Back to Journals » Infection and Drug Resistance » Volume 10
Fluconazole resistance in Candida species: a current perspective
Authors Berkow EL, Lockhart SR
Received 26 April 2017
Accepted for publication 13 July 2017
Published 31 July 2017 Volume 2017:10 Pages 237—245
DOI https://doi.org/10.2147/IDR.S118892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Elizabeth L Berkow, Shawn R Lockhart
Mycotic Diseases Branch, Centers for Disease Control and Prevention, Atlanta, GA, USA
Abstract: Candida albicans and the emerging non-albicans Candida spp. have significant clinical relevance among many patient populations. Current treatment guidelines include fluconazole as a primary therapeutic option for the treatment of these infections, but it is only fungistatic against Candida spp. and both inherent and acquired resistance to fluconazole have been reported. Such mechanisms of resistance include increased drug efflux, alteration or increase in the drug target, and development of compensatory pathways for producing the target sterol, ergosterol. While many mechanisms of resistance observed in C. albicans are also found in the non-albicans species, there are also important and unexpected differences between species. Furthermore, mechanisms of fluconazole resistance in emerging Candida spp., including the global health threat Candida auris, are largely unknown. In order to preserve the utility of one of our fundamental antifungal drugs, fluconazole, it is essential that we fully appreciate the manner by which Candida spp. manifest resistance to it.
Keywords: Candida, fluconazole resistance, ERG11, drug efflux, ergosterol
Introduction to Candida infections
Candida spp. exist as commensals of the skin, mouth, and gastrointestinal tract. Their growth and spread is kept in check by coexisting microbial flora, intact epithelial barriers, and defenses of the innate immune system. While Candida spp. are normal flora of the human body, they also possess the ability to transition to pathogens that cause a wide spectrum of conditions, ranging from superficial infections of the hair and nails to life-threatening systemic infections.1 In fact, Candida account for approximately 75–88% of all fungal infections in the US.2–4 Additionally, in both adult and pediatric populations, the presence of candidemia results in an increase in mortality and hospital length of stay, culminating in an annual attributable cost of $1.7 billion in the US alone, making Candida not only a clinical concern but an economic concern as well.5,6
The epidemiology of Candida infections has been in flux in recent decades, most likely due to our own medical practices. Risk factors for infection with Candida are similar to those of fungal infections in general and vary in cause, but are generally due to medical intervention or health status of the patient. Risk factors fall into three distinct categories: factors that promote colonization of Candida, factors that suppress the immune response to Candida, and factors that provide a direct route for Candida infection.7 Historically, 92–95% of all cases of Candida infection are a result of the five most common species: Candida albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis, and Candida krusei. And while this still holds true, there has been a shift in the species distribution among Candida infections. In past decades, C. albicans comprised >50% of all candidemia. However, the non-albicans Candida (NCAC) species are being encountered more frequently as human pathogens and, in some cases, NCAC species are more frequently isolated than C. albicans. In the US, C. glabrata is the second most common Candida spp., followed by C. tropicalis and C. parapsilosis.8 This rise in the NCAC species is possibly due to their inherently high levels of antifungal drug resistance (discussed later), but it is important to consider that improvements to laboratory detection and identification may simply provide more specific identification than in the past and account for emergence of less common species.
The global picture of candidemia has been recently expanded further with the emergence of the multidrug-resistant species Candida auris. Originally recognized in 2009 from an ear canal specimen in Japan, C. auris has since been reported from many countries including South Korea, South Africa, Kenya, India, Pakistan, Colombia, Venezuela, the UK, and the US.9–14 Further complicating our understanding of this newly emerging species are difficulties in identification – several identification platforms misidentify C. auris as other species of Candida – and difficulties in eradicating this organism from health care facilities.15,16
Fluconazole
One of the most commonly prescribed antifungal drugs for Candida infections is fluconazole, a triazole antifungal.17 The azoles function by inhibiting the cytochrome P450 enzyme lanosterol demethylase (14α-demethylase), encoded by ERG11, in the ergosterol biosynthesis pathway. More specifically, the free nitrogen atom of the azole ring binds an iron atom within the heme group of the enzyme.18 This prevents the activation of oxygen and in turn the demethylation of lanosterol, which inhibits the process of ergosterol biosynthesis.19 As ergosterol is an essential component of fungal cell membranes, this inhibition is toxic; methylated sterols accumulate in the fungal cellular membrane, and cell growth is arrested.20
Fluconazole is fungistatic rather than fungicidal, so treatment provides the opportunity for acquired resistance to develop in the presence of this antifungal. In the US, C. albicans has a low incidence of fluconazole resistance, approximately 0.5–2%. C. tropicalis, C. parapsilosis, and C. glabrata, on the other hand, have higher rates at 4–9%, 2–6%, and 11–13%, respectively.21,22 The emerging yeast C. auris can exhibit a rate of resistance to fluconazole as high as 93%.23 Alternatively, without prior introduction of the antifungal, fluconazole resistance may also be innate, as is seen with C. krusei.24,25 Understanding the mechanisms underlying fluconazole resistance is a crucial part of managing our limited antifungal repertoire and keeping fluconazole a possible option to treat many Candida infections.
Molecular mechanisms of fluconazole resistance
The development of fluconazole resistance in Candida spp. has been well characterized in C. albicans. As the epidemiology of candidemia shifts to include more instances of NCAC species, it has become crucial to characterize fluconazole resistance in these species as well. Several mechanisms have been noted to involve genes of the ergosterol biosynthesis pathway; others involve drug transporters, changes in ploidy, and loss of heterozygosity (LOH; Table 1).
