Back to Journals » Drug Design, Development and Therapy » Volume 10
Flurbiprofen–antioxidant mutual prodrugs as safer nonsteroidal anti-inflammatory drugs: synthesis, pharmacological investigation, and computational molecular modeling
Authors Ashraf Z, Alamgeer, Kanwal M, Hassan M, Abdullah S, Waheed M, Ahsan H, Kim SJ
Received 26 March 2016
Accepted for publication 8 May 2016
Published 27 July 2016 Volume 2016:10 Pages 2401—2419
DOI https://doi.org/10.2147/DDDT.S109318
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Zaman Ashraf,1,2 Alamgeer,3 Munazza Kanwal,1 Mubashir Hassan,2 Sahar Abdullah,3 Mamuna Waheed,3 Haseeb Ahsan,3 Song Ja Kim2
1Department of Chemistry, Allama Iqbal Open University, Islamabad, Pakistan; 2Department of Biological Sciences, College of Natural Sciences, Kongju National University, Gongju, Republic of Korea; 3Department of Pharmacology, Faculty of Pharmacy, University of Sargodha, Sargodha, Pakistan
Abstract: Flurbiprofen–antioxidant mutual prodrugs were synthesized to reduce the gastrointestinal (GI) effects associated with flurbiprofen. For reducing the GI toxicity, the free carboxylic group (–COOH) was temporarily masked by esterification with phenolic –OH of natural antioxidants vanillin, thymol, umbelliferone, and sesamol. The in vitro hydrolysis of synthesized prodrugs showed that they were stable in buffer solution at pH 1.2, indicating their stability in the stomach. The synthesized prodrugs undergo significant hydrolysis in 80% human plasma and thus release free flurbiprofen. The minimum reversion was observed at pH 1.2, suggesting that prodrugs are less irritating to the stomach than flurbiprofen. The anti-inflammatory, analgesic, antipyretic, and ulcerogenic activities of prodrugs were evaluated. All the synthesized prodrugs significantly (P<0.001) reduced the inflammation against carrageenan and egg albumin-induced paw edema at 4 hours of study. The reduction in the size of the inflamed paw showed that most of the compounds inhibited the later phase of inflammation. The prodrug 2-oxo-2H-chromen-7-yl-2-(2-fluorobiphenyl-4-yl)propanoate (4b) showed significant reduction in paw licking with percentage inhibition of 58%. It also exhibited higher analgesic activity, reducing the number of writhes with a percentage of 75%, whereas flurbiprofen showed 69% inhibition. Antipyretic activity was investigated using brewer’s yeast-induced pyrexia model, and significant (P<0.001) reduction in rectal temperature was shown by all prodrugs at all times of assessment. The results of ulcerogenic activity showed that all prodrugs produced less GI irritation than flurbiprofen. Molecular docking and simulation studies were carried out with cyclooxygenase (COX-1 and COX-2) proteins, and it was observed that our prodrugs have more potential to selectively bind to COX-2 than to COX-1. It is concluded that the synthesized prodrugs have promising pharmacological activities with reduced GI adverse effects than the parent drug.
Keywords: flurbiprofen prodrugs, natural antioxidants, molecular docking, molecular dynamic simulation, pharmacological investigation, NSAIDs
Introduction
The nonsteroidal anti-inflammatory drugs (NSAIDs) are one among the most commonly prescribed medications worldwide.1,2 Stomach ulceration, bleeding, perforation, and gastrointestinal (GI) irritations are the most frequently associated adverse effects of NSAIDs, including flurbiprofen.3,4 Shanbhag et al5 reported that gastric or duodenal ulcers are present in 15%–20% of patients taking NSAIDs chronically. These adverse effects of NSAIDs are believed to embrace two different mechanisms: a local action exerted by direct contact of drug with gastric mucosa6 and a general systemic action following absorption, now believed to be by inhibition of cyclooxygenase 1 (COX-1), a constitutive form of COX.7 The local contact effect, which is not related to the pharmacological efficacy, differs from drug to drug. This effect has been reported to be predominant in the case of acidic NSAIDs, and hence their GI tolerance can be improved by reducing the factors responsible for the local erosive effects. Flurbiprofen is one of the propionic acid derivatives that belong to the class of COX inhibitors of NSAIDs. Flurbiprofen can cause GI adverse effects due to the presence of free carboxylic acid group (–COOH). Its side effects such as gastric mucosal damage and modifying the platelet functions limit its safe use.
The concept of retrometabolic drug design of prodrug has been applied to design molecules in which a group is reversibly masked as a prodrug. The carboxylic group of NSAIDs can be temporarily masked and its direct effect on gastric mucosa can be reduced. A number of ester prodrugs of NSAIDs have been synthesized to reduce their GI tract irritation and improve bioavailability.8,9 A mutual prodrug consists of two pharmacologically active agents coupled together in such a way to provide a synergistic action, reducing the adverse effects, or some additional biological action that is lacking in the parent drug. Hence, there is a need to synthesize some new NSAIDs with no or reduced GI adverse effects and promising pharmacological activities. The objective of the present study is to reduce the gastric irritation caused by flurbiprofen due to its direct contact with the gastric mucosa.
The carboxylic group (–COOH) of flurbiprofen has been masked by synthesizing its ester analogs with naturally occurring phenolic antioxidants. The selected promoieties have traditional medicinal uses and flavoring properties with well-documented safety profiles.10 The synthesized mutual prodrugs thus do not involve the risk of unwanted effects after they are hydrolyzed. The stability of prodrugs in stomach was ascertained by checking their hydrolytic pattern in simulated gastric fluid (SGF), and regeneration of the active drugs was ensured using human plasma. The pharmacological activities such as anti-inflammatory, analgesic, antipyretic, and ulcerogenic activities were also evaluated, and the results were compared with those of standard flurbiprofen. Molecular docking and simulation studies were also carried out against COX-1 and COX-2 to confirm the selective affinity of the synthesized prodrugs against these targets.
Experimental
The characterization of drugs and anticipated structures of synthesized prodrugs were confirmed by various spectral analyses such as Fourier transform infrared spectroscopy (FTIR), 1H nuclear magnetic resonance (NMR), and 13C NMR spectral data. Melting points were determined using a Sanyo Gallenkamp melting point apparatus. Infrared spectra were recorded using a PerkinElmer spectrophotometer. 1H and 13C NMR spectra were recorded using a Bruker 300 MHz spectrometer with CDCl3 as solvent. Chemical shifts are given in δ scale (ppm) relative to tetramethylsilane. Mass spectra were recorded using an Agilent 6460 Series Triple Quadrupole instrument (Agilent Technologies, Santa Clara, CA, USA). The ionization was achieved by electrospray ionization in the positive ion mode (ESI+). The in silico docking studies were performed using Intel Core-i-7 workstation (Hewlett-Packard Company, Palo Alto, CA, USA). Formalin (EMD Millipore, Billerica, MA, USA), carrageenan (Sigma Chemicals, Perth, Australia), dried yeast (Sigma Chemicals), and normal saline (0.9% NaCl) were used.
Synthesis of flurbiprofen–antioxidant mutual prodrugs (4a–d)
Freshly distilled thionyl chloride (2.92 g, 24.58 mmol) was added to flurbiprofen (5.0 g, 20.49 mmol) and then refluxed for 3–4 hours, with continuous stirring on a magnetic stirrer. After the completion of reaction, the excess thionyl chloride was evaporated under reduced pressure. Acid chloride was collected at 100°C–110°C. A mixture of an appropriate antioxidant (3a–d; 0.01 mol), triethylamine (1.39 mL, 10.0 mmol) in dichloromethane (15 mL), was cooled in an ice–salt mixture to −5°C. To this mixture, flurbiprofen acid chloride (2.62 g, 10.0 mmol) in anhydrous dichloromethane (20 mL) was added dropwise. The reaction mixture was stirred overnight at 20°C. The reaction mixture was washed with 5% HCl, 5% sodium hydroxide, and finally with brine solution. The organic layer was dried over anhydrous sodium sulfate, filtered, and the solvent was removed under reduced pressure to afford the title prodrugs (4a–d).
Synthesis of 4-formyl-2-methoxyphenyl 2-(2-fluorobiphenyl-4-yl)propanoate (4a)
Yield 84%, viscous oil, retardation factor or ratio to front (Rf) 0.64 (petroleum ether:ethyl acetate, 3:1), FTIR νmax cm−1 (KBr): 2,980 (C–H), 1,757 (C=O ester), 1,150 (C–O, ester), and 1,697 (C=O, aldehyde); 1H NMR (CDCl3, δ ppm): 9.95 (s, 1H, H-5), 7.55 (d, J=3.6 Hz, 1H, H-5′′′), 7.54 (s, 1H, H-3′′′), 7.42 (d, J=2.4 Hz, 2H, H-2′′, H-6′′), 7.40 (d, J=3.6 Hz, 1H, H-6′′′), 7.36 (m, 3H, H-3′′, H-4′′, H-5′′), 7.26 (s, 1H, H-2′), 7.24 (d, J=2.0 Hz, 1H, H-6′), 7.14 (d, J=8.0 Hz, 1H, H-5′), 4.02 (q, J=6.8 Hz, 1H, H-2), 3.83 (s, 3H, H-4), and 1.67 (d, J=7.2 Hz, 3H, H-3); 13C NMR (CDCl3, δ ppm): 191.0 (C-5), 171.4 (C-5), 160.9 (C-3′), 145.0 (C-1′′′), 141.0 (C-1′′), 135.4 (C-1′), 135.3 (C-4′′′), 130.8 (C-2′′′), 130.8 (C-4′), 128.9 (C-5′), 128.5 (C-3′′), 128.5 (C-5′′), 127.7 (C-2′′), 127.7 (C-6′′), 124.6 (C-4′′), 123.8 (C-6′), 123.8 (C-6′′′), 123.2 (C-5′′′), 115.6 (C-2′), 115.4 (C-3′′′), 55.9 (C-4), 44.9 (C-2), and 18.5 (C-3); electrospray ionization mass spectrometry (ESI-MS): m/z 401 [M + 23] (M + Na).
Synthesis of 2-oxo-2H-chromen-7-yl 2-(2-fluorobiphenyl-4-yl)propanoate (4b)
Yield 74%, solid melting point (mp) 243°C–245°C, Rf=0.60 (petroleum ether:ethyl acetate 3:1), FTIR νmax cm−1 (KBr): 2,963 (C–H), 1,734 (C=O ester), and 1,085 (C–O, ester); 1H NMR (CDCl3, δ ppm): 7.47 (d, J=6.4 Hz, 2H, H-2′, H-6′), 7.40 (d, J=7.8 Hz, 1H, H-5), 7.36 (d, J=6.2 Hz, 1H, H-2′′), 7.29 (m, 3H, H-3′, H-4′, H-5′), 7.19 (d, J=9.2 Hz, 1H, H-3′′), 6.84 (d, J=7.6 Hz, 1H, H-6), 6.79 (d, J=5.3 Hz, 1H, H-2), 6.74 (s, 1H, H-6′′), 6.71 (d, J=7.6 Hz, 1H, H-4′′), 6.56 (d, J=7.6 Hz, 1H, H-1′′), 4.10 (q, J=5.4 Hz, 1H, –CH), and 1.45 (d, J=5.6 Hz, 3H, CH3); 13C NMR (CDCl3, δ ppm): 170.6 (C=O ester), 160.4 (C=O lactone), 159.2 (C-3), 151.0 (C-6′′), 148.3 (C-8′′), 142.1 (C-2′′), 138.4 (C-1′), 135.4 (C-1), 128.9 (C-4), 127.8 (C-5), 126.5 (C-6), 125.4 (C-2′, C-6′), 124.7 (C-3′, C-5′), 123.9 (C-4′), 121.2 (C-2), 119.1 (C-4′′), 118.6 (C-3′′), 117.2 (C-5′′), 42.2 (CH–), and 16.5 (CH3); ESI-MS: m/z 411 [M + 23] (M + Na).
