Back to Journals » Journal of Blood Medicine » Volume 12
Final Results of the Prospective ADVATE® Immune Tolerance Induction Registry (PAIR) Study with Plasma- and Albumin-Free Recombinant Factor VIII
Authors Shapiro AD, Fernandez A, Teitel J ![]() , Botha J, Khair K
, Botha J, Khair K ![]()
Received 5 August 2021
Accepted for publication 5 November 2021
Published 20 November 2021 Volume 2021:12 Pages 991—1001
DOI https://doi.org/10.2147/JBM.S329883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Amy D Shapiro,1 Alejandro Fernandez,2 Jerome Teitel,3 Jaco Botha,2 Kate Khair4
1Indiana Hemophilia & Thrombosis Center, Indianapolis, IN, USA; 2Takeda Pharmaceutical International AG, Zürich, Switzerland; 3St. Michael’s Hospital, University of Toronto, Toronto, ON, Canada; 4Centre for Outcomes and Experience Research in Children’s Health Illness and Disability (ORCHID), Great Ormond Street Hospital, London, UK
Correspondence: Jaco Botha
Takeda Pharmaceutical International AG, Thurgauerstrasse 130, Glattpark-Opfikon, Zürich, 8152, Switzerland
Tel +41 79 961 8561
Email [email protected]
Introduction: Neutralizing antibodies to coagulation factor VIII (FVIII) remain a major complication associated with FVIII replacement therapy.
Aim: To assess safety and efficacy of immune tolerance induction (ITI) therapy with ADVATE® (antihemophilic factor [recombinant] [rAHF]) in patients who participated in the Prospective ADVATE Immune Tolerance Induction Registry (PAIR) study.
Methods: The PAIR study was an international, multicenter, open-label, prospective, observational study in patients with hemophilia A and inhibitors, prescribed rAHF ITI therapy in clinical practice. The primary endpoint was adverse event (AE) reporting; the secondary endpoints included incidence of central venous access device-related complications and success rates of ITI therapy. Maintenance of immune tolerance was monitored for 12 months post-ITI therapy.
Results: Of 44 patients, 36 completed ITI therapy, including 31 completing the 12-month follow-up. Most patients received rAHF 90– 130 IU/kg/day (59.1%) and a mean of 6.0 doses/week; the median duration of rAHF ITI therapy during the PAIR study was 600 days. Overall, 284 AEs were reported; 56 AEs were serious, of which none were considered rAHF-related. Of 228 nonserious AEs, 14 (in six patients) were deemed rAHF-related: increase of FVIII inhibitors titer due to anamnestic response, nausea, catheter site pain, pyrexia, urticaria, upper respiratory tract infection, arthralgia, and hemarthrosis. None were severe or led to ITI discontinuation. Eighteen patients experienced ≥ 1 central venous access device-related complication, and 21 of 36 completers achieved a negative inhibitor titer. The Kaplan–Meier estimate of success for achievement of first negative titer at 18 months of ITI therapy was 68.3% (95% confidence interval 51.8– 83.6%) among completers. Of patients with partial or complete success post-ITI, 87% (20/23) maintained immune tolerance at 12-month follow-up.
Conclusion: Data suggest that rAHF ITI therapy in the PAIR study was effective, with no unexpected safety signals reported.
Keywords: hemophilia A, immune tolerance, post-marketing product surveillance, therapeutics, adverse effects
Introduction
Development of neutralizing inhibitory antibodies (inhibitors) remains a major risk associated with coagulation factor VIII (FVIII) replacement therapy. Estimates suggest that 8–44% of patients with severe hemophilia A and 3–13% of patients with mild or moderate hemophilia A develop inhibitory antibodies,1–4 with occurrence dependent on genetic- and patient-related factors, such as mutations, severity of hemophilia, ethnicity, age at first treatment, and family history of hemophilia.1,5–8 In the absence of intervention, inhibitors compromise the effectiveness of FVIII therapy and lead to increased morbidity and mortality due to poor bleed control.9,10
For patients with hemophilia A and inhibitors, induction of FVIII immune tolerance aims to restore FVIII pharmacokinetics (PK) and responsiveness to FVIII therapy, which is the standard of care.11,12 The success rate of immune tolerance induction (ITI) using various dosing regimens has been reported to range between 60 and 80% in patients with severe hemophilia A and inhibitors to FVIII.13–15 In a “good-risk” subgroup of ITI patients (inhibitor titer <10 Bethesda units (BU)/mL and historic peak titer of <200 BU/mL) and severe hemophilia A, data from a randomized, prospective study indicate that success rates were similar between low-dose (50 IU/kg FVIII, three times a week) and high-dose (200 IU/kg FVIII, daily) regimens; however, patients receiving low-dose ITI therapy took longer to achieve tolerance and had higher bleed rates compared with the high-dose group.14 The optimal ITI treatment and dosage regimen is unknown, and information on factors that may predict ITI therapy success or failure emanates from ITI therapy studies.16–21
Previous reports in small patient cohorts suggest that ITI therapy with ADVATE® (antihemophilic factor [recombinant] [rAHF] ; Baxalta US Inc., a Takeda company, Lexington, MA, USA) is effective and confirm an overall established record of safety.22,23 The international Prospective ADVATE Immune Tolerance Induction Registry (PAIR) study was designed to characterize the safety and tolerability of rAHF as a primary FVIII therapeutic agent in ITI therapy in patients with hemophilia A and inhibitors in clinical practices. The main objectives of the study were to assess the incidence of serious and nonserious adverse events (AEs) related to rAHF, incidence of central venous access device (CVAD)–related infections, and the success rates in patients with hemophilia A and inhibitors receiving ITI therapy with rAHF.
Here, we report the final analysis of the PAIR study, encompassing 7 years of prospective surveillance.
Materials and Methods
Study Design and Conduct
The PAIR study was an international, multicenter, open-label, prospective, observational study in patients with hemophilia A and inhibitors prescribed rAHF ITI therapy in clinical practice. The PAIR study was conducted at 27 sites in 10 countries (Belgium, Canada, Denmark, France, Germany, Greece, Italy, Spain, United Kingdom, and United States). Recruitment began in July 2007, and the study was completed in July 2015.
