Back to Journals » Journal of Blood Medicine » Volume 13
Five Challenging Cases of Hereditary Antithrombin Deficiency Characterized by Thrombosis or Complicated Pregnancy
Authors Roberts JC ![]() , von Drygalski A
, von Drygalski A ![]() , Zhou JY, Rodgers GM
, Zhou JY, Rodgers GM ![]() , Ansteatt K
, Ansteatt K ![]() , Tarantino MD
, Tarantino MD
Received 11 March 2022
Accepted for publication 23 August 2022
Published 21 October 2022 Volume 2022:13 Pages 611—618
DOI https://doi.org/10.2147/JBM.S365996
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Jonathan C Roberts,1 Annette von Drygalski,2 Jenny Y Zhou,2 George M Rodgers,3 Kristin Ansteatt,1 Michael D Tarantino1
1Bleeding & Clotting Disorders Institute, University of Illinois College of Medicine – Peoria, Peoria, IL, USA; 2Hemophilia and Thrombosis Treatment Center, University of California at San Diego, San Diego, CA, USA; 3Division of Hematology and Hematologic Malignancies, Department of Internal Medicine, University of Utah School of Medicine, Salt Lake City, UT, USA
Correspondence: Michael D Tarantino, The Bleeding and Clotting Disorders Institute, University of Illinois College of Medicine – Peoria, 427 West Northmoor Road, Peoria, IL, 61614, USA, Tel +1 309-692-5337, Email [email protected]
Abstract: Hereditary antithrombin deficiency (ATD) is a rare autosomal dominant condition (estimated prevalence 1:500– 1:5000). Most ATD patients have AT activity levels 40– 60% of normal. We present treatments for venous thromboembolism (VTE) in five cases of hereditary ATD. Four patients had a family history of ATD, and one had a de novo mutation. The majority of patients had a VTE while on prophylactic anticoagulation. AT concentrate augmentation was added in these cases to treat the VTE and for prophylaxis against further episodes. Two patients had significant bleeding events, one had permanent physical sequelae. Two of the patients were pregnant. VTE is a common cause of morbidity and mortality during pregnancy. Although low molecular weight heparins are the drugs of choice during pregnancy, this treatment was inadequate in one patient (developed VTE on therapy). These cases emphasize the need to screen for ATD in young patients (< 55 years) presenting with VTE. AT augmentation therapy may be necessary in patients inadequately treated with conventional anticoagulants. Careful monitoring and individualized care are needed in ATD patients, especially those with demonstrated bleeding tendencies.
Keywords: clotting disorders, venous thromboembolism, anticoagulation, bleeding tendencies
Graphical Abstract:

Introduction
Antithrombin (AT) is a serine protease inhibitor and natural anticoagulant that acts primarily through inactivation of the clotting factors thrombin and factor Xa and less effectively through factors IXa and XIa.1,2 In addition, the anticoagulant, heparin, depends on AT for its activity. Heparin binds to and induces a thousand-fold enhancement of the anticoagulant effect of AT.3 In addition, AT inhibits the tissue factor (TF)-factor VIIa (FVIIa) complex (as does TF pathway inhibitor: TFPI) preventing FVIIa activation of factor IX and X.4,5
Inherited AT deficiency is a rare autosomal dominant condition that was first described in 1965.6 AT deficiency makes affected individuals susceptible to the development of thrombosis. It has been estimated that 50% of individuals with congenital AT deficiency will have experienced a venous thromboembolism (VTE) by age 50.7–9
AT deficiency is classified as either a quantitative (Type I) or qualitative deficiency (Type II).10 Type I deficiency presents with reduced AT antigen and activity levels. Type II presents as normal AT antigen levels but reduced AT activity. Type I deficiency, commonly associated with heterozygous SERPINC1 gene variants (generally nonsense or frameshift mutations), is usually associated with a severe phenotype.11 Homozygous type I ATD has been found to be lethal in mice.12 Type II ATD is usually due to missense mutations and is generally associated with milder protein dysfunction.13 Type II is subdivided based on the site of the mutation: reactive site, heparin-binding site or pleiotropic.14–16Treatment and prevention of VTEs in patients with hereditary AT deficiency can be difficult due to heparin resistance. Treatment can also include administration of exogenous concentrated AT (either plasma-derived or recombinant). In this case series, the treatment of five cases of congenital AT deficiency with severe clotting tendencies will be described. A brief summary of patient and case characteristics is shown in Table 1.
