Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 12
FLT3 Inhibitors as Maintenance Therapy after Allogeneic Stem-Cell Transplantation
Authors Blackmon A, Aldoss I, Ball BJ
Received 18 May 2022
Accepted for publication 19 August 2022
Published 6 September 2022 Volume 2022:12 Pages 137—147
DOI https://doi.org/10.2147/BLCTT.S281252
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Wilson Gonsalves
Amanda Blackmon,* Ibrahim Aldoss,* Brian J Ball*
Department of Hematology and Hematopoietic Cell Transplantation, City of Hope National Medical Center, Duarte, CA, USA
*These authors contributed equally to this work
Correspondence: Brian J Ball, Division of Leukemia, Department of Hematology and HCT, City of Hope National Medical Center, 1500 E. Duarte Road, Duarte, CA, 91010, USA, Email [email protected]
Abstract: Mutations in the FLT3 gene are associated with poor prognosis in patients with AML, even after consolidation with allogeneic hematopoietic cell transplantation (alloHCT) in first remission. Treatment failure in FLT3-mutated AML is largely driven by excessive risk of relapse compared to other genetic subtypes, including in patients post-alloHCT. As a result, there is substantial interest in studying posttransplant maintenance therapy in FLT3-mutated AML as an approach to optimize disease control and improve long-term outcomes. Clinical trials utilizing posttransplant FLT3 inhibitors, such as sorafenib and midostaurin, have shown feasibility, safety, and encouraging posttransplant outcomes, and there are ongoing studies using newer-generation tyrosine-kinase inhibitors as posttransplant maintenance therapy. Here, we review the toxicities and efficacy of FLT3 inhibitors as posttransplant maintenance, recommendations on the use of FLT3 inhibitors by international consensus guidelines, and highlight key remaining questions.
Keywords: FLT3 inhibitor, posttransplantation, maintenance, stem-cell transplantation, midostaurin, sorafenib
Introduction
Acute myeloid leukemia is a biologically complex disease, often harboring a spectrum of diverse cytogenetic and mutational abnormalities, including the finding of different subclones in a single patient at diagnosis that could evolve during the disease course.1,2 This renders AML a challenging disease to target and eradicate, and a sole therapeutic targeted modality frequently lacks success in preventing relapse. At the time of diagnosis, patients with AML are stratified into favorable, intermediate, and adverse risk subgroups based on cytogenetic and mutational findings, which is employed to dictate initial therapy, identify eligibility for clinical studies, and guide postremission consolidation therapy.3
Mutations the FLT3 the gene are among the most common genetic aberrations, occurring in approximately 30% of patients with newly diagnosed AML.4,5 Activating mutations of the FLT3 gene occur most commonly as in-frame internal tandem duplications (ITDs) of between 3 and more than 100 amino acids located in the juxtamembrane region or as missense point mutations in the tyrosine-kinase domain (TKD), typically involving aspartic acid 835 residue. Of the two classes of mutations, FLT3-ITD abnormalities are more concerning, being associated with an increased risk of relapse and leading to the routine consideration of allogeneic transplantation in these patients.4,6 FLT3 encodes a membrane-bound receptor tyrosine kinase (RTK), which is expressed on normal hematopoietic progenitor cells involved in cell differentiation and proliferation.7,8 Upon binding of the FLT3 ligand, the receptor dimerizes and undergoes autophosphorylation, leading to activation of downstream pathways and promoting proliferation and preventing apoptosis.
FLT3-ITD mutations interfere with the regulatory function of the juxtamembrane region, and TKD mutations affect the activation loop and loss of autoinhibition, leading to constitutive activation of FLT3 (bypassing ligand activation), with downstream activation of pathways, including PI3K/Akt, MAPK, Ras, Mek, ERK, and STAT5 (Figure 1).9–15 The dependence of FLT3-mutated AML on FLT3 signaling provides a biological rationale for the development of FLT3-targeted therapies. Considerable effort has been dedicated to the development of potent and selective FLT3 TKIs, culminating in the approval of midostaurin in combination with upfront chemotherapy for newly diagnosed and gilteritinib for R/R FLT3-mutated AML.16,17 However, there remains an unacceptably high risk of relapse among those with AML harboring FLT3-ITD mutations, who undergo allogeneic stem-cell transplantation. The application of FLT3 inhibitors as maintenance therapy after allogeneic stem-cell transplantation may have the greatest potential for improving long-term survival for patients with FLT3 ITD–mutated AML. Here, we discuss the evidence, current recommendations, and remaining questions of FLT3-inhibitor maintenance after allogeneic hematopoietic cell transplantation (alloHCT).