Patients
Patients diagnosed at any age with hemophilia A of any severity, previously diagnosed with an inhibitor to FVIII (low-titer [<5 BU] and high-titer [≥5 BU]), and due to start rAHF ITI therapy as part of routine clinical practice were eligible for this study. The choice of rAHF ITI therapy was made before PAIR study participation. Previous failure of ITI therapy on rAHF and history of hypersensitivity reactions to FVIII were exclusion factors. The protocol for this study defined severe hemophilia A as a baseline level of FVIII ≤1% and non-severe hemophilia A as FVIII >1%, reflecting the classification of hemophilia severity at the time of the study. A FVIII baseline level of <1% has since been adopted for severe hemophilia A.12
Procedures
The rAHF ITI therapy dosing regimen and monitoring schedule were at the treating physician’s discretion, based on individual patient’s requirements. ITI therapy regimens described in the peer-reviewed literature14,16,19–21,24,25 or similar to those described in the International ITI Protocol26 were recommended.
The observation period for each patient was from the time ITI therapy was initiated through 33 months of ITI therapy, with an additional 12-month post-observation follow-up after ITI therapy was considered successful or partially successful. If ITI therapy (partial) success was not achieved within 33 months of ITI therapy initiation, participation in the study ended.
Study-specific patient diaries were used to record the following variables: AEs, ITI infusion administration details, treatment and cause of new bleeding episodes, response to bleed treatment, and concomitant medications taken (including vaccinations). Diary data were recorded by investigators on case report forms (CRFs).
Investigators monitored their patients for the occurrence of any AE. All AEs associated with rAHF during bleed management (not including bleed events during ITI therapy) were reported on the AE CRF. If an AE was serious, the event was recorded using separate serious AE (SAE) forms.
Laboratory testing for the quantitation of FVIII, FVIII inhibitor, and FVIII recovery and PK was performed at baseline, during ITI therapy, at the end of ITI therapy, and at a post-observation 12-month follow-up. All testing was conducted at the local laboratory of the participating site according to locally established procedures and methods. Laboratory tests were not mandated by the protocol, but investigators could utilize the procedure in the International Immune Tolerance Study14 to determine the disappearance of the inhibitor.
The approach to the management of bleeding episodes (ie, breakthrough bleeding) was determined by the treating physician. Bleed events and hemostatic agents received at home or in the clinic were recorded on the Bleeding Episode Treatment Record CRF.
Study Endpoints and Outcome Measures
The primary endpoint was the incidence of AEs possibly related to rAHF during ITI therapy, categorized according to the Medical Dictionary for Regulatory Activities (MedDRA, version 17.0). The study comprised a retrospective and prospective period, defined respectively as the period of time during which rAHF ITI therapy was administered prior to, and following, PAIR study enrollment.
Secondary endpoints were the incidence of CVAD-related infections, general success rate of ITI therapy, and success rate in patients with severe hemophilia A (baseline level of FVIII ≤1%) and receiving rAHF as their primary ITI therapy, with no previous ITI therapy attempt and any titer inhibitor to FVIII.
Consensus recommendations of the 2006 International ITI Workshop define the response to ITI (ie, success) as a combination of a decrease and absence of inhibitors (usually less than 0.6 BU) and a normalization of the PK of FVIII.13 Per the study protocol, precise ITI success was categorized as complete, partial, failure, unassessable, and relapse, based on inhibitor titers and PK assessments performed in the International Immune Tolerance Study.14 However, due to the observational nature of this study and as PK assessments were not available in all centers, success in this analysis was defined only on inhibitor titer. General success was defined as achieving a FVIII inhibitor titer <0.6 BU or cut-off limit of inhibitor detection per local laboratory standard. Partial success occurred within 9–33 months of ITI treatment and was an inhibitor titer <5 BU (ie, conversion from high to low titer). Failure was ≥33 months of treatment with titer ≥5 BU, or 9–33 months of treatment with failure to achieve a >20% reduction in titer. Relapse was a positive inhibitor titer during the 12-month follow-up after inhibitor disappearance.
Statistical Analyses
Safety data, including incidence of CVAD-related infections, were analyzed in the full analysis set (FAS), defined as all patients who enrolled and received at least one dose of rAHF ITI. The incidence of general success of rAHF ITI was analyzed using the completer analysis set (CAS), which comprised all patients who carried ITI therapy to completion, regardless of whether or not they were followed up for 12 months after completion. The nonparametric Kaplan–Meier product limit method was used to estimate time to ITI success (percentage of patients with inhibitor titers who achieved a negative FVIII inhibitor titer following ITI therapy) in the FAS, CAS, and per-protocol analysis set (PPS). The PPS was defined as the subset of patients in the CAS who completed ITI therapy with an assessable outcome (ie, all necessary inhibitor titer results were available in order to determine ITI therapy success or failure).
If the end date for a patient’s current/initial ITI regimen was not collected, the end date was assumed to be 1 day before the first recorded start date of a change in therapy or date of therapy completion, whichever came first. If there was a discrepancy between unique titer measurements reported in multiple places on the CRF, the result found in the patient’s titer log prevailed. To determine the end date of the initial ITI regimen, which was not collected on the CRF, if the initial ITI start date was not missing then the day before the start date of the current ITI regimen was used. If the initial ITI start date was missing, then the date of the end of the previous rAHF therapy was used.
Descriptive statistics were used to summarize continuous variables; frequencies were used for categorical variables. All data were analyzed with SAS® software package, version 9.4 (SAS Institute Inc., Cary, North Carolina, USA).
Results
The results reported in in the main text of this article are based on the protocol definitions of severe hemophilia A (baseline level of FVIII ≤1%) and non-severe hemophilia A (baseline FVIII >1%). Results based on the updated definition of severe hemophilia (baseline level of FVIII <1%)12 are provided in the supplementary materials where appropriate.
Patients
As of July 2015, all patients had completed study participation. Of the 44 patients who enrolled and were exposed to rAHF, 36 (81.8%) completed ITI therapy and 31 (70.5%) completed the 12-month follow-up (Figure 1).