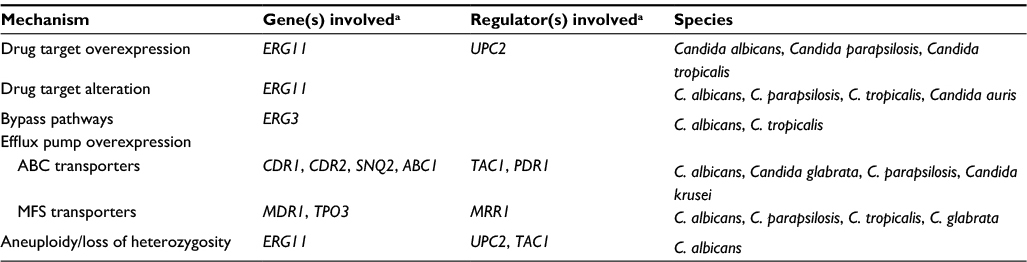 | Table 1 Summary of molecular mechanisms of fluconazole resistance in Candida spp. Note: aGenes and regulators that have been directly linked to the resistant phenotype in clinical isolates. Abbreviations: ABC, ATP-binding cassette; MFS, major facilitator superfamily. |
Increased drug target
Many C. albicans clinical isolates overexpress ERG11, the gene encoding the target of the azoles. In many cases, the level of overexpression is minimal or else observed in combination with other resistance mutations, making it difficult to assess the direct impact of such overexpression on the resistant phenotype. Studies have shown that this overexpression often involves Upc2p, a zinc cluster transcription factor that is induced upon ergosterol depletion.26 Mutations in Upc2p provide gain of function (GOF) for this regulator, resulting in constitutive transcriptional activity and increased production of Erg11p.27–29 One large study of C. albicans evaluated the impact of various Upc2p mutations on ERG11 expression as well as on the fluconazole-resistant phenotype. It identified several mutations that were associated with increased ERG11 expression as well as increased ergosterol production.30 This increased production of the azole target dilutes the activity of the fluconazole and results in resistance.
The significance of increased ERG11 expression in fluconazole-resistant NCAC is varied. While overexpression of ERG11 has been observed in several resistant C. parapsilosis isolates, these isolates also harbored additional resistance mechanisms which likely participated in their resistant phenotype.31,32 Moreover, when UPC2 was sequenced in such isolates, a single heterozygous mutation was identified in a single isolate – the remaining isolates contained no UPC2 mutation – indicating that the overexpression of ERG11 was UPC2-independent, a finding that has been observed in some C. albicans isolates as well.28,32 Likewise, several studies have identified higher expression of ERG11 in fluconazole-resistant clinical isolates of C. tropicalis as compared to fluconazole-susceptible isolates.33 However, sequencing of UPC2 revealed mutations that were present in both resistant and susceptible isolates. Further characterization of the cause(s) of increased ERG11 expression is needed in these NCAC species.
Unlike in other species of Candida, ERG11 does not appear to play a major role in fluconazole resistance in C. glabrata, and there are only two clinical isolates reported to display overexpression of ERG11.34,35 The contribution of this overexpression to fluconazole resistance in these isolates is unclear. The same is true for C. krusei; a single report of increased ERG11 expression observed in four clinical isolates can be found in the literature.36 The role of ERG11 overexpression in fluconazole resistance in C. auris is currently unknown.
Alteration in drug target
Point mutations in the coding region of the ERG11 gene impact susceptibility to fluconazole. These mutations lead to amino acid substitutions which alter the structure of the protein and render binding of the azoles less efficient.37,38 This reduced binding affinity decreases azole susceptibility. Over 140 substitutions have been described in C. albicans ERG11, indicating that this enzyme is highly permissive for structural changes.39 Rather than being found dispersed throughout the coding region, the majority of ERG11 amino acid substitutions fall into distinct “hot spot” regions within the protein: amino acids 105–165, 266–287, and 405–488.40 A handful of these substitutions have been conclusively linked to resistance (Table 2).39,41–46 One large study performed allelic transfer to determine the impact of 10 specific substitutions found in clinical isolates of C. albicans on fluconazole resistance. Protein modeling studies suggested that substitutions impact fluconazole-susceptibility cluster in the predicted enzyme catalytic site, a fungus-specific external loop, or a heme-interaction surface site.47 More commonly, substitutions are not conclusively linked to a resistant phenotype but, instead, are observed in isolates which are azole resistant but not in isolates that are azole susceptible.39,48 This is suggestive but not confirmatory of causing resistance.