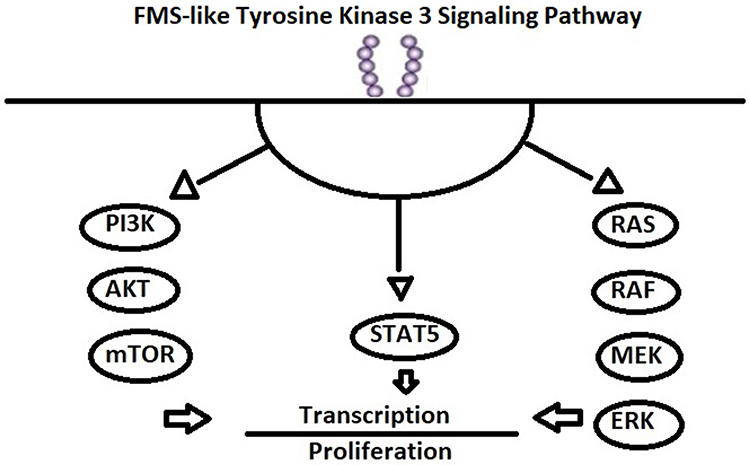 |
Figure 1 FLT3 signaling pathway. |
Prognostic Impact of FLT3-ITD AML
Although patients with newly diagnosed FLT3-mutated AML have comparable rates of complete remission at 65%, they are associated with a higher propensity for relapse, leading to shorter remission duration and survival, than their FLT3 wild-type (WT) counterparts.4,18,19 AlloHCT for patients in first complete remission has demonstrated an improvement in relapse-free survival (RFS) when compared to those receiving chemotherapy or autologous stem-cell transplant as consolidation.20,21 However, FLT3-ITD mutation remains a significant risk factor of relapse and death after alloHCT.22,23 A large international registry study conducted by the European Group for Blood and Marrow Transplantation (EBMT) of patients with de novo AML with normal cytogenetics who underwent myeloablative alloHCT from a matched unrelated donor demonstrated an increased incidence of relapse at 2 years (30% vs 16%, P=0.006) and decreased leukemia-free survival at 2 years (58% vs 71%, P=0.04) among those with FLT3-ITD mutations.22 The mutational burden or allelic ratio of FLT3-ITD mutations has important prognostic implications: those with high allelic ratio (>0.5, FLT3-ITDhigh), have worse outcomes with long-term disease-free survival of 20%–30% without allogeneic transplantation.22,24 While the data are less convincing for alloHCT in those with low allelic ratio (<0.5, FLT3-ITDlow), it is still common practice to perform allogeneic transplants following first remission in eligible patients, regardless of cytogenetic risk group, concurrent mutations, or negative minimal residual disease (MRD).25 Other factors impacting relapse risk and survival in patients with FLT3-ITD AML include the presence of increased ITD fragment length or measurable residual disease by FLT3 sequencing prior to alloHCT.26–29 FLT3-TKD mutations may have less influence on overall survival (OS) and disease-free survival; however, there is a suggestion this may differ among ethnicities, with more favorable outcomes in Asian patients compared to Caucasians.30,31
Development of FLT3 Inhibitors
Despite a strong biological rationale for inhibiting the FLT3 pathway in FLT3 ITD–mutated AML, translating these findings into the successful development of FLT3 inhibitors in the clinic has been challenging. First-generation FLT3 TKIs (sorafenib, sunitinib, lestaurtinib, and midostaurin) have lower specificity for FLT3 with more off-target activity. These agents inhibit various kinases, including Kit, VEGFR, PDGFR, and Ras/RAF, among others.32 The relatively broad inhibition can lead to toxicity, including fatigue, GI toxicity, HTN, and bleeding. In many cases, these inhibitors were initially developed for indications other than FLT3 inhibition. Sorafenib was originally developed as a B-Raf and multikinase inhibitor in renal and hepatocellular carcinoma before demonstrating potent inhibitory activity in FLT3-ITD AML.33 Midostaurin was initially developed as a protein kinase C inhibitor and was subsequently found to have inhibitory activity against multiple tyrosine kinases, including FLT3.34 These agents have demonstrated modest activity as single agents in the R/R AML setting. In phase I studies of sorafenib in R/R AML, there were transient reductions in blast counts; however, most patients had stable disease, and increase in dose was limited by toxicity.35,36 Lestaurtinib phase I/II data showed activity in 14 heavily pretreated patients with AML, with five having blast reduction and improvement in normal hematopoiesis; however, no patients had a CR.37 Similarly, midostaurin demonstrated blast reduction in phase I/II studies in R/R AML; however, no patients attained a CR, suggesting that midostaurin may not be sufficient as monotherapy.38,39
The first-generation FLT3 inhibitors have had more success when used in combination with induction and consolidation chemotherapy. The German SORAML trial was a phase II study evaluating sorafenib or placebo in combination with induction chemotherapy (7+3).40 The sorafenib arm had improved EFS (21 vs 9 months); however, there were significantly more grade 3 AEs, particularly fever, diarrhea, bleeding, cardiac events, and skin rash. The approval of midostaurin followed the multicenter phase III RATIFY trials, which showed a survival benefit when adding midostaurin to induction chemotherapy. In this study, younger patients with newly diagnosed FLT3-TKD or FLT3 ITD–mutated AML randomized to midostaurin in combination with induction and consolidation chemotherapy had significantly longer median OS (74.7 vs 25.6 months, P=0.009) when compared to placebo.17
Second-generation TKIs have higher potency and less off-target activity, with a lower half-maximal inhibitory concentration (IC50). These include gilteritinib, quizartinib, and crenolanib.32 Second-generation inhibitors have demonstrated greater single-agent activity in the R/R setting than salvage chemotherapy.16,41,42 Quizartinib demonstrated significantly longer survival (median OS 6.2 months vs 4.7 months, P=0.02) in a phase III study of patients with R/R AML with FLT3-ITD mutations when compared to patients receiving salvage chemotherapy.43 However, treatment with quizartinib was limited by the frequent emergence of resistance-conferring FLT3-TKD mutations at the time of relapse.44 Gilteritinib is an oral highly potent pan-FLT3 inhibitor and the only FLT3 inhibitor currently approved for patients with R/R AML harboring FLT3 mutations. Approval was based on the seminal phase III ADMIRAL trial, which demonstrated higher complete remission rates with full or partial hematologic recovery (34% vs 15%), prolonged survival (median OS 9.3 months vs 5.6 months, P<0.001), and a higher rate of alloHCT (26% vs 15%) among patients with FLT3-mutated R/R AML randomized to gilteritinib when compared to salvage chemotherapy.16 Crenolanib is a promising pan-FLT3 (ITD and TKD) inhibitor currently in phase III studies.
