Back to Journals » Drug Design, Development and Therapy » Volume 20
First-in-Human Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of Single and Multiple Ascending Oral Doses of BIIB091, an Oral BTK Inhibitor, in Healthy Adult Participants
Authors Tsai HH ![]() , Gu Y, Riester K, Chu SS, Bame E, Arefayene M, Kam JLYL, Coppell A, Hanna J, Hopkins BT, Scaramozza M
, Gu Y, Riester K, Chu SS, Bame E, Arefayene M, Kam JLYL, Coppell A, Hanna J, Hopkins BT, Scaramozza M
Received 4 October 2025
Accepted for publication 20 February 2026
Published 14 April 2026 Volume 2026:20 571780
DOI https://doi.org/10.2147/DDDT.S571780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Hui-Hsin Tsai,1,* Yi Gu,2,* Katherine Riester,3 Sherman S Chu,4 Eris Bame,5 Million Arefayene,6 Jeanelle LYL Kam,7 Alexander Coppell,8 Jerome Hanna,1 Brian T Hopkins,9 Matthew Scaramozza1
1Department of Multiple Sclerosis and Immunology Clinical Development, Biogen, Cambridge, MA, USA; 2Department of Clinical Pharmacology & Pharmacometrics, Biogen, Cambridge, MA, USA; 3Department of Biostatistics, Biogen, Cambridge, MA, USA; 4Department of Global Drug Safety, Biogen, Cambridge, MA, USA; 5Department of Biomarkers and Systems Biology, Biogen, Cambridge, MA, USA; 6Department of Clinical Pharmacology, Biogen, Cambridge, MA, USA; 7Department of CPS Medical & Scientific Affairs, Fortrea, Madison, WI, USA; 8Therapeutics Development Unit, Biogen, Maidenhead, UK; 9Department of Pharmaceutical Operations and Technology, Biogen, Cambridge, MA, USA
*These authors contributed equally to this work
Correspondence: Hui-Hsin Tsai, Department of Multiple Sclerosis and Immunology Clinical Development, Biogen, 225 Binney St, Cambridge, MA, 02142, USA, Tel +1-617-945-3799, Email [email protected]
Introduction: Multiple sclerosis (MS) affects 2.8 million people globally. Despite available disease-modifying therapies (DMTs), more effective treatments are needed to prevent/slow disability progression. BIIB091 is a selective, non-covalent oral Bruton’s tyrosine kinase (BTK) inhibitor. This Phase 1, first-in-human study evaluated safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of BIIB091.
Methods: Participants received single ascending doses (SAD; 50– 1200 mg) or multiple ascending doses (MAD; 50– 300 mg twice daily [BID] for 14 days) of BIIB091 or placebo. Safety assessments included labs, vitals, physical examinations, electrocardiograms, and adverse event (AE) recordings. PK was evaluated through blood and urine samples; PD was evaluated through blood samples, including BTK phosphorylation and B-cell receptor-mediated CD69 upregulation. Additional objectives assessed PK/PD relationship and food’s effect on PK.
Results: Sixty-four participants were randomized (SAD n=40; MAD n=24). BIIB091 showed rapid absorption, dose-proportional PK, and no significant accumulation. BIIB091 suppressed B cell activation, achieving > 90% inhibition of CD69 expression on CD19+ within 1 hour at all study doses; inhibition was sustained up to 24 hours at 1200 mg. A high-fat meal delayed Tmax by ~5.2 hours and increased AUC and Cmax about 2.046- and 1.420-fold. A single 300 mg dose prolonged the CD69 inhibition duration, maintaining > 90% 24 hours post-dose versus 42% in fasted state. MAD > 150 mg BID maintained rapid and consistent CD69 inhibition over 14 days. Mild AEs were reported in 14 participants, with no dose-limiting AEs, serious AEs, or withdrawals. Nausea and headache were the most common AEs.
Conclusion: BIIB091 was well tolerated at single doses up to 1200 mg and repeated doses up to 300 mg BID, showing dose-linear PK and sustained effective B cell activation suppression. BIIB091 PK appeared to be sensitive to food effect. These important insights support its continued development for MS.
Keywords: Bruton’s tyrosine kinase, non-covalent, reversible, Phase 1, BIIB091, multiple sclerosis dose-escalation study, CD69 inhibition
Introduction
Multiple sclerosis (MS) is a chronic inflammatory, demyelinating, and neurodegenerative disease. In 2020, MS affected an estimated 2.8 million people globally, which is a 30% increase since 2013.1 People of all ages can be affected by MS, but it is more common between the ages of 20 and 40, especially in females, making it one of the most common causes of neurological disability among young adults.1,2 Moreover, from the non-clinical aspect, living with MS impacts family planning, employment, and the quality of life of both patients and their caretakers.3 Despite the availability of numerous therapies, there remains a need for convenient and high-efficacy oral treatments to slow and/or prevent disability progression in MS.4
Since the approval of interferon beta therapies for the treatment of MS, there has been an insurgence in the number of disease-modifying therapies (DMTs), mostly targeting the T cell compartment.5 The recent success of B cell-depleting antibody therapies in treating MS validates the contribution of B cells to MS disease pathogenesis.6–10 Although T and B cells in adaptive immunity play critical roles in MS, nearly all the cell types in the innate immune system have shown involvement in MS pathogenesis, including monocytes, macrophages, mast cells, and neutrophils.11,12
In this regard, Bruton’s tyrosine kinase (BTK) has emerged as a promising therapeutic target for MS.13 Inhibiting BTK may target both adaptive and innate immune mechanisms based on its broad expression in hematopoietic cell lineage and microglia in the central nervous system.14 In B cells, BTK is a key node that connects the immunoreceptor tyrosine-based activation motif–containing B cell receptor (BCR) signaling complex with downstream signaling events. These events, including phospholipase Cγ activation, the Ca2+ response, and NF-kB activation, ultimately lead to B cell activation and promote antigen-dependent stimulation of T cells through B cell-mediated antigen presentation. In contrast to B cell-depleting therapies, BTK inhibition reduces B cell activation but preserves B cell viability and survival, which is important to avoid an increased risk of serious infection that might be associated with chronic B cell depletion.15,16 Additionally, BTK is involved in signaling events mediated by IgG-specific Fc receptor (FcγR) in macrophages and microglia and by the IgE-specific Fc receptor (FcεR) in mast cells. This regulates the release of pro-inflammatory cytokines, reactive oxygen species production, and inflammasome activation.17,18 The therapeutic relevance of BTK in CNS-resident microglia remains under investigation and has not been definitively quantified.
BIIB091 is a peripherally acting small molecule kinase inhibitor that selectively targets the orthosteric site of BTK, which is being developed as a reversible oral treatment option for MS.19 BIIB091 inhibits the phosphorylation of Tyr-551, a crucial residue for BTK activation. This is achieved by binding within the “H3” pocket, which contains 2 non-conserved backbone residues (Leu-552 and Ser-553), allowing for preferential binding to BTK compared to other members of the Tec family.20 The unique binding mode of BIIB091 provides high selectivity across the kinome and contributes to the favorable safety profile observed in preclinical toxicology studies (safety margins >10-fold).21 When tested in the DiscoverX KINOMscan against a panel of > 400 kinases, it exhibited > 500-fold selectivity for BTK over other kinases. Furthermore, in the Eurofins Cerep Panlabs lead profiling screen, BIIB091 exhibited no off-target activity against 68 physiologically relevant targets.19 Preclinically, BIIB091 demonstrated potent and on-target activity by inhibiting early signaling events and downstream cellular effector functions in both B cells and myeloid cells with IC50s ranging from 3 to 106 nM depending on species and assays.21 In addition to inhibiting B cell activation, proliferation, and differentiation, BIIB091 has been shown to block B cell receptor (BCR)–mediated antigen presentation to T cells, a recently validated mechanism for treating MS.22 BIIB091 is a reversible (non-covalent), and highly selective and potent oral BTK inhibitor that could robustly suppress peripheral inflammation by targeting pathways and cell types relevant to MS pathology, and differentiate from mechanism of actions of current approved DMTs.
This Phase 1, first-in-human study evaluated single and multiple ascending oral doses of BIIB091 in healthy adult participants.