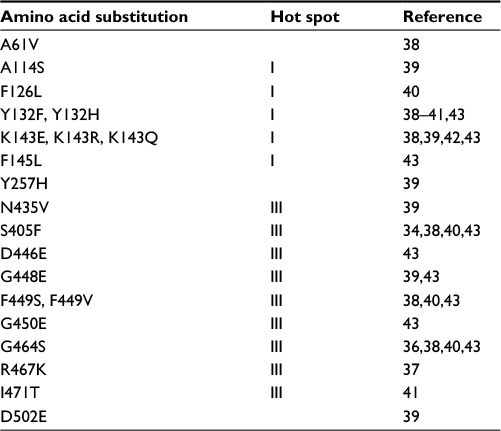 | Table 2 ERG11 amino acid substitutions linked to fluconazole resistance in clinical isolates of Candida albicans |
Studies in NCAC indicate far lower variability in ERG11 sequence. A single amino acid substitution, Y132F, either alone or in combination with an R398I substitution, has been observed exclusively in fluconazole-resistant clinical isolates of C. parapsilosis and not in susceptible isolates.31,32,49 The point mutation Y132F has also been noted in a single resistant isolate of C. tropicalis.50 Additionally, a single C. tropicalis isolate which contained a truncated Erg11p resulting from a deletion mutation and led to high-level fluconazole resistance was identified.51 A single instance of an ERG11 mutation has been reported in C. glabrata; a clinical isolate containing a missense mutation produced no membrane ergosterol and had high resistance to fluconazole.52
A chief mechanism underlying innate fluconazole resistance in C. krusei is a reduced susceptibility of Erg11p.53–55 This has not been concluded from changes in gene sequence but from the observation that a relatively high concentration of drug is required to inhibit the synthesis of ergosterol with C. krusei Erg11p compared to other fungal species. Other studies have detected point mutations in ERG11 and linked them to reduced susceptibility, but these studies are in relation to azoles other than fluconazole.56,57
As with many topics in C. auris, much is unknown concerning the contribution of ERG11 point mutations to fluconazole resistance. However, one recent study determined that a collection of C. auris isolates with elevated minimum inhibitory concentration (MIC) values to fluconazole carried point mutations which had been previously shown to impact drug resistance in other Candida spp.: Y132F, K143R, and F126T. These mutations were present in geographically distinct clades, with the isolates within a particular region having the same mutation. Isolates which did not carry one of these ERG11 mutations had lower MIC values to the azole drug class. All of this information culminates in the conclusion that the point mutations in the target protein play a role in the fluconazole-resistant phenotype of these isolates.23
Alterations in sterol biosynthesis
The development of bypass pathways within sterol biosynthesis may also occur and result in resistance to fluconazole. Considered an infrequent mechanism of resistance, this primarily results from loss-of-function mutations in ERG3. This inactivation of the enzyme Δ5,6-sterol desaturase permits the cell to bypass production of toxic methylated sterols in the presence of the azoles and minimizes the effect of fluconazole on the cell.58,59 This has been experimentally induced in the laboratory as well as observed in clinical isolates of C. albicans and, to a lesser extent, C. tropicalis.51,60–63
Increased drug efflux
In addition to mechanisms involving the ergosterol biosynthesis pathway, efflux of antifungal drug via transport proteins is found in a wide array of fungi. This results in a failure of the drug to accumulate intracellularly, resulting in resistance. In Candida spp., there are two main classes of efflux proteins – the major facilitator superfamily (MFS) class and the ATP-binding cassette (ABC) family. Each function to translocate compounds across the fungal cell membrane, and constitutive upregulation of members of each of these families can mediate resistance to fluconazole.
ABC transport proteins have a broad substrate specificity and rely on the hydrolysis of ATP for energy.64 Of the 28 individual ABC proteins predicted in C. albicans, two are well characterized and definitively linked to fluconazole resistance – the highly homologous Candida drug resistance 1 (Cdr1p) and Candida drug resistance 2 (Cdr2p).65,66 Only CDR1 is expressed at detectable levels under standard conditions in susceptible isolates, and its deletion can lead to hypersusceptibility. CDR2 is not usually expressed, and its deletion will not impact susceptibility in a drug-susceptible isolate.67–69 Many fluconazole-resistant isolates overexpress CDR1 and CDR2, and deletion of one or both genes results in reduction of this resistance.66 Constitutive upregulation of both CDR1 and CDR2 results from GOF mutations in the transcriptional regulator, TAC1. Currently, 19 different point mutations have been confirmed as GOF in TAC1; there are many candidate mutations still awaiting investigation.70,71
Homologous ABC transporters have also been identified in and linked to fluconazole resistance in NCAC species, including CgCdr1p, CgCdr2p, and CgSnq2p in C. glabrata (with corresponding transcriptional regulator, CgPdr1p), CpCdr1p in C. parapsilosis, and CkAbc1p in C. krusei.32,72–76 Increased expression of CtCDR1 has been detected in fluconazole-resistant isolates, although it is noteworthy that these isolates were passage-derived in the laboratory as opposed to clinically derived.77 Evaluation of clinical isolates of C. tropicalis has not reliably demonstrated this resistance mechanism.33,78 To date, there has been no investigation of the role of efflux pumps in fluconazole resistance in C. auris.
Compared to the ABC family of transporters, members of the MFS class have a more narrow range of substrate specificity and are powered by electrochemical proton-motive force.79 There are approximately 95 MFS transport proteins predicted in the C. albicans genome.80 Only one, Mdr1p, has been linked to fluconazole resistance; overexpression of MDR1 has been observed in resistant C. albicans clinical isolates, and deletion of MDR1 from overexpressing isolates reduces resistance.66,81 Regulation of Mdr1p expression is controlled by the transcription factor Mrr1p, and point mutations within this factor induce overexpression of its target gene.82 There are currently 15 different Mrr1p GOF mutations reported in C. albicans.83
MFS transporters play only a minor role in fluconazole resistance in the NCAC species. Loss of MDR1 in fluconazole-resistant C. parapsilosis isolates results in only a modest decrease in resistance.32 Likewise, no significant difference in expression of MDR1 has been observed in C. tropicalis fluconazole-resistant isolates as compared to fluconazole-susceptible.33 Recently, an MFS transporter in C. glabrata, Tpo3p, was shown to impact fluconazole susceptibility (upon deletion of this transporter, resistance to fluconazole decreased).84 Meaningful homologs of Mdr1p have not been identified in C. krusei or C. auris.