FLT3 inhibitors are further classified as type I or type II depending on their mechanism of action Type I inhibitors (midostaurin, lestaurtinib, crenolanib, and gilteritinib) work by competitive inhibition of the ATP-binding site of the intracellular TKD. Type II inhibitors (sorafenib, quizartinib, and ponatinib) interact with the inactive receptor at a hydrophobic region adjacent to the ATP-binding site, preventing activation.32 Type I inhibitors have activity against ITD and TKD mutations, while type II inhibitors have less activity in TKD mutations, as these mutations often lead to the active conformation of the receptor. Furthermore, the acquisition of TKD mutations is a major mechanism of secondary resistance for patients with ITD mutations receiving type II inhibitors.45–47We discuss sorafenib and midostaurin more in depth as they have been evaluated in the posttransplantation setting.
Sorafenib
Sorafenib was one of the earliest agents discovered to have activity in FLT3-mutated AML. It is an oral multikinase inhibitor approved for the treatment of advanced thyroid, hepatocellular, and renal cell carcinoma with potent inhibitory activity against RAF kinase, VEGF receptors, PDGF receptors, c-Kit, Ret kinase, and wild-type and ITD-mutated FLT3.48 In addition to targeting FLT3, sorafenib may enhance alloimmunity following transplantation. Treatment with sorafenib monotherapy leads to more durable responses among relapsed or refractory FLT3 ITD–mutated AML patients with a prior allogeneic stem-cell transplantation compared to conventional therapy.43,44,49 In a murine allogeneic stem-cell transplant model, maintenance sorafenib led to an increase in donor T cells and an increase in clinical graft-versus-host disease (GVHD) in T cell–replete donors.50 Sorafenib enhances the graft-versus-leukemia effect by inducing IL15 production in FLT3 ITD–mutated AML cells, which promotes the cytotoxicity and longevity of donor CD8+ T cells.51 Several retrospective studies have shown that compassionate use of sorafenib after alloHCT may reduce relapse and improve survival.52–56 In a phase I study of 22 patients, sorafenib given for 12 28-day cycles posttransplant was safe, and the maximum tolerated dose was found to be 400 mg twice daily. In this study, there were no episodes of significant acute GVHD after starting sorafenib and a comparable incidence of chronic GVHD to historical outcomes. Among the 19 patients in a conventional complete remission (CR1/CR2) before alloHCT, progression-free survival was 86% and OS at 2 years was 78%.57 A German randomized phase II study (SORMAIN trial) was conducted to evaluate sorafenib maintenance vs placebo in TKI-naïve patients with FLT3 ITD–mutated AML in CR after dose-reduced or myeloablative alloHCT from a 9/10 or 10/10 HLA-matched unrelated or sibling donor.58 Sorafenib was started at day +60 to day +100 at 400 mg daily, with the dose escalated every 2 weeks to a maximum of 800 mg daily and to be given continuously for 24 months until occurrence of relapse or intolerable toxicity. The trial was discontinued early due to slow accrual, but still met the primary end point by demonstrating longer RFS among patients receiving sorafenib vs placebo (median RFS: 30.9 months vs not reached, HR 0.39, 95% CI 0.18–0.85). The sorafenib arm also had superior 24-month RFS (85% vs 53%, HR 0.256, 95% CI 0.1–0.65) and OS (91% vs 66%, HR 0.241, 95% CI 0.08–0.74) compared to the placebo arm. Sorafenib was generally well tolerated, with dose reductions occurring in 49% and treatment discontinuation due to an adverse event occurring in 22% of patients in the sorafenib arm. The most common grade ≥3 adverse event was acute and/or chronic GVHD, which occurred in 32 of 42 patients (77%) in the sorafenib arm and 23 of 39 patients (60%) in the placebo arm. Other grade ≥3 adverse events that occurring at a higher frequency in the sorafenib than placebo arm included infections, GI toxicity, cardiotoxicity, renal insufficiency, skin toxicity, and electrolyte alterations.
A randomized phase III study compared maintenance therapy with sorafenib or placebo in patients aged 18–60 years with FLT3 ITD–mutated AML in composite CR after alloHCT.59 Patients enrolled received either a matched sibling donor, matched unrelated donor, or a haploidentical transplant with modified busulfan and cyclophosphamide myeloablative conditioning. Sorafenib was dosed at 400 mg twice daily from day 31–60 until day 180, but 60% of patients required dose reduction and the median dose was 200 mg twice daily. The study met the primary end point by demonstrating a reduced risk of relapse at 1 year compared to placebo (1-year cumulative incidence of relapse [CIR] 7% vs 24.5%, HR 0.25, 95% CI 0.11–0.57; P=0.001). Treatment with sorafenib also led to superior 2-year CIR and OS without adversely impacting 2-year NRM or rates of acute and chronic GVHD compared to placebo. Notably, there was no difference in rates of acute and chronic GVHD between treatment arms. Grade 3/4 adverse events occurring more commonly in the sorafenib arm than control included hematologic, skin-related, and gastrointestinal. Sorafenib required a dose modification due to adverse events in 60% of patients, and the median duration of treatment was 134 days. Treatment with sorafenib did not seem to increase the risk of acquiring FLT3-TKD mutations, which occurred in only one of eleven relapsing patients.