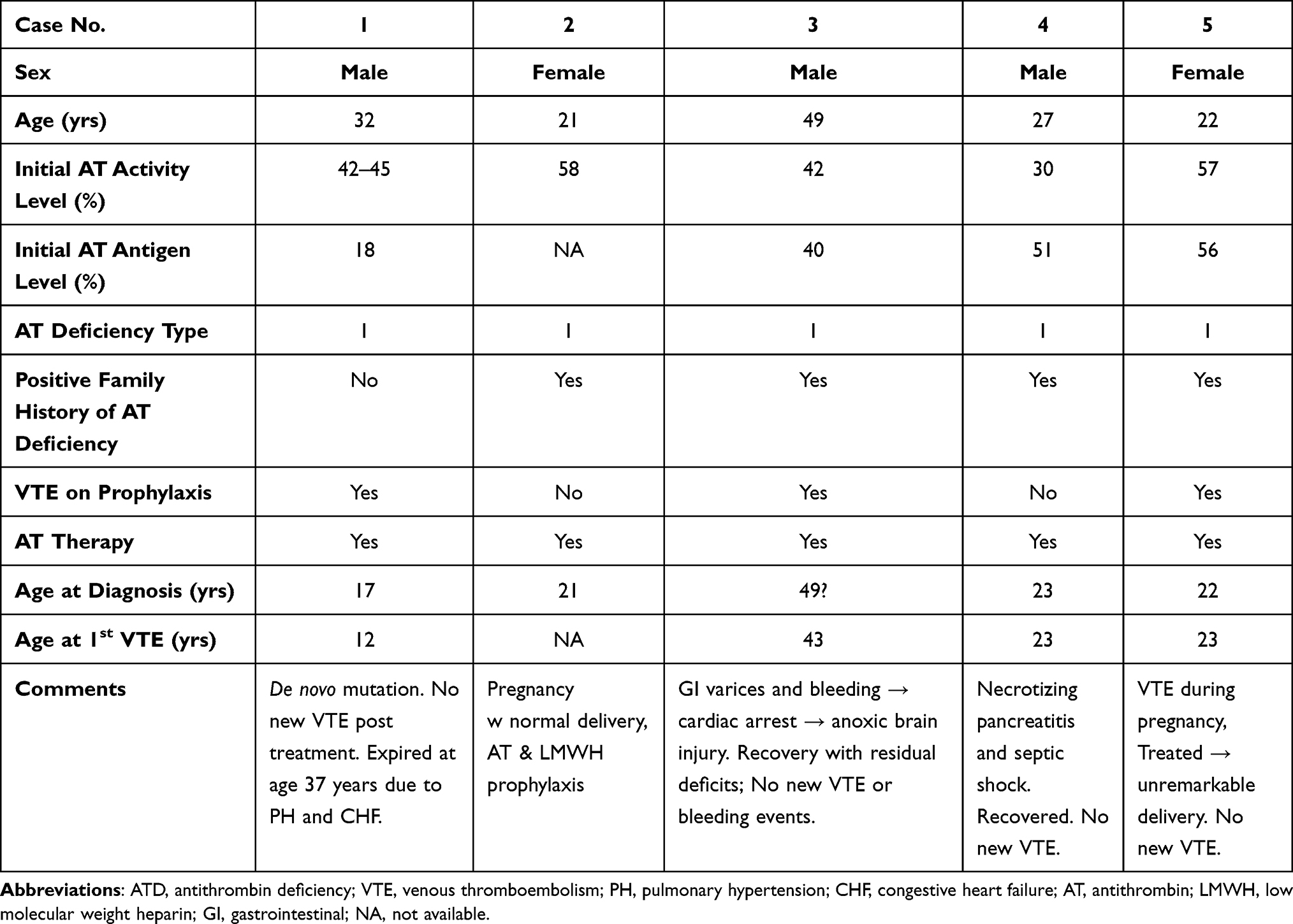 |
Table 1 Clinical Characteristics of Five Antithrombin Deficiency Cases |
Clinical Presentations
Case 1
A 32-year-old male had a known AT deficiency due to a de novo non-sense mutation 7522G>T, causing Q271X (GAG to TAG at codon 271 producing a stop codon) resulting in AT activity of 42–58% of normal and AT antigen of 18% of normal.17 The patient had his first thrombotic episode at age 12 years – a provoked DVT in a lower extremity that was treated with warfarin for three months (treatment goal prothrombin time two times baseline). At age 17 years, the patient had a recurrent lower extremity deep vein thrombosis (DVT) with a simultaneous retinal artery thrombosis. The latter resulted in a permanent reduction of the patient’s visual field.
