Back to Journals » Biologics: Targets and Therapy » Volume 12
Focus on biosimilar etanercept – bioequivalence and interchangeability
Received 10 May 2018
Accepted for publication 24 July 2018
Published 30 August 2018 Volume 2018:12 Pages 87—95
DOI https://doi.org/10.2147/BTT.S126854
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Fabrizio Cantini,1 Maurizio Benucci2
1Department of Rheumatology, Hospital of Prato, Prato, Italy; 2Rheumatology Unit, Hospital S. Giovanni di Dio, Florence, Italy
Background: The recent approval of reference etanercept (re-ETN) biosimilars SB4, GP2015, and HD203 produced relevant changes in the management of rheumatoid arthritis (RA), psoriatic arthritis, and ankylosing spondylitis due to the considerably lower cost of these products and the consequent savings.
Aims: To review the pharmacodynamics, pharmacokinetics, efficacy, and safety of ETN biosimilars when employed as first-line therapy or after transition from re-ETN. Patients’ acceptability was also addressed.
Evidence review: The available literature was reviewed through a search of PubMed database, and abstract books of the American College for Rheumatology and European League Against Rheumatism annual meetings. SB4, GP2015, and HD203 were licensed by the US, European and South Korea regulatory agencies after the bioequivalence to re-ETN was demonstrated through pharmacodynamic and pharmacokinetic studies, and randomized, head to head, controlled trials. Based on the evidence of efficacy and safety of SB4 and HD203 in RA, and of GP2015 in psoriasis, by the extrapolation principle, the three biosimilars were approved for all indications licensed for re-ETN, and the regulatory agencies introduced the interchangeability from the originator to the biosimilar. Extrapolation of indications, and particularly interchangeability raised relevant concerns among the rheumatologists due to the low level of evidence supporting the switching strategy (or transition). Rheumatologists’ concerns are oriented toward the relevant number of biosimilar discontinuations after the transition ranging from 7%–17% over a short-term follow-up period. As resulted from two studies, at least 20%–30% of the patients claimed more exhaustive information on the switching procedure.
Conclusion: Based on the available evidence, re-ETN biosimilars may be a good option as first-line therapy, while further data are needed to definitively establish the efficacy, safety, and the economic reflexes of transitioning from re-ETN.
Keywords: biosimilars, SB4, GP205, HD203, interchangeability, rheumatic diseases
Introduction
Over the past decade, the patent expiry of several branded biologics (termed as originators or reference products) employed in rheumatology, including infliximab (Remicade®), etanercept (Enbrel®), rituximab (Mabthera®), and adalimumab (Humira®), paved the way for the development and approval of biosimilars. As defined by the European Medicines Agency (EMA),1 a biosimilar is a biological medicine highly similar to another already approved biological medicine (the “reference medicine”), that should be comparable to the respective originator in terms of pharmacokinetic and pharmacodynamic properties, immunogenicity, efficacy, and safety, despite small differences in molecular composition. The US Food and Drug Administration (FDA) definition was less detailed indicating a biosimilar as a biologic product that is “highly similar to” an approved biologic product (the “reference”, “originator”, or “bio-originator” product) and that has “no clinically meaningful differences” in safety or effectiveness as compared to the reference product.2 To date, several biosimilars for the treatment of rheumatic diseases have been licensed in the European Union (EU) and US including biosimilars of reference infliximab, reference etanercept (re-ETN), reference adalimumab, and reference rituximab.1,3
The evidence of comparable efficacy and safety of currently licensed biosimilars with respect to the originators was provided by head to head, non-inferiority, randomized controlled trials (RCTs) of patients with rheumatoid arthritis (RA) naïve to biologics, with psoriasis (Pso), and with ankylosing spondylitis (AS).4 According to these data, by extrapolation, biosimilars were approved for all indications released for the reference product. After the RCT blinded phase, patients randomized to the originator arms were switched to the respective biosimilar with no significant loss of efficacy and no safety alerts.5
Biosimilars were created to reduce the heavy economic burden due to the high costs of originators, thus allowing treatment for a large number of patients,6 and to drive down the prices of the reference biologics in order to be competitive in the pharmaceutical market.
Re-ETN (Amgen/Pfizer’s Enbrel®) was approved by the FDA in November 1998 and by the EMA in February 2000.7 Due to its efficacy and safety in the treatment of inflammatory rheumatic disorders, including RA, AS, and psoriatic arthritis (PsA), during the last 20 years re-ETN conquered a large percentage of the market of biologics, reaching sales of US$8.7 billion in 2015, thus making it one of the most attractive targets for biosimilar producers.
The patents on Enbrel® will expire in the US in November 2028, and expired in Europe in August 2015.7,8
To date, through the years 2015–2017 both the FDA and EMA approved two biosimilars of re-ETN (bio-ETN), namely SB4 (trade name Benepali®, from Samsung Bioepis, Seoul, South Korea) and GP2015 (trade name Erelzi®, from Sandoz, Marburg, Germany).1,3 SB4 was also approved in Australia and Canada with the trade name Brenzys® (Samsung Bioepis, Seoul, South Korea).9,10 An adjunctive bio-ETN, HD203 (trade name Davictrel®, Hanwha Chemical Corporation, Seoul, South Korea/Merck KGaA, Darmstadt, Germany), was licensed in South Korea,11 while other biosimilars of re-ETN are under development but not yet approved, including LBEC0101 (LG Chem, Ltd, formerly LG Life Sciences, Ltd, Seoul, South Korea),12 and CHS-0214 (Coherus Biosciences, Inc., Redwood City, CA, USA).13,14
The aim of the present paper was to present the pharmacologic characteristics, the efficacy studies leading to the approval, the data on interchangeability, and the place in therapy of bio-ETN SB4, GP2015, and HD203, through a narrative review of the existing literature. The impact of bio-ETN on patient acceptance and satisfaction was also addressed.