Materials and Methods
Study Design and Participants
This was a 2-part, first-in-human, Phase 1, randomized, blinded, placebo-controlled, dose escalation study (NCT03943056) designed to evaluate the safety, tolerability, PK, and PD of SAD and MAD of orally administered BIIB091 (Figure 1) in healthy adult participants (Figure 2). The primary objective was to assess the safety and tolerability of single and multiple oral doses of BIIB091. The secondary objectives included characterizing the PK of BIIB091 for both single and multiple oral doses, as well as determining the effect of food on the PK of a single oral dose of BIIB091. Exploratory objectives were to evaluate the PD response to BIIB091 following single and multiple oral doses, assess the PK/PD relationship following single and multiple oral doses of BIIB091, identify major plasma and urine metabolites of BIIB091, and evaluate the effect of BIIB091 on QTc and other electrocardiogram (ECG) parameters in healthy participants. The study included healthy individuals aged 18–55 years with a body mass index of 18–30 kg/m2. Individuals unwilling to comply with protocol requirements or who had a history of any clinically significant disease were excluded. Participants were enrolled at a single clinical site in the United States.
 |
Figure 1 BIIB091 chemical structure. |
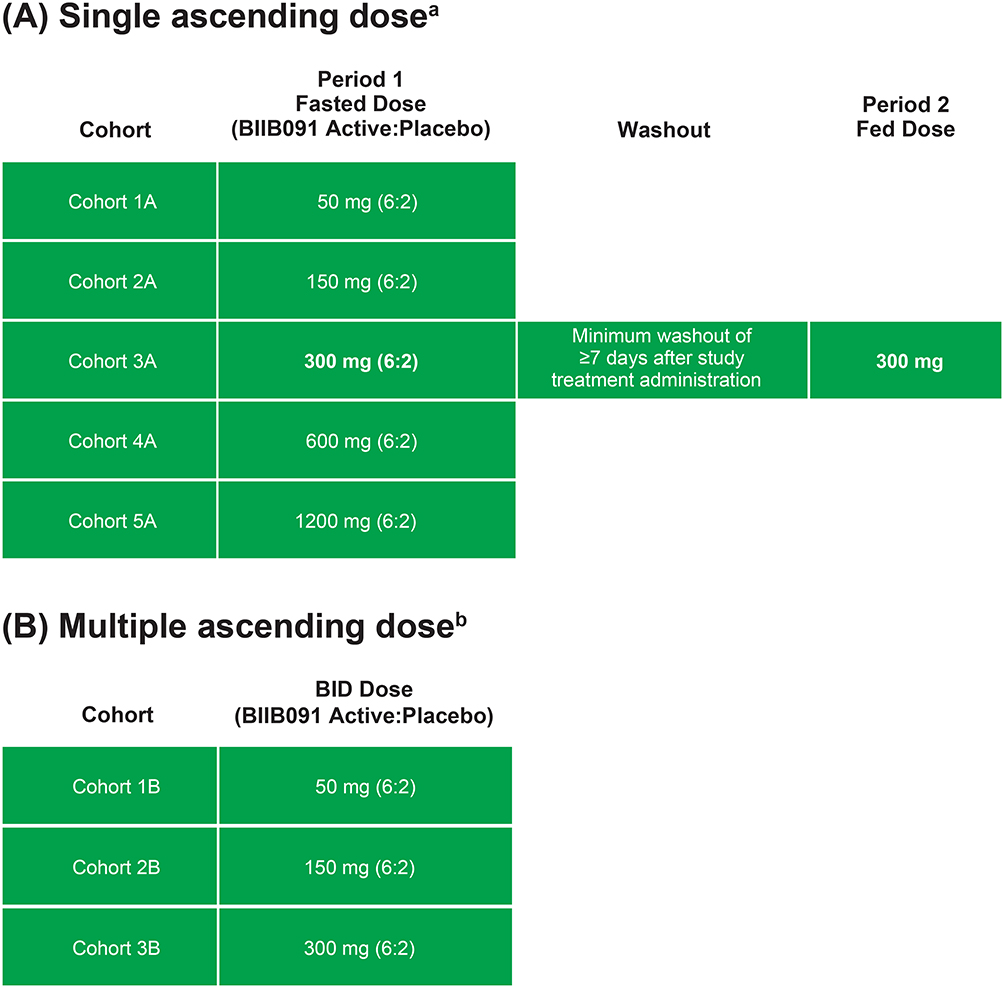 |
Figure 2 Study design. (A) Part 1: Single ascending dosea. (B) Part 2: Multiple ascending doseb. Abbreviation: BID, twice daily. Notes: aFor each Part, a single dose of study treatment was administered on study day 1. Participants remained housed in the clinical research unit through day 3 and returned for a single outpatient follow-up visit approximately 4 to 6 days after checkout. bStudy treatment was administered BID for 13 days, and once on the morning of study day 14. Participants remained housed in the clinical research unit though day 16 and returned for a single outpatient follow-up visit approximately 6 to 8 days after checkout. |
BIIB091 was an active pharmaceutical ingredient supplied as a crystalline powder form. BIIB091 drug product was prepared as a drug-in-capsule (DiC), immediate release dosage form for oral administration. The placebo was microcrystalline cellulose inside of a placebo capsule prepared to look identical to capsules containing BIIB091.
Single Ascending Dose (SAD): Part 1
In the SAD part of the study, participants were randomized in a 6:2 ratio to receive a single dose of the study treatment (BIIB091 at 50, 150, 300, 600, or 1200 mg) or placebo. Additionally, the study investigated the effect of food in a single 2-period arm in the SAD part of the study. In the first period, participants were dosed in the fasted state, and in the second period, they were dosed 30 minutes after consuming a standardized high-fat meal, with ≥50% of calories coming from fat, based on the FDA definition. The fed portion of the food effect evaluation was initiated at least 7 days after the first period to allow time for washout.
Multiple Ascending Dose (MAD): Part 2
In the MAD part of the study, participants were randomized to receive study treatment (BIIB091 at 50 mg, 150 mg, or 300 mg) or placebo in the fasted state twice daily (BID) for 14 days. Participants in the SAD part were not allowed to participate in the MAD part of the study.
Dose Escalation
Prior to each dose escalation decision in the SAD part of the study, the safety surveillance team reviewed blinded safety and PK data from at least 6 participants in the current cohort and all participants in the completed cohorts with the Investigator. Except for the fed portion of the food effect evaluation, a sentinel dosing approach was used. The first 2 participants in each cohort were randomized 1:1 to BIIB091 or placebo and administered study treatment. The remaining participants in each cohort were not dosed until the Investigator had reviewed safety data through 24 hours post-dose for the first 2 participants. The remaining participants in the cohort were randomized 5:1 BIIB091 to placebo and administered study treatment.
Prior to initiating the MAD part of the study, blinded safety and PK data were reviewed from all participants in the 50–300 mg cohort and at least 6 participants in the 600 mg cohort by the safety surveillance team with concurrence from the Investigator. Available PD data were also reviewed. Sentinel dosing was not used in the MAD part of the study because the expected exposures to be achieved on the first day of dosing were already evaluated in the SAD part of the study.
Assessments
Safety assessments included clinical laboratory assessments, vital signs, physical examinations, 12-lead safety ECGs, concomitant therapy and procedure recording, and adverse event (AE) and serious adverse event (SAE) recording. Blood and urine samples for measuring the plasma and urine concentrations of BIIB091 were collected for PK assessments, with the PK bioanalytical method described in the next section. PD was assessed through the expression of CD69 on the surface of B cells (total, naïve, and memory) upon ex vivo stimulation with anti-immunoglobulin (Ig)D in the SAD and MAD parts of the study and phosphorylated BTK in whole blood in the MAD part of the study. CD69 expression was assessed as an exploratory marker of B cell activation. Lymphocyte subsets were evaluated to assess BIIB091 treatment response and included total T cells, helper T cells, cytotoxic T cells, total natural killer cells, and total B cells. Additional B cell subsets evaluated in the MAD part of the study included naive, memory, and transitional B cell subsets. Continuous ECG recording was performed to assess the effect of BIIB091 on the QTc interval, including QTcF, QT, RR, and PR intervals, QRS duration, and heart rate for the first 24 hours post-dose in both the SAD and MAD parts of the study. Standard ECG morphology analysis, categorical analysis, and by-timepoint QT interval analysis were performed to assess the drug effect on cardiac repolarization.
Bioanalytical Methods for Pharmacokinetic Assessment
BIIB091 concentrations, used for the PK assessment, were determined using a validated LC/MS/MS method following protein precipitation. An ultra high-performance liquid chromatography was coupled with a SCIEX API4000 tandem mass spectrometry, via a turbo ion spray interface working in the positive mode. Mass spectrometry was scanned under the selected reaction monitoring mode. The concentration calibration range was established from 1.00 to 1000 ng/mL for BIIB091 concentration, with acceptable accuracy and precision. Sample dilution up to 100-fold was verified. Appropriate sample stability under various conditions was established.