Aneuploidy and other chromosomal abnormalities
Additional mechanisms of fluconazole resistance that involve gene amplification have also been identified. Aneuploidy may develop in laboratory strains or clinical isolates of C. albicans following routine culture and genetic manipulation. Such alterations in gene copy number provide a means for genetic variation without impairment of cell growth.85,86 One study analyzed gene copy number in clinical isolates of C. albicans and determined that aneuploid chromosomes were seven times more common in fluconazole-resistant isolates as compared to fluconazole susceptible. They also reported specifically isochromosome formation of chromosome 5 (chr5) to be associated with fluconazole resistance in C. albicans. Increases or reductions in resistance could be tied, respectively, with gain or loss of this chr5 isochromosome, as duplication of this segment accords additional copies of ERG11, its transcriptional regulator UPC2, as well as efflux pump regulator, TAC1.87
LOH of specific genomic regions is also implicated in fluconazole resistance. It is most commonly associated with genes involved in resistance mechanisms, including ERG11, TAC1, and MRR1, with point mutations arising in the heterozygous state, and evolving to homozygosity. Instances of this mechanism occurring in sequentially collected clinical isolates of C. albicans have been reported.87,88
There is little information regarding chromosomal abnormalities in the NCAC species. Ploidy changes do occur in C. tropicalis.89 One report showed that in response to fluconazole exposure, an intermediate trimera morphology associated with gene duplication forms in several NCAC including C. parapsilosis, C. tropicalis, and C. glabrata.90 Other reports observe fluconazole resistance in the presence of chromosomal abnormalities in both C. glabrata and C. krusei in the form of increased copy number of ERG11 and the formation of segmental aneuploidies.76,91,92 The causal link between these findings and fluconazole resistance however has not been demonstrated in the NCAC.
Additional considerations in fluconazole resistance
Beyond the traditionally recognized mechanisms of resistance against fluconazole, much is still unknown. Well-understood molecular mechanisms do not always completely account for the high-level resistance observed in many clinical isolates, and this highlights the importance of continued investigation. There are instances of ERG11 overexpression in several Candida spp. that cannot be linked to activating mutations within their transcriptional regulator, UPC2 – pointing to additional, unknown regulators of this key gene.30,32,33 It is also likely that alternate regulators of drug efflux pumps, such CaCAP1 and CaFCR1, can regulate the transport of drug from the cell but may be underappreciated or overlooked in studies which focus on the most commonly described regulators.93–95 Additionally, signal transduction pathways that promote response in Candida spp. to the stress imposed by fluconazole also likely play a role in fluconazole resistance, but the contributions of these pathways are not yet completely defined.96,97
Deficient drug import should be an additional consideration. Fluconazole uptake into the fungal cell is required for the drug to reach its target protein, Erg11p. The most comprehensive investigation into this process used biochemical analyses to determine that fluconazole is imported by facilitated diffusion mediated by an unknown transporter, at least in C. albicans and C. krusei.98 While this import is inadequately understood, it is certainly relevant to a discussion on resistance. It is possible that a mutation in a putative import transporter could impair the cell’s ability to uptake this antifungal and generate resistance.
Several studies have reported fluconazole-resistant isolates which simultaneously overexpress CDR1 and MDR1 and/or have multiple changes to genes involved in ergosterol biosynthesis.44,99–101 In fact, it is quite uncommon to find an isolate with high-level resistance attributable to a single mechanism (the exception being PDR1 mutation in C. glabrata).102 Most often, it is the combination of multiple mechanisms, acquired stepwise over time, that give rise to high-level resistance. This is observed clinically in patients undergoing long-term fluconazole treatment. In vitro, this additive effect of multiple mechanisms has been illustrated by the progressive introduction of point mutations in ERG11, MRR1, TAC1, and UPC2 in an otherwise drug-susceptible C. albicans and by the dissection of individual mutations appearing in a resistant clinical isolate of C. albicans.103,104 Moreover, the functional importance of individual mutations varies in terms of impact on resistance. For example, the change in fluconazole MIC imparted by CaErg11p point mutation K143R is higher than that of F449V.47
The expense paid by the cell for acquiring resistance mutations under antifungal stress is a fitness cost. This cost, resulting from altered membrane sterol content, structural changes in 14α-demethylase, or disregulated gene expression, may result in a competitive disadvantage against other, more “fit” organisms within the host in the absence of the drug. Multiple analyses demonstrate this to be accurate in both in vitro and in vivo models of Candida infection, although in vitro growth conditions do impact fitness assays.62,63,103,105,106 As a result, compensatory mechanisms arise which work to alleviate the fitness cost of such developed resistance.107,108 The methods by which Candida spp. develop compensatory mechanisms to alleviate the fitness cost of resistance is another area for consideration, but it is beyond the scope of this manuscript.
Finally, as the landscape of candidemia expands to include newly emerging species (C. auris for example), detecting resistance in these species will be of paramount importance. It could be hypothesized that mechanisms present in the extensively studied C. albicans would be operative in these species as well, but recent investigation into many NCAC species, C. glabrata for example, has shown that this may not tell the whole story. Moving forward, it will be essential to use a methodical approach of next-generation sequencing and global transcriptional analysis to identify the determinants of fluconazole resistance in newly emerging species.
Conclusion
The emergence of drug resistance can be considered an inevitable consequence of the selective pressures imposed by antifungal drugs. In the past two decades, several genes and mutations which increase resistance to fluconazole in clinical isolates, primarily in C. albicans, have been elucidated. In order to preserve the utility of fluconazole, a central antifungal option, it is important that we fully appreciate the manner by which all Candida spp. exhibit resistance to it.