Antithrombin deficiency was discovered at this second episode. Family history was negative for VTE and AT antigenic and functional tests were normal for both parents. Genetic studies, including linkage studies to confirm parentage, revealed a de novo nonsense mutation, and other markers of thrombophilia were unremarkable.17
The patient had a third DVT episode despite warfarin therapy with therapeutic prothrombin times. At that time, the patient received a permanent inferior vena cava filter and was started on AT therapy (Thrombate III, antithrombin III [human], Grifols, Research Triangle Park, NC). AT was administered daily initially (treatment goal = peak 120% of normal value based on the package insert formula for calculating dosage), then decreased to twice weekly with warfarin for VTE prophylaxis.
Approximately one year later, the patient voluntarily discontinued AT treatment and was lost to follow-up. The patient returned to the clinic at age 32 with a history of multiple VTE episodes including at least two pulmonary embolisms (PE) in the interim and an open-heart embolectomy at age 21. The patient also had a superior mesenteric vein thrombus treated by open laparotomy and small bowel resection at age 22 and pulmonary thromboendarterectomy at age 26 years. In the patient’s hiatus from the clinic, he continued to receive anticoagulant treatment with warfarin (target International Normalized Ratio (INR) = 2.3–3.5).
The sequelae of the multiple thromboembolic episodes included congestive heart failure, chronic pulmonary hypertension, atrial fibrillation, tachycardia-induced cardiomyopathy, hepatic cirrhosis and chronic severe venous stasis and post-thrombotic syndrome.
Upon his return to the clinic, due to the perceived high risk of VTE, the patient was restarted on prophylactic treatment with AT (twice weekly with the dose calculated to achieve trough AT activity of >75% of normal value). Central venous access to administer AT was extremely difficult due to radiographic and ultrasound evidence of occlusive thrombi in virtually every deep vein along with extensive, tortuous collateral venous circulation.
Over the following five years, the patient received either warfarin or rivaroxaban (Xarelto, Janssen Pharmaceuticals, Titusville, NJ, USA, 20mg daily: INR goal 2.5–3.5 measured monthly) along with twice weekly AT concentrate (peak activity level goal 120%). No new thromboembolic events occurred during this period. However, despite aggressive treatment, the patient died at age 37 years from complications of congestive heart failure and pulmonary hypertension.
Case 2
A 21-year-old female was evaluated for AT deficiency during family screening due to a strong family history of VTE. Her baseline AT activity was 58% (range of normal values 73–125% of the mean). The patient’s great-grandfather died of a VTE and her grandmother, father and aunt have all experienced VTE. Several females in the family also experienced pregnancy complications. At diagnosis, the patient had no history of VTE and had not been on any hormonal therapy.
The patient was tested monthly for pregnancy (beta-human chorionic gonadotropin (HCG)). After two months, she was found to be pregnant (six weeks, 1 day gestation). At that time, the patient was started on AT replacement therapy (Thrombate III, Grifols) and a low molecular weight heparin, enoxaparin (40 mg subcutaneously daily). AT was administered as a 50 units/kg loading dose followed (after 24 hr) by a daily maintenance dose of 30 units/kg. AT levels were monitored and the daily dose of AT adjusted to maintain AT activity levels in the range of 76–128% of normal. After 2 weeks of daily AT therapy, every other day infusions were started. During her pregnancy, the patient had the following adverse events: contact dermatitis in response to adhesive dressings, leg cramps (VTE excluded by ultrasound), two vessel umbilical cord with normal ultrasound, fetal growth restriction (<10th percentile), and mild hypertension and proteinuria at 37 weeks, 1 day.
The patient delivered at 37 weeks and 2 days by Caesarean section after forceps delivery failed. Epidural anesthesia was administered more than 24 hours after the last enoxaparin dose. The newborn received a “moderately abnormal” APGAR score of 5 at one min and a “reassuring” score of 7 at 5 min.18 AT infusions were continued every other day for six weeks post-partum and enoxaparin (40 mg subcutaneously daily) was resumed post-partum.
Case 3
A 49-year-old male with AT activity of 42% of normal at diagnosis (AT antigen levels were similar to activity levels with a nadir of 40% (reference range for this laboratory was 82–136%)), and several low repeat levels thereafter (~40% on warfarin) had extensive thromboses including the hepatic portal system which resulted in portal hypertension and gastroesophageal varices. His first occurrence of a distal VTE was at age 43 and was treated with warfarin.
