes further modified by tumor-active–targeted ligands such as antibodies, peptides, and small compounds like folate have been widely investigated.24,25 The application of liposomal formulations as carriers for anticancer drug modified by tumor-active–targeted ligand folate has been attempted and described in the literature in recent years.26–29 Folate and folate conjugates can bind to the folate receptor (FR) with high affinity and enter the FR elevated malignant cells by receptor-mediated endocytosis,30,31 and meanwhile avoid being endocytosed by those normal cells that express a low level of FRs.32 Thus, adding folic acid (FA) to the distal ends of liposome-anchored PEG molecules can further improve tumor cell selectivity and the uptake efficiency of liposomes.
The main aim of the present work was the development of a novel FR-targeted liposomal formulation that could exhibit efficient and stable encapsulation of imatinib and therefore mediate its delivery to the solid tumor effectively and specifically. Characterization of the liposomes, including transmission electron microscopy (TEM) morphology, size, and zeta potential, was carried out. Drug entrapment efficiency, drug loading capacity, release properties in vitro, and pharmacokinetic properties were tested. The in vitro growth inhibition activity, cellular uptake efficiency, and cell apoptosis of folate receptor-targeted liposomal formulations of imatinib were also investigated by a cervical cancer HeLa cell model which overexpresses PDGFR.9
Materials and methods
Materials
Imatinib mesylate with a purity of 99% was synthesized in the authors’ laboratory.33 Hydrogenated soy phosphatidylcholine (HSPC) and monomethoxy polyethylene glycol2000-distearoyl phosphatidylethanolamine (mPEG2000-DSPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). Cholesterol (CHOL) was obtained from J&K Chemical Ltd (Beijing, People’s Republic of China). Sephadex G-25 gel-filtration columns were obtained from Amersham Biosciences (Uppsala, Switzerland). Sepharose CL-4B chromatography media was purchased from Sigma-Aldrich (St Louis, MO, USA). FA was purchased from Huixing Biological and Chemical Reagents Co, Ltd (Shanghai, People’s Republic of China). Calcein was purchased from Aladdin Reagent Co (Shanghai, People’s Republic of China). Folate-PEG3350-CHEMS was synthesized in the authors’ laboratory.34 All reagents and solvents were of analytical or high-performance liquid chromatography (HPLC) grade and were used without further purification.
The human cervical carcinoma HeLa cell line and the human lung carcinoma A549 cell line were provided by Tongji Hospital (Tongji Medical College, Huazhong University of Science and Technology, Hubei, People’s Republic of China). L-glutamine, trypsin (1:250), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and EDTA-Na2 were purchased from Amresco (Solon, OH, USA). Penicillin-streptomycin (100,000 units/mL) was supplied by Hyclone (Logan, UT, USA). Fetal bovine serum (FBS) was a product of Hangzhou Sijiqing Biological Engineering Materials Co, Ltd (Hangzhou, People’s Republic of China). The Annexin-V/FITC (fluorescein isothiocyanate) and propidium iodide (PI) apoptosis detection kit was purchased from Molecular Probes, Inc. (Eugene, OR, USA). Roswell Park Memorial Institute medium (RPMI)−1640 10× medium without FA was purchased from Sigma-Aldrich.
Female Kunming mice (25 g, 8 weeks old) were purchased from the Animal Center of Tongji Medical College, Huazhong University of Science and Technology, People’s Republic of China.
Liposome preparation and drug loading
FR-targeted liposomes loaded with imatinib (F-L-imatinib) and non-targeted control liposomes loaded with imatinib (L-imatinib) were prepared by thin film hydration followed by polycarbonate membrane extrusion. Imatinib was remote-loaded into the liposomes using a transmembrane pH gradient.35 The lipid compositions of L-imatinib and F-L-imatinib were HSPC/CHOL/mPEG2000-DSPE at a molar ratio of 55:40:5, and HSPC/CHOL/mPEG2000-DSPE/folate-PEG3350-CHEMS at a molar ratio of 55:40:4:1, respectively. Briefly, the lipids (68 mg) in CHCl3 were dried by rotary evaporation at 40°C and further dried under vacuum to remove traces of CHCl3. The lipid film was hydrated with ammonium sulfate (2 mL, 300 mM) for 30 minutes at 60°C with occasional vortex mixing. The suspension of lipids was then extruded five times through a 100 nm pore-size polycarbonate membrane using a Lipex Extruder (Northern Lipids Inc., Vancouver, BC, Canada) driven by pressurized nitrogen at 60°C to produce unilamellar vesicles. Liposomes were then subjected to size exclusion chromatography on Sephadex G-25 columns to replace the extravesicular medium with phosphate-buffered saline (PBS, pH7.4) and establish a transmembrane pH gradient for active loading. Imatinib was dissolved in deionized H2O and added to the above prepared liposomes at different imatinib-to-total lipid ratios (1:2, 1:3, 1:4, 1:6, 1:8, mol:mol), followed by 30 minutes of incubation at 60°C. Residual-free imatinib in the liposomal preparation was removed by size exclusion chromatography on a Sepharose CL-4B column.
Fluorescent liposomes (calcein liposomes) were prepared using the above described procedures except that the lipid film was hydrated in 50 mM calcein instead of the ammonium sulfate solution. The lipid compositions of FR-targeted liposomes loaded with calcein (F-L-calcein) and non-targeted control liposomes loaded with calcein (L-calcein) were the same as the liposomes containing imatinib. Briefly, the lipids were dissolved in CHCl3 and dried into a thin film by rotary evaporation and then further dried under vacuum. The lipid film was hydrated with 2 mL of 50 mM calcein for 30 minutes at 60°C with occasional vortex mixing. The suspension of lipids was then extruded five times through a 100 nm pore-size polycarbonate membrane using a Lipex Extruder driven by pressurized nitrogen at 60°C. Residual-free calcein in the liposomal preparation was removed by size exclusion chromatography on a Sepharose CL-4B column.