Acknowledgments
The authors would like to extend their thanks to the staff of the Mycotic Diseases Branch at the Centers for Disease Control and Prevention. The findings and conclusions in this manuscript are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Disclosure
The authors report no conflicts of interest in this work.
References
Calderone RA, Clancy CJ. Candida and Candidiasis. 2nd ed. Washington, DC: American Society of Microbiology; 2012. | ||
Rees JR, Pinner RW, Hajjeh RA, Brandt ME, Reingold AL. The epidemiological features of invasive mycotic infections in the San Francisco Bay area, 1992–1993: results of population-based laboratory active surveillance. Clin Infect Dis. 1998;27(5):1138–1147. | ||
Jarvis WR. Epidemiology of nosocomial fungal infections, with emphasis on Candida species. Clin Infect Dis. 1995;20(6):1526–1530. | ||
Horn DL, Neofytos D, Anaissie EJ, et al. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin Infect Dis. 2009;48(12):1695–1703. | ||
Wilson LS, Reyes CM, Stolpman M, Speckman J, Allen K, Beney J. The direct cost and incidence of systemic fungal infections. Value Health. 2002;5(1):26–34. | ||
Zaoutis TE, Argon J, Chu J, Berlin JA, Walsh TJ, Feudtner C. The epidemiology and attributable outcomes of candidemia in adults and children hospitalized in the United States: a propensity analysis. Clin Infect Dis. 2005;41(9):1232–1239. | ||
7.Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clin Microbiol Rev. 2007;20(1):133–163. | ||
Cleveland AA, Harrison LH, Farley MM, et al. Declining incidence of candidemia and the shifting epidemiology of Candida resistance in two US metropolitan areas, 2008–2013: results from population-based surveillance. PLoS One. 2015;10(3):e0120452. | ||
Satoh K, Makimura K, Hasumi Y, Nishiyama Y, Uchida K, Yamaguchi H. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. Microbiol Immunol. 2009;53(1):41–44. | ||
Kim MN, Shin JH, Sung H, et al. Candida haemulonii and closely related species at 5 university hospitals in Korea: identification, antifungal susceptibility, and clinical features. Clin Infect Dis. 2009;48(6): e57–e61. | ||
Chowdhary A, Voss A, Meis JF. Multidrug-resistant Candida auris: ‘new kid on the block’ in hospital-associated infections? J Hosp Infect. 2016;94(3):209–212. | ||
Magobo RE, Corcoran C, Seetharam S, Govender NP. Candida auris-associated candidemia, South Africa. Emerg Infect Dis. 2014;20(7):1250–1251. | ||
Calvo B, Melo AS, Perozo-Mena A, et al. First report of Candida auris in America: clinical and microbiological aspects of 18 episodes of candidemia. J Infect. 2016;73(4):369–374. | ||
Borman AM, Szekely A, Johnson EM. Comparative pathogenicity of United Kingdom isolates of the emerging pathogen Candida auris and other key pathogenic Candida species. mSphere. 2016;1(4):e00189-16. | ||
Mizusawa M, Miller H, Green R, et al. Can multidrug-resistant Candida auris be reliably identified in clinical microbiology laboratories? J Clin Microbiol. 2017;55(2):638–640. | ||
Schelenz S, Hagen F, Rhodes JL, et al. First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrob Resist Infect Control. 2016;5:35. | ||
Pfaller MA, Diekema DJ, Gibbs DL, et al; Global Antifungal Surveillance Group. Results from the ARTEMIS DISK Global Antifungal Surveillance Study, 1997 to 2007: a 10.5-year analysis of susceptibilities of Candida species to fluconazole and voriconazole as determined by CLSI standardized disk diffusion. J Clin Microbiol. 2010;48(4):1366–1377. | ||
Hitchcock CA. Cytochrome P-450-dependent 14 alpha-sterol demethylase of Candida albicans and its interaction with azole antifungals. Biochem Soc Trans. 1991;19(3):782–787. | ||
Joseph-Horne T, Hollomon DW. Molecular mechanisms of azole resistance in fungi. FEMS Microbiol Lett. 1997;149(2):141–149. | ||
Heimark L, Shipkova P, Greene J, et al. Mechanism of azole antifungal activity as determined by liquid chromatographic/mass spectrometric monitoring of ergosterol biosynthesis. J Mass Spectrom. 2002;37(3):265–269. | ||
Cleveland AA, Farley MM, Harrison LH, et al. Changes in incidence and antifungal drug resistance in candidemia: results from population-based laboratory surveillance in Atlanta and Baltimore, 2008–2011. Clin Infect Dis. 2012;55(10):1352–1361. | ||
Pfaller MA, Rhomberg PR, Messer SA, Jones RN, Castanheira M. Isavuconazole, micafungin, and 8 comparator antifungal agents’ susceptibility profiles for common and uncommon opportunistic fungi collected in 2013: temporal analysis of antifungal drug resistance using CLSI species-specific clinical breakpoints and proposed epidemiological cutoff values. Diagn Microbiol Infect Dis. 2015;82(4):303–313. | ||
Lockhart SR, Etienne KA, Vallabhaneni S, et al. Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin Infect Dis. 2017;64(2):134–140. | ||
Marichal P, Gorrens J, Coene MC, Le Jeune L, Vanden Bossche H. Origin of differences in susceptibility of Candida krusei to azole antifungal agents. Mycoses. 1995;38(3–4):111–117. | ||