Synthesis of 2-(propan-2-yl)-5-methylphenyl 2-(2-fluorobiphenyl-4-yl)propanoate (4c)
Yield 77%, oil, Rf 0.66 (petroleum ether:ethyl acetate 3:1), FTIR νmax cm−1 (KBr): 2,978 (C–H), 1,719 (C=O ester), and 1,056 (C–O, ester); 1H NMR (CDCl3, δ ppm): 7.57 (d, J=8.4 Hz, 1H, H-5′), 7.47 (d, J=7.6 Hz, 2H, H-2′′, H-6′′), 7.27 (m, 3H, H-3′′, H-4′′, H-5′′), 7.09 (d, J=7.6 Hz, 2H, H-3′′′), 7.01 (d, J=7.6 Hz, 1H, H-6′), 6.77 (s, 1H, H-6′′′), 6.73 (d, J=7.6 Hz, 1H, H-4′′′), 6.56 (s, 1H, H-2′), 4.04 (q, J=6.8 Hz, 1H, H-2), 3.19 (septet, 1H, H-4), 2.63 (s, 3H, H-7), 1.72 (d, J=7.2 Hz, 3H, H-3), 1.25 (d, J=6.8 Hz, 6H, H-5); 13C NMR (CDCl3, δ ppm): 172.6 (C-1), 158.6 (C-3′), 147.8 (C-1′′′), 141.2 (C-2′′), 136.5 (C-1′′), 135.4 (C-1′), 135.4 (C-5′′′), 128.9 (C-4′), 128.6 (C-5′), 128.5 (C-6′), 128.4 (C-3′′), 128.4 (C-5′′), 127.7 (C-2′′), 127.7 (C-6′′), 127.1 (C-4′′), 126.3 (C-3′′′), 126.2 (C-4′′′), 123.7 (C-6′′′), 116.0 (C-2′), 45.2 (C-2), 26.7 (C-4), 22.9 (C-6), 22.7 (C-5), and 17.9 (C-3); ESI-MS: m/z 399 [M + 23] (M + Na).
Synthesis of 1,3-benzodioxal-5-yl 2-(2-fluorobiphenyl-4-yl)propanoate (4d)
Yield 72%, semisolid, Rf 0.54 (petroleum ether:ethyl acetate 3:1), FTIR νmax cm−1 (KBr): 2,945 (C–H), 1,726 (C=O ester), and 1,066 (C–O, ester); 1H NMR (CDCl3, δ ppm): 7.51 (d, J=9.2 Hz, 2H, H-2′, H-6′), 7.43 (d, J=6.9 Hz, 1H, H-5), 7.32 (m, 3H, H-3′, H-4′, H-5′), 7.10 (d, J=8.2 Hz, 1H, H-6), 6.92 (d, J=7.4 Hz, 1H, H-2), 6.71 (s, 1H, H-2′′), 6.62 (d, J=7.6 Hz, 1H, H-5′′), 6.59 (d, J=7.6 Hz, 1H, H-6′′), 4.87 (s, 4H, –CH2), 4.16 (q, J=6.1 Hz, 1H, –CH), and 1.56 (d, J=6.3 Hz, 3H, CH3); 13C NMR (CDCl3, δ ppm): 173.1 (C=O ester), 162.6 (C=O lactone), 159.2 (C-3), 150.2 (C-3′′), 147.6 (C-4′′), 144.5 (C-1′′), 136.4 (C-1′), 132.8 (C-1), 129.6 (C-4), 126.9 (C-5), 125.8 (C-6), 124.9 (C-2′, C-6′), 124.1 (C-3′, C-5′), 123.4 (C-4′), 120.6 (C-2), 117.6 (C-5′′), 116.1 (C-6′′), 108.2 (C-2′′), 79.2 (–CH2), 44.6 (CH–), and 17.5 (CH3); ESI-MS: m/z 387 [M + 23] (M + Na).
In vitro hydrolysis
In vitro hydrolysis of synthesized prodrugs was carried out in SGF (at pH 1.2), simulated intestinal fluid (SIF at pH 7.4), and in 80% human plasma (pH 7.4). SGF and SIF are widely used media in dissolution tests in the pharmaceutical industry. SGF is pH 1.2 HCl solution, while SIF is pH 7.4 monobasic potassium phosphate solution. The 0.1 N HCl solution (SGF) was prepared by combining a 0.2 M potassium chloride solution with a 0.2 M hydrochloric acid solution. The final pH was recorded, and then the solution was degassed with helium for at least 15 minutes. SIF is pH 6.8 monobasic potassium phosphate buffer solution. The phosphate buffers with pH from 3.0 to 7.8 were prepared with monobasic potassium phosphate in water. The final pH 7.4 was adjusted by the addition of potassium hydroxide solution. The final pH was recorded, and the solution was degassed with helium for 15 minutes. A solution of 10 mg of prodrug was prepared in 90 mL of SGF (pH 1.2) or SIF (pH 7.4). An aliquot of 15 mL of this solution was withdrawn repeatedly and kept in test tubes maintained at 37°C±0.5°C. At a definite interval of time (0.5 hour, 1 hour, 2 hours, and up to 8 hours), an aliquot was withdrawn from different test tubes and was transferred to microcentrifuge tubes. After this, the tubes were centrifuged at high speed (3,000 rpm) for 5 minutes. A total of 5 mL of clear supernatant obtained from each tube was measured using a ultraviolet spectrophotometer for the amount of free drug released after the hydrolysis in buffer solutions at 230–215 nm and 280–260 nm, respectively.
Pharmacology
Animals
Adult healthy mice of either sex weighing 20–30 g were used in this study. These mice were placed in polypropylene cages in the animal house of the Faculty of Pharmacy, University of Sargodha. Humidity and temperature of the animal house were maintained. Animals were allowed free access of water and were given palatable clean food. Dark and light cycle of 12/12 hours was preserved. The animals were treated according to the guidelines of National Institute of Health (NIH). The experimental protocol followed was according to internationally approved guidelines for animal use and care (EEC Directive of 1986; 86/09/EEC; National Institutes of Health publication 85-23, revised 1985) (Table S1). The acclimatization period lasted for 7 days. The study protocols were approved by the local ethical committee of the University of Sargodha.
Drug administration
The solutions/suspensions of drugs were administered to the animals by the following routes.
Oral administration
For administration of drugs, oral gavage was used. The needle of the 1 cc BD syringe was removed and Ryle tube #4 attached to needle no 17 was fixed at the place of the original needle. The animal was maintained in upright position, and the gavage was then passed from the side of mouth whilst ensuring that the tube entered into the esophagus and stomach. The solution was directly injected into the stomach by pressing the plunger of the syringe.
Subcutaneous administration
The skin of the animals between the shoulder blades was lifted up and a triangle was formed. The needle of the syringe was then introduced in the folds of the skin into the base of the triangle and the drug was advanced into subcutaneous tissue. The drug was ejected by pressing the plunger of the syringe.
Antiinflammatory activity
Carrageenan-induced paw edema in mice
Antiinflammatory activity of synthesized prodrugs (4a–d) against carrageenan-induced paw edema in mice was evaluated according to the reported methods.11 Briefly, mice of either sex weighing 20–30 g were distributed into six groups of five mice each. The mice used for the experiment were kept on fasting for 24 hours before starting the experiment. Group I that served as the control group was treated with 10 mL/kg po of normal saline, Group II was treated with 20 mg/kg po of standard flurbiprofen, whereas animals of Groups III–VI were treated with 20 mg/kg po of prodrugs 4a–d, respectively. One hour after the administration of prodrugs, the inflammation was induced by injecting 0.1 mL freshly prepared carrageenan suspension (1%) in normal saline into the sub planar surface of the right hind paw. The paw volume was measured at 0 hour, 1 hour, 2 hours, 3 hours, and 4 hours after the administration of carrageenan using a vernier caliper.
Percentage of inhibition (%) =100 (1 − X/Y) | (1) |
where, X = mean increase in paw thickness of treated mice and Y = mean increase in paw thickness of control mice.
Albumin-induced inflammation in mice
Anti-inflammatory activity of prodrugs (4a–d) against egg albumin-induced inflammation in mice was also evaluated for further verification by following the method described by Akah and Nwambie12 with some modification. Briefly, mice of either sex with a weight of 20–30 g were randomized into six groups consisting of five mice each. The animals were kept fasting for 24 hours before the beginning of the experiment. Group I (control) was treated with 10 mL/kg po of normal saline. Animals of Group II were treated with 20 mg/kg po of flurbiprofen, whereas Groups III–VI were treated with 20 mg/kg po of prodrugs 4a–d, respectively. After 1 hour of treatment, 0.1 mL of fresh egg albumin was injected into the paw for the induction of inflammation. The paw volume was calculated at 0 hour, 1 hour, 2 hours, and 3 hours after the administration of inflammatory agent using a vernier caliper.
Analgesic activity
The analgesic activity of the synthesized prodrugs was also evaluated by following two different protocols, ie, acetic acid-induced writhing in mice and formalin-induced licking in mice.
Acetic acid-induced writhing in mice
The method described by Koster et al13 with some modifications was followed for the evaluation of analgesic activity of the synthesized prodrugs (5a–c).13 Each of the mutual prodrugs (20 mg/kg po) was administered to 24-hour fasting mice divided into six groups of five mice each. Group I serving as control was treated with 10 mL/kg normal saline po. Group II was treated with 20 mg/kg po of flurbiprofen, while Groups III–VI were treated with 20 mg/kg po of prodrugs (4a–d), respectively. After 30 minutes of treatment, acetic acid solution (0.6% in normal saline, at the dose of 10 mL/kg body weight) was injected into every mouse to induce the writhing. The number of writhes was counted between 5 minutes and 15 minutes after injection. The responses of the prodrugs-treated groups and flurbiprofen-treated groups were compared with those of the animals in the control group.
Formalin-induced licking in mice
The analgesic activity of the synthesized prodrugs was also carried out according to the reported method with some modifications.14 Each of the prodrugs and standard flurbiprofen at a dose of 20 mg/kg po were administered to 24-hour fasting mice divided into six groups of five mice each. Group I serving as control was treated with 10 mL/kg normal saline po. Group II was treated with 20 mg/kg po of flurbiprofen, while Groups III–VI were treated with 20 mg/kg po of prodrugs (4a–d), respectively. Briefly, 0.05 mL of 2.5% formalin was injected into subplanter of right hind paw. The time the mice spent licking was noted and is indicative of pain. The responses were observed for 30 minutes.