Drug quantification
Imatinib concentration in the liposomes was determined by measuring absorption at 265 nm on an Agilent HPLC system (1100-series; Santa Clara, CA, USA) following liposome lysis in 10% Triton X-100. Calcein concentration in the liposomes was determined by measuring absorbance at 492 nm on a ultraviolet-visible spectrometer (Shanghai Spectrum Instruments Co, Ltd, Shanghai, People’s Republic of China) following liposome lysis in 10% Triton X-100. Drug encapsulation efficiency (EE%) was determined by running samples through a Sepharose CL-4B column, and calculated using the following equation:

Drug loading capacity (DL%) was calculated according to the following equation:

Evaluation of the physicochemical characteristics of liposomes
The particle size and zeta potential were determined using a Zetasizer 3000 laser particle size analyzer (Malvern Instruments, Malvern, UK). The morphology of liposomes was observed under TEM (Tecnai G2 20 TWIN; FEI Ltd, Hillsboro, OR, USA). A drop of the diluted liposome dispersion was applied onto a carbon-coated 300-mesh copper grid and left for 1.5 minutes to allow adherence between liposomes and the carbon substrate. After excess of dispersion was removed with filter paper, the grid was kept in 1% weight per volume phosphotungstic acid solution for 2 minutes and allowed to dry in air for 10 minutes before examination under the TEM.
Evaluation of in vitro stability of liposomes during storage
Stability of liposomes after storage at 4°C for a period of 30 days was analyzed by determining the remaining drug, the vesicle size, and zeta potential. Samples were withdrawn periodically and the remaining drug in the liposomes was measured as mentioned above in the determination of drug EE%. The sizes and size distributions were evaluated using a Zetasizer during this period. The zeta potential was determined using a Zetasizer at storage start and end points.
In vitro release study
Drug release from liposomes into PBS (pH 7.4 and pH 5.5, PBS) was monitored by a dialysis method. A prepared dialysis bag (molecular weight cut off 3,500 Da) containing 3 mL suspension of liposomes was incubated in the PBS (200 mL) at 37°C±1°C under mild stirring (120 rpm). At various time points during the dialysis process, an aliquot of 1 mL was taken from the supernatant of the PBS. After removal of the supernatant, the same volume of fresh PBS was replaced. The amount of the imatinib within the collected supernatant was measured by HPLC.
Cell culture
HeLa and A549 cells were grown in RPMI-1640 folate-free media, supplemented with 10% (volume per volume) heat-inactivated FBS, 100 U/mL of penicillin, and 100 μg/mL of streptomycin, and maintained in a humidified atmosphere containing 5% CO2 at 37°C. Cell lines were maintained within their exponential growth phase.
In vitro cytotoxicity assay
The cytotoxicity of liposomes was determined by the MTT assay. Approximately 6,000 cells (HeLa or A549 cells) per well were seeded in 96-well plates and allowed to attach overnight. The following day, 100 μL containing serial dilutions of drug formulations, including free imatinib, L-imatinib, or F-L-imatinib and blank liposomes were added to the cells. The plates were subsequently incubated at 37°C for 72 hours. At the end of this incubation period, 20 μL of MTT in media (5 mg/mL) was added to the cells and the plates were incubated for a further 4 hours. Finally, the precipitated formazan was solubilized in 150 μL of dimethyl sulfoxide and absorbance was read at 492 nm using a plate reader (Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA).
Cellular uptake
Fluorescence imaging of cells
Binding and internalization of the liposomes in HeLa cells were examined by fluorescence microscope. HeLa cells were plated in six-well plates at 37°C under 5% CO2 in folate-free RPMI 1640 medium containing 10% FBS. The cells were grown until 80% confluence was reached. The medium was then removed and replaced with fresh medium (without serum). L-Calcein or F-L-Calcein and F-L-Calcein plus 1 mM FA were added. All liposomes contained calcein at a concentration of 15 μM. FA was used as an FR-blocking agent. After incubation at 37°C for 1 hour, the cells were washed three times with cold PBS and were then examined on an Olympus IX71 fluorescence microscope (PerkinElmer, Waltham, MA, USA).
Quantitative analysis of cellular uptake
HeLa cells grown in a monolayer were suspended by brief treatment with trypsin, and then washed three times with folate-free RPMI 1640 medium (without serum). Aliquots of HeLa cell suspension were incubated with L-calcein or F-L-calcein for 1 hour at 37°C. To determine the role of FR binding, 1 mM FA was added to the incubation media as the FR-blocking group. After incubation at 37°C for 1 hour, the cells were washed three times with cold PBS to removed unbound liposomes. The fluorescence intensity of cells was analyzed by flow cytometry (FCM) on a BD-LSR cytometer (BD Biosciences, San Jose, CA, USA).
Cell apoptosis assay
HeLa cells were seeded in six-well plates at a density of 5×104 cells per well and allowed to attach overnight. On the following day, 2 mL drug formulations, including free imatinib, L-imatinib, or F-L-imatinib with or without 1 mM free folate were added to the cells. After 24 hours, cells were gently trypsinized and collected in tubes for FCM detection. Before detection, 5 μL of Annexin-V/FITC and 10 μL of PI were added to the binding-buffer–containing cells. Stained cells were analyzed by flow cytometry, using a BD-LSR cytometer, and detections were performed within 1 hour. The FITC+/PI- phase indicated early apoptosis, whereas the FITC+/PI+ phase indicated late apoptosis.
Pharmacokinetic studies
Experimental animals and study design
Female Kunming mice (25 g, 8 weeks old) were housed and handled according to the guidelines of the Experimental Animal Ethics Committee of Huazhong University of Science and Technology. Mice (four per group) were given intravenous injections of different drug formulations, including free imatinib, L-imatinib, or F-L-imatinib at a drug dose of 14.5 mg/kg via the tail vein. Blood samples were collected in heparin-treated tubes at various time points. Plasma was isolated by centrifugation (10 minutes, 5,000 rpm, 4°C) and stored at −35°C until HPLC analysis.