Bioanalytical Methods for Pharmacodynamic Assessment
Heparinized venous whole blood from subjects enrolled in the clinical study was aliquoted (100 μL) into appropriately labelled 15-mL conical tubes. One set of aliquots was left untreated while another set received 0.1 μg/mL of anti-human IgD conjugated to dextran (Fina Biosolutions, Rockville, MD). All samples were incubated at 37°C/5% CO2 for 18–24 h. Upon incubation, samples were transferred to 12×75 mm FACS tubes and stained for 30 min at room temperature with a cocktail of anti-human antibodies (CD19-PE [clone HIB19, BD Biosciences]; CD27-BV510; CD45-PerCP; CD69-APC [clone L78, BD Biosciences], CD86-BV421). Red blood cells were lysed by incubating samples for 10 min at room temperature with 2 mL of 1X FACSLyse solution (BD Biosciences, Franklin Lakes, NJ). Upon lysis, remaining cells were washed with 2 mL of Phosphate-Buffered Saline supplemented with 1% Bovine Serum Albumin, followed by fixation of pelleted cells with 500 μL of 1% Paraformaldehyde solution. Samples were acquired on a properly calibrated BD flow cytometer and analyzed for CD69 expression on CD19+, CD19+ CD27− or CD19+ CD27+-gated B cells, capturing activation of total, naïve, and memory B cells, respectively. Quantum™ APC MESF beads (Bangs Laboratories, Fishers, IN) were utilized to standardize the fluorescence intensity of CD69. The CD69 MESF expression difference between the anti-IgD stimulated and unstimulated sample conditions at post-dose timepoints was compared to that of pre-dose samples to calculate the extent of CD69 inhibition.
Statistical Analysis
Sample size was not based on statistical testing. Cohorts of 8 participants randomized 6:2 (BIIB091:placebo) were considered adequate for characterization of the single- and multiple-dose safety, tolerability, and PK profile of BIIB091 in this first-in-human study. PK parameters were calculated by Phoenix WinNonlin® (Certara, Radnor, PA, USA) version 8 or above.
Data for the SAD and MAD parts of the study were analyzed separately, with placebo participants pooled for each part. Descriptive statistics were used to summarize the data. Safety data were summarized in participants who received at least 1 dose of study treatment (BIIB091 or placebo). Concentration and PK parameters were summarized in participants who received at least 1 dose of BIIB091 with sufficient PK concentration data to allow calculation of PK parameters. PD data were summarized in participants who received at least 1 dose of study treatment with at least 1 PD measurement after baseline.
The dose proportionality of BIIB091 after administration of a single dose (day 1 in SAD and MAD) and multiple doses (day 14 in MAD) was assessed based on maximum concentration (Cmax) and area under the curve to infinity (AUCinf) separately, using both graphic presentations of the dose-normalized parameters and linear modeling approach for the log-transformed parameters versus log-transformed doses.
The natural log-transformed values of the last area under the curve (AUClast), AUCinf, and Cmax were analyzed using a mixed-effects model to estimate the effect of food on BIIB091 single-dose PK. Treatment (fasted versus fed) was considered a fixed effect and participant a random effect.
Results
Participants
A total of 64 participants were randomized in the study (Table 1). Forty participants were randomized in the SAD part of the study, of which 30 participants were randomized to receive a single dose of 50, 150, 300, 600, or 1200 mg BIIB091, and 10 were randomized to receive a single dose of placebo in the SAD part of the study. The 6 participants who were randomized to 300 mg BIIB091 and the 2 who were randomized to placebo received a single dose in the fasted state and a single dose in the fed state. Twenty-four participants were randomized in the MAD part of the study, of which 18 were randomized to 50, 150, or 300 mg BIIB091 BID for 14 days and 6 were randomized to placebo BID for 14 days. One participant in the MAD part of the study received placebo for 13 days but discontinued treatment prior to the last dose. All participants in the study treatment group received all doses and completed scheduled activities in the study.
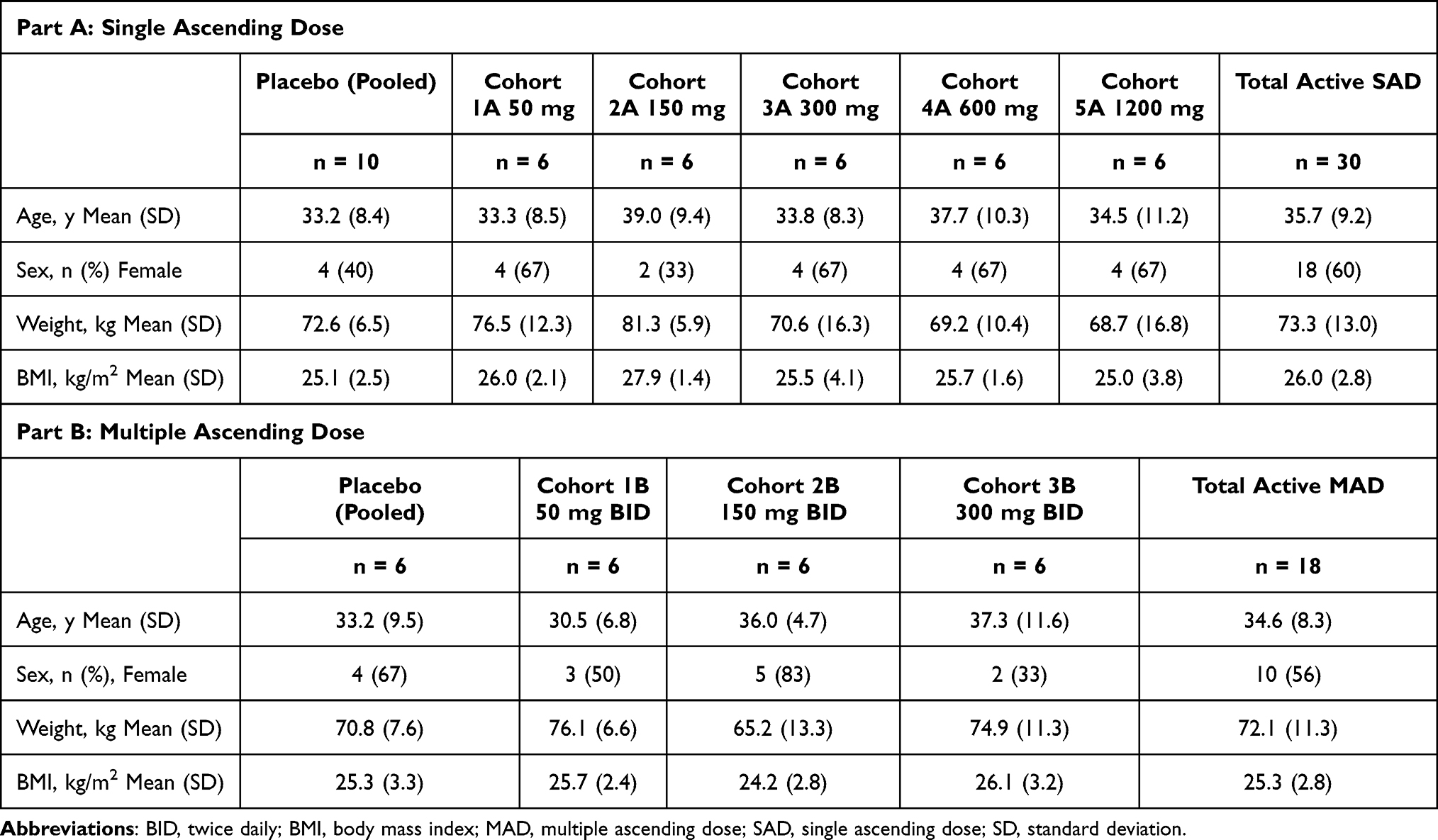 |
Table 1 Baseline Characteristics |
Eighteen male and 22 female participants aged 19–54 years were randomized in the SAD part of the study, of which most participants were Black or African American or White. The predominance of Black or African American and White participants reflects the demographics of the US study site; MS prevalence is known to vary by ancestry, with the highest prevalence among individuals of European descent.23 Ten male and 14 female participants aged 21–51 years were randomized in the MAD part of the study, of which the majority of participants were Black or African American or White. There were no significant differences in participant demographics between treatment groups in either part of the study. Additionally, no baseline medical histories or screening test data raised clinical concerns for any participant before dosing with the study treatment.
This study was conducted in accordance with the ethical principles outlined in the Declaration of Helsinki. Investigators obtained institutional review board (IRB) approval from Midlands IRB (Overland Park, KS) for the study protocol and all amendments. Written informed consent was obtained from each participant prior to any eligibility-related evaluations. Participants were given adequate time to review the informed consent information and were afforded the opportunity to ask questions, all of which were answered, regarding all aspects of the study. Each participant received a copy of the signed and dated informed consent form (approval number: 257HV101).
Safety
A total of 14 participants (4 in SAD and 10 in MAD) experienced AEs, all of which were mild in severity. No dose-limiting clinical AEs were identified in either part of the study. All AEs were reported as recovered/resolved, and no AEs led to withdrawal from the study drug or the study. There were no SAEs, hospitalizations, or deaths reported. There were no BIIB091 treatment-emergent clinical laboratory abnormalities resulting in AEs, and no treatment-emergent vital sign abnormalities except for 1 participant who had pyrexia 12 days after receiving a single dose of BIIB091, which resolved after 2 days. The Investigator deemed this unrelated to the study drug.