Methods
The literature review was done using PubMed database to identify English language articles related to the previously mentioned topics. Data were extracted from the Regulatory Agencies’ approval documents, available RCTs, national registries of biologics, national health care databases, and post-marketing surveys. When these source data were not available for specific topics, the evidence was derived from open-label studies on variable sample-size clinical series. Data from the abstract books of the American College for Rheumatology (ACR) and European League Against Rheumatism (EULAR) annual meetings were also included.
The search was performed by using the following key terms: biosimilar etanercept, SB4, GP2015, HD203, benepali, erelzi, brenzys, davictrel, rheumatoid arthritis, psoriatic arthritis, axial spondyloarthritis (ax-SpA), ankylosing spondylitis (AS), psoriasis (pso), pharmacodynamics, pharmacokinetics, efficacy, safety, immunogenicity, extrapolation of indications, interchangeability, automatic substitution, patient preference, patient acceptance, recommendations. The literature review was extended to June 30, 2018.
Molecular structure and pharmacokinetics of etanercept biosimilars
During the development of comparability exercise studies, it was observed that the molecular structure of SB4 is similar to re-ETN, in addition, its biological activity, post-translational modifications, and glycosylation were similar.15,16
Pharmacodynamic properties of SB4
TNFα is a cytokine that plays a pathogenic role in RA, AS, and PsA. Re-ETN is the recombinant human receptor of TNFα p75Fc that binds the soluble and transmembrane form of TNFα and prevents pathogenetic effects in the joints and skin. Studies have shown similarity between SB4 and re-ETN in terms of binding affinity and ability to inhibit TNFα signaling.15,16 Furthermore, similarity has also been demonstrated in the binding of the Fc region.15,16 Specific studies of pharmacodynamics for SB4 do not support the fact that biosimilarity was deduced from comparative clinical studies.16–18
Pharmacokinetic properties of SB4, GP2015, and HD203
The bioavailability of SB4 has not been specifically studied;16 re-ETN has an absolute bioavailability of 76%.16,19,20 SB4, after subcutaneous administration, reaches a peak of serum concentration (Cmax) obtained with a time (Tmax) of approximately 72 hours after the administration of 50 mg in healthy volunteers. The mean Tmax obtained after multiple subcutaneous doses of 50 mg in patients with RA for SB4 was 47.8 hours. The volume of distribution of SB4 has a range between 10.3 and 11.2 L in healthy volunteers, with a mean half-life of 106 hours measured after 4 days of administration.16 There are no studies evaluating SB4 pharmacokinetics in patients with hepatic or renal impairment or drug interactions.16
The pharmacokinetic profile comparison between SB4 and re-ETN was evaluated in a single-blind study, with a crossover period in 138 healthy volunteers. The crossover between the two drugs was performed after 28 days of washout. The single dose of SB4 was shown to be equivalent to the single dose of re-ETN in terms of area subtended under the concentration curve from time 0 (AUC0) to infinity (AUC∞) and from time 0 (AUC) to terminal concentration (AUC last).21 The 90% CI for the ratio of the geometric least squares means for AUC (94.71–103.58), AUC last (94.17–103.28), and Cmax (98.46–109.25) were contained within the accepted predefined bioequivalence range of 80%–125% (primary endpoint).21
GP2015’s biosimilarity was investigated in a double-blind crossover study that compared pharmacokinetics, safety, and immunogenicity in healthy volunteers. The patients received two separate treatments with a 35-day washout period. The study involved 57 patients and the equivalence margin was between 80% and 125% for AUC, AUC last, and Cmax.22,23
The South Korean Health Authorities requirements for biosimilar approval were developed according to the World Health Organization and EMA guidelines. Non clinical “in vitro” and “in vivo” studies to assess the receptor binding, pharmacokinetic/pharmacodynamic properties, and toxicity, and clinical studies to investigate the pharmacodynamic/pharmacokinetic comparability, the efficacy, safety, and immunogenicity are required to release the approval for a biosimilar product.24
In a double-blind, single dose, crossover study of 35 healthy volunteers, the pharmacokinetics and bioavailability of HD203 were comparable with re-ETN,25 and the biosimilarity and equivalence of the product was established by the HERA study.26 This 24-week RCT of 294 patients with RA demonstrated the equivalence of HD203, with no significant differences in comparison with re-ETN in terms of ACR responses, adverse events (AEs), and immunogenicity. In the same study the equivalence was confirmed by the comparable quality of life outcomes.27 Consequently, HD203 was licensed in South Korea in November 2014.
Efficacy studies including bioequivalence studies
The clinical efficacy of SB4 in comparison with re-ETN was evaluated in a randomized double-blind Phase III study in patients with RA. The first phase of the study was 52 weeks,28,29 followed by a 48-week extension where a subset of patients was evaluated after switching from re-ETN to SB4.30 Overall, 596 patients (SB4 299 patients and re-ETN 297 patients) were randomized and 505 patients (84.7%) completed the study. Patients from the Czech Republic and Poland entered the study extension at 48 weeks. At week 24, the ACR20 clinical response was equivalent in the per-protocol and in the intention-to-treat populations. The same response was observed for ACR50 and ACR70.