Sample processing and analysis
Imatinib concentration in the plasma was measured using HPLC. Plasma was added to an equal volume of methanol, vortex mixed, and centrifuged (15,000 rpm, 15 minutes, 4°C), after which 100 μL of the supernatant was transferred to a clean tube and evaporated to dryness under nitrogen. For HPLC sample loading, 100 μL of 0.1% phosphoric acid (volume per volume) was used to reconstitute the residue and a 20 μL aliquot was injected onto the C18 reverse phase column for analysis. The retention time for imatinib was about 7.6 minutes and linearity was obtained in the range of 0.5–300 μg/mL.
Pharmacokinetic analysis
Pharmacokinetic parameters were determined using the software WinNonlin (Pharsight, Mountain View, CA, USA), including area under the curve (AUC), mean residence time (MRT), total body clearance (CL), and plasma half-lives for the elimination phase (T1/2β).
Statistical analysis
All data are shown as means ± standard deviation (SD) unless specifically noted. Student’s t-test or one-way analyses of variance (ANOVA) were performed. A P-value <0.05 was considered to be statistically significant.
Results
Synthesis and characterization of liposomes
Imatinib was encapsulated into the FR-targeted (folate-PEG3350-CHEMS) and non-targeted liposomes using a remote-loading method with an ammonium sulfate gradient. In order to determine the influence of imatinib/lipid (I/L) incubation ratio on loading, several ratios (1:2, 1:3, 1:4, 1:6, 1:8, mol:mol) were tested on the F-L-imatinib (Figure 1). While a I/L incubation ratio of 1:4 or lower provided excellent EE% values (>90%), only the incubation ratio of 1:4 provided the highest loading capacity close to 0.23 mol imatinib/mol of total lipids.
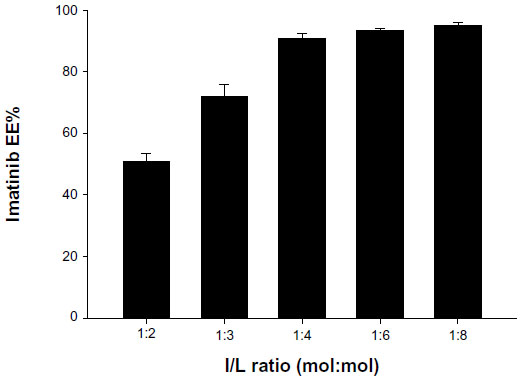 | Figure 1 Effect of initial imatinib-to-total lipid (I/L) ratio (mol:mol) on imatinib EE% of F-L-imatinib. |
Under the same loading conditions, non-targeted liposomes appeared to have a higher EE% value of 95.2%±1.26%; however, no relevant differences were observed in terms of encapsulation parameters between the two imatinib liposomes. The obtained optimal encapsulation parameters of the formulations are summarized in Table 1. The morphology of the FR-targeted imatinib liposomes was observed in a TEM image as shown in Figure 2. The F-L-imatinib formed discrete spherical vesicles at nanometer sizes. The average particle size of the prepared F-L-imatinib was further determined as shown in Figure 3. F-L-imatinib and L-imatinib presented similar size distributions with a mean diameter of around 140 nm. The zeta potentials of F-L-imatinib and L-imatinib were −15.97±1.89 mV and −24.13±2.43 mV, respectively. The negative zeta potentials might be due to the presence of the negatively charged lipid mPEG2000-DSPE in the formulation.36 A lower proportion of mPEG2000-DSPE in F-L-imatinib gave it a less negative charge on the surface than L-imatinib. These results indicate that imatinib can be successfully loaded into liposomes with a high EE% value. It appears that folate-PEG3350-CHEMS would not interfere with imatinib entrapment.
 | Table 1 Physicochemical properties of F-L-imatinib and L-imatinib |
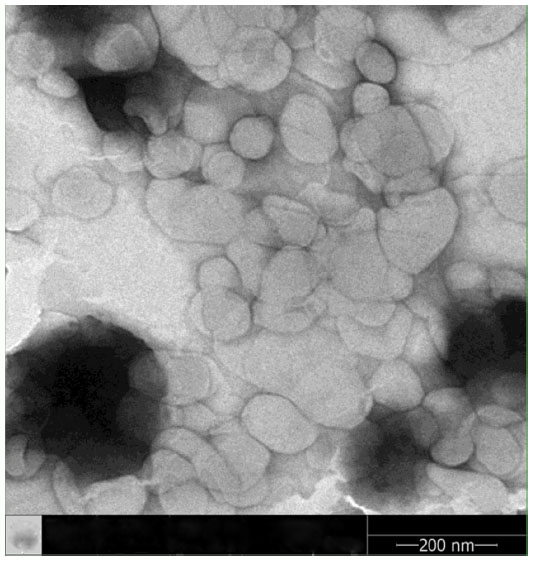 | Figure 2 Transmission electron microscopy images of F-L-imatinib. |
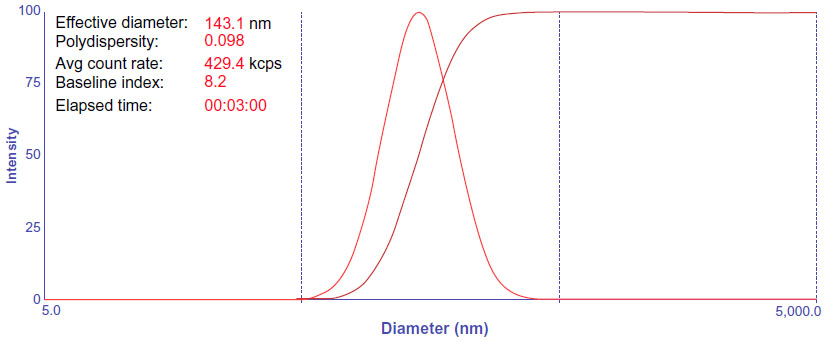 | Figure 3 Size distribution of F-L-imatinib. |
In vitro stability studies during storage
The stability of the imatinib liposome formulations was monitored by changes in zeta potential, particle size, and drug retention during storage at 4°C. The zeta potential at the start and end points was−15.97±1.89 mV and −17.47±3.06 mV, respectively. The zeta potential exhibited little change over the 1 month of storage, and reflects the stability of F-L-imatinib. As shown in Figure 4, the results also indicate that during storage at 4°C, no significant variation in liposome size and drug retention was detected. It appears that both liposomal formulations showed excellent stability during the storage and no drug precipitation or liposome aggregation was observed for at least 30 days.