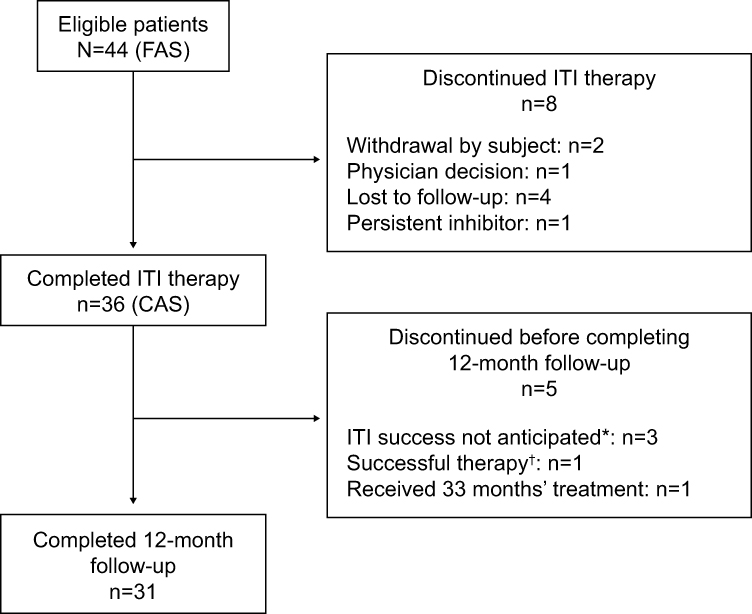 |
Figure 1 Flow of patients through the PAIR study. *Within 33 months of treatment. †As defined by the protocol. Abbreviations: CAS, completer analysis set; FAS, full analysis set; ITI, immune tolerance induction; PAIR, Prospective ADVATE Immune Tolerance Induction Registry. |
The median age at enrollment was 23.5 months (Table 1). Most patients (86.4%, 38/44) had severe hemophilia A (baseline FVIII level ≤1%); 13.6% (6/44) had non-severe hemophilia A (baseline FVIII level >1%). At initiation of ITI therapy (ie, baseline), high-titer inhibitors were present in 15 of 44 patients (≥10 BU in seven patients), low titers were present in 23 of 44 patients, and titers were unreported in six patients with any hemophilia severity. The most common type of FVIII mutation was an intron 22 inversion (43.2%, 19/44). Most of the remaining patients reported either unknown (27.3%, 12/44) or other (22.7%, 10/44) mutations; missense and nonsense mutations were reported in 4.5% of patients (2/44) and 2.3% of patients (1/44), respectively.
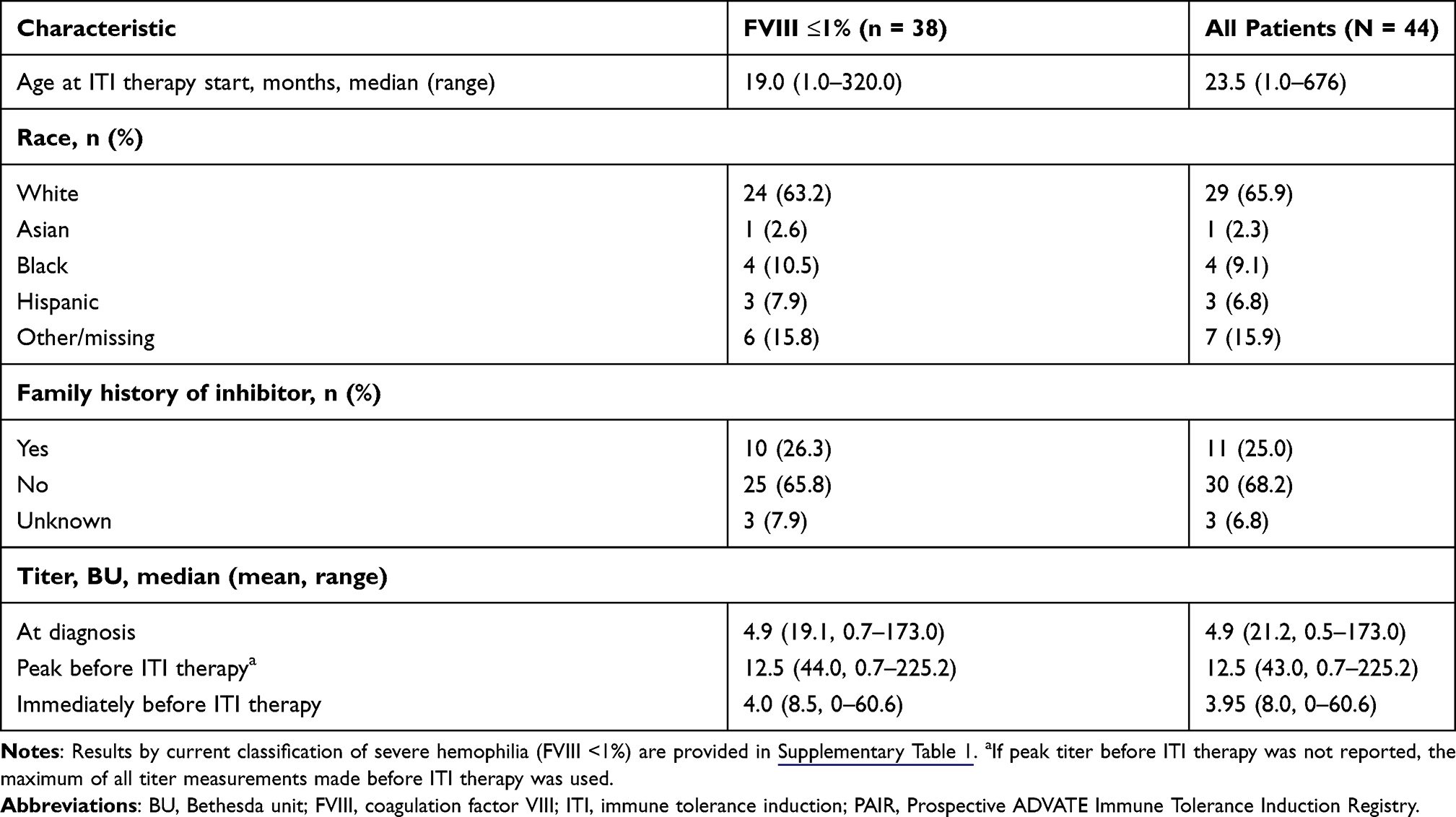 |
Table 1 Baseline Characteristics of Patients in the PAIR Study |
Use of rAHF as ITI Therapy
The median duration of rAHF ITI therapy administration before PAIR study enrollment was 122 days (n = 34), and during the PAIR study it was 600 days (n = 44).