Midostaurin
Midostaurin is a potent oral multikinase inhibitor of FLT3 TKD and ITD, as well as Kit, VEGF, Ret, Syk, and PDGFR.60 In the RATIFY trial, patients did not continue midostaurin maintenance therapy after alloHCT. However, among those undergoing alloHCT, midostaurin in combination with chemotherapy led to a significant decrease in CIR (HR 0.47, P=0.02). MRD testing was not performed in this study; however, the improved posttransplant outcomes in the midostaurin arm suggest that the FLT3 inhibitor with chemotherapy led to deeper remission prior to alloHCT.61
In contrast to the RATIFY trial, the phase II AMLSG 16–10 trial included maintenance midostaurin after alloHCT following induction and consolidation chemotherapy with midostaurin for younger patients with FLT3 ITD–mutated AML.62 Posttransplant midostaurin was administered at 50 mg twice daily between 30 and 100 days after transplantation for 1 year. The trial demonstrated improved 1-year EFS compared to that of historical controls with FLT3 ITD–mutated AML (HR 0.58, 95% CI 0.48–0.7; P<0.001]. Additionally, a landmark analysis in patients proceeding to alloHCT in CR1 who were event-free at day 100 after transplant demonstrated significantly longer EFS and OS among patients receiving midostaurin maintenance than the historical control patients who did not. The median time on maintenance therapy was 9 months after alloHCT, and early discontinuation from nonrelapse causes occurred in 86% of patients, most commonly due to nausea/vomiting, infection, cytopenia (mostly thrombocytopenia), elevated liver enzymes, pain, allergy, and dermatologic adverse events.
xThe RADIUS trial was a randomized phase II study comparing maintenance therapy with or without midostaurin 50 mg twice daily for 1 year after alloHCT for patients with FLT3 ITD–mutated AML in CR1.63 The trial showed no significant difference between treatment arms in RFS or OS. However, the trial was unable to detect differences between treatment arms due to being inadequately powered. (Table 1). The median midostaurin exposure was 10.5 months, and a majority of these patients had not had previous midostaurin with induction. Treatment discontinuation due to adverse events was less likely than AMLSG 16–10, at 27%, which may have been due to more stringent selection criteria. Notably, the addition of midostaurin did not increase rates of acute or chronic GVHD compared to standard of care (Table 1).
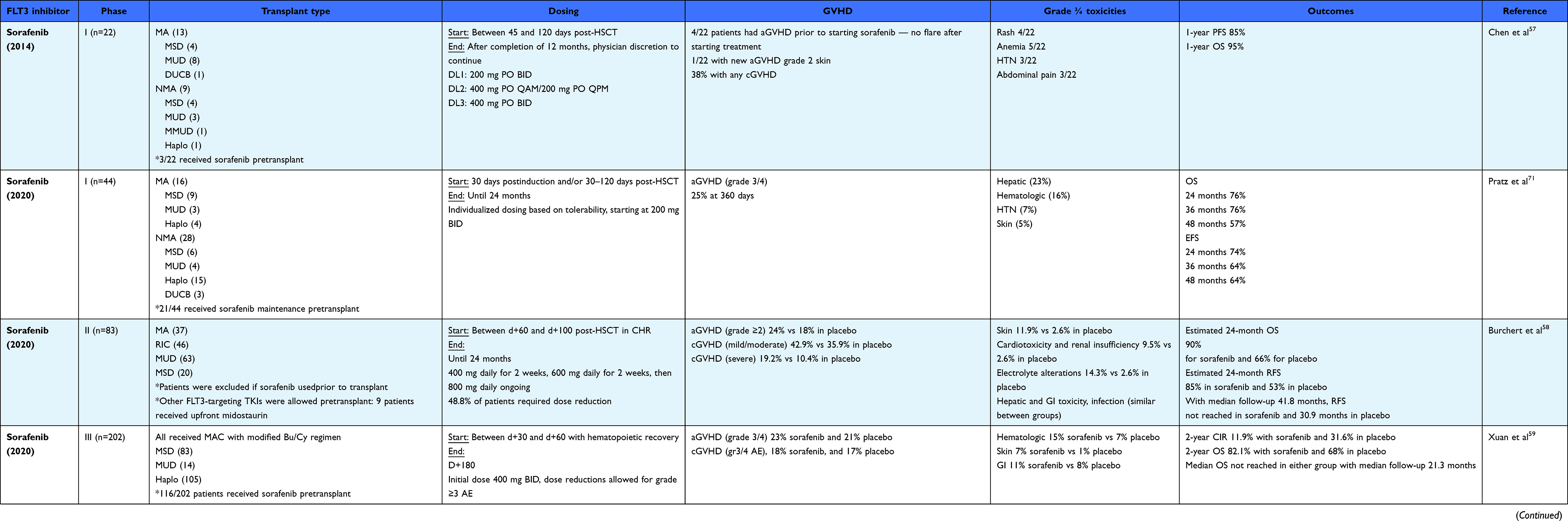 |
Table 1 Clinical trials evaluating FLT3 inhibitor maintenance after allogeneic hematopoietic cell transplantation |
Next-Generation FLT3-Inhibitor Posttransplant Maintenance
Based on promising results of early-phase trials, current phase IIII trials are evaluating next-generation FLT3 inhibitors vs placebo (quizartinib, QUANTUM-First trial) or midostaurin (gilteritinib, HOVON/AML-SG; crenolanib) in combination with induction and consolidation chemotherapy. Additionally, in several clinical trials evaluating FLT3 inhibitors in R/R AML, FLT3-inhibitor treatment was continued after alloHCT. In the ADMIRAL trial comparing gilteritinib to salvage chemotherapy in R/R FLT3-mutated AML, ~20% (49 of 247) of patients on the gilteritinib arm and ~10% (14 of 124) of patients on salvage chemotherapy arm were alive for ≥2 years.64 Of the patients who survived, 18 of 49 underwent alloHCT and 16 continued gilteritinib posttransplant.64 Gilteritinib was well tolerated, with the most common adverse event being elevated transaminases. Similarly, in the QUANTUM-R trial comparing quizartinib to salvage chemotherapy in R/R AML, more patients were able to undergo alloHCT on the quizartinib arm (32% [n=78] vs 11% [n=14]), and 62% of these patients continued quizartinib posttransplantation.65 There were no new safety signals in those continuing quizartinib posttransplant compared to the entire quizartinib arm.