All AEs in the SAD part of the study were reported at the highest dose tested. The most frequently reported AE in the SAD part of the study was nausea (2 active and 1 placebo participants; Table 2). The AEs in the MAD part of the study were seen at all dose levels. The most frequently reported AEs in the MAD part of the study were headache (2 participants on 150 mg BID and 1 placebo), polyuria (2 participants on 50 mg BID), and constipation (1 participant on 150 mg BID and 1 placebo). There was 1 AE of vomiting reported by 1 participant in the 300 mg BID group (Table 3).
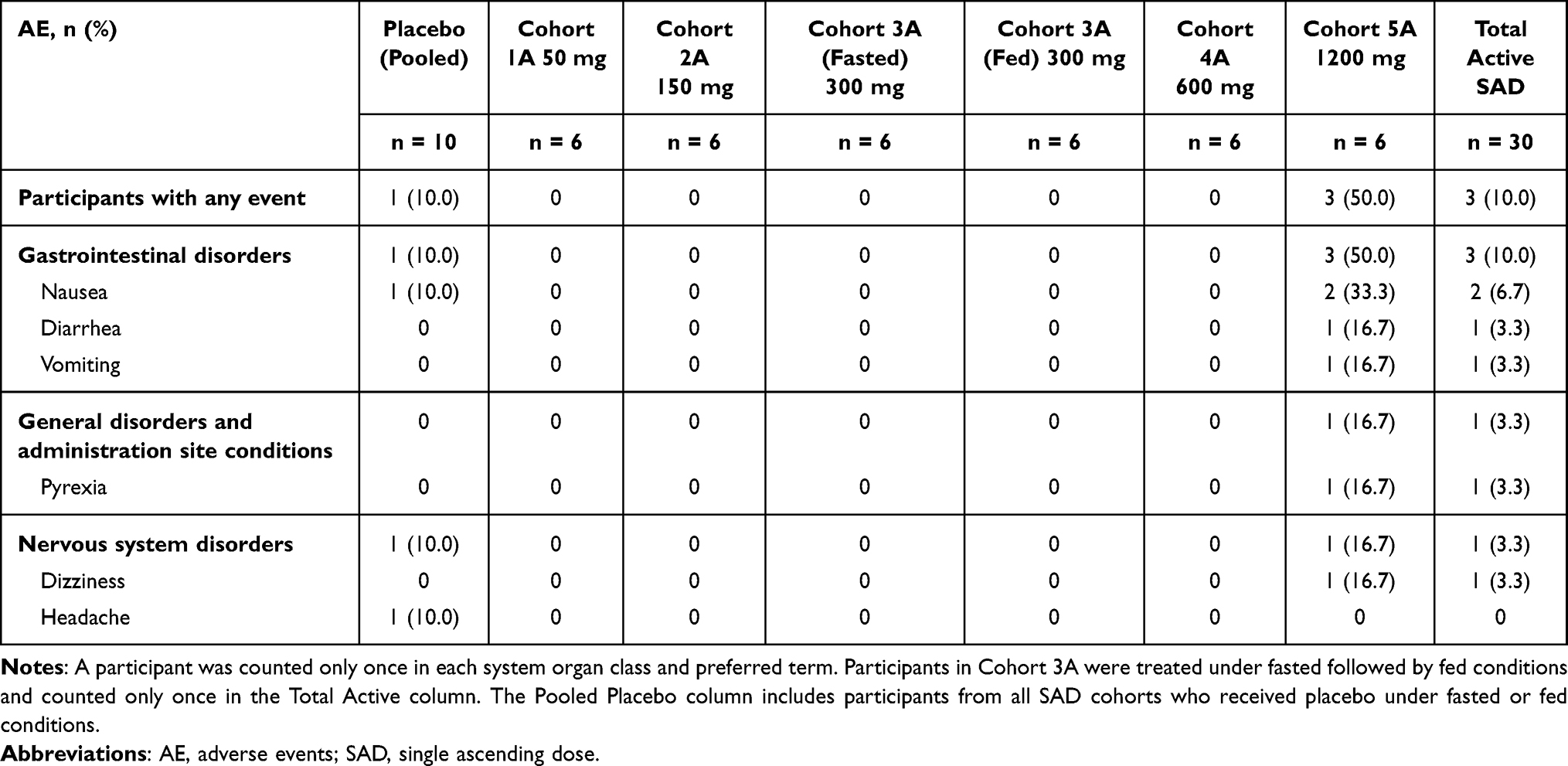 |
Table 2 Adverse Events by System Organ Class and Preferred Term: Single Ascending Dose |
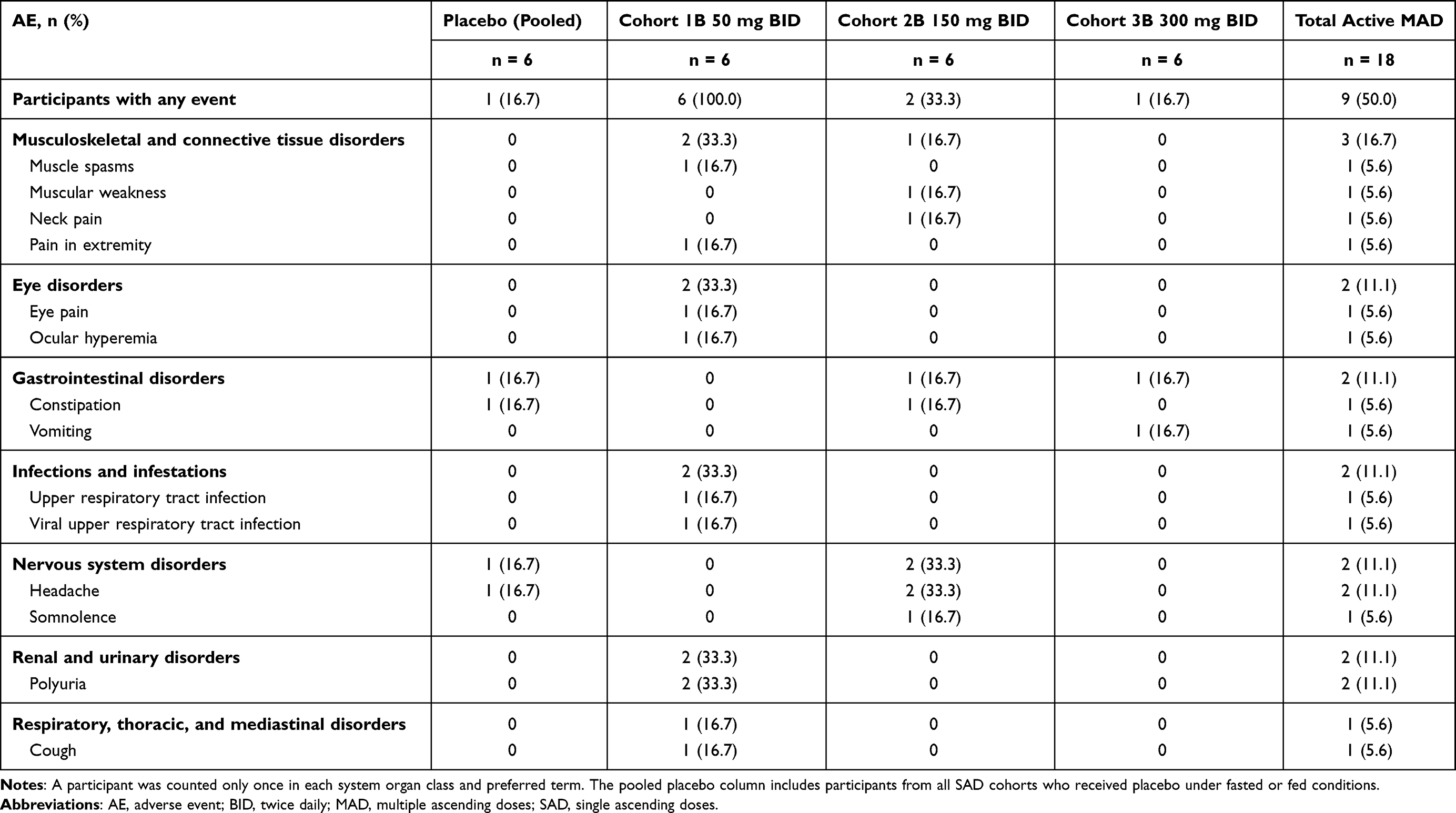 |
Table 3 AEs by System Organ Class and Preferred Term Part B: Multiple Ascending Doses |
Based on the continuous ECG data, BIIB091 at the studied doses did not have a clinically relevant effect on heart rate or the PR interval and QRS duration, and there were no cardiac AEs. No participant experienced QTcF >480 ms, although some QTc prolongations were noted at doses higher than 600 mg in SAD part of the study where the least square mean placebo-corrected QTcF changing from baseline (∆∆QTcF) was >10 ms but <20 ms at 600 mg and >20 ms at 1200 mg according to the by-timepoint analysis. In the MAD part of the study, small QTc prolongation was only noted on day 14 at the highest tested dose (300 mg BID), with the maximal least square mean ∆∆QTcF around 12 ms observed at 1 hour post-dose.