While on warfarin prophylaxis, the patient developed multiple recurrent DVTs in his femoral and popliteal veins as well as thromboses in his superior mesenteric, portal, and splenic veins. While the patient was on warfarin, his INR values fluctuated but was subtherapeutic (1.1) at this admission. The patient reported that one of his brothers and his mother died in their twenties and sixties, respectively, due to “abnormal blood clotting”, while another brother suffered from DVT in his youth.
The portal vein thromboses resulted in gastroesophageal varices and predisposed the patient to gastrointestinal bleeding. The patient suffered a devastating variceal bleeding episode in 2013 resulting in severe hemorrhagic shock with cardiac arrest, anoxic brain injury and a spinal cord infarction. Records regarding warfarin compliance and actual INRs were unavailable. Anticoagulant therapy was discontinued, and an inferior vena cava (IVC) filter was placed.
Subsequently, the patient was transferred to a tertiary care facility for further treatment. AT replacement was initiated with plasma-derived AT (Thrombate III, Grifols). The patient developed thrombus extension to the IVC filter on standard AT replacement dosing. AT treatment was intensified by increasing the dose and frequency of administration from 2100 units three times a week to 2200 units every other day with the goal to maintain AT trough activity levels as close to 80% as possible.
Over the past eight years, the patient gradually recovered from his neurological deficits but will likely remain wheelchair-bound with residual mentation deficits. However, there have been no new thrombotic or bleeding events since the initiation of the intensified AT regimen. Lifelong AT concentrate therapy will likely be necessary to prevent VTEs and anticoagulation should be avoided due to the patient’s history of catastrophic bleeding episodes.
Case 4
A 27-year-old male presented to the hospital with necrotizing pancreatitis and abdominal compartment syndrome. Hematology was consulted due to the patient’s personal and family history of type 1 AT deficiency. The patient’s brother was reported to have type 1 AT deficiency, diagnosed at 28 years of age subsequent to a cerebral vascular accident with a baseline AT activity 55% and AT antigen 51%.
The patient was diagnosed with type 1 AT deficiency by his primary care physician four years prior when he suffered bilateral lower extremity DVTs with a subsequent PE developing a few months after the initial thrombotic event. A permanent IVC filter was placed approximately five months prior to the presenting episode when he was being treated for acute pancreatitis. The patient had an elective cholecystectomy one month prior to admission. Subsequent to this surgery, he developed necrotizing pancreatitis with abdominal compartment syndrome.
The patient developed a duodenal hemorrhage after esophagogastroduodenoscopy and was subsequently intubated with central line placement. The patient developed septic shock secondary to necrotizing pancreatitis which was treated with broad spectrum antibiotics. An exploratory laparotomy was performed with placement of a wound vacuum. Abdominal CT revealed that the necrotizing pancreatitis involved 50% of the parenchyma.
At the initial hematology consult, the patient’s AT activity was 30%. Exogenous AT concentrate (Thrombate III, Grifols) was administered (5000 units) with a post-infusion peak of 111%. AT activity was then monitored daily with a target range of 80–120%. Unfractionated heparin was also administered with a PTT target of 50–70 seconds (institutional therapeutic PTT range is 70–100 sec), due to recent hemorrhage and temporal proximity to surgery. While an outpatient, he had been treated with warfarin with an INR target of 2.0–3, and no other family history of thrombophilia or bleeding disorders was reported.
During the patient’s hospitalization, he developed a vancomycin-resistance enterococcal infection in his abdominal compartment. Treatment included seven surgeries to drain and clean abdominal abscesses. The patient was placed on a continuous AT infusion during hospitalization to maintain AT activity in the target range. Other complications during his hospital course included superficial cephalic venous thrombosis during the first month and right popliteal vein thrombosis in July 2015 during the second month. After peripheral placement of a central catheter and continued vacuum wound management, the anticoagulation was transitioned to warfarin (goal INR 2.5–3.5) and the patient was discharged with completion of intravenous antibiotic therapy as an outpatient.
The patient was readmitted for abdominal wall reconstruction five months after the initial admission. During the perioperative period, he was administered AT infusions (goal 80–120%). The patient was again transitioned back to warfarin (INR goal 2.5–3.5). The patient has had no additional thrombotic complications since that time.