The proportion of patients satisfying the good or moderate EULAR response was 87.2% vs 88.3%, the low disease activity assessed with Disease Activity Score 28 was 31.4% vs 27.6%, and the remission was 16.7% vs 16.2%.28 The benefits of SB4 and re-ETN were maintained at 52 weeks of treatment.29 Despite the different response in the ACR70, the EMA concluded that there was sufficient evidence for the biosimilarity relationship.16 At 52 weeks, structural radiological damage was also evaluated using the van der Heijde modified Total Sharpe Score. SB4 and re-ETN had similar results with a score variation from baseline of 0.45 and 0.74.29 In the study extension, of 245 patients who had completed 52 weeks, 126 continued SB4 and 119 switched from re-ETN to SB4 for 48 weeks. A total of 94.4% and 95% of the two groups completed 100 weeks. The results obtained in terms of ACR20, ACR50, ACR70 at 52 weeks were comparable both in the SB4 group and in the group of switchers from re-ETN to SB4.30 An analysis of the patient subgroups indicated that anti-drugs antibodies ADA did not affect the ACR20 response, with no differences in the ADA group positivity compared to those with ADA negativity.
The efficacy of SB4 after switching from re-ETN was evaluated in the DANBIO registry.31 In this report, 1,548 patients (891 RA, 335 PsA, 322 SpA) had received re-ETN for 5.2 years (range 3.2–8.0) before switching, and the disease was stable during the 3 months preceding the switch. Methotrexate (MTX) was co-administered in 60% of RA, 49% of PsA, and 15% of SpA patients. The proportion of patients who stopped for disease flare was 13% in RA, 13% in PsA, and 5% in SpA, with 129 patients (9%) discontinuing the treatment over 5 months of follow-up.31
Biosimilar GP2015 equivalence was investigated in a 52-week RCT of 531 patients with Pso, the EGALITY study.32 The study design consisted of four periods. In the first 12-week period, 264 patients received GP2015 and 267 re-ETN at the same dose of 50 mg/once weekly. Then, patients of the GP2015 arm were switched to re-ETN, and those of the re-ETN arm to GP2015 for 12 weeks, and at week 24 patients were re-switched to the other treatment until week 30. An extension phase of the study up to week 52 was carried out thereafter. Besides the efficacy and safety, the study evaluated the immunogenicity and pharmacokinetics of GP2015. GP2015 results were equivalent to re-ETN with no significant differences in terms of efficacy, immunogenicity, safety, and pharmacokinetic assessments.33 The equivalent efficacy of GP2015 was also evaluated in the ongoing EQUIRA trial of patients with RA.34 Preliminary data of this study were presented at the 2017 ACR annual meeting.35 At week 24 GP2015 results were equivalent to re-ETN in terms of ACR and EULAR response criteria. As dictated by the study protocol, at week 24 all patients of the re-ETN arm were switched to GP2015 with further evaluation at week 48. After switching, results were presented at the 2018 EULAR annual meeting. No significant differences in terms of efficacy and safety emerged in “switchers” as compared with GP2015 “continuers”.
The HERA study was designed to assess the therapeutic equivalence of bio-ETN HD203 and re-ETN combined with MTX in 294 patients with active RA.26 At week 48, the two bio-drugs’ results were equivalent to comparable ACR20, 50, 70, EULAR, Clinical Disease Activity Index (CDAI), and Simplified Disease Activity Index (SDAI) responses. No differences were recorded regarding ADA development.
To date, no real-life studies of GP2015 and HD203 have been published.
Comparative safety and tolerability
Data from RCTs and their open long-term extension provided the evidence of comparable safety between re-ETN and biosimilars SB4 (Benepali®, Brenzys®),28–30 GP2015 (Erelzi®),32 and HD203 (Davictrel®). After the 24-week evaluation, the Phase III double-blind RCT of SB4 in patients with RA continued up to week 52.30 Of the 596 patients at baseline, 505 completed the 52-week follow up. Overall, 1,179 treatment emergent adverse events (TEAEs) were reported in 354 (59.4%) patients, Of these, 533 TEAEs were recorded in 175 (58.5%) patients in the SB4 treatment group and 646 in 179 (60.3%) patients in the re-ETN group, with no significant statistical differences. Similarly, the proportion of patients experiencing serious adverse events (SAEs) was not different between SB4 and re-ETN groups (SB4: 6%; re-ETN: 5.1%). Equally, no between-group differences were recorded relative to laboratory findings, ADA development, and injection site reactions, while a numerical imbalance with a greater number of malignancies and hepatobiliary disorders in the SB4 group as compared with re-ETN group (malignancy: 4 vs 1; hepatobiliary disorders: 17 vs 0) was underlined in the EMA approval procedure,37 and questioned by some researchers.38
In the EGALITY study,32 TEAEs, SAEs, and discontinuations due to AEs did not differ between GP2015 and re-ETN arms. Also, immunogenicity did not result in differences between the two treatment groups. However, even though not mentioned by the authors, according to findings of SB4 study,30 overall 22 (8.3%) out of 264 patients of GP2015 arm and seven (2.6%) of 267 in re-ETN arm had signs of hepatobiliary impairment (increased alanine aminotransferase, aspartate aminotransferase or gamma-glutamyltransferase).
In the EQUIRA study,35,36 no safety alerts occurred in the GP2015 treatment arm as compared with re-ETN arm, even though there was a trend of higher percentage of patients with increased alanine aminotransferase observed in the GP2015-exposed arm (4.3% vs 2.1%).
In the HERA trial,26 confirming its equivalence, HD203 safety results were comparable with re-ETN in terms of overall AEs, SAEs, and discontinuation rate.