Pharmacokinetics
As shown in Figure 3A, following administration of single doses of 50–1200 mg under fasted conditions, the absorption of BIIB091 was rapid, with a median time to maximum concentration (Tmax) occurring between 0.8 and 2.0 hours post-dose. After reaching Cmax following a single dose, plasma concentrations of BIIB091 appeared to decline in a biphasic manner. The geometric mean values of the terminal half-life (t1/2) ranged from 6.0–14.4 hours across all dose levels, with the individual t1/2 ranging from 4.2–33.1 hours. Over the dose range of 50–1200 mg, single-dosing AUCinf and Cmax increased in an approximately dose-proportional manner, with estimates of the slopes (90% CI) from the statistical dose proportionality analysis of 1.014 (0.795–1.233) and 0.847 (0.609–1.085), respectively, which approximated unity (Figure 3B and Table 4). At the starting dose of 50 mg, the geometric mean (CV%) of AUCinf and Cmax was 2098.4 (18.4%) h*ng/mL and 512.3 (51.5%) ng/mL, respectively. At the maximal dose of 1200 mg, the geometric mean (CV%) of AUCinf and Cmax was 55706.8 (25.5%) h*ng/mL and 8400.4 (10.7%) ng/mL, respectively, with the maximal individual exposure achieved in this study being 67885.19 h*ng/mL for AUCinf and 9740.00 ng/mL for Cmax. Food effect of BIIB091 was investigated in the SAD part of this study at 300 mg with high-fat meals (Figure 3C). With the prototype DiC formulation, the fed state showed a delayed Tmax by approximately 5.2 hours and a positive food effect with AUCinf, AUClast, and Cmax increased by 2.046-, 2.646-, and 1.420-fold, respectively, when compared with administration in the fasted state. Food also decreased the inter-participant variability, although it remained moderately high.
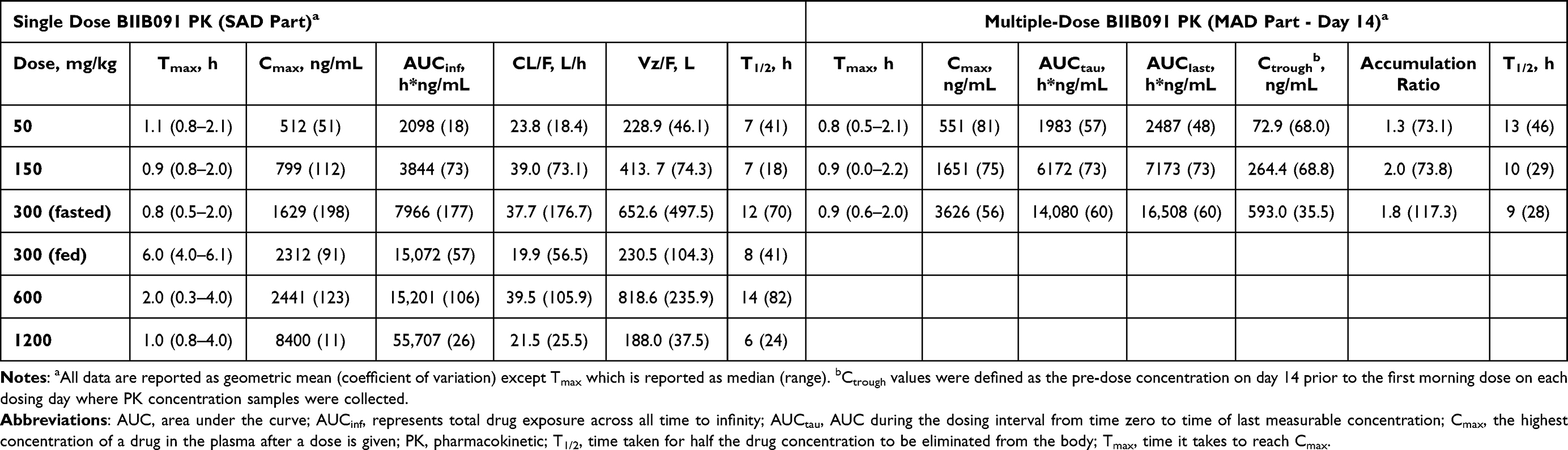 |
Table 4 Single- and Multiple-Dose Pharmacokinetics |
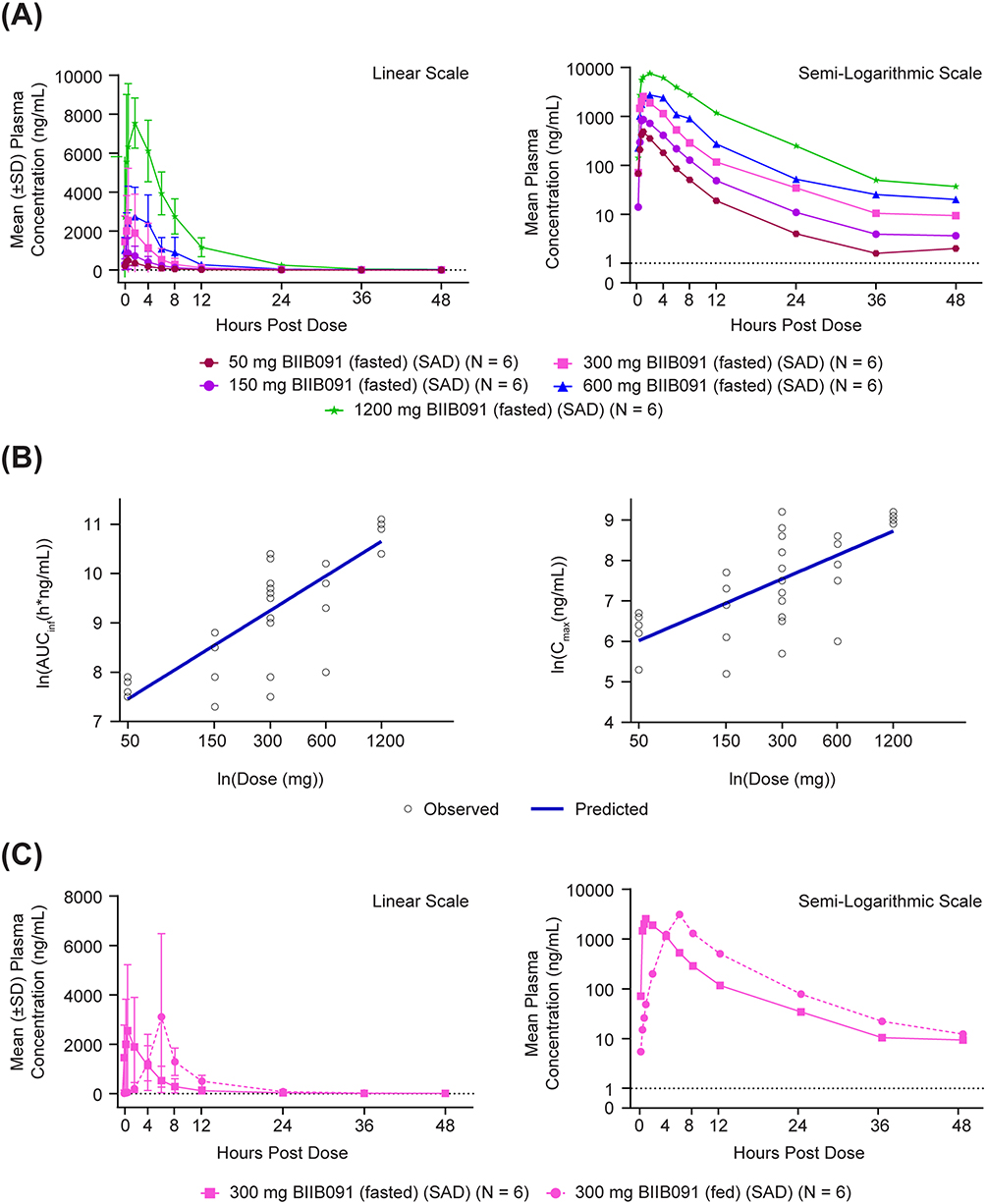 |
Figure 3 Single dose pharmacokinetics of BIIB091. (A) PK profiles across dose levels. (B) Dose proportionality, C1: Ln (AUCinf) versus Ln (dose), C2: Ln (Cmax) versus Ln (dose). (C) Food effect on PK. Abbreviations: PK, pharmacokinetics; SAD, single ascending dose; SD, standard deviation; Cmax, maximum concentration; PK, pharmacokinetics; AUC, area under the curve. Notes: Lower limit of quantification = 1 ng/mL and is represented by the axis line. Pre-dose measurements below the level of quantification (BLQ) were set to zero before statistics were calculated. Post-dose BLQ measurements were set to missing before statistics were calculated. X-axis is displayed as [ln(dose)] with actual dose below for interpretation. |
Similar to the SAD part of the study, the PK profiles (Figure 4A) after 50–300 mg of BIIB091 BID for 14 days showed rapid absorption, with a median Tmax across dose levels that ranged from 0.7–1.5 hours on day 1 and 0.8–0.9 hours on day 14, followed by a biphasic decline on day 14 with plasma concentrations measured up to 48 hours post-dose. Visual inspection (Figure 4B) of geometric mean values of Ctrough indicated that steady state was achieved by approximately day 5 across the tested dose level. The geometric mean values of the terminal t1/2 ranged from 8.9–12.8 hours on day 14, similar to the single-dosing values. The geometric mean values of the accumulation ratio on day 14 were calculated to be 1.3, 2.0, and 1.8 for the dose levels of 50, 150, and 300 mg, respectively. The exposures also increased in an approximately dose-proportional manner in the MAD part of the study. Therefore, it was considered there was no significant accumulation for BIIB091 at the steady state, and multiple dosing would not significantly alter BIIB091 PK. Generally, BIIB091 displayed moderately high inter-participant variability for the PK parameters in both SAD and MAD parts, with this prototype DiC formulation.