Case 5
A 22-year-old patient with AT deficiency (baseline AT activity 57%; antigen level 56%) presented with an intrauterine pregnancy. Her family history was remarkable for type 1 AT deficiency in her father and sister. Her father had a history of multiple thrombotic events beginning at the age of 23. At 31 years old, he died from refractory progressive VTE related to small cell carcinoma of the lung.
The patient’s first pregnancy was uncomplicated. She received enoxaparin (40 mg subcutaneously daily) during her pregnancy and post-partum. Prior to her scheduled induction of labor, the patient received AT concentrate (Thrombate III, Grifols) dosed to maintain AT activity at 80–120% of normal levels. After delivery, her AT activity was 63%, so she received an additional dose of AT. The patient was scheduled to receive prophylactic anticoagulation for 12 weeks post-partum, but she voluntarily discontinued therapy at six weeks. Despite her non-compliance, the patient did not experience any thrombotic events.
This patient became pregnant again less than one year later. During this second pregnancy, she was prescribed prophylactic enoxaparin (40 mg subcutaneously daily). Despite receiving enoxaparin, the patient developed a DVT in her right lower extremity at gestational week 13. The DVT extended from the common femoral vein to the popliteal vein. Her enoxaparin dose was increased to therapeutic levels (1 mg/kg, subcutaneously, every 12 hr) and she received AT concentrate, targeting a peak AT activity of 120%. Despite the increased dose of enoxaparin, she experienced progression of the DVT in her right lower extremity to involve the common femoral vein, popliteal vein, and calf veins. The patient underwent pharmaco-mechanical thrombolysis and was given ticagrelor, 180 mg × 1 day, 90 mg on the second day, then discontinued. Anticoagulation management was escalated by the addition of AT concentrate to therapeutic dosing of enoxaparin. AT was administered three times a week with a peak AT activity target of 120%. Trough AT activity was measured at least weekly and low-molecular weight heparin anti-factor Xa activity was monitored monthly. Enoxaparin was discontinued 24 hrs prior to delivery and resumed 12 hrs after delivery. There was no further progression of the DVT, and the baby’s vaginal delivery was unremarkable.
Discussion
Hereditary AT deficiency is a rare autosomal dominant disorder. The prevalence is estimated to be between 1 in 500 to 1 in 5000.19,20 Normal plasma levels of AT range from 112 to 140 µg/mL. Due to variability between laboratories, AT activity levels are usually expressed as a percentage of the normal level for that laboratory with the usual normal range being 80–120%.21 Most patients with AT deficiency have AT activity levels in the range of 40–60%.22 The five patients included in this paper were mostly in this range: 30–58%.
The five case summaries presented here represent the more clinically complicated and severe phenotype of HATD and illustrate important commonalities that could influence the approach to diagnosis and management of AT deficiency. Four of the five patients presented had a positive family history for AT deficiency. This emphasizes the need for obtaining a thorough family history regarding the occurrence of DVTs or diagnosed AT deficiency. This is especially true of DVT patients younger than 55 years. In the general population, the vast majority of thromboembolic events (DVT or PE) occur in patients over 55.23–25
In addition, most of these patients experienced a DVT episode while on prophylactic therapy with anti-coagulants. This observation demonstrates the need for careful monitoring of AT deficiency patients to determine their thrombosis risk and assess their compliance with therapy. Due to the occurrence of DVTs in these patients while on prophylactic anticoagulation, AT supplementation therapy was added to help prevent these episodes.
One patient (Case 1) was treated with rivaroxaban, a direct oral anticoagulant (DOAC) in the course of his treatment for AT deficiency. Although studies directly evaluating the use of DOACs in AT deficiency are limited. Two recent review articles on the use of DOACs in inherited thrombophilic disorders (including AT deficiency) concluded that, although there was some evidence of efficacy, current data were insufficient to recommend the use of DOACs in inherited thrombophilic conditions.26,27
Two of the cases presented were in pregnant women. Pregnancy is a documented risk factor for development of DVT and PE. The risk of DVT in pregnant women has been estimated to be four times greater than non-pregnant women of the same age.28 In fact, thromboembolism is a leading cause of death in pregnant women in developed countries.29,30
Both of these patients were treated with a low molecular weight heparin (enoxaparin) and AT concentrate augmentation therapy. Low molecular weight heparins are the drugs of choice in pregnancy as they do not cross the placenta and are not excreted in breast milk.31 It is unknown whether pregnant women with AT deficiency have heparin resistance and whether a low molecular weight heparin will be adequate for prophylaxis of thromboembolism. In one of the cases presented here, prophylaxis with a low molecular weight heparin was clearly inadequate as a thrombotic event occurred while on this therapy. Both pregnant women presented here received successful treatment or prophylaxis from a combination of AT and a low molecular weight heparin. These cases illustrate that ante- and/or post-partum prophylaxis as now recommended by guidelines of the American Society of Hematology32 may not always be sufficient to prevent pregnancy-related VTE in HATD.