Safety data from real-life practice are available only for SB4. All studies were focused on non-medical switching from re-ETN to SB4. Overall, the maintenance of response after switching was satisfactory, but, as shown in Table 1, some concerns emerged regarding the safety and discontinuation rates. All but one study were presented at the ACR and EULAR 2017 annual meetings. The BIO-SPAN study from the Netherlands was recently published.39 Non-medical switching from re-ETN to SB4 was done in 635 consenting patients (433 RA, 128 PsA, 64 AS) during the year 2016. A historical cohort of 600 patients treated with re-ETN over the year 2014 served as controls. The primary outcome measure of the study was the discontinuation rate over a 6-month follow-up period. In the switcher cohort a significantly higher rate of discontinuation compared to re-ETN group was recorded (60 vs 46; HR 1.57, 95% CI 1.05–2.36), with a significantly higher number of overall AEs and AEs leading to discontinuation. Of note, 46 (76.6%) out of 60 AEs recorded in the transition cohort were related to subjective health complaints, possibly related to patients’ mistrust toward the new therapy.
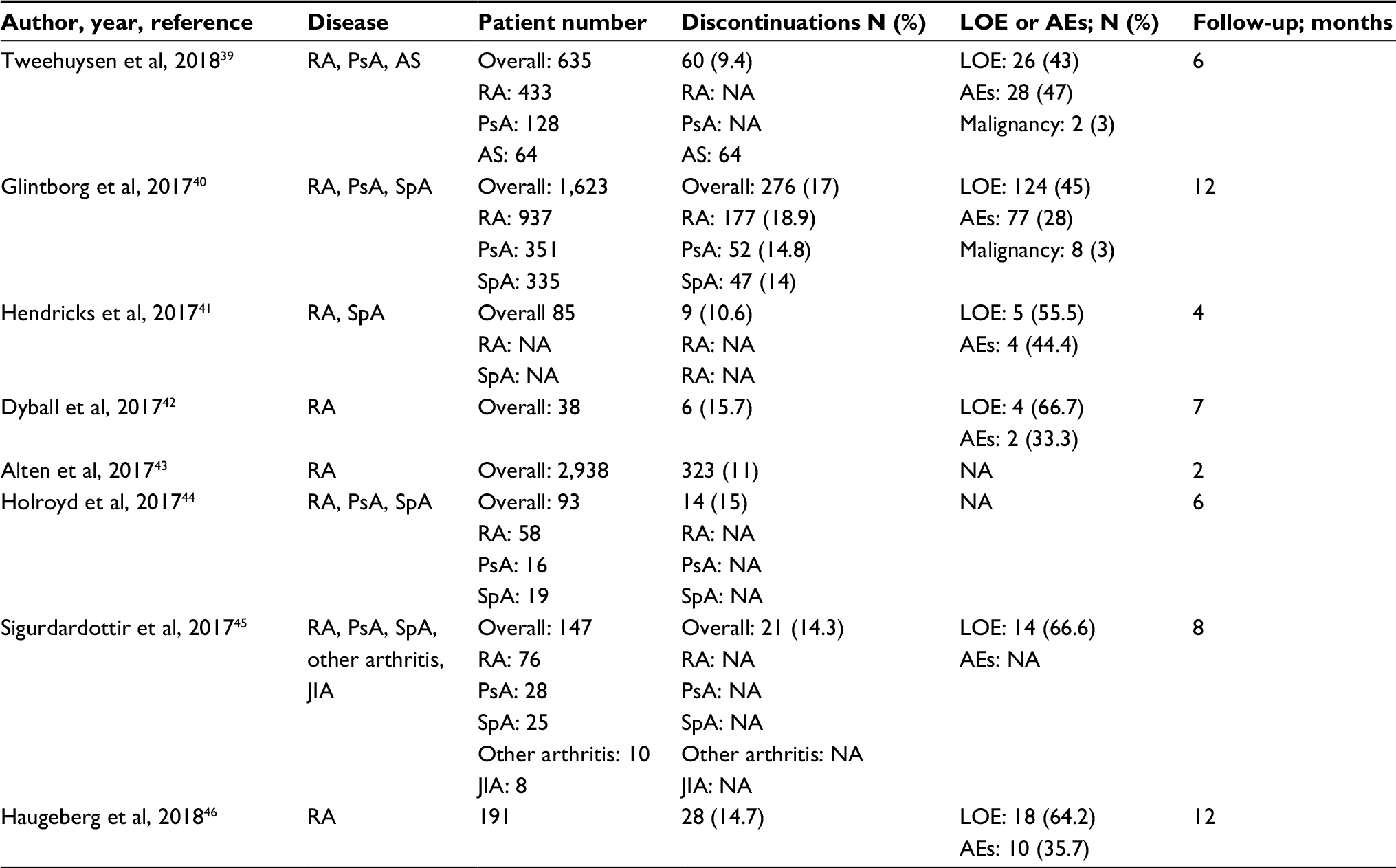 | Table 1 Reported studies on non-medical switching from re-ETN to SB4 biosimilar in real-life clinical studies Abbreviations: AEs, adverse events; AS, ankylosing spondylitis; JIA, juvenile idiopathic arthritis; LOE, loss of efficacy; NA, not available; PsA, psoriatic arthritis; RA, rheumatoid arthritis; re-ETN, reference etanercept; SpA, spondyloarthritis. |
Extrapolation of indications and interchangeability with re-ETN.