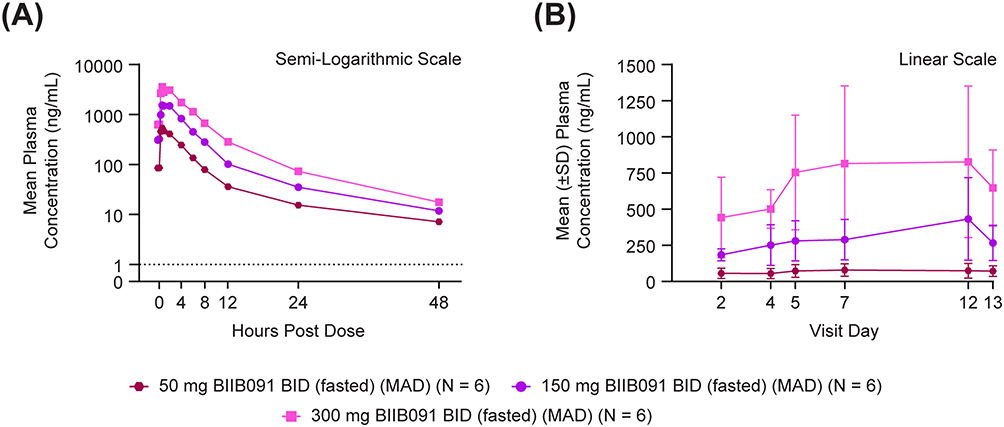 |
Figure 4 Multiple-dose pharmacokinetics of BIIB091. (A) Day 14. (B) Baseline to Day 13. Abbreviations: BID, twice daily; MAD, multiple ascending dose; SD, standard deviation. Notes: Lower limit of quantification = 1 ng/mL and is represented by the dashed line. Pre-dose measurements below the level of quantification (BLQ) were set to zero before statistics were calculated. Post-dose BLQ measurements were set to missing before statistics were calculated. |
The renal excretion of BIIB091 was assessed in the SAD part of the study. Across all dose cohorts, 2.9–9.8% of BIIB091 were excreted unchanged in urine up to 48 hours post-dose, with the majority (>90%) excreted within the first 24 hours. The clearance (CLr) ranged from 0.9–2.2 L/h for doses between 50 and 1200 mg, with the geometric coefficient of variation ranging from 18.9–62.7%. This CLr level is close to the product of the unbound fraction in human plasma (fu=0.197) multiplying the glomerular filtration rate (GFR) in humans (5.4 L/h for a typical adult with a body surface area of 1.73 m2), suggesting that BIIB091 renal excretion is primarily driven by glomerular filtration with minimal active secretion or reabsorption.
Pharmacodynamics
BIIB091 suppressed B cell activation, as measured by CD69 expression on anti-IgD–stimulated CD19+ B cells. CD69 was chosen as the surrogate biomarker of B cell activation because the upregulation of CD69 is BTK dependent24–27 and correlated with the broader transcriptional program that is induced upon BCR-mediated B cell activation. Following the administration of single doses of 50–1200 mg BIIB091 in the SAD part of the study and multiple doses of 50 to 300 mg BIIB091 BID in the MAD part of the study, there was a mean inhibition of over 90% in CD69 expression on CD19+ B cells (measured by molecules of equivalent soluble fluorochrome), occurring 1–2 hours post-dose (Figure 5A and B). At 24 hours post-dose, the mean (SD) percentages of CD69 inhibition on anti-IgD-stimulated CD19+ B cells for the 1200, 600, 300, and 150 mg BIIB091 cohorts were 94% (6.2), 70% (19), 42% (49), and 41% (21), respectively. Mean CD69 inhibition for the 50 mg BIIB091 cohort at 24 hours post-dose had returned to similar levels observed following administration of placebo.
 |
Figure 5 B cell receptor-mediated CD69 expression on CD19+ B cells from placebo-controlled (A) single dose BIIB091 or (B) multiple-dose BIIB091. (A) BCR-induced CD69 expression on CD19+ B cells from single dose BIIB091- or Placebo-treated patients. (B) BCR-induced CD69 expression on CD19+ B cells from multiple dose BIIB091- or Placebo-treated patients. Abbreviations: BID, twice daily; MESF, molecules of equivalent soluble fluorochrome; SD, standard deviation. Notes: CD19 (cluster differentiation 19) is a human transmembrane protein that is a biomarker for B cell development. CD69 (cluster differentiation 69) is a human transmembrane; C-type lectin protein is an early marker of lymphocyte activation. |
In the SAD part of the study, the overall duration of inhibition suggested a dose dependency, with complete inhibition maintained at 24 hours post-dose following a single dose of 1200 mg BIIB091 and relatively lower levels of mean CD69 inhibition observed at the same timepoint following administration of single doses of 50, 150, 300, and 600 mg BIIB091. BIIB091 dosing with a high-fat meal (single dose 300 mg fed cohort) appeared to prolong the duration of CD69 inhibition, compared with dose administration in the fasted state. Following administration of 300 mg BIIB091 in the fed state, >90% mean CD69 inhibition on CD19+ B cells was maintained at 24 hours post-dose; however, following administration of 300 mg BIIB091 in the fasted state, the mean (SD) percentage of inhibition of CD69 at 24 hours post-dose was markedly less (42% [49]).
To assess the reversibility of the inhibitory pharmacodynamic effect, CD69 inhibition was measured following administration of the final BIIB091 dose on day 14 in the MAD part of the study. For all BIIB091 dose levels, >90% mean CD69 inhibition on CD19+ B cells was observed by 1 hour post final dose on day 14. The extent of the mean CD69 inhibition returned to the levels observed following administration of placebo by 8 hours post final dose for the 50 mg BIIB091 cohort (N=2) and by 24 hours post final dose for the 150 and 300 mg cohorts (N=6, respectively) (Figure 5B). For the 50 mg cohort, mean CD69 expression levels at 12 hours and 24 hours post final BIIB091 dose were higher than the mean CD69 levels at baseline, however, these data should be interpreted with caution given the small size of evaluated samples (N=2) at these timepoints. The trends in CD69 inhibition observed for total CD19+ B cells were similar for CD19+ CD27- (naïve) B cells and CD69 inhibition was also observed in the CD19+ CD27+ (memory) B cells.21
Multiple doses between 50 and 300 mg BIIB091 BID inhibited BTK phosphorylation, with peak inhibition generally occurring by 2 hours post-dose. This inhibition was maintained throughout the dosing interval and up to 12 hours post final dose. After 12 hours, inhibition decreased, and by 48 hours after the final dose, BTK phosphorylation levels were similar to those observed following administration of the placebo. BIIB091 inhibited the constitutive phosphorylation of BTK. There were no discernable decreasing trends in circulating lymphocyte subsets following administration of any dose level of BIIB091.21
Discussion
BIIB091 was well tolerated at single doses up to 1200 mg and multiple doses up to 300 mg BID, with all reported AEs being mild in severity using the DiC immediate release formulation. BIIB091 was shown to be potent in inhibiting B cell activation, as shown by CD69 inhibition. The results from this first-in-human study support further development of BIIB091 and are being assessed in a 2-part, randomized, blinded, active-controlled Phase 2 trial (FUSION) as a monotherapy or in combination with diroximel fumarate in patients with relapsing MS (NCT05798520).