One of the pregnant patients was treated with ticagrelor per her physician prior to hematology’s involvement in her care. Her obstetrician preferred ticagrelor over alternative therapy based on their teratogenic risk versus benefit.
In both cases, enoxaparin was discontinued 24 hrs prior to delivery and resumed post-partum. Despite an extensive DVT during pregnancy in one of the patients, both of the deliveries (one Cesarean and one vaginal) were uneventful and resulted in healthy babies.
A recent retrospective study analyzed 64 pregnancies in 28 women with AT deficiency.33 This study showed that risk of pregnancy-related VTE was similar between patients with type I and type II AT deficiency. However, risk of an adverse pregnancy outcome was related only to type II deficiency especially the homozygous AT Budapest 3 variant.
Another retrospective study investigated 219 pregnancies in 87 women with type 1 AT deficiency.34 This study showed a lower risk of VTE in the women treated with LMWH (3 events/43 pregnancies; 7.0%) compared to women not treated with LMWH (17 events/146 pregnancies: 11.6%), but the difference was not statistically significant (p = 0.57). However, the incidence of late complications was increased in the LMWH-treated group (24%) compared to the untreated group (6% p = 0.0006). This study also showed that the risk of pregnancy-related VTE was highest among women with a positive family history of VTE suggesting LMWH therapy should be considered in these patients.
A study of the clinical characteristics of AT deficiencies found that the type II heparin-binding site mutation, AT Padua I, was more frequently associated with pregnancy complications than type I or other type II mutations.35 Taken together, these studies indicate that genetic information on the mutation underlying AT deficiency, if available, can help guide the course of prophylaxis and treatment.
There are some distinct differences among the cases as well. One patient had a de novo mutation resulting in significant AT deficiency (42–45%),17 suggesting measurement of AT activity may be called for in patients with DVTs without a family history of AT deficiency especially patients younger than 55 years old. VTE is a relatively rare occurrence in people under 55 years old.23–25 For patients under 55 without known risk factors for VTE (eg, cancer, surgery, chronic obstructive pulmonary disease, some cardiovascular diseases), AT deficiency is certainly worth considering as the cause of their thrombotic event.
Two of these patients suffered significant bleeding events – one was catastrophic with significant clinical sequelae. These cases emphasize the need for careful monitoring of coagulation parameters especially in patients with a history of bleeding episodes. Both of these patients were initially managed with AT and without anticoagulants (warfarin or low molecular weight heparins). The patient with less severe bleeding was eventually transitioned to warfarin monotherapy. The patient with the severe bleeding episode has been maintained on AT therapy without the use of anticoagulants to avoid another potentially catastrophic bleeding event. Management of patients with AT deficiency and bleeding tendencies can be especially challenging.
Conclusion
These cases of AT deficiency demonstrate a wide range of manifestations, some commonalities, but also important differences necessitating an individualized approach to management. All of these patients received AT augmentation and, in most cases, after anticoagulant therapy proved inadequate. This emphasizes that in some instances AT replacement therapy, although costly, is a necessary component of successful treatment. Treatment of patients with AT deficiency in combination with bleeding tendencies can be particularly difficult.
Abbreviations
AT, antithrombin; ATD, antithrombin deficiency; DVT, deep vein thrombosis; HCG, human chorionic gonadotropin; INR, international normalized ratio; IVC, inferior vena cava; PE, pulmonary embolism; VTE, venous thromboembolism.
Data Sharing Statement
All the relevant data for this study are available within the article. Complementary data can be made available by the corresponding author upon reasonable request.
Ethics Approval and Informed Consent
Ethics committee approval was not sought for this study because the institutions involved do not require approval for reporting individual cases. This study was completed in accordance with the Helsinki Declaration as revised in 2013. Written informed consent was obtained from the patients described below or their authorized representatives for their anonymized information to be published in this article.
Consent for Publication
There are no images, videos, recordings, or other materials requiring consent to be published other than the informed consent described above.