Concerning the biosimilar approval procedures and their use in clinical practice, extrapolation of indications and interchangeability caused some perplexities among rheumatologists.47 Once the biosimilar was demonstrated equivalent to the respective originator by a Phase III RCT in a single disease, both the FDA and EMA accepted the concept to approve the product for all indications licensed for the reference biologic.1 Based on this principle, SB4 and GP2015 were licensed for all indications approved for re-ETN,36,48,49 despite the fact that their equivalence was only demonstrated in one trial of RA and one of Pso, respectively.28,32 The same procedure was applied for HD203.11 Even though rheumatologists may regard this procedure as anomalous and a source of debate, the EMA has stated that extrapolation is a well-known concept that is supported by the comparability exercise and by the RCT results of equivalence of the biosimilar as compared with the originator.1
Interchangeability and substitution were defined, as stated by the EMA, as follows: “interchangeability refers to the possibility to switch from the originator to the respective biosimilar or vice versa or to the automatic substitution at pharmacy level without consulting the prescriber”.1 To date, automatic substitution is not permitted both in most EU states and the US. Pushed by the opportunity of relevant savings, health authorities of different countries recommended the switching (or transition) from re-ETN to SB4, and GP2015, despite the absence of controlled studies. In a recent consensus document of the EULAR organization, in recommendation 6, switching from originator to the respective biosimilar was defined as effective and safe, but clinicians should consider the patient’s perspective.50 However, as recently questioned, available evidence on the efficacy and safety of non-medical switching from re-ETN and the respective biosimilars is mostly based on a few observational, short-term studies on a small number of participants, thus seeming largely insufficient to recommend this procedure.47,51 In addition, as reported in Table 1, the proportion of discontinuation rates, ranging from 9%–17%, related to AEs or loss of efficacy in patients who were in clinical remission or had low disease activity at baseline, seems to confirm the concerns regarding non-medical switching. The lower percentage of discontinuations after switching from re-ETN recorded in the RCTs of SB430 and GP201532 (5% and 9%, respectively) as compared with real-life trials is not unexpected. Indeed, it has long been recognized that patients treated in the setting of real world practice greatly differ from those of clinical trials. Two analyses from the German RABBIT registry,52 and from the National Register for Biologic Treatment in Finland,53 showed that only 7.6%–44% of the included patients would have been eligible for RCTs.
In addition, SB4 and GP2015 RCTs raised some alarm bells related to the occurrence of hepatobiliary disorders.37,49 Probably, the alterations in the liver test results were slight and not of particular clinical relevance, however, long-term monitoring of hepatic function is advisable in bio-ETN-exposed subjects. Confirming the previously mentioned concerns, the FDA,54 ACR,55 and other national societies for rheumatology,56 have suggested controlled switching studies to better address the efficacy and safety of the transition strategy. However, the attitude of some national societies of rheumatology is changing toward the assumption of a favorable position.57,58
Patient and biosimilars: perception, attitude, and acceptance
The rapid introduction of SB4 and GP2015, and more generally of biosimilars, for the therapy of rheumatic inflammatory disorders, has created a new scenario which has probably caught both physicians and patients by surprise. A recent survey from Germany investigated the prescribing behavior and the attitude of 50 rheumatologists toward biosimilars, and the acceptance and satisfaction of 261 patients.59 In unrestricted circumstances, more than 95% of the rheumatologists would prescribe the bio-originators for first-, second-, and third-line treatment. Specifically asked, rheumatologists indicated the following reasons for prescribing biosimilars: desire to get experience, 86%; belief that biosimilars were equivalent to the bio-originator, 64%; lower cost, 64%. Regarding the patients, 71%, 61%, and 56% accepted, without reluctance, the biosimilar as first biologic, switching for medical and non-medical reasons, respectively, and only a minority (7%–18%) refused the biosimilar and accepted the originator. With respect to switching from the originator to the biosimilar, 60% of the patients were indifferent toward the procedure and around 20% declared being unhappy or somewhat unhappy. Regarding the patients’ perception, 30% had no concerns and approximately 60% declared a lack of information or fear of potential long-term AEs.59 In another survey from Belgium, 35% of 121 patients with RA had concerns regarding the biosimilar safety and around half of 41 rheumatologists questioned the equivalence of biosimilars.60 More knowledge and acceptance of biosimilars was reported in 182 patients with RA or AS in a web-based survey from the UK.61 In the same survey, lack of communication regarding biosimilars was underlined by the patients. The insufficient information may, at least in part, explain the subjective reactions (nocebo effect) observed after switching to SB4 from re-ETN. As suggested by Tweehuysen et al,39 in the BIO-SPAN study the nocebo effect was the cause of discontinuation in a consistent percentage of 60 switchers from re-ETN to SB4. However, the nocebo effect is an undefined condition that, in absence of validated classification/diagnostic criteria, is challenging to address.62 Nevertheless, complete and exhaustive information may improve patient acceptance and contribute to reduce biosimilar withdrawals. Indeed, in a study of 92 patients with RA, PsA, and AS a lower rate of discontinuation was recorded after a proper education program prior to switching,45 and in a further survey from the UK,63 21% of switchers from re-ETN to SB4 expressed the need for more detailed information before the transition.