Pfaller MA, Diekema DJ, Gibbs DL, et al; Global Antifungal Surveillance Group. Candida krusei, a multidrug-resistant opportunistic fungal pathogen: geographic and temporal trends from the ARTEMIS DISK Antifungal Surveillance Program, 2001 to 2005. J Clin Microbiol. 2008;46(2):515–521. | ||
MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother. 2005;49(5):1745–1752. | ||
Hoot SJ, Smith AR, Brown RP, White TC. An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob Agents Chemother. 2011;55(2):940–942. | ||
Heilmann CJ, Schneider S, Barker KS, Rogers PD, Morschhäuser J. An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob Agents Chemother. 2010;54(1):353–359. | ||
Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, Rogers PD. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell. 2008;7(7):1180–1190. | ||
Flowers SA, Barker KS, Berkow EL, et al. Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot Cell. 2012;11(10):1289–1299. | ||
Souza AC, Fuchs BB, Pinhati HM, et al. Candida parapsilosis resistance to fluconazole: molecular mechanisms and in vivo impact in infected Galleria mellonella larvae. Antimicrob Agents Chemother. 2015;59(10):6581–6587. | ||
Berkow EL, Manigaba K, Parker JE, Barker KS, Kelly SL, Rogers PD. Multidrug transporters and alterations in sterol biosynthesis contribute to azole antifungal resistance in Candida parapsilosis. Antimicrob Agents Chemother. 2015;59(10):5942–5950. | ||
Jiang C, Dong D, Yu B, et al. Mechanisms of azole resistance in 52 clinical isolates of Candida tropicalis in China. J Antimicrob Chemother. 2013;68(4):778–785. | ||
Redding SW, Kirkpatrick WR, Saville S, et al. Multiple patterns of resistance to fluconazole in Candida glabrata isolates from a patient with oropharyngeal candidiasis receiving head and neck radiation. J Clin Microbiol. 2003;41(2):619–622. | ||
vanden Bossche H, Marichal P, Odds FC, Le Jeune L, Coene MC. Characterization of an azole-resistant Candida glabrata isolate. Antimicrob Agents Chemother. 1992;36(12):2602–2610. | ||
Tavakoli M, Zaini F, Kordbacheh M, Safara M, Raoofian R, Heidari M. Upregulation of the ERG11 gene in Candida krusei by azoles. Daru. 2010;18(4):276–280. | ||
Warrilow AG, Martel CM, Parker JE, et al. Azole binding properties of Candida albicans sterol 14-alpha demethylase (CaCYP51). Antimicrob Agents Chemother. 2010;54(10):4235–4245. | ||
Sanglard D, Ischer F, Koymans L, Bille J. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother. 1998;42(2):241–253. | ||
Morio F, Loge C, Besse B, Hennequin C, Le Pape P. Screening for amino acid substitutions in the Candida albicans Erg11 protein of azole-susceptible and azole-resistant clinical isolates: new substitutions and a review of the literature. Diagn Microbiol Infect Dis. 2010;66(4):373–384. | ||
Marichal P, Koymans L, Willemsens S, et al. Contribution of mutations in the cytochrome P450 14alpha-demethylase (Erg11p, Cyp51p) to azole resistance in Candida albicans. Microbiology. 1999;145 (Pt 10):2701–2713. | ||
Lamb DC, Kelly DE, White TC, Kelly SL. The R467K amino acid substitution in Candida albicans sterol 14alpha-demethylase causes drug resistance through reduced affinity. Antimicrob Agents Chemother. 2000;44(1):63–67. | ||
Chau AS, Mendrick CA, Sabatelli FJ, Loebenberg D, McNicholas PM. Application of real-time quantitative PCR to molecular analysis of Candida albicans strains exhibiting reduced susceptibility to azoles. Antimicrob Agents Chemother. 2004;48(6):2124–2131. | ||
Xiang MJ, Liu JY, Ni PH, et al. Erg11 mutations associated with azole resistance in clinical isolates of Candida albicans. FEMS Yeast Res. 2013;13(4):386–393. | ||
Perea S, López-Ribot JL, Kirkpatrick WR, et al. Prevalence of molecular mechanisms of resistance to azole antifungal agents in Candida albicans strains displaying high-level fluconazole resistance isolated from human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2001;45(10):2676–2684. | ||
Xu Y, Chen L, Li C. Susceptibility of clinical isolates of Candida species to fluconazole and detection of Candida albicans ERG11 mutations. J Antimicrob Chemother. 2008;61(4):798–804. | ||
Goldman GH, da Silva Ferreira ME, dos Reis Marques E, et al. Evaluation of fluconazole resistance mechanisms in Candida albicans clinical isolates from HIV-infected patients in Brazil. Diagn Microbiol Infect Dis. 2004;50(1):25–32. | ||
Flowers SA, Colón B, Whaley SG, Schuler MA, Rogers PD. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother. 2015;59(1):450–460. | ||
Kakeya H, Miyazaki Y, Miyazaki H, Nyswaner K, Grimberg B, Bennett JE. Genetic analysis of azole resistance in the Darlington strain of Candida albicans. Antimicrob Agents Chemother. 2000;44(11):2985–2990. | ||
Grossman NT, Pham CD, Cleveland AA, Lockhart SR. Molecular mechanisms of fluconazole resistance in Candida parapsilosis isolates from a U.S. surveillance system. Antimicrob Agents Chemother. 2015;59(2):1030–1037. | ||
Tan J, Zhang J, Chen W, et al. The A395T mutation in ERG11 gene confers fluconazole resistance in Candida tropicalis causing candidemia. Mycopathologia. 2015;179(3–4):213–218. | ||