The most frequently initiated rAHF dose range (59.1%, 26/44 patients) was 90–130 IU/kg/day (Table 2), which was initiated in patients from Germany (n = 1), Denmark (n = 1), Spain (n = 1), Greece (n = 1), France (n = 3/5), Italy (n = 3/5), the United Kingdom (n = 6/9), and United States (n = 9/17). In Belgium, one patient was initiated on 90–130 IU/kg/day and one patient on ≥200 IU/kg/day rAHF. In Canada, two of two patients were initiated on <90 IU/kg/day rAHF. The mean (SD) dose per infusion of rAHF ITI therapy was 113.4 (66.6) IU/kg; patients with severe hemophilia A had a higher mean (SD) dose per infusion than those with non-severe hemophilia A (119.3 [69.4] versus 77.3 [26.9] IU/kg, respectively). Results using the current classification of severe hemophilia are provided in Supplementary Table 2. The total mean (median, range) number of rAHF doses per week was 6.0 (5.2, 0.6–14.0).
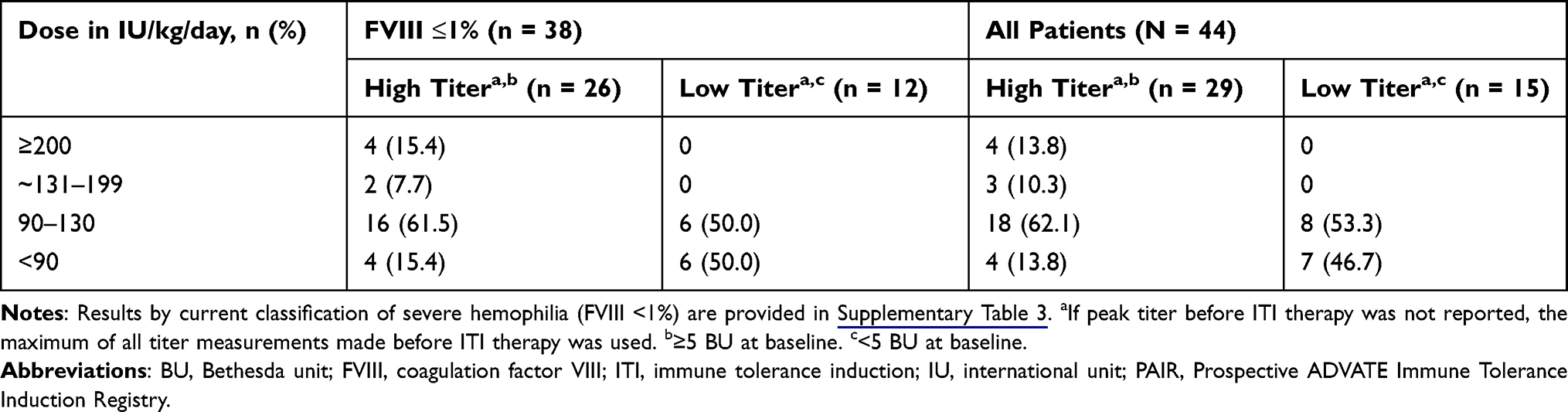 |
Table 2 ITI Therapy Dosage Regimens Utilized in the PAIR Study |
Following the initial dose, 47.7% of all patients (21/44) and 47.4% of patients with severe hemophilia (18/38) experienced at least one dose increase during the study. Overall, the percentage of patients receiving dose increases during the study was similar for those with low- (<5 BU, 53.3%) and high-inhibitor titers (≥5 BU, 44.8%) at baseline. Information on whether inhibitors were low- or high-responding was not available in this study.
Safety
A total of 284 AEs were reported in 44 patients before (retrospective period) and after (prospective period) PAIR study enrollment; 56 AEs (19.7%) were serious and not considered related to rAHF ITI therapy (Table 3). Before study enrollment, 31 AEs were reported in 10 of 34 patients, of which eight were SAEs reported in five patients. During the study, 253 AEs were reported in 32 of 44 patients, of which 48 were SAEs reported in 21 patients. A total of 14 nonserious AEs considered related to rAHF ITI therapy occurred in six patients (13.6%), and none of these AEs were severe. These were pyrexia (n = 1), urticaria (n = 1), nausea (n = 2), catheter site pain (n = 1), upper respiratory tract infection (n = 1), arthralgia (n = 2), hemarthrosis (n = 2), and increase of FVIII inhibitors titer due to anamnestic response (n = 4).