Of note, two studies of 191 and 220 RA patients reported an overall preference for SB4 autoinjector compared to re-ETN Myclic injector due to its ease of use and simplicity, thus making administration easier.64,65
Conclusion
To date, three biosimilars of re-ETN, namely SB4, GP2015, and HD203, have been licensed for the treatment of RA, PsA, and AS. The results of RCTs demonstrating comparable efficacy and safety of biosimilars with respect to the originator ETN, have led to their approval by health regulatory agencies worldwide. The lower price of SB4, GP2015, and HD203 seems to ensure elevated cost savings, and health authorities of all countries vigorously promote their use in patients with inflammatory rheumatic diseases. However, rheumatologists have raised some concerns regarding the extrapolation of indications, some aspects of their safety profile, and interchangeability. To date, the latter issue does not seem to be supported by adequate scientific evidence of efficacy and safety, and the consistent rate of discontinuations after switching in the short-term period might negatively impact on the expected reduction of economic burden. Hence, based on the available evidence, re-ETN biosimilars’ place in therapy can be recommended as first-line intervention, while further evidence on the efficacy, safety, and the economic reflexes of transitioning from re-ETN would be preferable. Another important issue is related to the need to create adequate information strategies that would improve patients’ acceptance and adherence to therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
European Medicines Agency (EMA). Biosimilars in EU. Information guide for healthcare professionals. London: EMA; 2017. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf. Accessed August 1, 2018. | ||
US Food and Drug Administration. Biosimilars. Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580419.htm#top. Accessed October 23, 2017. | ||
US Food and Drug Administration. Background information: Lists of licensed biological products with reference product exclusivity and biosimilarity or interchangeability evaluations (purple book). Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm. Accessed October 23, 2017. | ||
Schulze-Koops H, Skapenko A. Biosimilars in rheumatology: A review of the evidence and their place in the treatment algorithm. Rheumatology. 2017;56(Suppl 4):iv30–iv48. | ||
Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching Reference Medicines to Biosimilars: A Systematic Literature Review of Clinical Outcomes. Drugs. 2018;78(4):463–478. | ||
Ruff L, Rezk MF, Uhlig T, Gommers JW. Budget impact analysis of an etanercept biosimilar for the treatment of rheumatoid arthritis in Europe. Value Health. 2015;18A:639. | ||
Derbyshire M. Patent expiry dates for biologicals: 2016 update. Gen Biosimilars Init J. 2017;6(1):27–30. | ||
GaBI Online - Generics and Biosimilars Initiative [homepage on the Internet]. New Amgen Enbrel patent could block biosimilars until 2028. Mol, Belgium: Pro Pharma Communications International; 2011. Available from: http://www.gabionline.net/Biosimilars/News/New-Amgen-Enbrel-patent-could-block-biosimilars-until-2028. Accessed August 1, 2018. | ||
GaBI Online - Generics and Biosimilars Initiative [homepage on the Internet]. Biosimilars approved in Australia. Mol, Belgium: Pro Pharma Communications International; 2018. Available from: www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Australia. Accessed August 1, 2018. | ||
GaBI Online - Generics and Biosimilars Initiative [homepage on the Internet]. Canadian approval for etanercept biosimilar. Mol, Belgium: Pro Pharma Communications International; 2016. Available from: http://www.gabionline.net/Biosimilars/News/Canadian-approval-for-etanercept-biosimilar. Accessed August 1, 2018. | ||
GaBI Online - Generics and Biosimilars Initiative [homepage on the Internet]. Merck enters biosimilars deal with Korea’s Hanwha. Mol, Belgium: Pro Pharma Communications International; 2011. Available from: www.gabionline.net/Biosimilars/News/Merck-enters-biosimilars-deal-with-Korea-s-Hanwha. Accessed August 1, 2018. | ||
Lee H, Chung H, Lee S, et al. LBEC0101, A Proposed Etanercept Biosimilar: Pharmacokinetics, Immunogenicity, and Tolerability Profiles Compared with a Reference Biologic Product in Healthy Male Subjects. BioDrugs. 2017;31(4):349–355. | ||
Coherus Biosciences, Inc. Comparison of CHS-0214 to Enbrel (Etanercept) in Patients With Rheumatoid Arthritis (RA) (CHS-0214-02). Available from: https://clinicaltrials.gov/ct2/show/NCT02115750. ClinicalTrials.gov identifier: NCT02115750. Accessed August 1, 2018. | ||
Coherus Biosciences, Inc. Comparison of CHS-0214 to Enbrel (Etanercept) in Patients With Chronic Plaque Psoriasis (PsO). Available from: https://clinicaltrials.gov/ct2/show/NCT02134210. ClinicalTrials.gov identifier: NCT02134210. Accessed August 1, 2018. | ||
Cho IH, Lee N, Song D, et al. Evaluation of the structural, physicochemical, and biological characteristics of SB4, a biosimilar of etanercept. MAbs. 2016;8(6):1136–1155. | ||
European public assessment report (EPAR). Benepali (etanercept). European Medicines Agency; 2016. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004007/WC500200380.pdf. Accessed November 19, 2015. | ||
Scott LJ. Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs. 2014;74(12):1379–1410. | ||
European Medicines Agency. Similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues: EMEA/CHMP/BMWP/42832/2005 Rev. 1. 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001378.jsp&mid=. Accessed July 4, 2016. | ||