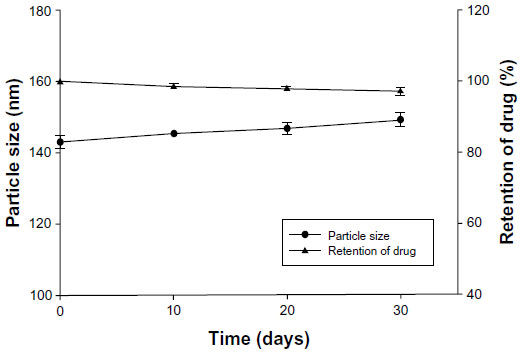 | Figure 4 Stability of F-L-imatinib as measured by the particle size (n=3) and the drug retention (n=3). |
In vitro release
The in vitro stability studies were performed in PBS buffer at 37°C±1°C to ascertain the effect of pH on the drug release at pH 5.5 and 7.4 and to predict the in vivo stability profile of F-L-imatinib and L-imatinib. The releasing rate was calculated at several time points within 72 hours and drug leakage profiles were plotted (Figure 5). At the physiological pH 7.4 in PBS at 37°C±1°C, F-L-imatinib and L-imatinib showed very similar imatinib release profiles. Both types of imatinib-encapsulated liposomes showed minimal imatinib leakage in PBS within 72 hours of incubation, and more than 95% of the encapsulated imatinib was still retained in liposomes. However, when incubating at pH 5.5, corresponding to the pH in endosomal compartments, the imatinib release from both targeted and non-targeted liposomes increased significantly. At 72 hours of incubation, the cumulative release percentages of F-L-imatinib and L-imatinib reached 29.5% and 10.9%, respectively, at pH 5.5. Clearly, the findings indicate that the incubation pH has an impact on the stability profile of the liposomes. The reason for the increased drug release is due to the collapse of the transmembrane pH gradient of these liposomes at the lower pH value. Thus, it can be concluded that F-L-imatinib and L-imatinib are stable under the physiological pH, and that these liposomes exhibit pH-sensitive properties.
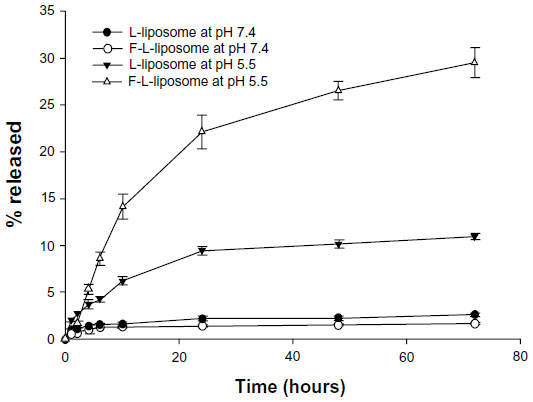 | Figure 5 In vitro release of imatinib from F-L-imatinib or L-imatinib in pH 7.4 PBS or pH 5.5 PBS at 37°C±1°C. |
In vitro cytotoxicity assay
The cytotoxicity of F-L-imatinib, L-imatinib, and imatinib solution was determined in HeLa cells and A549 cells using an MTT assay. The results are summarized in Table 2. In FR-positive and PDGFR-positive HeLa cells, both the F-L-imatinib and imatinib solutions induced cell death in a dose-dependent manner. L-imatinib did not demonstrate significant antitumor efficacy, with an IC50 of 906 μM. When imatinib was loaded into FR-targeted liposomes, it revealed growth inhibition activity (IC50 =151 μM) that was six times higher than that of L-imatinib. Free imatinib solution showed a much lower IC50 of 15.7 μM, as was expected. Both the liposomal drug formulations exhibited at least ten times lower growth inhibition activity as compared to the free drug. This is in agreement with results obtained from previous studies.27,28 Empty liposomes were tested as controls and did not induce significant cell death (data not shown). In contrast, in FR-negative and PDGFR-positive A549 cells,27,37 neither F-L-imatinib nor L-imatinib showed significant antitumor efficacy. Only free imatinib solution showed dose-dependent inhibition to A549 cells with an IC50 of 34.6 μM. All the results showed that the FR-targeted liposomal imatinib led to a significant increase in antitumor activity via folate-mediated endocytosis in HeLa cells as compared to simple liposomes (P<0.05).
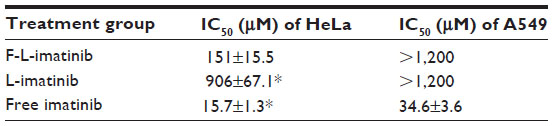 | Table 2 IC50 of F-L-imatinib, L-imatinib, and free imatinib against HeLa cells and A549 cells at 72 hours |
Cellular uptake of F-L-calcein compared to L-calcein
In order to demonstrate that F-L-imatinib can be taken up efficiently by HeLa cells via folate-mediated endocytosis, the calcein-labeled liposomes were prepared and the cellular uptake of liposomes by HeLa cells was visualized by fluorescence microscopy and quantitatively analyzed using FCM. Green fluorescence was associated with liposomal calcein. As shown in Figure 6, the fluorescence intensity of the F-L-calcein group was significantly higher than that of the L-calcein group, and intensity decreased when free FA was added. The study indicates that FR-targeted liposomes can be more efficiently taken up by the FR-positive cells compared to the non-targeted liposomes and the FR-mediated endocytosis can be blocked by free FA. Meanwhile, FCM results indicated that the cellular uptake of FR-targeted liposomal calcein was about 40 times higher than that of non-targeted liposomal calcein (Figure 7). These data demonstrate that FR-targeted liposomes could effectively target the HeLa cells through the FR-mediated endocytosis process.