Antipyretic activity
Antipyretic activity against yeast-induced pyrexia in mice was evaluated according to the methods described by Sakande et al.15 Mice were administered 10 mg/kg of aqueous suspension of brewer’s yeast subcutaneously (20% in distilled water). After 16 hours of administration of brewer’s yeast, rectal temperatures of mice were noted (pretreatment values). Only, those animals in which the pyrexia had been induced were used further. The animals were divided into six groups of five mice each. Control group received 10 mL/kg po of normal saline; standard group received 20 mg/kg po of flurbiprofen; and treated groups received 20 mg/kg po of prodrugs (4a–d), respectively. Then animals were treated as described earlier with control, standard, and prodrugs (4a–d). Rectal temperatures of animals were recorded at 1 hour, 2 hours, 3 hours, and 4 hours after drug administration.
Ulcerogenic studies
The fasted rat model has been used for comparing ulcerogenic potential of flurbiprofen and synthesized mutual prodrugs (4a–d) in laboratory animals. The animals were divided into six groups with five animals in each group. Control group was given only 0.5% carboxymethyl cellulose suspension. The mutual prodrugs and flurbiprofen were administered orally as a 1 mg/kg suspension in 0.5% carboxymethyl cellulose. The animals were fasted for 24 hours prior to dosing. However, water was given ad libitum. The animals were sacrificed 4 hours after dosing. Stomachs were removed and preserved on saline-soaked filter paper until inspection. The stomach was cut open along larger curvature and washed with distilled water. The mucus was wiped off, and the areas of ulcers and stomachs were noted.16,17 Ulcer index (UI) was calculated using the following formula:18
|
|
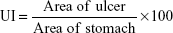
Statistical analysis
The results are expressed as mean ± standard error of the mean. Parametric data were compared with those of the control group and were assessed by one-way analysis of variance followed by Dunnett’s test and two-way analysis of variance followed by Bonferroni post-test. Values of P<0.05 were considered to be statistically significant.
Computational molecular modeling
Molecular modeling of COX-1 and COX-2
Current study, sequence comparison, structure prediction, chemoinformatics analysis, and docking studies were performed on Intel Core-i7 workstation. The crystal structures of human COX-1 and COX-2 were not reported in Protein Data Bank (PDB).19,20 Therefore, homology modeling approach was employed to predict three-dimensional (3D) structures of COX-1 and COX-2 proteins. The amino acid sequences of COX-1 and COX-2 in FASTA format having accession numbers (P23219 and P35354) were obtained from Uniprot Knowledge database (http://www.uniprot.org/). Two different templates (401Z and 4FM5) from sheep (Ovis aries) and mouse (Mus musculus) were selected for COX-1 and COX-2 on the basis of sequence identity (92.97% and 87.12%) and query coverage (31-583 aa and 18-569 aa), respectively. The reliable 3D crystal structures of both COX-1 and COX-2 were modeled using Modeller 9v8.21 Furthermore, the validity of predicted structures was confirmed using various evaluation tools such as ERRAT, Anolea, ProCheck, and Rampage.22–25 The energy minimization of these structures was performed using Vega-ZZ.26 UCSF Chimera 1.6 was used to visualize these predicted structures.27 The overall stereochemical properties, hydrophobicity, and Ramachandran values of COX-1 and COX-2 were assessed using Discovery Studio 4.0 and Molprobity server, respectively.28,29 Finally, the poor rotamers and outliers were removed using WinCoot tool to obtain the best optimized and reliable structures for further computational analysis.30
Ligand evaluation
The structures of flurbiprofen and synthesized prodrugs (4a–d) in 2D format have been drawn using ChemSketch. These ligand molecules were minimized using UCSF Chimera 1.6 and VegaZZ, respectively. Multiple tools and servers such as ChemDraw 8, Molinspiration, and Osiris Property Explorer were employed to check the chemoinformatics and biological properties of these novel designed ligand compounds.31,32 The number of rotatable bonds, hydrogen bond acceptors, and hydrogen bond donors were obtained using Molinspiration and PubChem.33 Similarly, molecular properties like molar refractivity, density, surface tension, and polarizability were accessed using ChemSketch. The Osiris Property Explorer (http://www.organic-chemistry.org/prog/peo/) an online tool was used to evaluate the possible tumorigenic or mutagenic risks and to estimate the drug-like properties. Lipinski’s rule of five was analyzed using the Cresset (www.cresset-group.com/tag/drug-discovery) server and Molsoft (www.molsoft.com).
Molecular docking
Molecular docking study was employed on ligands against protein (COX-1 and COX-2) molecules in Linux operating (OpenSuse 13.2) system to obtain different docked complexes using the Pyrex 9.0 docking tool.34 The grid parameter values were adjusted by default separately for both COX-1 (X=246.523, Y=99.180, and Z=4.4308) and COX-2 (X=22.874, Y=22.0301, and Z=−60.8588). Multiple docked complexes were obtained for both COX-1 and COX-2 and further evaluated on the basis of energy values and compared separately.
Molecular dynamics simulations
The residual backbone flexibility and stability of COX-1 and COX-2 were confirmed by molecular dynamics (MD) simulations using Groningen Machine for Chemicals Simulations (GROMACS) 4.5.4 package35 with GROMOS 53A6 force field and water model SPC216.36 The receptor and ligand topology files were generated using GROMOS 53A6 force-field and online PRODRG Server, respectively.37 Furthermore, both receptor–ligand complexes were solvated and placed in the middle of the cubic box with an adjusted distance of 0.8 Å. The overall system charge was neutralized by adding ions. The energy minimization (nsteps =50,000) was done by steepest descent approach (1,000 ps) and energy calculation was done by the particle mesh Ewald method.38 The final MD run was adjusted to 10,000 ps with n-steps 5,000,000 and analyzed using Xmgrace software (http://plasma-gate.weizmann.ac.il/Grace/).
Results
Chemistry
Mutual prodrugs of flurbiprofen having ester moieties instead of free carboxylic acid that are involved in GI side effects have been synthesized. The free carboxylic acid group of the flurbiprofen was temporarily masked by a promoiety so as not to expose stomach mucosa to this free acidic group. Acid chloride was synthesized by treating flurbiprofen with freshly distilled thionyl chloride. The natural antioxidants, vanillin, umbelliferone, thymol, and sesamol, were then condensed with acid chloride to provide ester prodrugs (4a–d); Figure 1. The natural antioxidants have been selected as promoiety because they are an important part of human diet and their safety profile is well known.
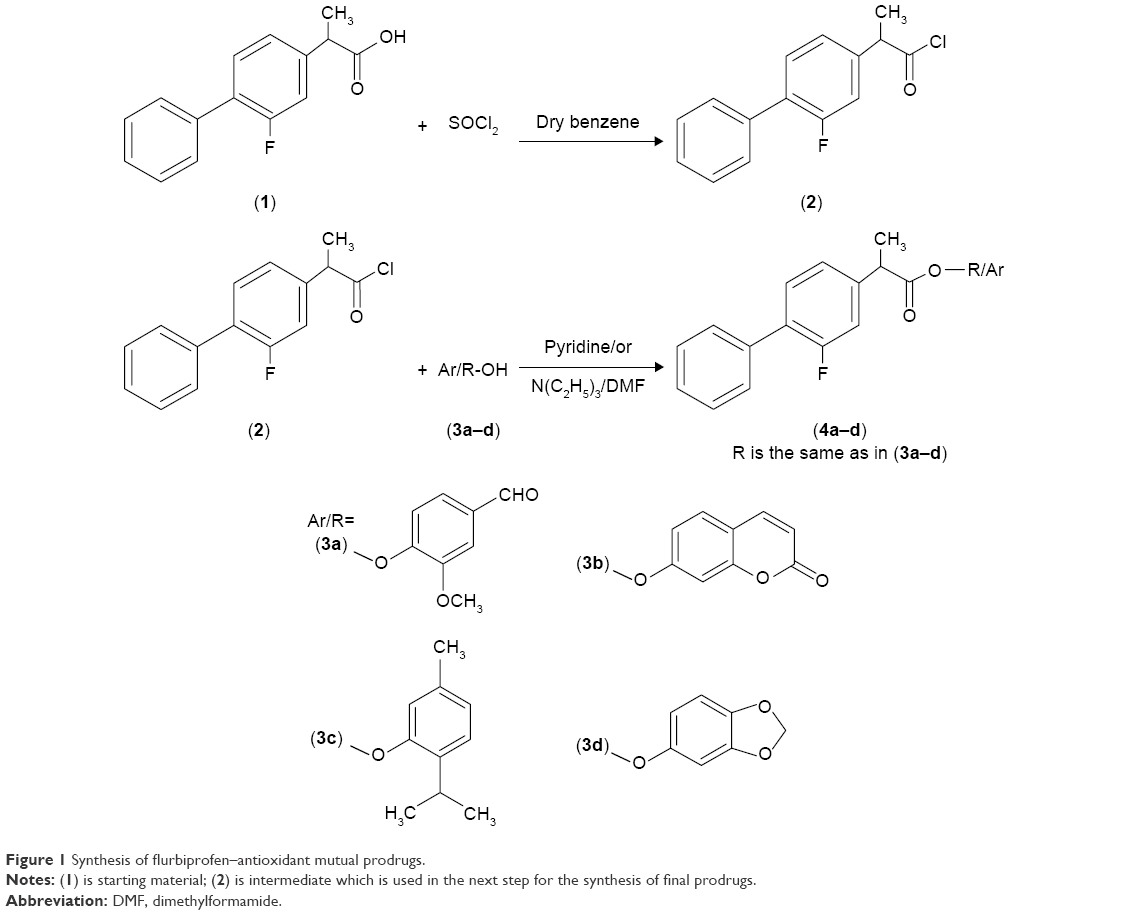 | Figure 1 Synthesis of flurbiprofen–antioxidant mutual prodrugs. |
In vitro hydrolysis
The in vitro hydrolysis studies were designed to mimic the entire GI tract pH; hence, as a primary requirement, a calibration curve in the experimental pH value related to the study was plotted. The hydrolysis studies were carried out in SGF at pH 1.2 to mimic the stomach environment, in SIF at pH 7.4, and in 80% human plasma at pH 7.4 to simulate the conditions of physiological pH of blood. The minimum reversion was observed at gastric pH (SGF, pH 1.2) suggesting the stability of synthesized prodrugs in gastric pH. However, at higher pH values, ie, in SIF representing intestine, the percentage reversion was significantly higher thereby making the free drug available for absorption in the intestine. The amount of flurbiprofen regenerated on hydrolysis after 1 hour (in SIF, pH 7.4) in prodrugs 4b and 4a was found to be from 6.34% to 17.56%, whereas for prodrugs 4c and 4d from 19.03% to 21.16%, respectively (Figure 2).