Eddouzi J, Parker JE, Vale-Silva LA, et al. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother. 2013;57(7):3182–3193. | ||
Hull CM, Parker JE, Bader O, et al. Facultative sterol uptake in an ergosterol-deficient clinical isolate of Candida glabrata harboring a missense mutation in ERG11 and exhibiting cross-resistance to azoles and amphotericin B. Antimicrob Agents Chemother. 2012;56(8):4223–4232. | ||
Orozco AS, Higginbotham LM, Hitchcock CA, et al. Mechanism of fluconazole resistance in Candida krusei. Antimicrob Agents Chemother. 1998;42(10):2645–2649. | ||
Fukuoka T, Johnston DA, Winslow CA, et al. Genetic basis for differential activities of fluconazole and voriconazole against Candida krusei. Antimicrob Agents Chemother. 2003;47(4):1213–1219. | ||
Venkateswarlu K, Denning DW, Kelly SL. Inhibition and interaction of cytochrome P450 of Candida krusei with azole antifungal drugs. J Med Vet Mycol. 1997;35(1):19–25. | ||
Silva DB, Rodrigues LM, Almeida AA, Oliveira KM, Grisolia AB. Novel point mutations in the ERG11 gene in clinical isolates of azole resistant Candida species. Mem Inst Oswaldo Cruz. 2016;111(3):192–199. | ||
Ricardo E, Miranda IM, Faria-Ramos I, Silva RM, Rodrigues AG, Pina-Vaz C. In vivo and in vitro acquisition of resistance to voriconazole by Candida krusei. Antimicrob Agents Chemother. 2014;58(8):4604–4611. | ||
Kelly SL, Lamb DC, Kelly DE, et al. Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol delta5,6-desaturation. FEBS Lett. 1997;400(1):80–82. | ||
Martel CM, Parker JE, Bader O, et al. Identification and characterization of four azole-resistant erg3 mutants of Candida albicans. Antimicrob Agents Chemother. 2010;54(11):4527–4533. | ||
Miyazaki T, Miyazaki Y, Izumikawa K, et al. Fluconazole treatment is effective against a Candida albicans erg3/erg3 mutant in vivo despite in vitro resistance. Antimicrob Agents Chemother. 2006;50(2):580–586. | ||
Sanglard D, Ischer F, Parkinson T, Falconer D, Bille J. Candida albicans mutations in the ergosterol biosynthetic pathway and resistance to several antifungal agents. Antimicrob Agents Chemother. 2003; 47(8):2404–2412. | ||
Chau AS, Gurnani M, Hawkinson R, Laverdiere M, Cacciapuoti A, McNicholas PM. Inactivation of sterol Delta5,6-desaturase attenuates virulence in Candida albicans. Antimicrob Agents Chemother. 2005;49(9):3646–3651. | ||
Miyazaki Y, Geber A, Miyazaki H, et al. Cloning, sequencing, expression and allelic sequence diversity of ERG3 (C-5 sterol desaturase gene) in Candida albicans. Gene. 1999;236(1):43–51. | ||
Rees DC, Johnson E, Lewinson O. ABC transporters: the power to change. Nat Rev Mol Cell Biol. 2009;10(3):218–227. | ||
Prasad R, Goffeau A. Yeast ATP-binding cassette transporters conferring multidrug resistance. Annu Rev Microbiol. 2012;66:39–63. | ||
Tsao S, Rahkhoodaee F, Raymond M. Relative contributions of the Candida albicans ABC transporters Cdr1p and Cdr2p to clinical azole resistance. Antimicrob Agents Chemother. 2009;53(4):1344–1352. | ||
Morschhäuser J, Michel S, Staib P. Sequential gene disruption in Candida albicans by FLP-mediated site-specific recombination. Mol Microbiol. 1999;32(3):547–556. | ||
Sanglard D, Ischer F, Monod M, Bille J. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob Agents Chemother. 1996;40(10):2300–2305. | ||
Schillig R, Morschhäuser J. Analysis of a fungus-specific transcription factor family, the Candida albicans zinc cluster proteins, by artificial activation. Mol Microbiol. 2013;89(5):1003–1017. | ||
Coste AT, Crittin J, Bauser C, Rohde B, Sanglard D. Functional analysis of cis- and trans-acting elements of the Candida albicans CDR2 promoter with a novel promoter reporter system. Eukaryot Cell. 2009;8(8):1250–1267. | ||
Siikala E, Rautemaa R, Richardson M, Saxen H, Bowyer P, Sanglard D. Persistent Candida albicans colonization and molecular mechanisms of azole resistance in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) patients. J Antimicrob Chemother. 2010;65(12):2505–2513. | ||
Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, Rogers PD. Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot Cell. 2011;10(3):373–383. | ||
Katiyar SK, Edlind TD. Identification and expression of multidrug resistance-related ABC transporter genes in Candida krusei. Med Mycol. 2001;39(1):109–116. | ||
Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, Bille J. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother. 1999;43(11):2753–2765. | ||
Torelli R, Posteraro B, Ferrari S, et al. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributing to the CgPDR1-dependent azole resistance of Candida glabrata. Mol Microbiol. 2008;68(1):186–201. | ||
Lamping E, Ranchod A, Nakamura K, et al. Abc1p is a multidrug efflux transporter that tips the balance in favor of innate azole resistance in Candida krusei. Antimicrob Agents Chemother. 2009;53(2):354–369. | ||
Barchiesi F, Calabrese D, Sanglard D, et al. Experimental induction of fluconazole resistance in Candida tropicalis ATCC 750. Antimicrob Agents Chemother. 2000;44(6):1578–1584. | ||