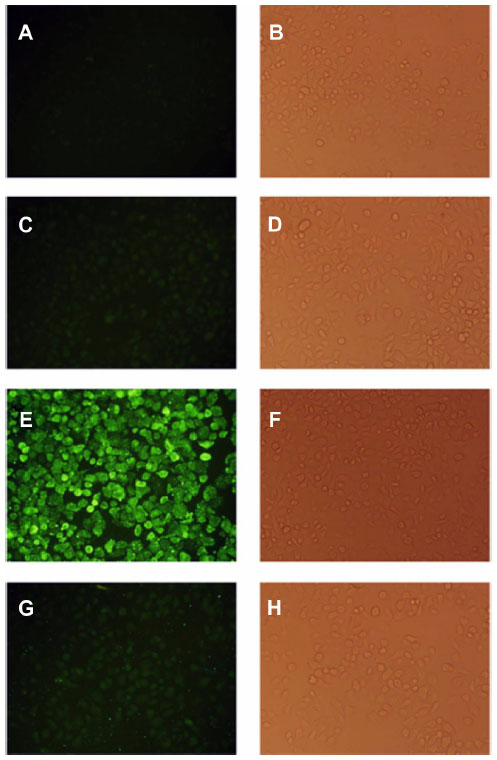 | Figure 6 Uptake of F-L-calcein and L-calcein by HeLa cells. |
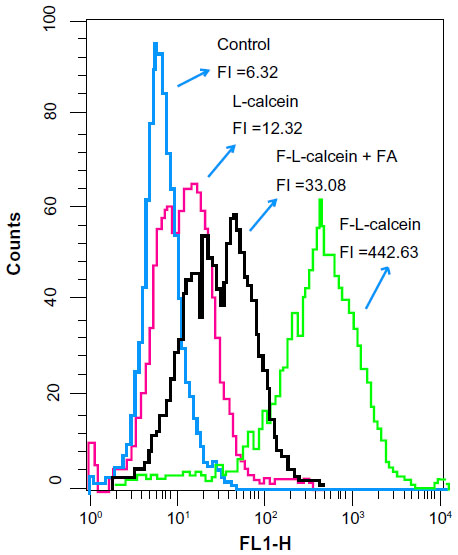 | Figure 7 Uptake of liposomal calcein by cultured HeLa cells measured by flow cytometry. |
Cellular apoptosis assay
To assess the apoptosis of tumor cells induced by various imatinib formulations, FCM analysis using HeLa cells was performed. As shown in Figures 8 and 9, incubation of HeLa cells with L-imatinib induced apoptosis in 10.9% of cells after 24 hours, whereas cells incubated with F-L-imatinib at the same concentration showed a significantly higher apoptotic rate of 26.4% (P<0.05). When the free FA was added, the percentage of cell apoptosis induced by F-L-imatinib fell to 16.5%. The result can be explained by the fact that the excess free FA competitively binds to FRs, resulting in lower binding of folate-linked liposomes. These results are entirely consistent with the report for a folate-linked liposomal curcumin formulation in the previous study.31 Both F-L-imatinib and F-L-imatinib plus 1mM of free FA showed dose-dependent apoptosis to HeLa cells (Figure 10). However, cells treated with L-imatinib still showed little apoptosis when the concentration was raised to 350 μM. It appears that L-imatinib is stable in cell culture medium at pH 7.4 and internalizing of the drug is limited in the tumor cells.
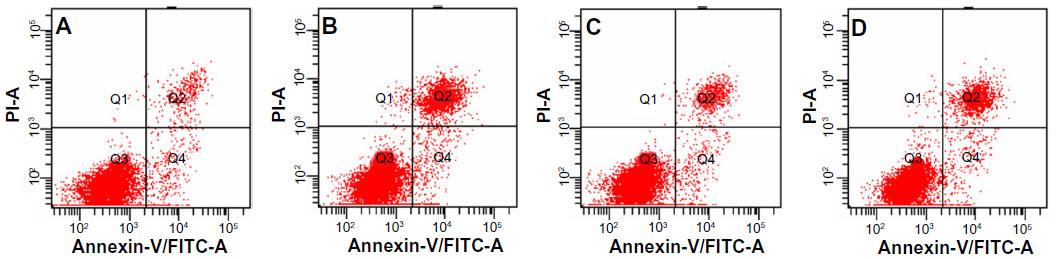 | Figure 8 Typical pictures of cellular apoptosis analyzed by flow cytometry. |
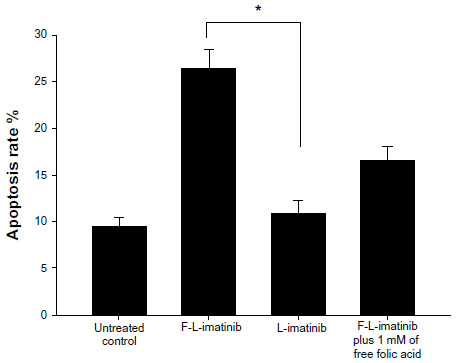 | Figure 9 Mean apoptosis rate of each group. |
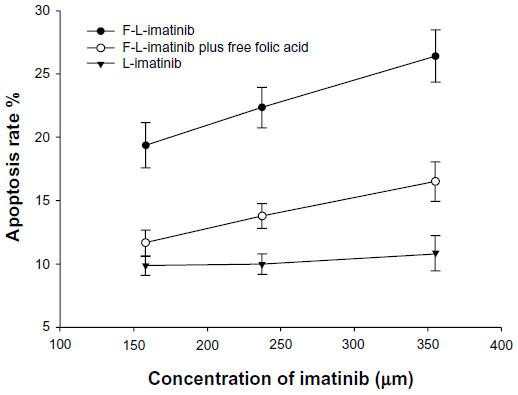 | Figure 10 Dose–response curves obtained for HeLa cells treated with different imatinib formulations. |
Pharmacokinetic properties of imatinib-loaded liposomes
In addition to drug release properties, the circulation lifetime of liposomes following intravenous administration is an important parameter in the evaluation of the performance of liposome–drug formulations. This is because long-circulating systems accumulate more effectively at tumor sites, leading to improved therapeutic benefits. The circulation lifetimes of F-L-imatinib and L-imatinib were examined in Kunming mice and the plasma clearance kinetics are shown in Figure 11 and Table 3. As expected, free imatinib showed rapid clearance from the plasma. Meanwhile, the circulation time data demonstrates that both liposomal imatinib formulations showed much longer circulation times than the free imatinib (P<0.05). The shorter T1/2β value observed for F-L-imatinib compared with L-imatinib reflects that the presence of folate-PEG3350-CHEMS lipid increases liposome clearance, which is consistent with the findings in other studies,28,34 although the ANOVA results show that the difference is not statistically significant (P>0.05).