BMT CTN 1506 is a randomized, double-blinded phase III trial evaluating posttransplant maintenance with gilteritinib vs placebo in patients with FLT3 ITD–mutated AML in CR1 that is currently enrolling patients (NCT02997202). Gilteritinib will be administered between days 30 to 90 after alloHCT at 120 mg daily for 2 years.66 The study includes a deep-sequencing assay that is highly sensitive and specific for FLT3-ITD mutations for MRD testing that will determine patients most likely to benefit from maintenance therapy.66,67 A key objective of BMT CTN 1506 is defining the impact of MRD on outcomes with post-HCT maintenance.
Key Remaining Questions
Is FLT3-Inhibitor Maintenance Necessary for All FLT3 ITD–Mutated AML Patients Undergoing AlloHCT?
The detection of MRD at the time of allogeneic stem-cell transplantation is well established as one of the most significant factors in determining relapse risk.68,69 However, patients with MRD-negative disease prior to transplantation have a lower risk of relapse, and thus the risks of treatment toxicity need to be considered in addition to potential benefit from FLT3-inhibitor treatment. Additionally, mutations in FLT3 alone do not result in leukemogenesis, and preclinical studies have identified leukemic stem cells that do not harbor FLT3 mutations, suggesting this may not always be enough for higher-risk patients.70 In the SORMAIN trial, patients with MRD negativity prior to transplantation benefited from posttransplant maintenance with sorafenib; however, MRD- positive patients derived the greatest benefit.58 In the phase III trial of Xuan et al, both MRD-positive and -negative patients after alloHCT randomized to sorafenib had a reduced risk when compared to placebo on subgroup analysis.59 These results suggest that all patients, regardless of MRD status, should receive FLT3 inhibitors for posttransplant maintenance. However, midostaurin was only recently approved as frontline therapy, and most patients enrolled in the SORMAIN, RADIUS, or the phase III trial of Xuan et al did not receive prior FLT3-inhibitor treatment with chemotherapy.66 This is notable, since patients randomized to midostaurin with induction and consolidation that proceeded to alloHCT in the RATIFY trial also had a reduction in relapse risk, likely as a result of MRD eradication.17 The phase III BMT-CTN 1506 study with gilteritinib vs placebo as posttransplant maintenance will be essential in determining the role of FLT3-inhibitor maintenance posttransplant for patients who have previously received FLT3 inhibitors in combination with chemotherapy. Additionally, the BMT-CTN study includes a novel highly sensitive and specific FLT3-ITD MRD assay to further determine patients most likely to benefit from FLT3-inhibitor maintenance therapy after alloHCT.
What is the Optimal Duration of FLT3-Inhibitor Therapy?
Although EBMT guidelines recommend 2 years of maintenance therapy, the duration of FLT3-inhibitor treatment is often limited by tolerability. In the SORMAIN trial, ~50% of patients required dose reduction, 21% of patients discontinued treatment due to toxicity, and the median duration of treatment was ~9 months.58 In the phase III trial of Xuan et al, the intended duration of sorafenib maintenance was shorter until day 180 to reduce the risk of developing drug resistance.59 Although the duration of treatment was half as long in the phase III study, the rates of relapse in each study seemed similar (SORMAIN — 2-year RFS, 85%; Xuan et al — 2-year CIR 12%).58,59 Therefore, further studies are needed to determine the optimal duration of FLT3-inhibitor maintenance. Pratz et al demonstrated that individualized dosing of sorafenib titrated to tolerability allowed for a longer duration of treatment of around ~22 months without impacting levels of FLT3 inhibition on pharmacodynamic studies.71 Next-generation FLT3 inhibitors, such as gilteritinib, crenolanib, and quizartinib, are more selective and reduce off-target toxicities. This may enhance tolerability and enable a longer duration of FLT3-inhibitor treatment after alloHCT.
What is the Optimal FLT3 Inhibitor for Post-AlloHCT Maintenance?
Currently, only sorafenib has demonstrated a decreased risk of relapse in phase II and phase III trials of maintenance therapy after transplant. The efficacy of sorafenib in the posttransplant setting may be due to FLT3-ITD targeting and other FLT3-independent immunomechanisms. It is not known if other more potent and selective FLT3 inhibitors will have similar benefit in randomized prospective trials. The RADIUS trial did not show an improvement in median RFS or OS for patients randomized to posttransplant maintenance with midostaurin, but the study was not adequately powered. Enrollment in clinical trials evaluating FLT3 inhibitors is encouraged. For patients unable to enroll in a clinical trial, sorafenib would be the treatment of choice, but may be more difficult to obtain than other current FLT3 inhibitors FDA-approved for AML, such as midostaurin and gilteritinib. Another consideration for choosing an FLT3 inhibitor would the co-occurrence of a, FLT3 ITD and FLTTKD mutation, which would be associated with resistance to sorafenib, but not other type 1 inhibitors, such as gilteritinib, crenolanib, and midostaurin. Lastly, as we have more patients who now are failing sequential FLT3 inhibitors, this will be important in choosing the best posttransplant maintenance option.