Acknowledgments
Michael K. James, Ph.D. is acknowledged for medical writing and Jordi Bozzo, Ph.D., CMPP, for editorial assistance.
Author Contributions
All authors made significant contributions to the work including conception, execution, acquisition of data, and analysis. All authors participated in the drafting and revision of the manuscript and have agreed to submission to this journal. All authors have agreed to revisions and final form of the manuscript for submission and take responsibility and accountability for the contents therein.
Funding
This study was supported by Grifols (Barcelona, Spain).
Disclosure
Dr Annette von Drygalski reports that the manuscript was developed by a medical writer with funding support from Grifols. Dr Michael Tarantino is an Advisory Board Consultant and member of Speaker Bureau for Amgen, BioMarin, Dova, Genentech, Grifols, Octapharma, Principia, Sobi, Takeda, and UCB; in addition Grant Reviewer, Clinical Trial PI for Pfizer and Spark Therapeutics outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Rodgers GM. Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency. Thromb Haemost. 2017;101(05):806–812. doi:10.1160/th08-10-0672
2. Levy JH, Sniecinski RM, Welsby IJ, Levi M. Antithrombin: anti-inflammatory properties and clinical applications. Thromb Haemost. 2016;115(4):712–728. doi:10.1160/TH15-08-0687
3. Roemisch J, Gray E, Hoffmann JN, Wiedermann CJ. Antithrombin: a new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis. 2002;13(8):657–670. doi:10.1097/00001721-200212000-00001
4. Kondo S, Kisiel W. Regulation of factor VIIa activity in plasma: evidence that antithrombin III is the sole plasma protease inhibitor of human factor VIIa. Thromb Res. 1987;46(2):325–335. doi:10.1016/0049-3848(87)90294-5
5. Broze GJ
6. Egeberg O. Inherited antithrombin deficiency causing thrombophilia. Thromb Diath Haemorrh. 1965;13:516–530.
7. Martinelli I, Mannucci PM, De Stefano V, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. 1998;92(7):2353–2358. doi:10.1182/blood.V92.7.2353
8. Bucciarelli P, Rosendaal FR, Tripodi A, et al. Risk of venous thromboembolism and clinical manifestations in carriers of antithrombin, protein C, protein S deficiency, or activated protein C resistance: a multicenter collaborative family study. Arterioscler Thromb Vasc Biol. 1999;19(4):1026–1033. doi:10.1161/01.atv.19.4.1026
9. Crowther MA, Kelton JG. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138(2):128–134. doi:10.7326/0003-4819-138-2-200301210-00014
10. Mannucci PM, Tripodi A, Mari D. Congenital deficiencies of anticoagulant proteins (antithrombin III and protein C). Haematologica. 1984;69(6):730–746.
11. Cooper PC, Coath F, Daly ME, Makris M. The phenotypic and genetic assessment of antithrombin deficiency. Int J Lab Hematol. 2011;33(3):227–237. doi:10.1111/j.1751-553X.2011.01307.x
12. Ishiguro K, Kojima T, Kadomatsu K, et al. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. 2000;106(7):873–878. doi:10.1172/JCI10489
13. Bravo-Perez C, Vicente V, Corral J. Management of antithrombin deficiency: an update for clinicians. Expert Rev Hematol. 2019;12(6):397–405. doi:10.1080/17474086.2019.1611424
14. Lane DA, Caso R. 9 Antithrombin: structure, genomic organization, function and inherited deficiency. Baillières Clin Haematol. 1989;2(4):961–998. doi:10.1016/S0950-3536(89)80054-X
15. Luxembourg B, Delev D, Geisen C, et al. Molecular basis of antithrombin deficiency. Thromb Haemost. 2011;105(4):635–646. doi:10.1160/TH10-08-0538
16. van Boven HH, Lane DA. Antithrombin and its inherited deficiency states. Semin Hematol. 1997;34(3):188–204.
17. Tarantino MD, Curtis SM, Johnson GS, Waye JS, Blajchman MA. A novel and de novo spontaneous point mutation (Glu271STOP) of the antithrombin gene results in a type I deficiency and thrombophilia. Am J Hematol. 1999;60(2):126–129. doi:10.1002/(sici)1096-8652(199902)60:2<126::aid-ajh7>3.0.co;2-l
18. Committee on Obstetric Practice: American College of Obstetricians and Gynecologists AAoP-CoFaN. The Apgar Score. American College of Obstetricians and Gynecologists; 2021. Available from: https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2015/10/the-apgar-score.