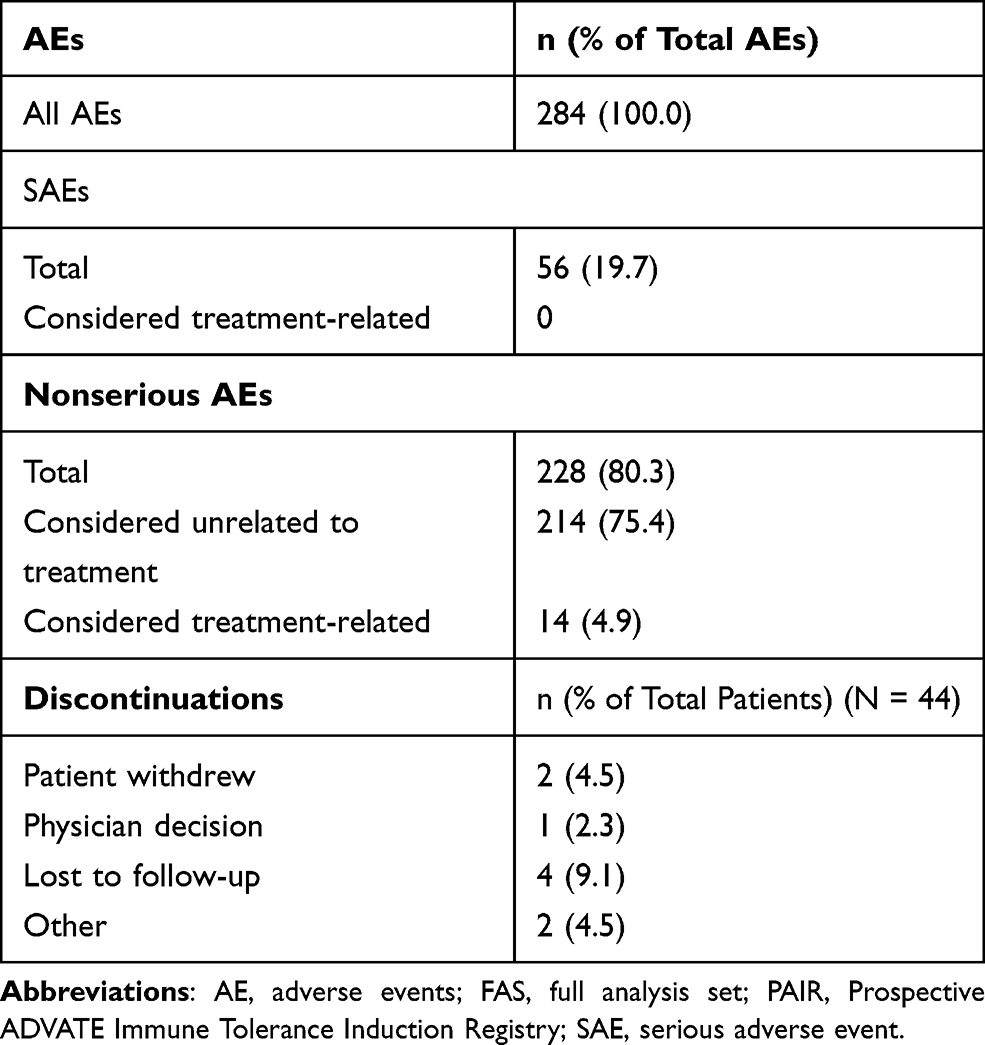 |
Table 3 Summary of AEs in the PAIR Study (FAS) |
Of 44 patients, 18 (40.9%) had a CVAD placed before rAHF ITI therapy initiation, 10 patients (22.7%) had their first CVAD placed during ITI, six patients (13.6%) had their CVAD replaced during ITI, and 18 patients (40.9%) had a CVAD placed due to complications during ITI therapy. In total, 18 patients experienced at least one CVAD-related complication. The most frequently reported CVAD complications included 18 episodes of local site line infection in five patients (11.0%), nine episodes of line insertion site bleed in three patients (6.8%), and eight episodes of systemic/septic line infection in five patients (11.0%). Other CVAD-associated complications occurring in more than one patient were line insertions (five occurrences in four patients) and line removal (two occurrences in two patients).
Efficacy of ITI Therapy
Of the 36 patients who completed ITI treatment (ie, those in the CAS), 21 (58.3%) achieved a negative titer during the study, two (5.6%) converted from a high- to a low-titer inhibitor, and eight (22.2%) experienced treatment failure (Table 4). Six of 13 patients (46.2%) in the CAS with severe hemophilia (baseline level of FVIII ≤1%) and high titer at baseline achieved a negative titer. Of 23 patients with partial or complete success after ITI therapy, 20 (87%) maintained immune tolerance at 12-month follow-up.
 |
Table 4 Outcomes in Patients Who Completed rAHF ITI Therapy (CAS) |
The estimated cumulative success rate of rAHF ITI therapy at approximately 18 months (516 days) was 72.4% (95% confidence interval [CI] 55.5–87.0%) in the PPS (n = 31) and 68.3% (95% CI 51.8–83.6%) in the CAS (n = 36) (Figure 2). These rates were in patients with high- and low-titer inhibitors (PPS patients: 20 with high-titer and 11 with low-titer inhibitors; CAS patients: 23 with high-titer and 13 with low-titer inhibitors), and success was defined as the percentage of patients with inhibitors who achieved a negative FVIII inhibitor titer following ITI therapy. The median times to first and second negative titers for all patients were 4.3 months and 5.8 months, respectively. For those patients with a high-titer inhibitor at baseline, the median times to first and second negative titer test were 4.8 and 6.7 months, respectively. Specifically in children (<18 years of age), the estimated cumulative success rate of rAHF ITI therapy at approximately 18 months was 69.4% (95% CI 51.5–85.6%) in the PPS and 65.4% (95% CI 48.1–82.1%) in the CAS.
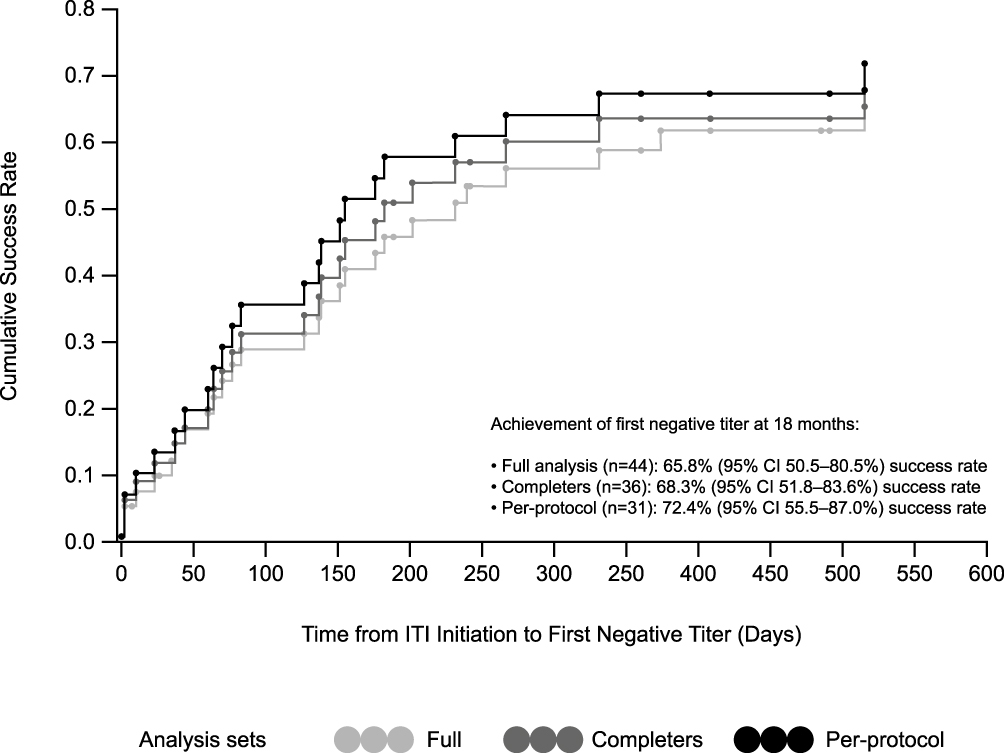 |
Figure 2 Kaplan–Meier success curves for achievement of first negative titer. Abbreviations: CI, confidence interval; ITI, immune tolerance induction. |
Maximum inhibitor titers ≥5 BU, ≥10 BU, and ≥100 BU were detected in 25, 22, and 17 patients, respectively, during ITI therapy. Of the patients who achieved a negative inhibitor titer (61.5%, 16/26), three patients (18.8%) reported a relapse at their 12-month follow-up appointment. The first of these patients was 56 years of age at ITI therapy initiation and had a peak inhibitor titer before ITI therapy of 1.6 BU and an inhibitor titer of 1.1 BU at the 12-month follow-up. The second patient was 3 years of age at ITI therapy initiation, with a peak inhibitor titer before ITI therapy of 196.0 BU; although relapse was indicated in the report, the most recent titer at 12 months was 0 BU. The third patient with relapse was 4 months old at ITI initiation and had a peak inhibitor titer of 4.3 BU before ITI therapy and an inhibitor titer of 0.5 BU at follow-up (equal to the local laboratory cut-off for inhibitor detection). The first patient had been receiving a follow-up prophylaxis regimen of 35 IU/kg every other day; the second patient had continued ITI therapy (50 IU/kg every day). The third patient had received 71 IU/kg twice a week and 89 IU/kg once a week.