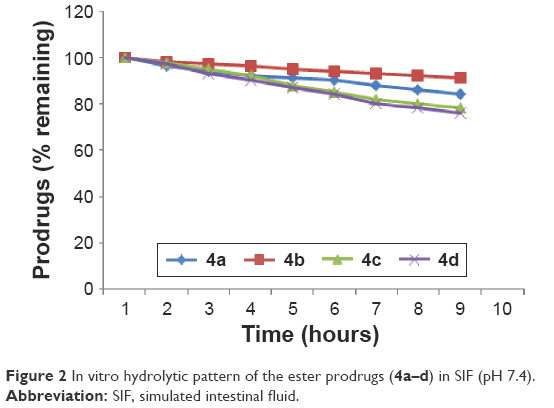 | Figure 2 In vitro hydrolytic pattern of the ester prodrugs (4a–d) in SIF (pH 7.4). |
The amount of flurbiprofen regenerated on hydrolysis after 1 hour (in 80% human plasma, pH 7.4) was found to be from 51.6% to 36.41% in prodrugs 4a and 4b. The amount of regenerated drug was found to be from 69.4% to 64.7% for prodrug 4d, and in prodrug 4c the amount of regenerated drug was found to be from 61.5% to 58.8%. None of the prodrugs showed hydrolysis in SGF (pH 1.2). Satisfactory hydrolysis was observed in SIF (pH 7.4) and all the mutual prodrugs showed very encouraging hydrolysis rate in 80% human plasma (pH 7.4), and the regeneration of active drug was found to be from 61% to 92% Figure 3.
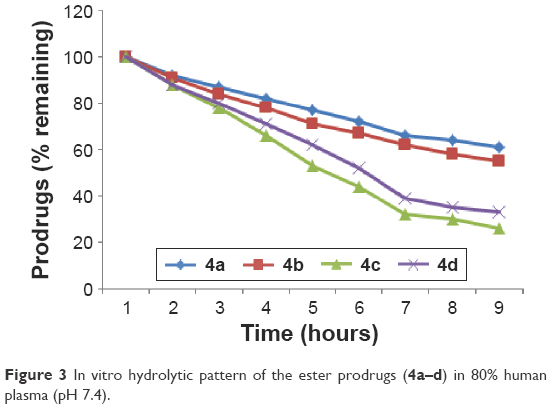 | Figure 3 In vitro hydrolytic pattern of the ester prodrugs (4a–d) in 80% human plasma (pH 7.4). |
Pharmacology
The pharmacological investigation indicated that all of the synthesized flurbiprofen prodrugs (4a–d) showed comparable reduction in paw edema with standard drug. The results obtained showed that there was a gradual increase in paw volume of mice in the control group (Table 1). However, in the treated groups, all selective derivatives produced a significant reduction in edema formation. The title prodrugs exhibited anti-inflammatory effect by significantly (P<0.001) inhibiting the paw edema at 4 hours of postcarrageenan-induced inflammation. The synthesized prodrugs showed comparable anti-inflammatory effect to standard flurbiprofen, which also produced significant inhibition at 4 hours after the administration of carrageenan. It is reported that edema induced by carrageenan occurs in two phases; the first phase that occurs between 0 hour and 2.5 hours of injection of agent is mediated by release of inflammatory mediators such as histamine, serotonin, and bradykinin.39 The second phase is attributed to release of prostaglandins (PGs). The results revealed that all the synthetic prodrugs showed very significant inhibitory effects (P<0.001) in second phase. These results showed that anti-inflammatory effect of these mutual prodrugs in carrageenan-induced paw edema could be due to their inhibitory effect on COX, leading to decrease in the production of PGs. Standard flurbiprofen also produced anti-inflammatory effect due to inhibition of both COX-1 and COX-2, which leads to the inhibition of PG synthesis.40
 | Table 1 Determination of anti-inflammatory activity of different derivatives of flurbiprofen by the carrageenan hind paw edema method |
The anti-inflammatory activity of mutual prodrugs was also investigated by the albumin-induced method, and it was confirmed that significant (P<0.001) anti-inflammatory effects shown by prodrugs (4a–d) at 4 hours postalbumin induced inflammation during the studies conducted to evaluate anti-inflammatory effects in egg-induced inflammation. Flurbiprofen prodrugs showed comparable results with standard flurbiprofen, which also produced significant reduction at 4 hours after the administration of egg albumin (Table 2). Previously, it has been reported that injection of egg albumin caused release of histamine and serotonin, which leads to inflammation. Histamine is a vasodilator and during vascular events it increases vascular permeability.41,42 The synthesized prodrugs displayed that anti-inflammatory effects might be due to inhibition of the release of mediators, such as histamine, which causes swelling that is the sign of inflammation.43
 | Table 2 Determination of anti-inflammatory activity of different derivatives of flurbiprofen by egg albumin-induced paw edema in mice |
During the studies conducted to evaluate analgesic effects, all of the synthesized prodrugs exhibited a significant (P<0.001) reduction in writhings induced by acetic acid. The mean% inhibition indicated that 4b was more active in decreasing the number of writhes induced by acetic acid than standard flurbiprofen, whereas 4d showed comparable analgesic activity to flurbiprofen. Similarly in formalin-induced licking in mice, the mutual prodrugs 4b and 4d have higher analgesic activity than standard flurbiprofen. The data further indicated that prodrugs 4b and 4d produced a significant increase in percentage inhibition of analgesic activity. The analgesic response of synthetic prodrugs (4a–d) in formalin-induced licking in mice was in the order of 4b>4d>4a>4c (Table 3).
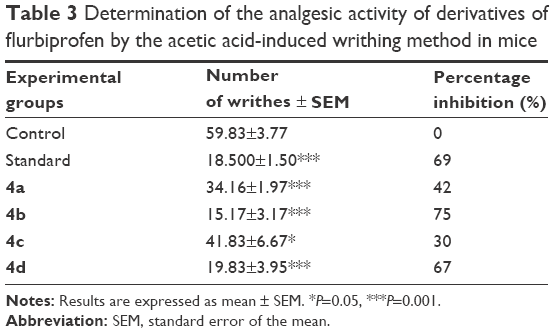 | Table 3 Determination of the analgesic activity of derivatives of flurbiprofen by the acetic acid-induced writhing method in mice |
The abdominal constriction response induced by acetic acid is a sensitive procedure to calculate peripherally acting analgesics.44 In the broad sense, acetic acid induces pain by releasing endogenous substances such as serotonin, histamine, PGs, bradykinins, and substance P from nerve fiber endings. Local peritoneal receptors are postulated to be involved in the abdominal constriction response.45 The response has also been associated with prostanoids in general, that is, increased levels of PGE2 and PGF2α in peritoneal fluids as well as lipoxygenase products.46 The synthesized prodrugs produced significant decrease in pain threshold in these models, suggesting the involvement of central pain pathways (Table 4). Pain is centrally modulated via a number of complicated processes such as opiate, dopaminergic descending noradrenergic, and serotonergic systems.47–49 The synthesized prodrugs produced analgesic effect via central mechanisms involving these receptor systems or via peripheral mechanisms involved in the inhibition of PGs, leukotrienes, and other endogenous substances that are key players in pain. Formalin test is a biphasic protocol. Neurogenic and inflammatory phases are related to pain mechanism. Substance P, histamine, serotonin, bradykinin PGs, and leukotrienes were released in neurogenic phase.50 Results indicated the significant antineurogenic effect of synthesized mutual prodrugs (4a–d). The mechanism of action of prodrugs could be due to inhibition of lipoxygenase or cyclooxygenase. Inhibition of inflammatory phase showed the peripheral effect of test compounds.
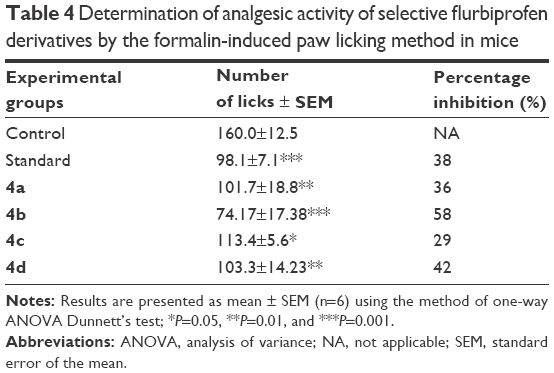 | Table 4 Determination of analgesic activity of selective flurbiprofen derivatives by the formalin-induced paw licking method in mice |
Antipyretic effects of the synthesized flurbiprofen prodrugs (4a–d) in mice indicated that all prodrugs showed significant inhibition of pyrexia (P<0.001) comparable with the standard flurbiprofen (Table 5). It has been well established that yeast induces pyrexia by increasing the production of PGs, mainly PGE2, which elevates the set point of the thermoregulatory center in the hypothalamus region of brain.51 Therefore, it is anticipated that all derivatives of flurbiprofen may likely reduce pyrexia by reducing PGE2 concentration in brain predominantly in the hypothalamus by acting on COX-3 or by increasing the production of the body’s own antipyretic substances such as vasopressin and arginine.52,53
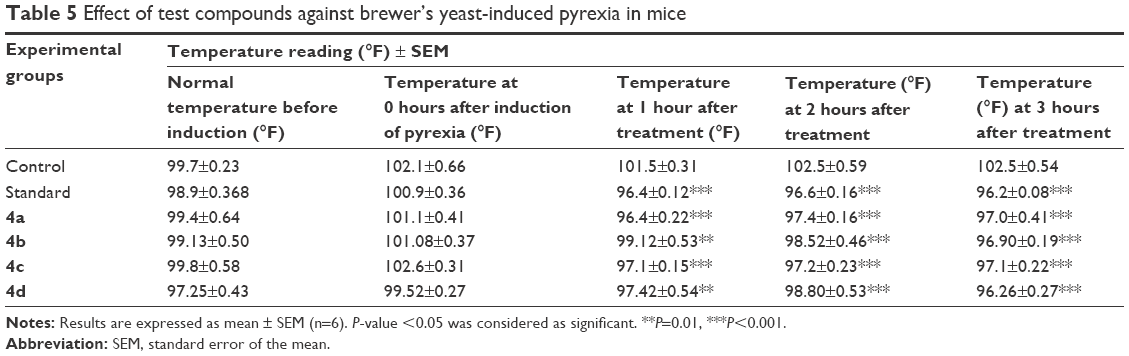 | Table 5 Effect of test compounds against brewer’s yeast-induced pyrexia in mice |
The ulcerogenic activity of the synthesized prodrugs was also determined to identify any undesirable effects on GI mucosa. UI of animals administered with synthesized prodrugs was compared with that of flurbiprofen, and the results were evaluated using unpaired t-test (Table 6). UI of flurbiprofen-treated animals is 3.07 and is due to direct contact mechanism as well as PG inhibition. All of the synthesized prodrugs significantly reduce the GI toxicity of flurbiprofen as the UI caused by synthesized prodrugs is less than that of the parent drug.