Preclinical studies demonstrated that BIIB091 has favorable pharmacokinetic and pharmacodynamic properties, including potent in vitro inhibition of BTK and robust suppression of B-cell activation, with wide toxicology margins observed in nonclinical species over the human projected efficacious exposure. These findings, described in Hopkins et al, supported the dose selection rationale applied in the present study.19 Single doses of 50 to 1200 mg BIIB091 and multiple doses of 50 to 300 mg BIIB091 BID exhibited approximately dose-proportional PK, with median Tmax occurring within the first 2 hours of dosing. For single BIIB091 doses of 50 to 1200 mg, geometric mean T1/2 ranged from 6.0 to 14.4 hours. T1/2 ranged from 8.9 to 12.8 hours on day 14 following multiple doses of 50 to 300 mg BIIB091 BID. Geometric mean accumulation ratios for BIIB091 over 14-day dosing were 1.3 to 2.0, with steady state achieved between study days 2 and 5. High between-participant variability in area under the curve (AUC0-12h) and Cmax on day 14 at 300 mg BID was observed across all cohorts in the MAD part of the study. Administration of a single dose of 300 mg BIIB091 with food resulted in a delay of Tmax by approximately 5.2 hours with increases in AUCinf and Cmax by 2.046- and 1.420-fold, respectively. Food also slightly decreased the between-participant variability. The excretion of BIIB091 was assessed in the SAD part of the study and the data suggested that BIIB091 excretion is primarily driven by glomerular filtration with minimal active secretion or reabsorption. BIIB091 metabolite profiles were explored in this study and showed that unchanged BIIB091 was the major drug-related component in plasma. Detailed metabolite profiling will be reported elsewhere. Overall, BIIB091 demonstrates the PK is favorable for further development.
The most frequently reported AE in the SAD part of the study was nausea, while in the MAD part of the study, the most frequent was headache, polyuria, and constipation. There were no dose-limiting clinical AEs identified, AEs leading to discontinuation from treatment or study withdrawal, or SAEs. BIIB091 at the studied doses did not have a clinically relevant effect on heart rate or on the PR interval and QRS duration and there were no cardiac-related clinical AEs reported. Although some events with ∆∆QTcF >10 ms were noted with high dose levels (single dosing ≥ 600 mg or on day 14 after 300 mg BID multiple dosing), no subject in the entire study experienced QTcF >480 ms. Due to the small sample size of this study in terms of the QT effect assessment, no conclusion can be drawn and the QT prolongation potential of BIIB091 is being further investigated in other studies. Hepatic events have been reported with other BTK inhibitors after repeated dosing. Specifically, there were 2 cases of drug-induced hepatic injury in the evobrutinib Phase 3 study, and 4.0% of participants in the tolebrutinib Phase 3 trial experienced liver enzymes levels at least 3 times the upper limit of normal, with one participant requiring a liver transplant.28,29 In this first-in-human study, no hepatic events were identified. Furthermore, while bleeding events have been observed upon treatment with some BTK inhibitors (reversible and nonreversible),30,31 no obvious trends in coagulation data across the dose levels were seen in this study. Although values for some coagulation parameters were outside of the clinical reference ranges at some timepoints, there were no bleeding-related AEs reported. Overall, there were no dose-dependent changes, including liver enzyme and coagulation values.
BIIB091 suppression of B cell activation was measured by CD69 expression on anti-IgD–stimulated CD19+ B cells. In both SAD and MAD parts of the study, BIIB091 has a rapid onset of >90% mean CD69 inhibition as early as 1 hour in all dose levels coinciding with the median Tmax. Administering a single dose of 300 mg BIIB091 with a high-fat meal appeared to prolong the duration of CD69 inhibition, as compared with dose administration in the fasted state, and >90% mean CD69 inhibition on CD19+ B cells was maintained at 24 hours post-dose. Following administration of 150 or 300 mg BIIB091 BID for 14 days, >90% mean inhibition of CD69 expression on CD19+ B cells was maintained from day 2 through to 2 hours post-dose on day 14. The overall duration of inhibition suggested a dose dependency. In parallel, BTK inhibition was shown by inhibition of BTK phosphorylation. Multiple doses of BIIB091 between 50 and 300 mg BID demonstrated peak BTK inhibition by 2 hours post-dose that was maintained during the dosing interval and up to 12 hours post final dose across all dose levels. In addition to quick action, there was evidence of reversibility of BTK inhibition and B cell inhibition following the dosing period. At 24 hours post-dose, mean CD69 inhibition for the single 50 mg BIIB091 cohort had returned to similar levels observed following administration of placebo. By day 15 in the MAD part of the study (corresponding to 24 hours post final dose), mean CD69 expression levels on B cells increased and were similar to those of placebo-treated participants, indicating rapid reversibility of B cell inhibitory effect. By 48 hours post last dose of multiple doses of between 50 and 300 mg BIIB091 BID, the levels of inhibition of BTK phosphorylation were similar to that observed following administration of placebo. The fact that CD69 inhibition had reverted to almost the placebo level 24 hours after the last dose of BIIB091 in the MAD part suggests that stopping BIIB091 dosing could have an immediate effect on AE management.
In addition to BIIB091 blocking functions downstream of BCR and Fc receptor (FcR) in vitro cellular assays with high potency, samples from 300 mg BID cohort in the MAD part of this first-in-human study have shown that in vivo BIIB091 treatment reduces circulating B cell proliferation and B cells’ mitochondria respiration.32 BIIB091 also reduces activation-induced costimulatory molecule expression, such as CD80 and CD86, and mediates an anti-inflammatory shift in B cell responses which in turn reduces T cell pro-inflammatory responses via B and T cell interaction.22 Immunophenotyping of in vivo samples exposed to BIIB091 found an increased proportion of circulating transitional B cells and a decreased proportion of memory B cells, suggesting that B cell subsets are differentially affected by BIIB091 toward an anti-inflammatory phenotype.22 The in vitro and in vivo results suggest that BIIB091 could efficiently mitigate pathological processes downstream of BTK that are implicated in MS. During this study, there were no discernable decreasing trends in changes in circulating lymphocyte subsets following administration of BIIB091.
Multiple compounds targeting BTK inhibition for MS have been developed and some of them have ongoing Phase 3 studies, including fenebrutinib and remibrutinib. Evobrutinib Phase 3 trials (evolutionRMS1 [NCT04338022] and evolutionRMS2 [NCT04338061])28 and tolebrutinib Phase 3 trials (GEMINI 1 [NCT04410978] and GEMINI 2 [NCT04410991])33 did not demonstrate superior efficacy versus teriflunomide in reducing annualized relapse rate (ARR) in relapsing MS population. On the other hand, fenebrutinib sharing similar binding mechanisms to BTK with BIIB091 has reported strong ARR repression at 0.04 for all participants analyzed at week 48.34 Evobrutinib and tolebrutinib covalently reacted with C481 residue on BTK to gain the benefit of longer-lasting inhibitory activity. In contrast, BIIB091 is a reversible BTK inhibitor binding to the H3 pocket to achieve potency and selectivity without extensive binding and is highly selective for BTK.19 Furthermore, BIIB091 sequesters Y551, which is thought to be an important determinant of potency against FcR signaling. That is, BTK inhibitors that sequester Y551 are equally potent in inhibiting BCR and FcR signaling, whereas BTK inhibitors that do not sequester Y551 showed reduced activity against basophils and B cells as measured in whole blood.35 The biological hypothesis that the differentiation of the binding mechanism of BIIB091 can be translated into superior clinical benefits will be evaluated in the Phase 2 study.
Conclusions
BIIB091 was well tolerated at single doses up to 1200 mg and repeat doses up to 300 mg BID, with all reported AEs being mild in severity. No dose-limiting clinical AEs were identified in the single or multiple ascending dose parts of the study. BIIB091 PK showed an approximately dose-linear exposure following the administration of single and multiple doses. BIIB091 doses >300 mg suppressed B cell activation >90% over the dosing interval as measured by CD69 expression on anti-IgD–stimulated CD19+ B cells. This study demonstrated that BIIB091, with the current prototype DiC immediate release formulation, was sensitive to food effect, where 2.046-fold and 1.420-fold increases in AUCinf and Cmax, respectively, were observed after dosing with high-fat meal. The positive food effect on the PK of BIIB091 prototype DiC formulation translated into a more sustained PD response. Therefore, food effects will be a key factor for dose regimen selection, and further food-effect assessment is needed pending formulation development to support Phase 2 development. The safety, PK, and PD profiles presented here support continued development of BIIB091, an orally active, selective, reversible, small molecule noncovalent inhibitor of BTK, for the treatment of MS. Currently, BIIB091 is being evaluated in patients with relapsing MS in a Phase 2 study FUSION (NCT05798520).
Data Sharing Statement
Individual participant data collected during the study may be shared after anonymization and upon approval of a research proposal. Biogen commits to sharing patient-level data, study-level data, and relevant study documents (such as clinical study reports and study protocols) with qualified scientific researchers who submit a methodologically sound proposal.
All data requests are reviewed by Biogen in accordance with its Clinical Trial Transparency and Data Sharing Policy. Deidentified data and documents will be shared under data-sharing agreements designed to protect against participant reidentification.
Data may be made available after publication of the study results and for a period determined by Biogen. Requests for data access should be submitted through Biogen’s established data-sharing request process.