The median time from inhibitor diagnosis to ITI therapy initiation for all patients with available data (n = 42) was 2.5 months; the median time for patients with severe hemophilia (baseline FVIII level ≤1%) (n = 36) was 2.2 months, and for patients with FVIII level >1% (n = 6) it was 3.7 months.
The median time from inhibitor diagnosis to ITI therapy initiation was 0.5 months for patients with an ITI therapy outcome of general success (n = 20), 34.0 months for patients with partial success (n = 2, high to low titer conversion), and 12.48 months for patients with ITI therapy failure (n = 7).
Treatment of Bleeding Episodes
A total of 339 bleeding episodes occurred in 33 of 36 patients in the CAS on rAHF during ITI therapy. Most bleeding episodes (n = 308) occurred in 29 of 31 patients with severe hemophilia (FVIII ≤ 1%), and the remaining 31 bleeding episodes occurred in four of five patients with non-severe hemophilia (FVIII >1%). The results using the current classification of severe hemophilia are provided in Supplementary Table 5. The most frequent cause of bleeding episodes was trauma-related (156/339), whereas 94 of 339 bleeding episodes were spontaneous. The etiologies of a further 89 bleeding episodes were either unknown or missing.
Most bleeding episodes that occurred during ITI therapy were treated exclusively with recombinant activated factor VII (rFVIIa) products (n = 177), whereas three were exclusively treated with activated prothrombin complex concentrates (aPCC) and 37 with rAHF. For 84 bleeding episodes, treatment began with rAHF and alternated with rFVIIa products, and for five bleeding episodes, treatment began with rFVIIa products and alternated with rAHF.
Discussion
The PAIR study was an international, non-interventional prospective study of rAHF as ITI therapy in a real-world setting. The observational, non-interventional nature of the study enabled the assessment of safety, tolerability and efficacy of rAHF ITI therapy under routine conditions in common clinical practice. In this setting, patients had independently chosen to receive ITI therapy with rAHF and were treated by the investigators according to their clinical judgement and experience. The PAIR study collected and analyzed this real-life experience and summarized the most salient findings. Overall, the findings from the PAIR study add to previous experiences with rAHF ITI therapy in other clinical studies utilizing different designs and treatment regimens.14,22,23,27
The safety results were consistent with the known safety profile of rAHF, and there were no unknown or unexpected safety signals for treatment of bleeding episodes or for ITI therapy.28–30 Most complications that occurred in the PAIR study related to CVAD use. These are known issues associated with the use of these devices, and education on best practices may help to mitigate them. Of note, there were no reports of CVAD-related thromboses, which are a relatively common complication of CVAD insertion.31
The present investigation supports the efficacy conclusion of previous ITI therapy studies.22,23,32 rAHF was effective in a variety of ITI dosing regimens currently used in clinical practice. The estimated PPS and CAS cumulative success rates at 18 months were 72.4% and 68.3%, respectively, in patients who had both high- and low-titer inhibitors at baseline (information on whether inhibitors were low- or high-responding was not available). In the International Immune Tolerance Study, patients with severe hemophilia (defined as FVIII <1%) who were defined as good risk and had high-titer and high-responding inhibitors achieved a success rate of 69.7% (n = 46/66).14 This compares with 46.2% of patients with severe hemophilia (baseline FVIII ≤1%) and high titer in the present study. However, this percentage is based on a sample of 13 patients. In addition, the apparently lower success rate may be a consequence of how severe hemophilia was defined in our study, as patients with moderate hemophilia generally have a lower success rate versus those with severe disease.
Separate retrospective analyses from two US centers suggested that an interval of <1 month from inhibitor detection to ITI therapy start was associated with improved outcome.15 In the present study, although the numbers were small, 20 patients with general success had an inhibitor-diagnosis-to-ITI-therapy-initiation interval of 0.48 months versus 12.48 months for patients with ITI therapy failure (n = 7) and 33.95 months for patients with high to low titer conversion (n = 2). The overall rate of relapse in the present study (13%) is in line with that reported by previous ITI therapy studies (2.3% to 29.7%).33–35
The PAIR study was conducted in eight European countries, as well as the United States and Canada, without the limitation of eligibility criteria used in interventional clinical trials. The number of patients enrolled in all sites and followed up was adequate for a reliable assessment of ITI therapy across multiple geographies, though country-specific comparisons were not possible. The PAIR study provides important information on the administered doses of rAHF for ITI therapy in real-world conditions and may indicate patterns of dosing based on the state of disease. Due to the non-interventional design of the study, it is possible that not all data available were entered into the database. Of note, more than half of the patients had missing bleeding episode data. In addition, the subgroup data may have been skewed by the different sample sizes of patients with severe or moderately severe hemophilia and the fact that the patient age range was wide. Another potential limitation is that the study used the protocol definition of ≤1% for severe hemophilia, which differs from the current definition of <1%.12 However, the results using both definitions are similar and do not change the conclusions in this report. Finally, the number of patients in the study was too low for conclusions about differences between countries.