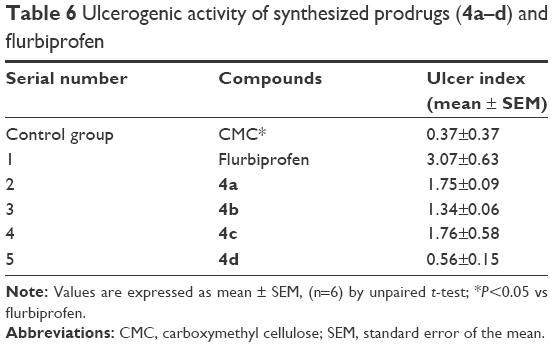 | Table 6 Ulcerogenic activity of synthesized prodrugs (4a–d) and flurbiprofen |
Receptor and ligand analysis
There are two isozymes of COX-1 and COX-2 that catalyze the first committed step in PG synthesis. The residual length of COX-1 is 599 AA, whereas that of COX-2 is 604 AA. It has been observed that crystal structure of both COX-1 and COX-2 has not been reported; therefore, in silico approach was employed to predict 3D structures of both proteins separately. Superimposed structures of COX-1 and COX-2 with binding pocket are presented in Figure S1. The reliability and efficacy of top-ranked modeled structure were confirmed by Ramachandran plots and hydrophobicity graphs. The hydrophobicity graphs are shown in Figures S2 and S3, and Ramachandran plots are shown in Figures S4 and S5. The Ramachandran plots indicated that the COX-1 95.01% and COX-2 94.7% residues are in favored regions for GS1 and GS2, respectively. Subsequently, outliers and all the poor rotamers were corrected to refine the predicted model of COX-1 and COX-2. Ramachandran graphs for COX-1 and COX-2 protein also showed the good accuracy of phi (φ) and psi (ψ) angles among the coordinates and most of the residues of COX-1 and COX-2 domains plummeted in the acceptable region. The hydrophobicity graphs for both domains have shown residues within the favored region. The chemoinformatics evaluation including molecular weight, logP, logS, number of hydrogen bond donors, number of hydrogen bond acceptors, density, polar surface area, and drug score values were calculated for all ligand molecules (Table 7). This detailed computational analysis suggested that all inhibitors follow the Lipinski rule and have inhibitory potential to target the COX protein.
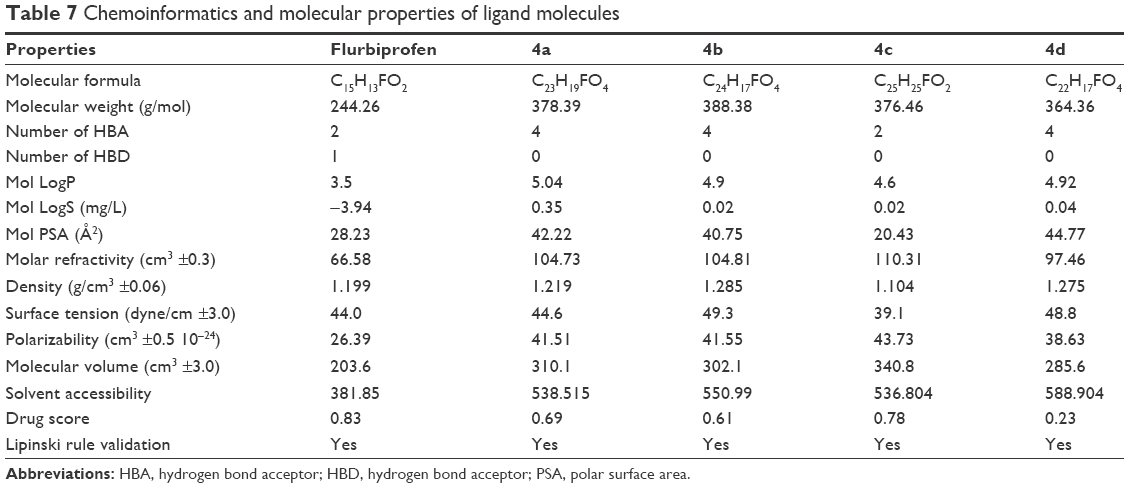 | Table 7 Chemoinformatics and molecular properties of ligand molecules |
COX-1 and COX-2 docking analysis
Docking studies of flurbiprofen and synthesized prodrugs (4a–d) were performed against COX-1 and COX-2 proteins separately to find the binding affinities of these ligands against COX proteins. The evaluation of different docked complexes was performed on the basis of binding energy values for both COX-1 and COX-2 against ligands using the PyRx 9.0 docking tool. The predicted energy values for COX-1 and COX-2 protein against flurbiprofen and synthesized prodrugs are listed in Table 8. The overall binding energy values of COX-2 protein are much better than the binding energy values of COX-1. COX-2 has the best binding value of −11.3 kcal/mol, while COX-1 has −10.1 kcal/mol. The comparative docking analysis shows that all of the synthesized prodrugs (4a–d) have good binding affinity to COX-2 when compared with COX-1 protein. This docking study suggested that the synthesized prodrugs have greater ability to inhibit the COX-2 protein when compared with COX-1 thus producing less gastric irritation. The COX-1 and COX-2 docking complexes with interacting residues are mentioned in Figures 4 and 5, respectively.
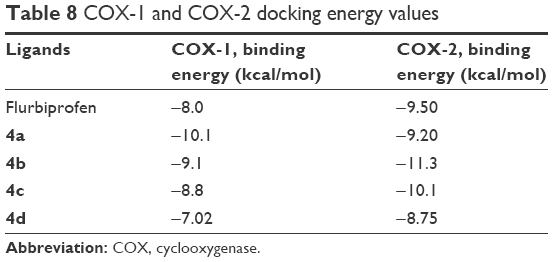 | Table 8 COX-1 and COX-2 docking energy values |
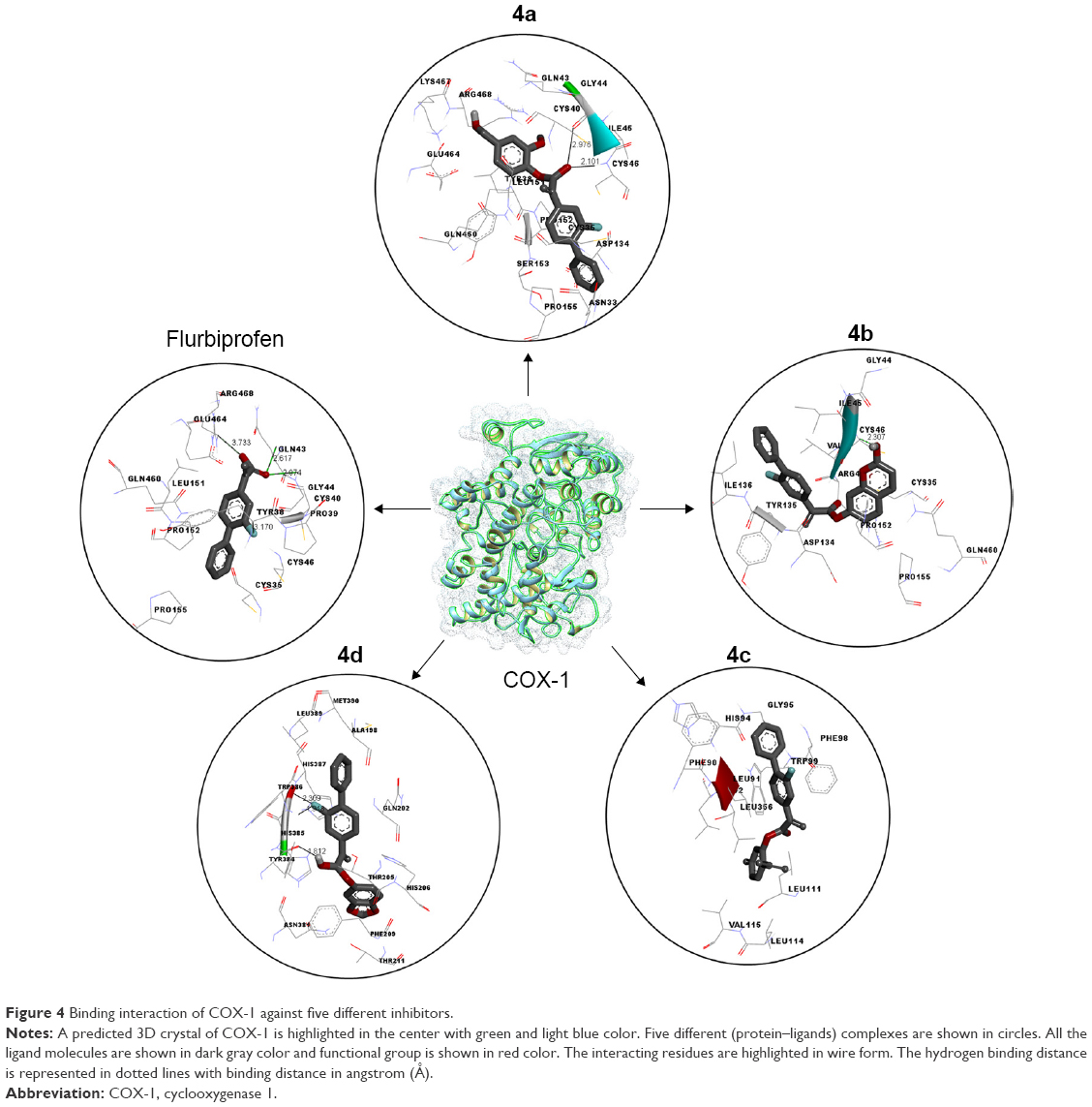 | Figure 4 Binding interaction of COX-1 against five different inhibitors. |
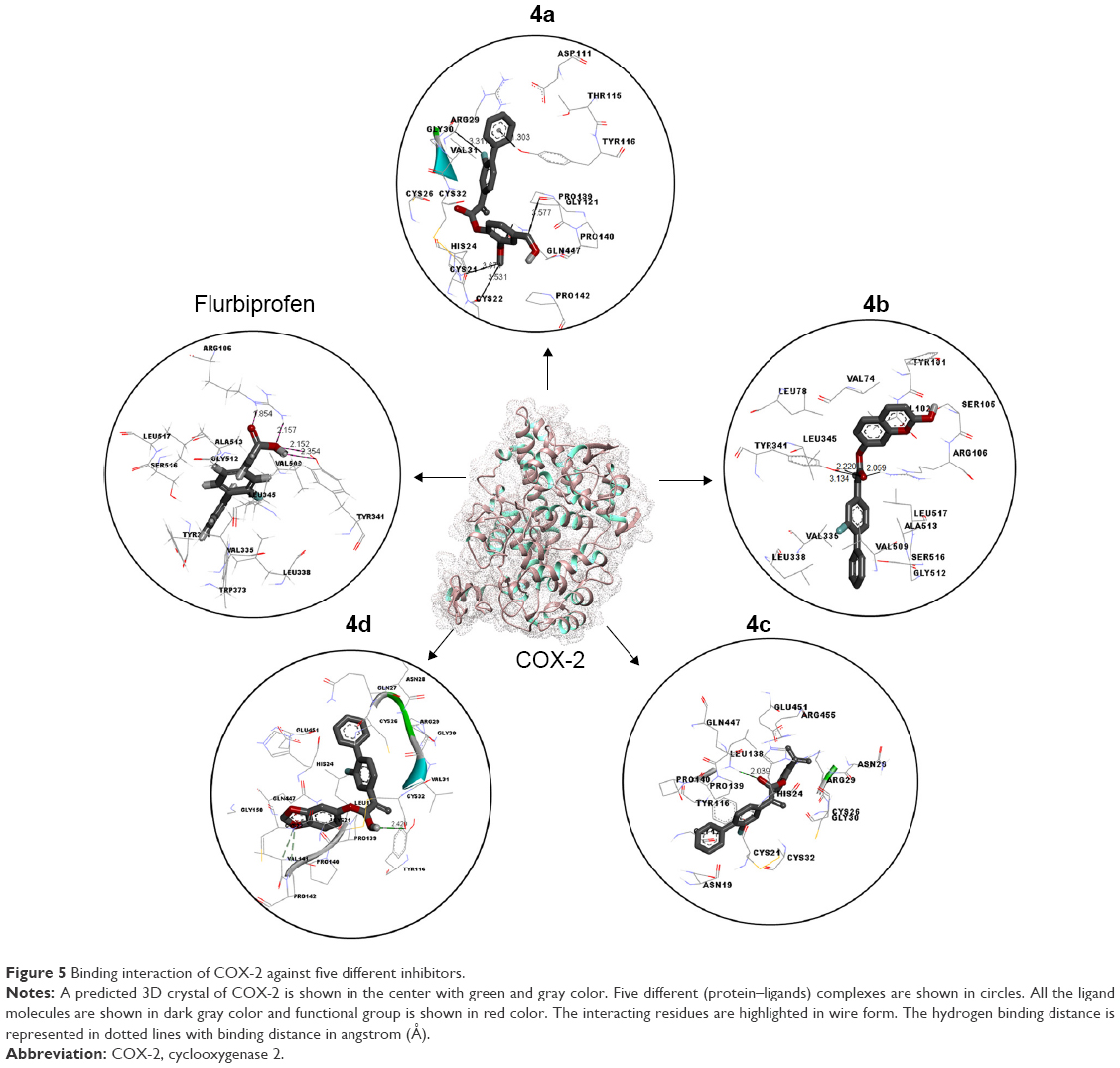 | Figure 5 Binding interaction of COX-2 against five different inhibitors. |
Discussion
The carboxylic acid group (–COOH) in flurbiprofen was masked by the ester group (–COOR) in the synthesized prodrugs. All of the synthesized prodrugs have similar structures with dissimilar R groups. The formation of the title prodrugs was ascertained by their spectroscopic data. The FTIR spectra of prodrugs showed absorption bands at 1,733–1,757 cm−1 (C=O stretching of ester) and 1,085–1,150 cm−1 (C–O stretching of ester), which confirms the presence of ester group, whereas in the FTIR spectrum of flurbiprofen the above ester peaks were absent. Instead, bands were observed at 1,255 cm−1 (CO stretching of –COOH), 1,417 cm−1 (OH bending of –COOH), 2,900 cm−1 (OH stretching of COOH), and 926 cm−1 (OH bending out of plane of –COOH), which showed the acidic nature of flurbiprofen due to the presence of free carboxylic acid group. The results of in vitro hydrolysis studies revealed that the synthesized prodrugs have different stability levels in the SIF, SGF, and in human plasma. All of the synthesized prodrugs possess ester groups, and because of the presence of esterase enzymes in human plasma, the prodrugs are easily hydrolyzed. The difference in the hydrolysis rate among the synthesized prodrugs is due to different R groups, as we know that the stability of different ester groups can be determined by R groups. The prodrug (4b) possesses the fused ring heterocyclic moiety, which may increase the stability of ester group.