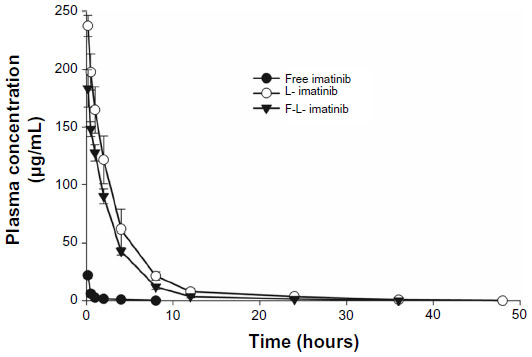 | Figure 11 Pharmacokinetic profiles of various imatinib formulations. |
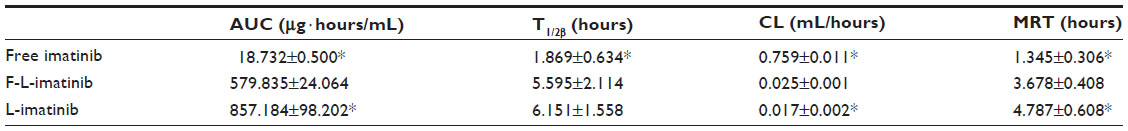 | Table 3 Pharmacokinetic parameters of three imatinib formations in Kunming mice following intravenous administration at a dose of 14.5 mg/kg via the tail vein |
Discussion
The presence of PDGFR in a number of human solid tumors, such as cervical cancer, is correlated with tumor metastasis and poor prognosis.38 Thus, PDGFR provides a novel therapeutic target for solid tumor treatment. In a cervical carcinoma chemosensitivity assay, 66.6% of cases were sensitive to imatinib, the rate of which was only lower than paclitaxel (93.8%), and far higher than cisplatin (13.3%) and carboplatin (7.6%).11 The research prompted a clinical study to evaluate the efficacy of imatinib for patients with cervical carcinoma as second-line treatment of recurrent or metastatic cervical cancer expressing PDGFR-α. The results, however, demonstrated that imatinib has poor efficacy in patients with cervical cancer at doses up to 600 mg a day.12 We hypothesized that loading of imatinib in a drug delivery system to reduce nonspecific drug binding with AGP and increase imatinib cancer cellular concentration would be a promising therapeutic approach in the treatment of cervical cancer. In our current study, we designed FR-targeted sterically stabilized liposomes encapsulating imatinib to improve the therapeutic effect of imatinib in the treatment of cervical cancer via the combination of active targeting and molecular targeting. A systematic approach was undertaken to develop and characterize preparation methods and liposome formulations in order to control drug leakage, and thus maintain optimal drug loading inside the liposomes to increase the delivery of the drug to tumor tissues. Characterization of the developed formulations was performed in terms of loading parameters, mean size diameter, drug retention, and growth inhibition activity against a HeLa cell line.
Few studies have reported imatinib encapsulation into liposomes.39–41 Since imatinib has four protonizable amine functional groups and is uncharged as a neutral form at physiological pH, it is possible to perform an active entrapment approach, in which imatinib can cross the liposomal membrane and be entrapped in the liposomal lumen due to its protonation at acidic pH values. Active drug loading is known for enhanced in vitro and in vivo drug retention when compared to passive methods. Using the active procedure, the ammonium sulfate gradient method was selected, and imatinib was encapsulated inside the liposomes at very high efficiency (>90%) and drug loading (I/L molar ratio, 1:4.4). In the preparation, F-L-imatinib had an average particle size of 140 nm with a narrow size distribution (low polydispersity index values), which was verified in TEM images. The uniform nanoscale sizes of the liposomes could give these nanoparticles a consistent kinetic profile in vivo, which could help the liposomes to exhibit the EPR effect. As we all know, passive targeting of liposomes can occur through the EPR effect. Because blood vessels in tumors are abnormal and leaky (gaps about 400–600 nm), drug carriers loaded with anticancer drugs of an average size of ~200 nm or less are appropriate for the EPR effect.42 Meanwhile, both L-imatinib and F-L-imatinib possessed negative zeta potentials, which might be due to the presence of the negatively charged lipid mPEG2000-DSPE in the formulation. With an increasing amount of negative charge on the surface of liposomes, the physical stability of liposomes against aggregation and fusion can be increased.43
In vitro and in vivo release of liposomes is another important factor in evaluating the properties of drug-loaded liposomes. To simulate the drug release profile in the bloodstream and in endosomes, PBS (pH =7.4 and pH =5.5) was chosen. The results in PBS (pH =7.4) showed that less than 5% of both loaded drug was released from F-L-imatinib and L-imatinib over 72 hours, suggesting that imatinib could remain stable in the internal water phase of the liposomes during blood circulation before the drug carrier targets the tumor site. Meanwhile, when incubated in PBS (pH =5.5), the imatinib release from both targeted and non-targeted liposomes increased significantly, suggesting that these liposomes possess pH sensitivity and can release the drug in the acidic environment of endosomes after being internalized into tumor cells. The increased drug release is due to the transmembrane pH gradient collapse of these liposomes on the lower pH. The result is consistent with a previous report by Lee et al.44 In their study, the collapse of the transliposomal pH gradient indirectly drove much more leakage of doxorubicin from liposomes. Compared with L-imatinib, F-L-imatinib exhibits much more drug release at pH 5.5, because folate-PEG3350-CHEMS decreases the rigidity and stability of the lipid bilayer membrane of liposomes at an acidic pH. The addition of folate-PEG3350-CHEMS made the lipid membranes more heterogeneous due to the poor lipid packing, and thus increased membrane permeability.45 Pharmacokinetic tests in vivo echoed the results of the drug release in vitro. Only when the drug release is slower than the liposome clearance will the liposome control the drug pharmacokinetics and bio-distribution and express the EPR effect for passive targeting.22 From the results of the test, liposomal imatinib showed a markedly different pharmacokinetic profile from free imatinib. Compared with free imatinib, liposomal imatinib can achieve a much higher plasma concentration over time, larger AUC, longer half-life and mean retention time, and lower clearance rate from the body. These data suggest that the process of imatinib in vivo could be changed by drug carriers, and liposomal imatinib is stable in the blood circulation with greatly prolonged blood circulation time. Therefore, with the longer blood circulation time and higher plasma concentration, the liposomal drug has more chance to perfuse into tumor tissues.