Current Recommendations
As the data on FLT3-mutated AML continues to expand, the guidelines are evolving. Currently, the European Society for Blood and Marrow Transplantation (EBMT) recommends transplantation in CR1 in patients with FLT3-ITD AML. In patients with an NPM1-mutated disease and low FLT3-ITD allelic ratio disease, several European cooperative groups do not recommend allogeneic transplant in CR1, while the National Comprehensive Cancer Network (NCCN) does recommend transplant in CR1 for this group.72 The acute leukemia working party (ALWP) of EBMT also recommends posttransplant maintenance therapy in those without acute GVHD with sorafenib at a dose of 400 mg daily (or 800 mg daily in those with MRD-positive disease) to begin at the time of hematologic reconstitution and to be continued for a minimum of 2 years, depending on tolerance. The NCCN also recommends the use of sorafenib post-alloHCT in patients with a history of FLT3 ITD–mutated AML.58,59,73
Conclusion
As FLT3 ITD–mutant AML has a striking propensity for relapse after allogeneic stem-cell transplantation, FLT3 inhibitors as posttransplant maintenance represent a very promising approach for both reducing relapse risk and increasing long-term survival. In accordance with retrospective studies, prospective randomized clinical trials of the FLT3 inhibitor sorafenib as posttransplant maintenance have demonstrated a survival benefit. Additional randomized trials with more selective and less toxic FLT3 inhibitors as posttransplant maintenance are ongoing. Although the practice of administering posttransplant TKI maintenance therapy has gained popularity lately and is recommended by international consensus guidelines, challenges remain regarding management related to TKI toxicities, and many unanswered questions, including which TKI to administer, for how long, and to which patient population, remain.
Disclosure
Dr Brian Ball reports an NIH K12 grant and personal fees from Oncovalent outside the submitted work. Ibrahim Aldoss serves on advisory boards for Amgen, Pfizer, Kite pharmaceuticals, Jazz, AbbVie and Agios Pharmaceuticals, and is a consultant for Autolus Therapeutics, Pfizer and Amgen, research support by MacroGenics and Abbvie. The authors report no other conflicts of interest in this work.
References
1. Miles LA, Bowman RL, Merlinsky TR, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477–482. doi:10.1038/s41586-020-2864-x
2. Ball BJ, Hsu M, Devlin SM, et al. The prognosis and durable clearance of RAS mutations in patients with acute myeloid leukemia receiving induction chemotherapy. Am J Hematol. 2021;96(5):E171–E175. doi:10.1002/ajh.26146
3. Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi:10.1182/blood-2016-08-733196
4. Fröhling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100(13):4372–4380. doi:10.1182/blood-2002-05-1440
5. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. New Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
6. Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752–1759. doi:10.1182/blood.v98.6.1752
7. McKenna HJ, Stocking KL, Miller RE, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95(11):3489–3497. doi:10.1182/blood.V95.11.3489
8. Lyman SD, Jacobsen SEW. c-kit Ligand and Flt3 Ligand: stem/progenitor cell factors with overlapping yet distinct activities. Blood. 1998;91(4):1101–1134. doi:10.1182/blood.V91.4.1101
9. Zhang S, Fukuda S, Lee Y, et al. Essential role of signal transducer and activator of transcription (Stat)5a but not Stat5b for Flt3-dependent signaling. J Exp Med. 2000;192(5):719–728. doi:10.1084/jem.192.5.719
10. Dosil M, Wang S, Lemischka IR. Mitogenic signalling and substrate specificity of the Flk2/Flt3 receptor tyrosine kinase in fibroblasts and interleukin 3-dependent hematopoietic cells. Mol Cell Biol. 1993;13(10):6572–6585. doi:10.1128/mcb.13.10.6572-6585.1993
11. Rosnet O, Bühring HJ, deLapeyrière O, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematol. 1996;95(3–4):218–223. doi:10.1159/000203881
12. Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089–5102. doi:10.1182/blood-2010-04-261867
13. Pratz KW, Levis M. How I treat FLT3-mutated AML. Blood. 2017;129(5):565–571. doi:10.1182/blood-2016-09-693648
14. Kayser S, Levis MJ. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: clinical implications and limitations. Leuk Lymphoma. 2014;55(2):243–255. doi:10.3109/10428194.2013.800198
15. Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96(12):3907–3914. doi:10.1182/blood.V96.12.3907
16. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. New Engl J Med. 2019;381(18):1728–1740. doi:10.1056/NEJMoa1902688
17. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New Engl J Med. 2017;377(5):454–464. doi:10.1056/NEJMoa1614359
18. Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335. doi:10.1182/blood.v99.12.4326
19. Schiller GJ, Tuttle P, Desai P. Allogeneic hematopoietic stem cell transplantation in FLT3-ITD-positive acute myelogenous leukemia: the role for FLT3 tyrosine kinase inhibitors post-transplantation. Biol Blood Marrow Transplant. 2016;22(6):982–990. doi:10.1016/j.bbmt.2016.01.013
20. Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. doi:10.1056/NEJMoa074306
21. Schlenk RF, Kayser S, Bullinger L, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124(23):3441–3449. doi:10.1182/blood-2014-05-578070
22. Brunet S, Labopin M, Esteve J, et al. Impact of FLT3 internal tandem duplication on the outcome of related and unrelated hematopoietic transplantation for adult acute myeloid leukemia in first remission: a retrospective analysis. JCO. 2012;30(7):735–741. doi:10.1200/JCO.2011.36.9868
23. Deol A, Sengsayadeth S, Jagasia M, et al. FLT3 mutation increases relapse risk after allogeneic hematopoietic cell transplant for acute myeloid leukemia in first or second complete remission: a Center for International Blood and Marrow Transplant Research (CIBMTR) analysis. Blood. 2014;124(21):322. doi:10.1182/blood.V124.21.322.322
24. Ho AD, Schetelig J, Bochtler T, et al. Allogeneic stem cell transplantation improves survival in patients with acute myeloid leukemia characterized by a high allelic ratio of mutant FLT3-ITD. Biol Blood Marrow Transpl. 2016;22(3):462–469. doi:10.1016/j.bbmt.2015.10.023
25. Tien FM, Tsai CH, Huang SC, et al. Distinct clinico-biological features in AML patients with low allelic ratio FLT3-ITD: role of allogeneic stem cell transplantation in first remission. Bone Marrow Transplant. 2022;57(1):95–105. doi:10.1038/s41409-021-01454-z
26. Gaballa S, Saliba R, Oran B, et al. Relapse risk and survival in patients with FLT3 mutated acute myeloid leukemia undergoing stem cell transplantation. Am J Hematol. 2017;92(4):331–337. doi:10.1002/ajh.24632
27. Liu SB, Dong HJ, Bao XB, et al. Impact of FLT3-ITD length on prognosis of acute myeloid leukemia. Haematologica. 2019;104(1):e9–e12. doi:10.3324/haematol.2018.191809
28. Stirewalt DL, Kopecky KJ, Meshinchi S, et al. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood. 2006;107(9):3724–3726. doi:10.1182/blood-2005-08-3453
29. Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386–2392. doi:10.1182/blood-2009-03-209999
30. Li S, Wang X, Yang M, et al. FLT3-TKD in the prognosis of patients with acute myeloid leukemia: a meta-analysis. Soc Sci Res Network. 2021. doi:10.2139/ssrn.3804793
31. Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters—an analysis of 3082 patients. Blood. 2008;111(5):2527–2537. doi:10.1182/blood-2007-05-091215
32. Antar AI, Otrock ZK, Jabbour E, Mohty M, Bazarbachi A. FLT3 inhibitors in acute myeloid leukemia: ten frequently asked questions. Leukemia. 2020;34(3):682–696. doi:10.1038/s41375-019-0694-3
33. Zhang W, Konopleva M, Shi YX, et al. Sorafenib (BAY 43-9006) directly targets FLT3-ITD in acute myelogenous leukemia. Blood. 2006;108(11):255. doi:10.1182/blood.V108.11.255.255
34. Propper DJ, McDonald AC, Man A, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein Kinase C. J Clin Oncol. 2016. doi:10.1200/JCO.2001.19.5.1485
35. Pratz KW, Cho E, Karp J, et al. Phase I dose escalation trial of sorafenib as a single agent for adults with relapsed and refractory acute leukemias. JCO. 2009;27(15_suppl):7065. doi:10.1200/jco.2009.27.15_suppl.7065
36. Borthakur G, Kantarjian H, Ravandi F, et al. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011;96(1):62–68. doi:10.3324/haematol.2010.030452
37. Smith BD, Levis M, Beran M, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–3676. doi:10.1182/blood-2003-11-3775
38. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–4345. doi:10.1200/JCO.2010.28.9678
39. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. doi:10.1182/blood-2004-03-0891
40. Röllig C, Serve H, Hüttmann A. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. doi:10.1016/S1470-2045(15)00362-9
41. Garcia JS, Stone RM. The development of FLT3 inhibitors in acute myeloid leukemia. Hematol Oncol Clin North Am. 2017;31(4):663–680. doi:10.1016/j.hoc.2017.03.002
42. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2019;20(7):984–997. doi:10.1016/S1470-2045(19)30150-0
43. Cortes J, Perl AE, Döhner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2018;19(7):889–903. doi:10.1016/S1470-2045(18)30240-7
44. Smith CC, Paguirigan A, Jeschke GR, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood. 2017;130(1):48–58. doi:10.1182/blood-2016-04-711820
45. Wodicka LM, Ciceri P, Davis MI, et al. Activation state-dependent binding of small molecule kinase inhibitors: structural insights from biochemistry. Chem Biol. 2010;17(11):1241–1249. doi:10.1016/j.chembiol.2010.09.010
46. Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi:10.1038/nature11016
47. Ke YY, Singh VK, Coumar MS, et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci Rep. 2015;5:11702. doi:10.1038/srep11702
48. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
49. Metzelder SK, Schroeder T, Finck A, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26(11):2353–2359. doi:10.1038/leu.2012.105
50. Yokoyama H, Lundqvist A, Su S, Childs R. Toxic effects of sorafenib when given early after allogeneic hematopoietic stem cell transplantation. Blood. 2010;116(15):2858–2859. doi:10.1182/blood-2010-06-291104
51. Mathew NR, Baumgartner F, Braun L, et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat Med. 2018;24(3):282–291. doi:10.1038/nm.4484