The anti-inflammatory activity of the synthesized prodrugs was investigated using carrageenan as well as albumin-induced inflammation method. The ester group in the synthesized prodrugs is responsible for the pharmacological activity difference between flurbiprofen and its prodrugs. The prodrug (4b) showed most potent anti-inflammatory activity among the synthesized prodrugs due to the presence of coumarin moiety as the R group. This fused ring heterocyclic moiety is also responsible for most potent analgesic activity of the prodrug (4b) when compared with other prodrugs. The prodrugs (4b) and (4d) showed higher analgesic activity than prodrugs (4a) and (4c), the latter possesses homocyclic R groups, while the former possesses heterocyclic R groups. Antipyretic effects of flurbiprofen prodrugs may be due to the action on the hypothalamus, resulting in an increased peripheral blood flow, vasodilation, and subsequent heat dissipation. The temporary masking of the –COOH group of flurbiprofen with ester –COOR by natural oxidants has been proved to be useful in decreasing its GI toxicity. Based upon our results it is proposed that the ester group is less irritant to GI mucosa than the carboxylic acid group. The difference in ulcerogenic activity among synthesized prodrugs is again due to different R groups.
Computational structure prediction and its reliability were confirmed by Ramachandran plots and Molprobity values for COX-1 (95.01%) and COX-2 (94.7%). These predicted values justified that COX-2 is more reliable and can be considered as a good receptor for all synthesized prodrugs (4a–d). The flurbiprofen and its prodrugs were docked against both COX-1 and COX-2; the results indicated that prodrugs have greater binding affinity to COX-2 compared with COX-1. The prodrug (4b) has highest value of binding energy, −11.3 kcal/mol against COX-2, among all of the synthesized prodrugs. The ester carbonyl oxygen in the case of prodrug (4b) formed hydrogen bond with residue at position ARG106 of COX-2 with a bond length 2.05Å. The same carbonyl oxygen also interacts with amino acid TYR341 of the COX-2 with a bond length 2.22Å. The bond distance values are <5Å, which indicated that there are enough stronger binding interactions between prodrugs and receptor proteins.
The residual flexibility of receptors COX-1 and COX-2 was evaluated by MD simulation study at 10 ns. The backbone residual fluctuations in target proteins (COX-1 and COX-2) were determined by root mean square deviation (RMSD) and root mean square fluctuation (RMSF) graphs. Furthermore, radius of gyration (Rg) and solvent accessible surface area (A2) were also determined to check the compactness of protein. The RMSD graphs showed the residual fluctuations of targeted proteins COX-1 and COX-2. Initially, both lines showed an increasing trend with RMSD values ranging from 0.1 to 0.3 (nm) at time 0–1,000 ps. After that, both COX-1 and COX-2 showed steady graphs with very little fluctuations from 1,000 ps to 4,000 ps. From 4,000 ps to 8,000 ps time frame, COX-1 showed increased fluctuations and RMSD value when compared with COX-2. The results confirmed the greater stability of COX-2 when compared with COX-1. At time 8,000 ps both COX-1 and COX-2 again showed the same RMSD value of 0.3 nm. After that, an increasing trend was observed for COX-1, whereas COX-2 remained stable with little fluctuations. The comparative analysis justified that the simulation graph of COX-2 was more stable when compared with that of COX-1 throughout the simulation time period (Figure 6). The predicted RMSF graph lines of both COX-1 and COX-2 also reflected the C- and N-terminal lobes throughout the simulation period. The predicted results of RMSF graph showed that COX-2 was more stable and depicts less fluctuated peaks for loop conformation compared with COX-1 (Figure 7).
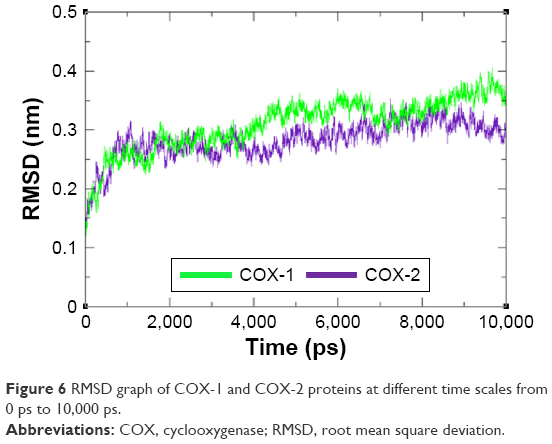 | Figure 6 RMSD graph of COX-1 and COX-2 proteins at different time scales from 0 ps to 10,000 ps. |
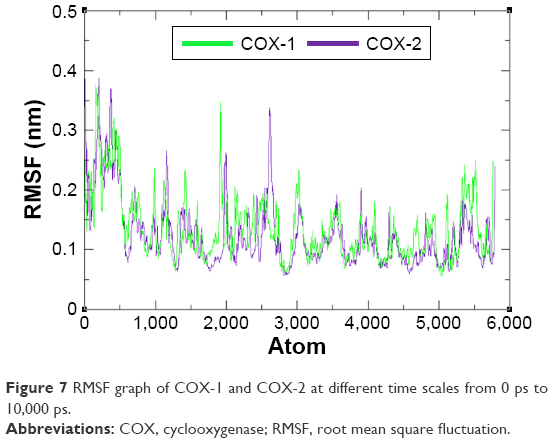 | Figure 7 RMSF graph of COX-1 and COX-2 at different time scales from 0 ps to 10,000 ps. |
The compactness of protein was evaluated by Rg and solvent accessible surface area as mentioned in Figures 8 and 9. The predicted results showed that Rg value for COX-2 is much better and remained stable between 2.4 nm and 2.35 nm values when compared with COX-1 throughout the simulation time frame of 0 ps to 10,000 ps. The comparative results showed that residual backbone and folding of target protein was steadily stable for COX-2 when compared with COX-1.
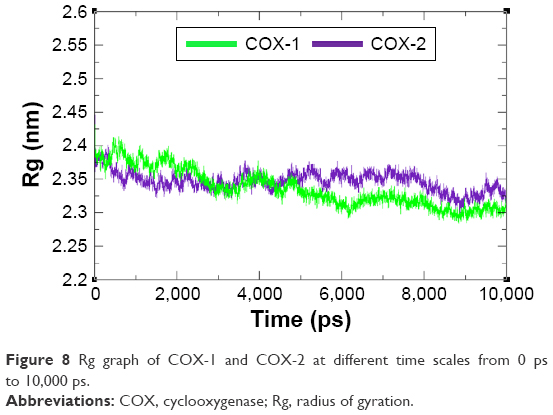 | Figure 8 Rg graph of COX-1 and COX-2 at different time scales from 0 ps to 10,000 ps. |
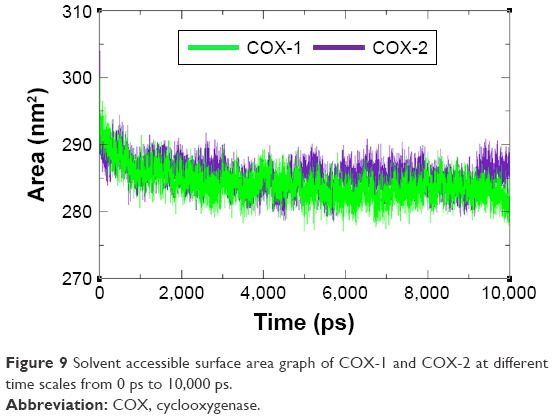 | Figure 9 Solvent accessible surface area graph of COX-1 and COX-2 at different time scales from 0 ps to 10,000 ps. |
Conclusion
The mutual prodrugs of flurbiprofen (4a–d) with natural phenolic antioxidants have been synthesized efficiently with good yields. The free –COOH group in flurbiprofen was temporarily masked by esterification with antioxidants to reduce its GI irritation. The in vitro hydrolysis studies showed that all prodrugs are quite stable at GI pH and are cleaved in SIF + 80% human plasma by esterase enzymes in plasma. The pharmacological investigation data proved that all of the synthesized prodrugs produced less GI irritation when compared with flurbiprofen. Computational molecular modeling and simulation studies results revealed that all prodrugs showed good binding affinities to both COX-1 and COX-2. These prodrugs also possess greater affinity to bind at COX-2 when compared with COX-1.