Conclusions
The observational PAIR study provides important real-world data on a widely used recombinant factor VIII concentrate. The results of the PAIR study are consistent with rAHF as an effective therapy for ITI in patients with hemophilia A and inhibitors. No SAEs related to treatment were reported. Future prospective studies may further define factors associated with ITI therapy success and failure in patients with hemophilia A.
Abbreviations
AE, adverse event; BU, Bethesda unit; CAS, completer analysis set; CRF, case report form; CVAD, central venous access device; FAS, full analysis set; FVIII, coagulation factor VIII; ITI, immune tolerance induction; IU, international unit; PAIR, Prospective ADVATE Immune Tolerance Induction Registry; PK, pharmacokinetic; PPS, per-protocol analysis set; rAHF, antihemophilic factor (recombinant); SAE, serious adverse event.
Study Registration
The PASS-INT-004 registry study titled “Prospective ADVATE immune tolerance induction registry (PAIR)” was a non-interventional post-authorization study initiated in 2007. At the time of initiation, there was no legal requirement to register such types of studies in the clinicaltrials.gov, EudraCT, and EUPAS registers.
Data Sharing Statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results reported in this article, will be made available within 3 months from initial request, to researchers who provide a methodologically sound proposal. The data will be provided after their de-identification, in compliance with applicable privacy laws, data protection, and requirements for consent and anonymization. Data requests should follow the process outlined in the Data Sharing section on:
www.takeda.com/what-we-do/research-and-development/takeda-clinical-trial-transparency/.
Ethics Approval and Informed Consent
The PAIR study conforms to the Declaration of Helsinki aspects of ethical considerations (ethics committee/institutional review board approval and participants’ informed consent). Before enrollment, the study protocol and informed consent form were reviewed and approved by the independent ethics committees/institutional review boards of all participating sites in accordance with local requirements (see Supplementary Table 6). Written consent before enrollment was obtained from all adult patients and caregivers of patients <18 years of age.
Acknowledgments
The authors thank all patients and their caregivers who took part in the PAIR study. The authors acknowledge the expertise and participation of all principal investigators and personnel at the study sites.
This article was presented in abstract form at the World Federation of Hemophilia (WFH) World Congress; July 24–28, 2016. Medical writing support was provided by Nasser Malik, PhD, CMPP, of Excel Medical Affairs (Southport, CT, USA) and was funded by Takeda Development Center Americas Inc., a Takeda Company, Lexington, MA, USA.
Author Contributions
Amy D. Shapiro, Kate Khair, and Jerome Teitel were principal investigators of the PAIR study and recruited and treated patients; they also contributed to the study concept and design. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The PAIR study was funded by Baxalta US Inc., a Takeda company, Lexington, MA, USA. Medical writing support was provided by Nasser Malik, PhD, CMPP, of Excel Medical Affairs (Southport, CT, USA), and was funded by Takeda Development Center Americas Inc., a Takeda Company, Lexington, MA, USA.
Disclosure
Amy D. Shapiro received grant/research support from Novo Nordisk, Bayer Healthcare, Bioverativ, Genentech, Prometic Life Sciences, Kedrion, Sangamo Biosciences, Bio Products Laboratory, Octapharma, OPKO, Daiichi Sankyo, Chugai Pharmaceutical, Glover Blood Therapeutics, and Shire (a Takeda company). Kate Khair received grant/research support from Baxalta/Shire (a Takeda company), CSL Behring, Novo Nordisk, Pfizer, Sobi, and uniQure; and is on the speaker bureau of Bayer, Novo Nordisk, Octapharma, Pfizer, Roche, Shire (a Takeda company), and Sobi. Jerome Teitel has served as a consultant and participated on advisory boards for Baxalta US Inc., Bayer, Octapharma, Biogen, CSL Behring, Roche, and Takeda. He is also the data monitoring committee for BioMarin. Jaco Botha is an employee of Takeda Pharmaceuticals International AG and Takeda stock owner. Alejandro Fernandez was an employee of Takeda Pharmaceuticals International AG at the time of this work. The authors report no other conflicts of interest in this work.
References
1. Kreuz W, Ettingshausen CE, Zyschka A, et al. Inhibitor development in previously untreated patients with hemophilia A: a prospective long-term follow-up comparing plasma-derived and recombinant products. Semin Thromb Hemost. 2002;28(3):285–290. doi:10.1055/s-2002-32664
2. Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. doi:10.1182/blood-2012-09-457036
3. Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9(4):418–435. doi:10.1046/j.1365-2516.2003.00780.x
4. Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4(4):558–563. doi:10.1046/j.1365-2516.1998.440558.x
5. Scharrer I, Bray GL, Neutzling O. Incidence of inhibitors in haemophilia A patients – a review of recent studies of recombinant and plasma-derived factor VIII concentrates. Haemophilia. 1999;5(3):145–154. doi:10.1046/j.1365-2516.1999.00300.x
6. Lorenzo JI, López A, Altisent C, Aznar JA. Incidence of factor VIII inhibitors in severe haemophilia: the importance of patient age. Br J Haematol. 2001;113(3):600–603. doi:10.1046/j.1365-2141.2001.02828.x
7. Astermark J. Why do inhibitors develop? Principles of and factors influencing the risk for inhibitor development in haemophilia. Haemophilia. 2006;12(s3):52–60. doi:10.1111/j.1365-2516.2006.01261.x
8. Astermark J, Altisent C, Batorova A, et al. Non-genetic risk factors and the development of inhibitors in haemophilia: a comprehensive review and consensus report. Haemophilia. 2010;16(5):747–766. doi:10.1111/j.1365-2516.2010.02231.x
9. Scalone L, Mantovani LG, Mannucci PM, Gringeri A; The COCIS Study Investigators. Quality of life is associated to the orthopaedic status in haemophilic patients with inhibitors. Haemophilia. 2006;12(2):154–162. doi:10.1111/j.1365-2516.2006.01204.x
10. Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13(5):606–612. doi:10.1111/j.1365-2516.2007.01518.x
11. Valentino LA, Kempton CL, Kruse-Jarres R, et al. US guidelines for immune tolerance induction in patients with haemophilia A and inhibitors. Haemophilia. 2015;21(5):559–567. doi:10.1111/hae.12730
12. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26(Suppl 6):1–158. doi:10.1111/hae.14046
13. DiMichele DM, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recommendations. Haemophilia. 2007;13(s1):1–22. doi:10.1111/j.1365-2516.2007.01497.x
14. Hay CRM, DiMichele DM; International Immune Tolerance Study. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119(6):1335–1344. doi:10.1182/blood-2011-08-369132
15. Nakar C, Manco-Johnson MJ, Lail A, et al. Prompt immune tolerance induction at inhibitor diagnosis regardless of titre may increase overall success in haemophilia A complicated by inhibitors: experience of two U.S. centres. Haemophilia. 2015;21(3):365–373. doi:10.1111/hae.12608
16. DiMichele DM, Kroner BL; The North American Immune Tolerance Study Group. The North American Immune Tolerance Registry: practices, outcomes, outcome predictors. Thromb Haemost. 2002;87(1):52–57. doi:10.1055/s-0037-1612943
17. Freiburghaus C, Berntorp E, Ekman M, Gunnarsson M, Kjellberg B, Nilsson IM. Tolerance induction using the Malmö treatment model 1982–1995. Haemophilia. 1999;5(1):32–39. doi:10.1046/j.1365-2516.1999.00195.x
18. Lenk H; ITT Study Group. The German registry of immune tolerance treatment in hemophilia—1999 update. Haematologica. 2000;85(10 Suppl):45–47.