52. Metzelder S, Wang Y, Wollmer E, et al. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009;113(26):6567–6571. doi:10.1182/blood-2009-03-208298
53. Antar A, Kharfan-Dabaja MA, Mahfouz R, Bazarbachi A. Sorafenib maintenance appears safe and improves clinical outcomes in FLT3-ITD acute myeloid leukemia after allogeneic hematopoietic cell transplantation. Clin Lymphoma Myeloma Leuk. 2015;15(5):298–302. doi:10.1016/j.clml.2014.12.005
54. Brunner AM, Li S, Fathi AT, et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol. 2016;175(3):496–504. doi:10.1111/bjh.14260
55. Battipaglia G, Ruggeri A, Massoud R, et al. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for Fms-like tyrosine kinase 3-mutated acute myeloid leukemia. Cancer. 2017;123(15):2867–2874. doi:10.1002/cncr.30680
56. Battipaglia G, Massoud R, Ahmed SO, et al. Efficacy and feasibility of sorafenib as a maintenance agent after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 mutated acute myeloid leukemia: an update. Clin Lymphoma Myeloma Leuk. 2019;19(8):506–508. doi:10.1016/j.clml.2019.04.004
57. Chen YB, Li S, Lane AA, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant. 2014;20(12):2042–2048. doi:10.1016/j.bbmt.2014.09.007
58. Burchert A, Bug G, Fritz LV, et al. Sorafenib maintenance after allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with FLT3 –internal tandem duplication mutation (SORMAIN). J Clin Oncol. 2020;38:2993–3002. doi:10.1200/JCO.19.03345
59. Xuan L, Wang Y, Huang F, et al. Sorafenib maintenance in patients with FLT3-ITD acute myeloid leukaemia undergoing allogeneic haematopoietic stem-cell transplantation: an open-label, multicentre, randomised phase 3 trial. Lancet Oncol. 2020;21(9):1201–1212. doi:10.1016/S1470-2045(20)30455-1
60. Manley PW, Caravatti G, Furet P, et al. Comparison of the kinase profile of midostaurin (Rydapt) with that of its predominant metabolites and the potential relevance of some newly identified targets to leukemia therapy. Biochemistry. 2018;57(38):5576–5590. doi:10.1021/acs.biochem.8b00727
61. Levis M, Shi W, Chang K, et al. FLT3 inhibitors added to induction therapy induce deeper remissions. Blood. 2020;135(1):75–78. doi:10.1182/blood.2019002180
62. Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133(8):840–851. doi:10.1182/blood-2018-08-869453
63. Maziarz RT, Levis M, Patnaik MM, et al. Midostaurin after allogeneic stem cell transplant in patients with FLT3-internal tandem duplication-positive acute myeloid leukemia. Bone Marrow Transplant. 2021;56(5):1180–1189. doi:10.1038/s41409-020-01153-1
64. Perl AE, Larson RA, Podoltsev NA, et al. Follow-up of patients with R/R FLT3-mutation-positive AML treated with gilteritinib in the phase 3 ADMIRAL trial. Blood. 2022. doi:10.1182/blood.2021011583
65. Ganguly S, Cortes JE, Krämer A, et al. Clinical outcomes in patients with FLT3-ITD-Mutated Relapsed/Refractory acute myelogenous leukemia undergoing hematopoietic stem cell transplantation after quizartinib or salvage chemotherapy in the QuANTUM-R trial. Transpl Cell Ther. 2021;27(2):153–162. doi:10.1016/j.bbmt.2020.09.036
66. Levis MJ, Chen YB, Hamadani M, Horowitz MM, Jones RJ. Blood and marrow transplant clinical trials network. FLT3 inhibitor maintenance after allogeneic transplantation: is a placebo-controlled, randomized trial ethical? J Clin Oncol. 2019;37(19):1604–1607. doi:10.1200/JCO.19.00321
67. Levis MJ, Perl AE, Altman JK, et al. A next-generation sequencing–based assay for minimal residual disease assessment in AML patients with FLT3-ITD mutations. Blood Adv. 2018;2(8):825–831. doi:10.1182/bloodadvances.2018015925
68. Ravandi F, Jorgensen J, Borthakur G, et al. Persistence of minimal residual disease assessed by multiparameter flow cytometry is highly prognostic in younger patients with acute myeloid leukemia. Cancer. 2017;123(3):426–435. doi:10.1002/cncr.30361
69. Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–1199. doi:10.1056/NEJMoa1716863
70. Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal Evolution of Pre-Leukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci Transl Med. 2012;4(149):149ra118. doi:10.1126/scitranslmed.3004315
71. Pratz KW, Rudek MA, Smith BD, et al. A prospective study of peritransplant sorafenib for patients with FLT3-ITD acute myeloid leukemia undergoing allogeneic transplantation. Biol Blood Marrow Transplant. 2019;26(2):300–306. doi:10.1016/j.bbmt.2019.09.023
72. Bazarbachi A, Bug G, Baron F, et al. Clinical practice recommendation on hematopoietic stem cell transplantation for acute myeloid leukemia patients with FLT3-internal tandem duplication: a position statement from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2020;105(6):1507–1516. doi:10.3324/haematol.2019.243410
73. Guidelines detail. NCCN. Available from: https://www.nccn.org/guidelines/guidelines-detail.
74. Döhner H, Weber D, Krzykalla J, et al. Midostaurin plus intensive chemotherapy for younger and older patients with AML and FLT3 internal tandem duplications. Blood Adv. 2022. doi:10.1182/bloodadvances.2022007223
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.