Acknowledgments
This work was supported by a research grant from the Kongju National University in 2015.
Author contributions
ZA, A, and SJK supervised the study, designed the experiment, and wrote the manuscript. MK performed the experiment to synthesize the title compounds and ZA characterized the compounds. MW and HA performed the in vivo studies. MH and SA designed the in silico experiment and helped in performing the docking studies of the title compounds. MH performed the molecular dynamic simulation studies. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
Khan MSY, Khan RM. Synthesis and biological evaluation of glycolamide esters as potential prodrugs of some non-steroidal anti-inflammatory drugs. Indian J Chem Sect B. 2002;41(10):2172–2175. | ||
Edelson JT, Tosteson AN, Sax P. Cost-effectiveness of misoprostol for prophylaxis against nonsteroidal anti-inflammatory drug-induced gastrointestinal tract bleeding. JAMA. 1990;264(1):41–47. | ||
Manon B, Sharma PD. Design, synthesis and evaluation of diclofenacantioxidant mutual prodrugs as safer NSAIDs. Indian J Chem Sect B. 2009;48(9):1279–1287. | ||
Polónia J. Interaction of antihypertensive drugs with anti-inflammatory drugs. Cardiology. 1997;88(suppl 3):47–51. | ||
Shanbhag VR, Crider AM, Gokhale R, Harpalani A, Dick RM. Ester and amide prodrugs of ibuprofen and naproxen: synthesis, anti-inflammatory activity and gastrointestinal toxicity. J Pharm Sci. 1992;81(2):149–154. | ||
Guyton CA, Hall JE. Textbook of Medical Physiology. ninth ed. Singapore: Harcourt Asia Pte. Ltd; 1998:846. | ||
Vane JR, Bakhle YS, Bolting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. | ||
Mahfouz NM, Omar FA, Aboul-Fadl T. Cyclic amide derivatives as potential prodrugs II: N-hydroxymethylsuccinimide-/isatin esters of some NSAIDs as prodrugs with an improved therapeutic index. Eur J Med Chem. 1999;34(7):551–562. | ||
Ingram MJ, Moynihan HA, Powell MW, Rostron C. Synthesis and hydrolytic behaviour of glycerol-1, 2-diibuprofenate-3-nitrate, a putative pro-drug of ibuprofen and glycerol-1-nitrate. J Pharm Pharmacol. 2001;53(3):345–350. | ||
Sharma P, Kaur G, Kansal S, Chandiran SK. Mutual prodrugs of 4-biphenylacetic acid and phytophenolics as safer NSAIDs: synthetic and spectral studies. Indian J Chem. 2004;43(10):2159–2164. | ||
Nwafor PA, Okwuasaba FK. Anti-nociceptive and anti-inflammatory effects of methanolic extract of Asparagus pubescens root in rodents. J Ethnopharmacol. 2003;84(2–3):125–129. | ||
Akah PA, Nwambie AI. Evaluation of Nigerian traditional medicines: 1. Plants used for rheumatic (inflammatory) disorders. J Ethnopharmacol. 1994;42(3):179–182. | ||
Koster R, Anderson M, Debeer JM. Acetic acid used for analgesic screening. Fed Proc. 1959;18:412–416. | ||
Correa CR, Calixto JB. Evidence for participation of B1 and B2 kinin receptors in formalin-induced nociceptive response in the mouse. Br J Pharmacol. 1993;110(1):193–198. | ||
Sakande J, Nacoulma OG, Nikiema JB, Lompo M, Bassene E, Guissou IP. Etude de l’effetantipyrétiqued’extraits des inflorescences mâles du ronierBorassusaethiopum mart (Arecaceae). Médecined’Afriquenoire. 2004;51(5):280–282. | ||
Ezer E, Palosi E, Hajos G, Szporny L. Antagonism of the gastrointestinal ulcerogenic effect of some nonsteroidal anti-inflammatory agents by sodium salicylate. J Pharm Pharmacol. 1976;28(8):655–656. | ||
Vogel WH. Analgesic, anti-inflammatory, and antipyretic activity. Drug Discovery and Evaluation. Hock FJ. Berlin Heidelberg: Springer; 1997:360–420. | ||
Majumdar B, Ray Chaudhuri SG, Ray A, Bandyopadhyay SK. Effect of ethanol extract of Piper betle Linn leaf on healing of NSAID-induced experimental ulcer – a novel role of free radical scavenging action. Indian J Exp Biol. 2003;41(4):311–315. | ||
Marjan MN, Hamzeh MT, Rahman E, Sadeq V. A computational prospect to aspirin side effects: aspirin and COX-1 interaction analysis based on non-synonymous SNPs. Comput Biol Chem. 2014;51:57–62. | ||
Amaravani M, Prasad NK, Ramakrishna V. COX-2 structural analysis and docking studies with gallic acid structural analogues. Springerplus. 2012;1(1):58. | ||
Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Structural Proteomics. Kobe B, Guss M, Huber T. New York, NY: Humana Press; 2008:145–159. | ||
Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. | ||
Melo F, Devos D, Depiereux E, Feytmans E. ANOLEA: a www server to assess protein structures. Proc Int Conf Intell Syst Mol Biol. 1997;5:187–190. | ||
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283–291. | ||
Lovell SC, Davis IW, Arendall WB, et al. Structure validation by Calpha geometry: φ, ψ and Cβ deviation. Proteins. 2002;50(3):437–450. | ||
Pedretti A, Villa L, Vistoli G. VEGA-an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des. 2004;18(3):167–173. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Accelrys Software Inc. D. Studio, Version 4.1. San Diego, CA: Accelrys Software Inc; 2007. | ||
Chen VB, Arendall WB, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 1):12–21. | ||
Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 4):486–501. | ||
Mendelsohn LD. ChemDraw 8 ultra, windows and macintosh versions. J Chem Inf Comput Sci. 2004;44(6):2225–2226. | ||
Mishra NK, Raghava GP. Prediction of specificity and cross-reactivity of kinase inhibitors. Lett Drug Des Discov. 2011;8:223–228. | ||
Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–241. | ||
Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–250. | ||
Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. | ||
Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J Comput Chem. 2004;25(13):1656–1676. | ||
Schüttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(pt 8):1355–1363. | ||
Wang H, Dommert F, Holm C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J Chem Phys. 2010;133(3):034117. | ||
Kasahara YK, Yasukawa S, Kitanaka M, Khan T, Evans FJ. Effect of methanol extract from flower petals of Tagetes patella L. on acute and chronic inflammation model. Phytother Res. 2002;16(3):217–222. | ||
Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971;231(25):232–235. | ||
Linardi A, Costa SKP, Desilva GR, Antunes E. Involvement of kinins, mast cells and sensory neurons in the plasma exudation and paw edema induced by staphylococcal entrotoxin B in the mouse. Eur J Pharmacol. 2002;399(2–3):235–242. | ||
Vasudevan M, Gunman KK, Parle M. Antinociceptive and anti-inflammatory effects of the Thespesia populnea bark extract. J Ethnopharmacol. 2007;109(2):264–270. | ||
Rang HP. Local hormones, inflammation and immune reactions: Eicosanoids I. In: Rang HP, Dale MM, Ritter JM, Flower RJ, editors. Rang & Dale’s Pharmacology. 6th ed. Edinburgh: Churchill Livingstone Elsevier; 2007:215. | ||
Gene RM, Segura L, Adzet T. Heterothecainuloides: anti-inflammatory and analgesic effects. J Ethnopharmacol. 1989;60(2):157–162. | ||
Bentley GA, Newton SH, Starr J. Studies on the anti-nociceptive action of agonist drugs and their interaction with opioid mechanisms. Br J Pharmacol. 1983;79:125–134. | ||
Deraedt R, Jongney S, Delevalcee F, Falhout M. Release of prostaglandin E and F in an analgesic reaction and its inhibition. Eur J Pharmacol. 1980;61(1):17–24. | ||
Bensreti MM, Sewell RD. Selective effects of dopaminergic modifiers on antinociception produced by different opioid receptor agonists. Pro Br Pharmacol Soc. 1983;28:70. | ||
Headley PM, Shaughnessy CT. Evidence for opiate and dopamine interaction in striatum. Br J Pharmacol. 1985;86:700. | ||
Wigdor S, Wilcox GL. Central and systemic morphine-induced antinociception in mice: of contribution descending serotonergic and noradrenergic pathways. J Pharmacol Exp Ther. 1987;242(1):90. | ||
Sharma A, Bhatia S, Kharya MD, et al. Anti-inflammatory and analgesic activity of different fractions of Boswelliaserrata. Int J Phytomed. 2010;2:94–99. | ||
Rititid W, Ruangsang P, Reanmongkol W, Wongnawa M. Studies of the anti-inflammatory and antipyretic activities of the methanolicextract of Piper sarmentosumRoxb. Leaves in rats. Songklanakarin J Sci Techol. 2007;6:1519–1526. | ||
Ayoub SS, Colville NPR, Willoughby DA, Botting RM. Theinvolvement of a cyclooxygenase-1 gene-derived protein in the antinociceptive action of paracetamol in mice. Eur J Pharmacol. 2006;538(1–3):57–65. | ||
Chandrasekharan DV, Roos H, Evanson KI, Tomsik NK, Elton J, Simmons T. Cox-3, a cox-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure and expression. Proc Natl Acad Sci U S A. 2002;139:26–31. |
Supplementary materials
Animal care and use
The proper care and use of laboratory animals in research, testing, teaching, and production (animal use) require scientific and professional judgment based on the animals’ needs and their intended use.
Temperature and humidity
The animals were housed within the temperature range of 26°C–34°C. The relative humidity was kept in a controlled range of 30%–70%.
Ventilation and air quality
The aim of proper ventilation is to provide the animals with quality air and stable environment. In the present experiment, animals were protected from direct exposure to high velocity air (drafts). In accordance with the guidelines provision, 10–15 fresh air changes per hour in the animal house were carried out to maintain macroenvironmental air quality by constant volume systems and may also ensure microenvironmental air quality. Modern heating, ventilation, and air conditioning systems were used to maintain the ventilation rates in accordance with heat load and other variables. Heating, ventilation, and air conditioning system were regularly evaluated for proper functioning.
Illumination
As light can affect the physiology, morphology, and behavior of various animals, light used in the animal house was between 130 lux and 325 lux in the room at the cage level. Lighting was diffused in the whole area used for animal handling and provided sufficient illumination for the animals’ well-being. To maintain the good housekeeping practices, the bottom-most cages in racks were inspected regularly.
Noise and vibration
Noise produced by animals and animal care activities is inherent in the operation of an animal facility. An environment was designed to accommodate animals’ exposure to sound louder than 85 dB, as it can have both auditory and nonauditory effects; hence, it is kept below this level.