19. Mariani G, Kroner B. International immune tolerance registry, 1997 update. Vox Sang. 1999;77(Suppl 1):25–27. doi:10.1159/000056710
20. Mauser-Bunschoten EP, Nieuwenhuis HK, Roosendaal G, van den Berg HM. Low-dose immune tolerance induction in hemophilia A patients with inhibitors. Blood. 1995;86(3):983–988. doi:10.1182/blood.V86.3.983.983
21. Oldenburg J, Schwaab R, Brackmann HH. Induction of immune tolerance in haemophilia A inhibitor patients by the ‘Bonn Protocol’: predictive parameter for therapy duration and outcome. Vox Sang. 1999;77(Suppl 1):49–54. doi:10.1159/000056717
22. Auerswald G, Thompson AA, Recht M, et al. Experience of Advate rAHF-PFM in previously untreated patients and minimally treated patients with haemophilia A. Thromb Haemost. 2012;107(6):1072–1082. doi:10.1160/TH11-09-0642
23. Valentino LA, Recht M, Dipaola J, et al. Experience with a third generation recombinant factor VIII concentrate (Advate®) for immune tolerance induction in patients with haemophilia A. Haemophilia. 2009;15(3):718–726. doi:10.1111/j.1365-2516.2008.01960.x
24. Rocino A, de Biasi R. Successful immune tolerance treatment with monoclonal or recombinant factor VIII concentrates in high responding inhibitor patients. Vox Sang. 1999;77(Suppl 1):65–69. doi:10.1159/000056720
25. Scheibel E, Ingerslev J, Dalsgaard-Nielsen J, Stenbjerg S, Knudsen JB. Continuous high-dose factor VIII for the induction of immune tolerance in haemophilia A patients with high responder state: a description of eleven patients treated. Thromb Haemost. 1987;58(4):1049–1052. doi:10.1055/s-0038-1646054
26. Dimichele DM, Hay CR. The international immune tolerance study: a multicenter prospective randomized trial in progress. J Thromb Haemost. 2006;4(10):2271–2273. doi:10.1111/j.1538-7836.2006.02127.x
27. Yoshioka A, Ishii E, Ueno T, et al. The international immune tolerance induction study and its follow-up study on Japanese hemophilia A patients with inhibitors. Int J Hematol. 2015;101(4):362–368. doi:10.1007/s12185-015-1734-z
28. Advate [antihemophilic factor (recombinant)] lyophilized powder for reconstitution, for intravenous injection [prescribing information]. Lexington, MA: Baxalta US Inc.; 2018. Available from: https://www.shirecontent.com/PI/PDFs/ADVATE_USA_ENG.pdf. Accessed November 15, 2021.
29. Iorio A, Marcucci M, Cheng J, et al. Patient data meta-analysis of Post-Authorization Safety Surveillance (PASS) studies of haemophilia A patients treated with rAHF-PFM. Haemophilia. 2014;20(6):777–783. doi:10.1111/hae.12480
30. Romanov V, Marcucci M, Cheng J, Thabane L, Iorio A. Evaluation of safety and effectiveness of factor VIII treatment in haemophilia A patients with low titre inhibitors or a personal history of inhibitor. Patient data meta-analysis of rAFH-PFM Post-Authorization Safety Studies. Thromb Haemost. 2015;114(1):56–64. doi:10.1160/TH14-10-0882
31. Wall C, Moore J, Thachil J. Catheter-related thrombosis: a practical approach. J Intensive Care Soc. 2016;17(2):160–167. doi:10.1177/1751143715618683
32. Spotts G, Abbuehl BE, Luu HD, et al. Safety and efficacy profile of rAHF-PFM for immune tolerance induction as assessed in 3 clinical trials (PUP ITI, PRE-PAIR, and PAIR). Blood. 2010;116(21):3670. doi:10.1182/blood.V116.21.3670.3670
33. Antun A, Monahan PE, Manco-Johnson MJ, et al. Inhibitor recurrence after immune tolerance induction: a multicenter retrospective cohort study. J Thromb Haemost. 2015;13(11):1980–1988. doi:10.1111/jth.13143
34. Coppola A, Margaglione M, Santagostino E, et al. Factor VIII gene (F8) mutations as predictors of outcome in immune tolerance induction of hemophilia A patients with high-responding inhibitors. J Thromb Haemost. 2009;7(11):1809–1815. doi:10.1111/j.1538-7836.2009.03615.x
35. Mariani G, Kroner B; Immune Tolerance Study G. Immune tolerance in hemophilia with factor VIII inhibitors: predictors of success. Haematologica. 2001;86(11):1186–1193.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.