19. Tait RC, Walker ID, Perry DJ, et al. Prevalence of antithrombin deficiency in the healthy population. Br J Haematol. 1994;87(1):106–112. doi:10.1111/j.1365-2141.1994.tb04878.x
20. Wells PS, Blajchman MA, Henderson P, et al. Prevalence of antithrombin deficiency in healthy blood donors: a cross-sectional study. Am J Hematol. 1994;45(4):321–324. doi:10.1002/ajh.2830450409
21. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008;14(6):1229–1239. doi:10.1111/j.1365-2516.2008.01830.x
22. Lane DA, Bayston T, Olds RJ, et al. Antithrombin mutation database: 2nd (1997) update. For the plasma coagulation inhibitors subcommittee of the scientific and standardization committee of the international society on thrombosis and haemostasis. Thromb Haemost. 1997;77(1):197–211. doi:10.1055/s-0038-1655930
23. Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ. Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med. 1998;158(6):585. doi:10.1001/archinte.158.6.585
24. Anderson FA
25. Heit JA, Spencer FA, White RH. The epidemiology of venous thromboembolism. J Thromb Thrombolysis. 2016;41(1):3–14. doi:10.1007/s11239-015-1311-6
26. Skelley JW, White CW, Thomason AR. The use of direct oral anticoagulants in inherited thrombophilia. J Thromb Thrombolysis. 2017;43(1):24–30. doi:10.1007/s11239-016-1428-2
27. Valanejad SM, Davis KA. Direct oral anticoagulants in select patients with hypercoagulable disorders. Ann Pharmacother. 2021;55(7):891–901. doi:10.1177/1060028020968551
28. Barsoum MK, Heit JA, Ashrani AA, Leibson CL, Petterson TM, Bailey KR. Is progestin an independent risk factor for incident venous thromboembolism? A population-based case-control study. Thromb Res. 2010;126(5):373–378. doi:10.1016/j.thromres.2010.08.010
29. Khan F, Vaillancourt C, Bourjeily G. Diagnosis and management of deep vein thrombosis in pregnancy. BMJ. 2017;357:j2344. doi:10.1136/bmj.j2344
30. Nair M, Knight M. Chapter 2. Maternal mortality in the UK 2011–13: surveillance and epidemiology. In: Saving Lives, Improving Mothers’ Care - Surveillance of Maternal Deaths in the UK 2011–13 and Lessons Learned to Inform Maternity Care Form the UK and Ireland Confidential Enquiries into Maternal Deaths and Morbidity 2009–2013; 2015.
31. Greer IA, Solomon CG. Clinical practice: pregnancy complicated by venous thrombosis. N Engl J Med. 2015;373(6):540–547. doi:10.1056/NEJMcp1407434
32. Bates SM, Rajasekhar A, Middeldorp S, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: venous thromboembolism in the context of pregnancy. Blood Adv. 2018;2(22):3317–3359. doi:10.1182/bloodadvances.2018024802
33. Kovac M, Mitic G, Mikovic Z, et al. The influence of specific mutations in the AT gene (SERPINC1) on the type of pregnancy related complications. Thromb Res. 2019;173:12–19. doi:10.1016/j.thromres.2018.11.006
34. Abbattista M, Gianniello F, Novembrino C, et al. Risk of pregnancy-related venous thromboembolism and obstetrical complications in women with inherited type I antithrombin deficiency: a retrospective, single-centre, cohort study. Lancet Haematol. 2020;7(4):e320–e328. doi:10.1016/S2352-3026(20)30007-7
35. Gindele R, Selmeczi A, Olah Z, et al. Clinical and laboratory characteristics of antithrombin deficiencies: a large cohort study from a single diagnostic center. Thromb Res. 2017;160:119–128. doi:10.1016/j.thromres.2017.10.023
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Antithrombin Deficiency and Thrombosis: A Wide Clinical Scenario Reported in a Single Institution
Marco-Rico A, Marco-Vera P
Journal of Blood Medicine 2023, 14:499-506
Published Date: 1 September 2023
Efficacy and Safety of Rivaroxaban for Extremely Aged Patients with Venous Thromboembolism: A Retrospective, Cross-Sectional Real-World Study
Wang C, Fan X, Nie L, Wang Q, Li S, Zheng W, Zhang W, Dai W, Chen M
Clinical Interventions in Aging 2024, 19:1103-1116
Published Date: 18 June 2024