The HeLa cell line used in our study is a FR(+) cell line derived from a human cervical cancer. As expected, our FR-targeted liposomal calcein was more efficiently taken up by the HeLa cells, which was confirmed by the liposomal calcein uptake study. The uptake could be blocked by 1 mM free FA, suggesting that internalization of F-L-calcein was specifically mediated by an FR-mediated endocytosis process. The higher internalization efficiency demonstrated that liposomes conjugated with folate-PEG3350-CHEMS could effectively target the HeLa cells through the FR, which had previously been proved by reports on different lipid compositions or different cell lines.29,31,34 Consistent with the cellular uptake test, an FR-dependence cytotoxicity was demonstrated in a cell-killing test. F-L-imatinib was able to induce cell death more efficiently in HeLa cells compared to L-imatinib. L-imatinib showed low cytotoxicity to HeLa cells, because very little imatinib leaked from L-imatinib in the cell culture medium at pH 7.4 and liposomes without an internalizing ligand resulted in little chance to be endocytosed by tumor cells.46 F-L-imatinib exhibited an efficient killing effect on HeLa cells, although it has a similar drug release profile to L-imatinib at pH 7.4. The reason is that F-L-imatinib can be taken up efficiently by HeLa cells via FR-mediated endocytosis and releases imatinib at an acidic pH as in endosomes, which had been proved in the cellular uptake test and in vitro drug release study. Free imatinib can be directly taken up by HeLa cells through diffusion, so it had a lower IC50 value compared to both liposomal formulations. Apoptosis induced by F-L-imatinib was significantly greater than that seen in cells treated with L-imatinib and the result is consistent with the cytotoxicity assay. The results of the apoptosis test showed that F-L-imatinib can induce more apoptosis in HeLa cells through the combination of active targeting and molecular targeting.
We built a delivery system of FR-targeted liposomes with encapsulated imatinib in this study. From the results, the drug delivery system in our study is in the appropriate nanoscale size range and exhibits a longer systemic circulation time to allow it to concentrate in tumor tissues through the passive-targeting effect; it can specifically target to tumor cells and induce more cell death and cell apoptosis and also has the characteristic of pH sensitivity to make the drug stable in blood circulation and only release the drug in endosomes after uptake by tumor cells. These results suggest that F-L-imatinib possesses the basic characteristics of targeted delivery systems and has the potency to improve the antitumor effects of imatinib. The actual effect of F-L-imatinib will be confirmed in vivo by animal models and clinical research in future.
Conclusion
In conclusion, we have successfully encapsulated imatinib in folate-linked liposomes and demonstrated that F-L-imatinib could specifically target HeLa cells, and thus induce specific and efficient death and apoptosis of HeLa cells. This new therapeutic approach may be useful for the treatment of cervical cancer with an enhanced therapeutic potential and decreased toxicity when compared to free imatinib.
Acknowledgments
This work was supported by a Natural Science Foundation of China grant (No 81072596, No 81301235) and a Key Project of Natural Science Foundation of Hubei Province grant (No 2011CDA029).