European Medicines Agency. Similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues: EMEA/CHMP/BMWP/42832/2005 Rev. 1. 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_001281.jsp&mid=WC0b01ac0580032ec5. Accessed July 4, 2016. | ||
European Medicines Agency. Enbrel powder and solvent for solution for injection: summary of product characteristics; 2016. Available from: https://ec.europa.eu/health/documents/community-register/2016/2016 0919135935/anx_135935_en.pdf. Accessed August 19, 2018. | ||
Samsung Bioepis. Samsung Bioepis, etanercept biosimilar, BRENZYS (SB4) approved in South Korea [press release]. Samsung Bioepis; 2015 [Sept]. Available from: http://www.samsungbioepis.com/en/newsroom/detail/Samsung-Bioepis-Etanercept-Biosimilar-BRENZYS-(SB4)-approved-in-South-Korea.html. Accessed September 12, 2016. | ||
Afonso M, Sanguino Heinrich S, Poetzl J, et al. Pharmacokinetics and safety of GP2015, a proposed etanercept biosimilar, and etanercept originator product in healthy male subjects: a randomised two-way crossover study. Ann Rheum Dis. 2016;75(Suppl 2):234. | ||
von Richter O, Skerjanec A, Afonso M, et al. GP2015, a proposed etanercept biosimilar: Pharmacokinetic similarity to its reference product and comparison of its auto-injector device with pre-filled syringes. Br J Clin Pharmacol. 2017;83(4):732–741. | ||
GaBI Online – Generics and Biosimilars Initiative [homepage on the Internet]. South Korean guidelines for biosimilars. Mol, Belgium: Pro Pharma Communications International; 2015. Available from: www.gabionline.net/Guidelines/South-Korean-guidelines-for-biosimilars. Accessed August 1, 2018. | ||
Yi S, Kim SE, Park MK, et al. Comparative pharmacokinetics of HD203, a biosimilar of etanercept, with marketed etanercept (Enbrel®): a double-blind, single-dose, crossover study in healthy volunteers. BioDrugs. 2012;26(3):177–184. | ||
Bae SC, Kim J, Choe JY, et al. A phase III, multicentre, randomised, double-blind, active-controlled, parallel-group trial comparing safety and efficacy of HD203, with innovator etanercept, in combination with methotrexate, in patients with rheumatoid arthritis: the HERA study. Ann Rheum Dis. 2017;76(1):65–71. | ||
Bae SC, Lee SR, Ahn Y. Quality Of Life Assessments In Korean Patients With Rheumatoid Arthritis (Ra): An Analysis From The Phase Iii Trial To Evaluate Equivalence Of The Etanercept Biosimilar Hd203 And Enbrel® In Combination With Methotrexate (Mtx) In Patients With Ra; The Hera Study. Value Health. 2014;17(7):A374. | ||
Emery P, Vencovský J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76(1):51–57. | ||
Emery P, Vencovský J, Sylwestrzak A, et al. 52-week results of the phase 3 randomized study comparing SB4 with reference etanercept in patients with active rheumatoid arthritis. Rheumatology. 2017;56(12):2093–2101. | ||
Emery P, Vencovský J, Sylwestrzak A, et al. Long-term efficacy and safety in patients with rheumatoid arthritis continuing on SB4 or switching from reference etanercept to SB4pii. Ann Rheum Dis. 20172017211591. Epub 2017 Aug 9. | ||
Glintborg B, Sørensen I, Loft A, et al. FRI0190 Clinical outcomes from a nationwide non-medical switch from originator to biosimilar etanercept in patients with inflammatory arthritis after 5 months follow-up. results from the danbio registry. Ann Rheum Dis. 2017;76:553–554. | ||
Griffiths CEM, Thaçi D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176(4):928–938. | ||
Gerdes S, Thaçi D, Griffiths CEM, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32(3):420–427. | ||
EU Clinical Trials Register. A randomized, double-blind, parallel group phase III study to demonstrate equivalent efficacy and to compare safety and immunogenicity of GP2015 and Enbrel® (EU-authorized) in patients with moderate to severe, active rheumatoid arthritis. Available from: https://clinicaltrials.gov/ct2/show/NCT02638259. Accessed December 23, 2015. | ||
Kavanaugh A, Allanore Y, Kucharz EJ, Babic G. Etanercept Biosimilar GP2015 Has Equivalent Efficacy and Safety to Etanercept Originator in Patients with Moderate to Severe Rheumatoid Arthritis: The Phase 3 Equira Study [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). | ||
Matucci-Cerinic M, Schulze-Koops H, Buch M, et al. Switch between reference etanercept (ETN) and GP2015, an etanercept biosimilar, did not impact efficacy and safety in patients with moderate-to-severe rheumatoid arthritis: 48-week results from the phase 3 equira study. Ann Rheum Dis. 2018;77(Suppl):A609. | ||
EMA/CHMP/819219/2015 Committee for Medicinal Products for Human Use (CHMP). Assessment report. Benepali International non-proprietary name: etanercept. Procedure No. EMEA/H/C/004007/0000. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004007/WC500200380.pdf. Accessed November 19, 2015. | ||
Scheinberg M, Azevedo V. Difference between Enbrel and Benepali treatment groups in ‘hepatobiliary disorders’. Ann Rheum Dis. 2016;75(10):e64. | ||
Tweehuysen L, Huiskes VJ, van den Bemt BJ, et al. Open-Label Non-Mandatory Transitioning From Originator Etanercept to Biosimilar SB4: 6-Month Results From a Controlled Cohort. Study Arthritis Rheumatol. Epub 2018 Apr 2. | ||
Glintborg B, Omerovic E, Danebod K, et al. One-Year Clinical Outcomes in 1623 Patients with Inflammatory Arthritis Who Switched from Originator to Biosimilar Etanercept – an Observational Study from the Danish Danbio Registry [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). | ||