Guidelines followed during experiments
- Swiss albino mice with a weight of 22–25 g (mean weight, 23.1 g) were obtained from the animal house, University of Sargodha, Sargodha.
- The animals were housed in stainless metabolic cages (each cage had four mice according to the National Institutes of Health (NIH) guidelines that recommends that animals occupy space according to their size) and observed under a 12-hour/12-hour light/dark cycle to maintain illumination, in a well-ventilated room at 26°C–27°C, because according to NIH guidelines this is a thermo neutral zone for mice.
- Standard diet was given to these mice (Ladokun Feeds, Ibadan, Nigeria) and water ad libitum.
- The experimental protocol was according to internationally approved guidelines for animal use and care (EEC Directive of 1986; 86/09/EEC; National Institutes of Health publication 85-23, revised 1985). The acclimatization period lasted for 7 days.
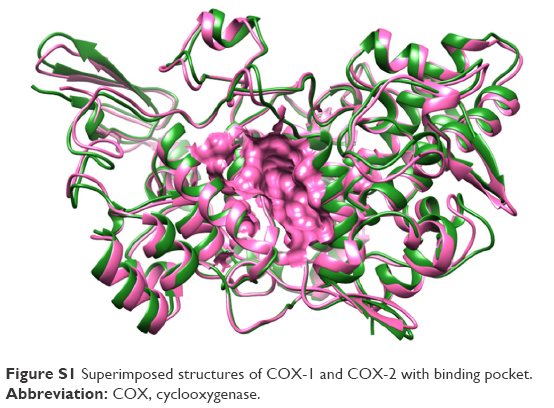 | Figure S1 Superimposed structures of COX-1 and COX-2 with binding pocket. |
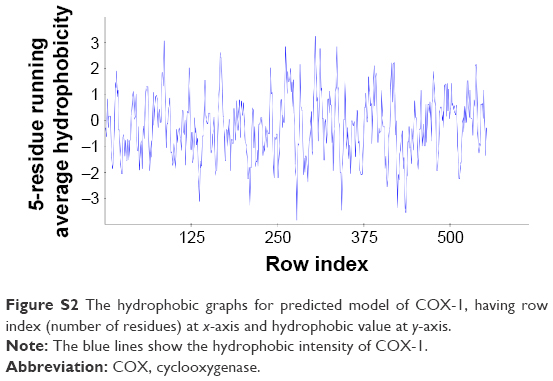 | Figure S2 The hydrophobic graphs for predicted model of COX-1, having row index (number of residues) at x-axis and hydrophobic value at y-axis. |
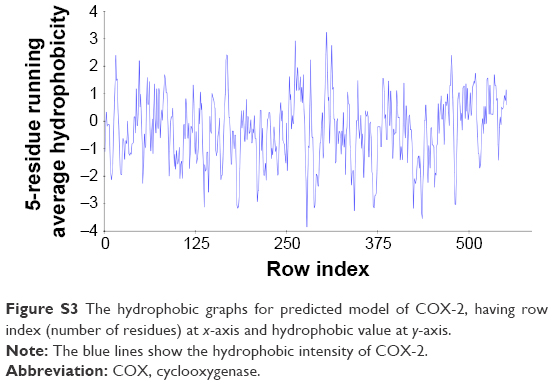 | Figure S3 The hydrophobic graphs for predicted model of COX-2, having row index (number of residues) at x-axis and hydrophobic value at y-axis. |
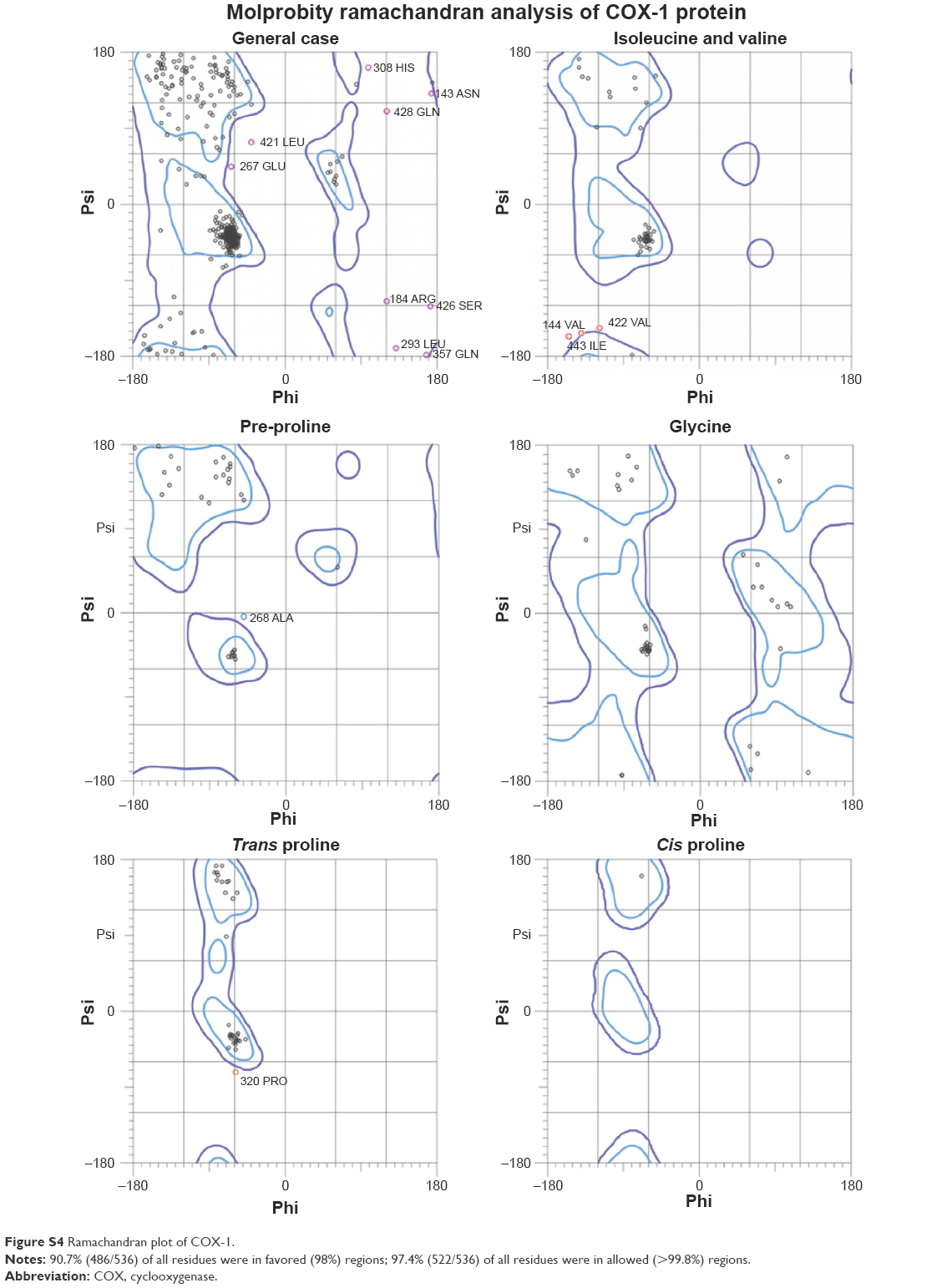 | Figure S4 Ramachandran plot of COX-1. |
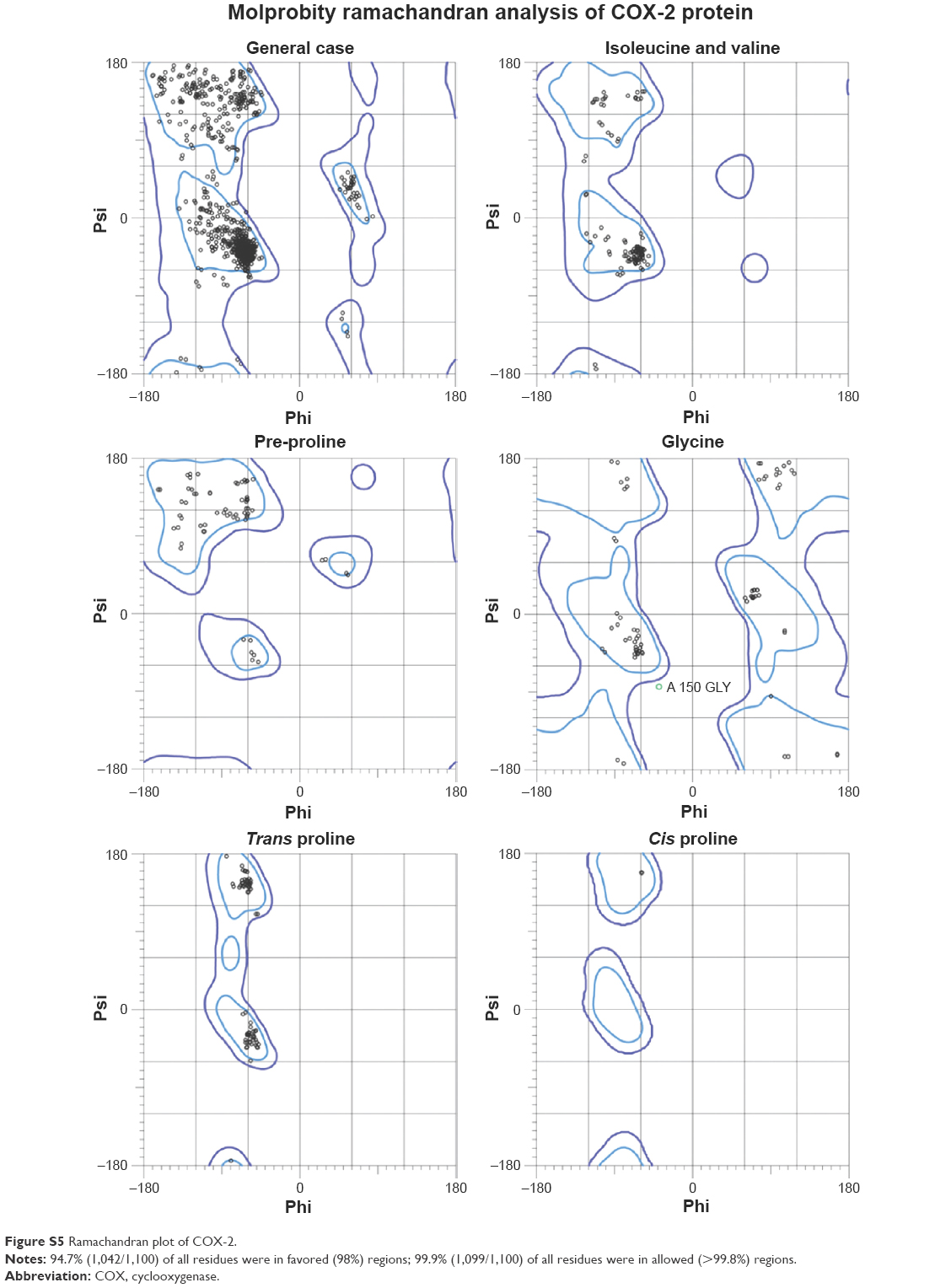 | Figure S5 Ramachandran plot of COX-2. |
 | Table S1 The clinical effects on animals during the experiments |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.