Ethics Approval and Informed Consent
Investigators obtained IRB approval (Midlands IRB; Overland Park, KS) for the study protocol and all amendments. Written informed consent was obtained from each participant prior to evaluations performed for eligibility. Participants were given adequate time to review the information in the informed consent and could ask, and have answered, questions concerning all portions of the conduct of the study. Participants were provided with a copy of the signed and dated ICF (approval number: 257HV101).
Consent for Publication
All authors consent to the publication of this manuscript.
Acknowledgments
Biogen provided funding for medical writing support in the development of this paper; Lauren Ramsey, PharmD, CMPP (Excel Scientific Solutions, Inc., Fairfield, CT) provided writing assistance with the manuscript based on input from the authors, and Cara Farrell (Excel Scientific Solutions, Inc., Fairfield, CT) copyedited and styled the manuscript per journal requirements. We thank trial participants in the study and colleagues involved in the trial conduction.
Author Contributions
*Hui-Hsin Tsai and Yi Gu are co-first authors.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by Biogen.
Disclosure
HHT, YG, KR, EB, BH, JH, and SC are employees of and hold stock in Biogen.
MS was an employee of Biogen at the time of the study and may hold stock in the company. MS is an employee of Nimbus Therapeutics at the time of this manuscript submission and may hold stock/stock options in the company.
MA was an employee of Biogen at the time of the study and may hold stock in the company. MA is an employee of Alexion Pharmaceuticals at the time of manuscript submission and may hold stock/stock options in the company.
JK was an employee of Covance Clinical Research Unit, Inc., at the time of the study and have held stock/stock options in the company. JK is an employee of Fortrea at the time of manuscript submission and may hold stock/stock options in the company.
AC was an employee of Biogen at the time of the study and may hold stock in the company.
Dr Eris Bame reports a patent WO/2024/233379 issued to Biogen MA Inc.
Dr Jerome Hanna reports other from Biogen, outside the submitted work; and I am an employee of Biogen (the company which sponsored the study). Dr Brian Hopkins reports a patent WO2018191577 issued to Biogen, a patent 20240216330 issued to Biogen. Dr Matthew Scaramozza reports other from Biogen, other from Nimbus Therapeutics, outside the submitted work; In addition, Dr Matthew Scaramozza has a patent WO202433374 A1 licensed to Biogen.
The authors report no other conflicts of interest in this work.
References
1. Walton C, King R, Rechtman L, et al. Rising prevalence of multiple sclerosis worldwide: insights from the Atlas of MS, third edition. Mult Scler. 2020;26(14):1816–18. doi:10.1177/1352458520970841
2. Didonna A, Oksenberg JR. The genetics of multiple sclerosis. In: Zagon IS, McLaughlin PJ, editors. Multiple Sclerosis: Perspectives in Treatment and Pathogenesis. Brisbane (AU): Exon Publications; 2017.
3. Hakim EA, Bakheit AM, Bryant TN, et al. The social impact of multiple sclerosis--a study of 305 patients and their relatives. Disabil Rehabil. 2000;22(6):288–293. doi:10.1080/096382800296755
4. Jakimovski D, Bittner S, Zivadinov R, et al. Multiple sclerosis. Lancet. 2024;403(10422):183–202. doi:10.1016/S0140-6736(23)01473-3
5. Filipi M, Jack S. Interferons in the treatment of multiple sclerosis: a clinical efficacy, safety, and tolerability update. Int J MS Care. 2020;22(4):165–172. doi:10.7224/1537-2073.2018-063
6. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–234. doi:10.1056/NEJMoa1601277
7. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–220. doi:10.1056/NEJMoa1606468
8. Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–557. doi:10.1056/NEJMoa1917246
9. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–471. doi:10.1002/ana.21867
10. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–688. doi:10.1056/NEJMoa0706383
11. Brunner C, Müller B, Wirth T. Bruton’s tyrosine kinase is involved in innate and adaptive immunity. Histol Histopathol. 2005;20(3):945–955. doi:10.14670/HH-20.945
12. Bar-Or A, Li R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 2021;20(6):470–483. doi:10.1016/S1474-4422(21)00063-6
13. Saberi D, Geladaris A, Dybowski S, Weber MS. Bruton’s tyrosine kinase as a promising therapeutic target for multiple sclerosis. Expert Opin Ther Targets. 2023;27(4–5):347–359. doi:10.1080/14728222.2023.2218615
14. Geladaris A, Torke S, Weber MS. Bruton’s tyrosine kinase inhibitors in multiple sclerosis: pioneering the path towards treatment of progression? CNS Drugs. 2022;36(10):1019–1030. doi:10.1007/s40263-022-00951-z
15. Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics. 2017;14(4):835–841. doi:10.1007/s13311-017-0557-4
16. Nyhoff LE, Clark ES, Barron BL, Bonami RH, Khan WN, Kendall PL. Bruton’s tyrosine kinase is not essential for B cell survival beyond early developmental stages. J Immunol. 2018;200(7):2352–2361. doi:10.4049/jimmunol.1701489
17. Rip J, Van Der Ploeg EK, Hendriks RW, Corneth OBJ. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit Rev Immunol. 2018;38(1):17–62. doi:10.1615/CritRevImmunol.2018025184
18. Krämer J, Bar-Or A, Turner TJ, Wiendl H. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat Rev Neurol. 2023;19(5):289–304. doi:10.1038/s41582-023-00800-7
19. Hopkins BT, Bame E, Bajrami B, et al. Discovery and preclinical characterization of BIIB091, a reversible, selective BTK inhibitor for the treatment of multiple sclerosis. J Med Chem. 2022;65(2):1206–1224. doi:10.1021/acs.jmedchem.1c00926
20. Mano H. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 1999;10(3–4):267–280. doi:10.1016/S1359-6101(99)00019-2
21. Bame E, Tang H, Burns JC, et al. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and Fc receptor signaling and downstream functions in B cells and myeloid cells. Clin Transl Immunology. 2021;10(6):e1295. doi:10.1002/cti2.1295
22. Li R, Tang H, Burns JC, et al. BTK inhibition limits B-cell-T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 2022;143(4):505–521. doi:10.1007/s00401-022-02411-w
23. Hittle M, Culpepper WJ, Langer-Gould A, et al. Population-based estimates for the prevalence of multiple sclerosis in the united states by race, ethnicity, age, sex, and geographic region. JAMA Neurol. 2023;80(7):693–701. doi:10.1001/jamaneurol.2023.1135
24. Benson MJ, Rodriguez V, von Schack D, et al. Modeling the clinical phenotype of BTK inhibition in the mature murine immune system. J Immunol. 2014;193(1):185–197. doi:10.4049/jimmunol.1302570
25. Corneth OB, Klein Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. 2016;393:67–105. doi:10.1007/82_2015_478
26. Rankin AL, Seth N, Keegan S, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191(9):4540–4550. doi:10.4049/jimmunol.1301553
27. Xu D, Kim Y, Postelnek J, et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther. 2012;341(1):90–103. doi:10.1124/jpet.111.187740
28. Montalban X, Vermersch P, Arnold DL, et al. Safety and efficacy of evobrutinib in relapsing multiple sclerosis (evolutionRMS1 and evolutionRMS2): two multicentre, randomised, double-blind, active-controlled, phase 3 trials. Lancet Neurol. 2024;23(11):1119–1132. doi:10.1016/S1474-4422(24)00328-4
29. Fox RJ, Bar-Or A, Traboulsee A, et al. Tolebrutinib in nonrelapsing secondary progressive multiple sclerosis. N Engl J Med. 2025;392(19):1883–1892. doi:10.1056/NEJMoa2415988
30. Lamanna N, Tam CS, Woyach JA, et al. Evaluation of bleeding risk in patients who received pirtobrutinib in the presence or absence of antithrombotic therapy. EJHaem. 2024;5(5):929–939. doi:10.1002/jha2.1013
31. von Hundelshausen P, Siess W. Bleeding by bruton tyrosine kinase-inhibitors: dependency on drug type and disease. Cancers. 2021;13(5):1103. doi:10.3390/cancers13051103
32. Li R, Lei Y, Rezk A, et al. Oxidative phosphorylation regulates B cell effector cytokines and promotes inflammation in multiple sclerosis. Sci Immunol. 2024;9(95):eadk0865. doi:10.1126/sciimmunol.adk0865
33. Oh J, Arnold DL, Cree BAC, et al. Tolebrutinib versus teriflunomide in relapsing multiple sclerosis. N Engl J Med. 2025;392(19):1893–1904. doi:10.1056/NEJMoa2415985
34. Bar-Or A, Oh J, Dufek M, et al. Fenebrutinib maintains low disease activity in P1612 relapsing multiple sclerosis: results from the FENopta trial open-label extension.
35. Bender AT, Gardberg A, Pereira A, et al. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of Fc receptor but not B-cell receptor signaling. Mol Pharmacol. 2017;91(3):208–219. doi:10.1124/mol.116.107037
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.