Hendricks O, Hørslev-Petersen K. When Etanercept Switch Fails – Clinical Considerations [abstract]. Arthritis Rheumatol. 2017;69(Suppl 10). | ||
Dyball S, Hoskins V, Christy-Kilner S, Haque S. Effectiveness and Tolerability of Benepali in Rheumatoid Arthritis Patients Switched from Enbrel [abstract]. Arthritis Rheumatol. 2017;69(Suppl 10). | ||
Alten R, Neregard P, Jones H, et al. Preliminary Real World Data on Switching Patterns between Etanercept, Its Recently Marketed Biosimilar Counterpart and Its Competitor Adalimumab, Using Swedish Prescription Registry [abstract]. Arthritis Rheumatol. 2017; 69(Suppl 10). | ||
Holroyd C, Wallis D, Bennett S, Clayton P, Edwards CJ. Switching from original etanercept to biosimilar etanercept SB4: patient acceptability and outcomes in the real world. Ann Rheum Dis. 2017;76(2):1180. | ||
Sigurdardottir V, Husmark T, Svärd A. Switching from reference product etanercept to the biosimilar sb4 in a real-life setting: follow-up of 147 patients. Ann Rheum Dis. 2017;76(2):835. | ||
Haugeberg G, Bakland G, Rødevand E, Fevang BT, Hansen IJW, Diamantopoulos A. Drug survival and reason for drop-out in rheumatoid arthritis patients with a non-medical switch from originator to biosimilar etanercept – preliminary data from a norwegian multicenter study. Ann Rheum Dis. 2018;77(Suppl):A1383. | ||
Fleischmann R. Editorial: The American College of Rheumatology White Paper on Biosimilars: It Isn’t All White-There Is Some Gray and Black. Arthritis Rheumatol. 2018;70(3):323–325. | ||
Food and Drug Administration. Center for Drug Evaluation and Research. Application number 761042Orig1s000. GP2015 approval. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/761042Orig1s000Approv.pdf. Accessed August 29, 2016. | ||
European Medicines Agency (EMA). EMA/CHMP/302222/2017 Committee for Medicinal Products for Human Use (CHMP). Assessment report Erelzi International non-proprietary name: etanercept Procedure No. EMEA/H/C/004192/0000. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/004192/WC500230144.pdf. Accessed April 21, 2017. | ||
Kay J, Schoels MM, Dörner T, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2018;77(2):165–174. | ||
Cantini F, Benucci M. Switching from the bio-originators to biosimilar: is it premature to recommend this procedure? Ann Rheum Dis. Epub 2017 Dec 29. | ||
Zink A, Strangfeld A, Schneider M, et al. Effectiveness of tumor necrosis factor inhibitors in rheumatoid arthritis in an observational cohort study: comparison of patients according to their eligibility for major randomized clinical trials. Arthritis Rheum. 2006;54(11):3399–3407. | ||
Aaltonen KJ, Ylikylä S, Tuulikki Joensuu J, et al. Efficacy and effectiveness of tumour necrosis factor inhibitors in the treatment of rheumatoid arthritis in randomized controlled trials and routine clinical practice. Rheumatology. 2017;56(5):725–735. | ||
US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product: guidance for industry. FDA; 2017. Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM537135.pdf. Accessed August 1, 2018. | ||
American College for Rheumatology. David I. Daikh. Feedback to [FDA-2017-N-5092] Review of Existing Center for Biologics Evaluation and Research Regulatory and Information Collection Requirement. 2017. Available from: https://www.rheumatology.org/Portals/0/Files/ACR-Comments-FDA-Regulation-Reduction.pdf. Accessed December 7, 2017. | ||
Araújo FC, Sepriano A, Teixeira F, et al. The Portuguese Society of Rheumatology position paper on the use of biosimilars - 2017 update. Acta Reumatol Port. 2017;42(3):219–228. | ||
Spanish Society of Rheumatology. Updating 2018. Stance of the Spanish Society of Rheumatology on biosimilars. Spanish Society of Rheumatology; 2018. Available from: https://www.ser.es/wp-content/uploads/2018/02/Position-Statement-on-Biosimilars-Drugs.pdf. Accessed August 1, 2018. | ||
Bridges SL Jr, White DW, Worthing AB, et al. American College of Rheumatology. The Science Behind Biosimilars: Entering a New Era of Biologic Therapy. Arthritis Rheumatol. 2018;70(3):334–344. | ||
Waller J, Sullivan E, Piercy J, Black CM, Kachroo S. Assessing physician and patient acceptance of infliximab biosimilars in rheumatoid arthritis, ankylosing spondyloarthritis and psoriatic arthritis across Germany. Patient Prefer Adherence. 2017;11:519–530. | ||
van Overbeeke E, de Beleyr B, de Hoon J, Westhovens R, Huys I. Perception of Originator Biologics and Biosimilars: A Survey Among Belgian Rheumatoid Arthritis Patients and Rheumatologists. BioDrugs. 2017;31(5):447–459. | ||
Aladul MI, Fitzpatrick RW, Chapman SR. Patients’ Understanding and Attitudes Towards Infliximab and Etanercept Biosimilars: Result of a UK Web-Based Survey. BioDrugs. 2017;31(5):439–446. | ||
Planès S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4(2):e00208. | ||
Attipoe L, Patel S, Birt R, et al. What Factors Predict Good Patient Experiences of Switching from Reference Etanercept (EnbrelTM) to an Etanercept Biosimilar (BenepaliTM) in a South West London General Hospital? [abstract]. Arthritis Rheumatol. 2017;69(Suppl 10). | ||
Egeth M, Soosaar J, Nash P, et al. Patient and Healthcare Professionals Preference for Brenzys vs. Enbrel Autoinjector for Rheumatoid Arthritis: A Randomized Crossover Simulated-Use Study. Adv Ther. 2017;34(5):1157–1172. | ||
Thakur K, Biberger A, Handrich A, Rezk MF. Patient Perceptions and Preferences of Two Etanercept Autoinjectors for Rheumatoid Arthritis: Findings from a Patient Survey in Europe. Rheumatol Ther. 2016;3(2):245–256. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.