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | |
Kong TW, Chang SJ, Paek J, et al. Comparison of concurrent chemoradiation therapy with weekly cisplatin versus monthly fluorouracil plus cisplatin in FIGO stage IIB-IVA cervical cancer. J Gynecol Oncol. 2012;23(4):235–241. | |
Rose PG. Chemoradiotherapy for cervical cancer. Eur J Cancer. 2002;38(2):270–278. | |
Steinert DM, McAuliffe JC, Trent JC. Imatinib mesylate in the treatment of gastrointestinal stromal tumour. Expert Opin Pharmacother. 2005;6(1):105–113. | |
Savage DG, Antman KH. Imatinib mesylate – a new oral targeted therapy. N Engl J Med. 2002;346(9):683–693. | |
Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1(7):493–502. | |
Buchdunger E, O’Reilly T, Wood J. Pharmacology of imatinib (STI571). Eur J Cancer. 2002;38 Suppl 5:S28–S36. | |
Östman A, Heldin CH. PDGF receptors as targets in tumor treatment. 2007;97:247–274. | |
Taja-Chayeb L, Chavez-Blanco A, Martinez-Tlahuel J, et al. Expression of platelet derived growth factor family members and the potential role of imatinib mesylate for cervical cancer. Cancer Cell Int. 2006;6:22. | |
Mayer TJ, Frauenhoffer EE, Meyers AC. Expression of epidermal growth factor and platelet-derived growth factor receptors during cervical carcinogenesis. In Vitro Cell Dev Biol Anim. 2000;36(10):667–676. | |
Kummel S, Heidecke H, Brock B, et al. [Imatinib – a possible therapeutic option for cervical carcinoma: results of a preclinical phase I study]. Gynakol Geburtshilfliche Rundsch. 2008;48(2):94–100. German. | |
Candelaria M, Arias-Bonfill D, Chavez-Blanco A, et al. Lack in efficacy for imatinib mesylate as second-line treatment of recurrent or metastatic cervical cancer expressing platelet-derived growth factor receptor alpha. Int J Gynecol Cancer. 2009;19(9):1632–1637. | |
Rao K, Goodin S, Levitt MJ, et al. A phase II trial of imatinib mesylate in patients with prostate specific antigen progression after local therapy for prostate cancer. Prostate. 2005;62(2):115–122. | |
Alberts DS, Liu PY, Wilczynski SP, et al. Phase II trial of imatinib mesylate in recurrent, biomarker positive, ovarian cancer (Southwest Oncology Group Protocol S0211). Int J Gynecol Cancer. 2007;17(4):784–788. | |
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. | |
Kubler HR, van Randenborgh H, Treiber U, et al. In vitro cytotoxic effects of imatinib in combination with anticancer drugs in human prostate cancer cell lines. Prostate. 2005;63(4):385–394. | |
Mundhenke C, Weigel MT, Sturner KH, et al. Novel treatment of ovarian cancer cell lines with Imatinib mesylate combined with Paclitaxel and Carboplatin leads to receptor-mediated antiproliferative effects. J Cancer Res Clin Oncol. 2008;134(12):1397–1405. | |
Gambacorti-Passerini C, Zucchetti M, Russo D, et al. Alpha1 acid glycoprotein binds to imatinib (STI571) and substantially alters its pharmacokinetics in chronic myeloid leukemia patients. Clin Cancer Res. 2003;9(2):625–632. | |
Slingerland M, GucHeLaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today. 2012;17(3–4):160–166. | |
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. | |
Wang X, Yang L, Chen ZG, Shin DM. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58(2):97–110. | |
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | |
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. | |
Chen X, Wang X, Wang Y, et al. Improved tumor-targeting drug delivery and therapeutic efficacy by cationic liposome modified with truncated bFGF peptide. J Control Release. 2010;145(1):17–25. | |
Mamot C, Drummond DC, Noble CO, et al. Epidermal growth factor receptor-targeted immunoliposomes significantly enhance the efficacy of multiple anticancer drugs in vivo. Cancer Res. 2005;65(24):11631–11638. | |
Chaudhury A, Tan BJ, Das S, Chiu GN. Increased ERK activation and cellular drug accumulation in the enhanced cytotoxicity of folate receptor-targeted liposomal carboplatin. Int J Oncol. 2012;40(3):703–710. | |
Watanabe K, Kaneko M, Maitani Y. Functional coating of liposomes using a folate-polymer conjugate to target folate receptors. Int J Nanomedicine. 2012;7:3679–3688. | |
Liu Y, Xu S, Teng L, et al. Synthesis and evaluation of a novel lipophilic folate receptor targeting ligand. Anticancer Res. 2011;31(5):1521–1525. | |
Xiong S, Yu B, Wu J, Li H, Lee RJ. Preparation, therapeutic efficacy and intratumoral localization of targeted daunorubicin liposomes conjugating folate-PEG-CHEMS. Biomed Pharmacother. 2011;65(1):2–8. | |
Hilgenbrink AR, Low PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94(10):2135–2146. | |
Lu Y, Ding N, Yang C, Huang L, Liu J, Xiang G. Preparation and in vitro evaluation of a folate-linked liposomal curcumin formulation. J Liposome Res. 2012;22(2):110–119. | |
Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv Drug Deliv Rev. 2004;56(8):1099–1109. | |
Liu YF, Wang CL, Bai YJ, Han N, Jiao JP, Qi XL. A facile total synthesis of imatinib base and its analogues. Org Process Res Dev. 2008;12(3):490–495. | |
Xiang G, Wu J, Lu Y, Liu Z, Lee RJ. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharm. 2008;356(1–2):29–36. | |
Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. | |
Hinrichs WL, Mancenido FA, Sanders NN, et al. The choice of a suitable oligosaccharide to prevent aggregation of PEGylated nanoparticles during freeze thawing and freeze drying. Int J Pharm. 2006;311(1–2):237–244. | |
Zhang P, Gao WY, Turner S, Ducatman BS. Gleevec (STI-571) inhibits lung cancer cell growth (A549) and potentiates the cisplatin effect in vitro. Mol Cancer. 2003;2:1. | |
Ehnman M, östman A. Therapeutic targeting of platelet-derived growth factor receptors in solid tumors. Exp Opin Invest Drug. 2014;23(2):211–226. | |
Mendonca LS, Moreira JN, de Lima MC, Simoes S. Co-encapsulation of anti-BCR-ABL siRNA and imatinib mesylate in transferrin receptor-targeted sterically stabilized liposomes for chronic myeloid leukemia treatment. Biotechnol Bioeng. 2010;107(5):884–893. | |
Harata M, Soda Y, Tani K, et al. CD19-targeting liposomes containing imatinib efficiently kill Philadelphia chromosome-positive acute lymphoblastic leukemia cells. Blood. 2004;104(5):1442–1449. | |
Beni S, Budai M, Noszal B, Grof P. Molecular interactions in imatinib-DPPC liposomes. Eur J Pharm Sci. 2006;27(2–3):205–211. | |
Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond). 2012;7(10):1577–1590. | |
Riaz M. Review article: stability and uses of liposomes. Pak J Pharm Sci. 1995;8(2):69–79. | |
Lee RJ, Wang S, Turk MJ, Low PS. The effects of pH and intraliposomal buffer strength on the rate of liposome content release and intracellular drug delivery. Biosci Rep. 1998;18(2):69–78. | |
Karve S, Bajagur Kempegowda G, Sofou S. Heterogeneous domains and membrane permeability in phosphatidylcholine-phosphatidic acid rigid vesicles as a function of pH and lipid chain mismatch. Langmuir. 2008;24(11):5679–5688. | |
Xiong XB, Huang Y, Lu WL, et al. Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release. 2005;107(2):262–275. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.