Vandeputte P, Larcher G, Bergès T, Renier G, Chabasse D, Bouchara JP. Mechanisms of azole resistance in a clinical isolate of Candida tropicalis. Antimicrob Agents Chemother. 2005;49(11):4608–4615. | ||
Pao SS, Paulsen IT, Saier MH Jr. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62(1):1–34. | ||
Gaur M, Puri N, Manoharlal R, et al. MFS transportome of the human pathogenic yeast Candida albicans. BMC Genomics. 2008;9:579. | ||
Wirsching S, Michel S, Morschhäuser J. Targeted gene disruption in Candida albicans wild-type strains: the role of the MDR1 gene in fluconazole resistance of clinical Candida albicans isolates. Mol Microbiol. 2000;36(4):856–865. | ||
Morschhäuser J, Barker KS, Liu TT, BlaB-Warmuth J, Homayouni R, Rogers PD. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog. 2007;3(11):e164. | ||
Dunkel N, Blass J, Rogers PD, Morschhäuser J. Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol. 2008;69(4):827–840. | ||
Costa C, Ribeiro J, Miranda IM, et al. Clotrimazole drug resistance in Candida glabrata clinical isolates correlates with increased expression of the drug:H(+) antiporters CgAqr1, CgTpo1_1, CgTpo3, and CgQdr2. Front Microbiol. 2016;7:526. | ||
Chen X, Magee BB, Dawson D, Magee PT, Kumamoto CA. Chromosome 1 trisomy compromises the virulence of Candida albicans. Mol Microbiol. 2004;51(2):551–565. | ||
Rustchenko E. Chromosome instability in Candida albicans. FEMS Yeast Res. 2007;7(1):2–11. | ||
Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313(5785):367–370. | ||
Coste A, Selmecki A, Forche A, et al. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell. 2007;6(10):1889–1904. | ||
Seervai RN, Jones SK Jr, Hirakawa MP, Porman AM, Bennett RJ. Parasexuality and ploidy change in Candida tropicalis. Eukaryot Cell. 2013;12(12):1629–1640. | ||
Harrison BD, Hashemi J, Bibi M, et al. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol. 2014;12(3):e1001815. | ||
Poláková S, Blume C, Zárate JA, et al. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci U S A. 2009;106(8):2688–2693. | ||
Marichal P, Vanden Bossche H, Odds FC, et al. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob Agents Chemother. 1997;41(10):2229–2237. | ||
Alarco AM, Raymond M. The bZip transcription factor Cap1p is involved in multidrug resistance and oxidative stress response in Candida albicans. J Bacteriol. 1999;181(3):700–708. | ||
Schubert S, Barker KS, Znaidi S, et al. Regulation of efflux pump expression and drug resistance by the transcription factors Mrr1, Upc2, and Cap1 in Candida albicans. Antimicrob Agents Chemother. 2011;55(5):2212–2223. | ||
Talibi D, Raymond M. Isolation of a putative Candida albicans transcriptional regulator involved in pleiotropic drug resistance by functional complementation of a pdr1 pdr3 mutation in Saccharomyces cerevisiae. J Bacteriol. 1999;181(1):231–240. | ||
Sanglard D, Ischer F, Marchetti O, Entenza J, Bille J. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Mol Microbiol. 2003;48(4):959–976. | ||
Cruz MC, Goldstein AL, Blankenship JR, et al. Calcineurin is essential for survival during membrane stress in Candida albicans. EMBO J. 2002;21(4):546–559. | ||
Mansfield BE, Oltean HN, Oliver BG, et al. Azole drugs are imported by facilitated diffusion in Candida albicans and other pathogenic fungi. PLoS Pathog. 2010;6(9):e1001126. | ||
Franz R, Kelly SL, Lamb DC, Kelly DE, Ruhnke M, Morschhäuser J. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob Agents Chemother. 1998;42(12):3065–3072. | ||
White TC, Pfaller MA, Rinaldi MG, Smith J, Redding SW. Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis. 1997;3 Suppl 1:S102–S109. | ||
Martínez M, López-Ribot JL, Kirkpatrick WR, et al. Heterogeneous mechanisms of azole resistance in Candida albicans clinical isolates from an HIV-infected patient on continuous fluconazole therapy for oropharyngeal candidosis. J Antimicrob Chemother. 2002;49(3):515–524. | ||
Ferrari S, Ischer F, Calabrese D, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009;5:e1000268. | ||
Sasse C, Dunkel N, Schäfer T, et al. The stepwise acquisition of fluconazole resistance mutations causes a gradual loss of fitness in Candida albicans. Mol Microbiol. 2012;86(3):539–556. | ||
MacCallum DM, Coste A, Ischer F, Jacobsen MD, Odds FC, Sanglard D. Genetic dissection of azole resistance mechanisms in Candida albicans and their validation in a mouse model of disseminated infection. Antimicrob Agents Chemother. 2010;54(4):1476–1483. | ||
Calvet HM, Yeaman MR, Filler SG. Reversible fluconazole resistance in Candida albicans: a potential in vitro model. Antimicrob Agents Chemother. 1997;41(3):535–539. | ||
Huang M, McClellan M, Berman J, Kao KC. Evolutionary dynamics of Candida albicans during in vitro evolution. Eukaryot Cell. 2011;10(11):1413–1421. | ||
Cowen LE. Predicting the emergence of resistance to antifungal drugs. FEMS Microbiol Lett. 2001;204(1):1–7. | ||
Anderson JB. Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol. 2005;3(7):547–556. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.