Back to Journals » Research and Reports in Tropical Medicine » Volume 14
Field-Deployable Treatments For Leishmaniasis: Intrinsic Challenges, Recent Developments and Next Steps
Authors Pacheco-Fernandez T ![]() , Markle H, Verma C
, Markle H, Verma C ![]() , Huston R
, Huston R ![]() , Gannavaram S, Nakhasi HL, Satoskar AR
, Gannavaram S, Nakhasi HL, Satoskar AR
Received 1 March 2023
Accepted for publication 8 June 2023
Published 20 July 2023 Volume 2023:14 Pages 61—85
DOI https://doi.org/10.2147/RRTM.S392606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mario Rodríguez-Pérez
Thalia Pacheco-Fernandez,1,* Hannah Markle,1,* Chaitenya Verma,2 Ryan Huston,2,3 Sreenivas Gannavaram,1 Hira L Nakhasi,1 Abhay R Satoskar2,3
1Division of Emerging and Transfusion Transmitted Disease, Center for Biologics Evaluation and Research Food and Drug Administration, Silver Spring, MD, 20993, USA; 2Department of Pathology, Wexner Medical Center, The Ohio State University, Columbus, OH, 43201, USA; 3Department of Microbiology, Wexner Medical Center, The Ohio State University, Columbus, OH, 43201, USA
*These authors contributed equally to this work
Correspondence: Abhay R Satoskar, Email [email protected]
Abstract: Leishmaniasis is a neglected tropical disease endemic primarily to low- and middle-income countries, for which there has been inadequate development of affordable, safe, and efficacious therapies. Clinical manifestations of leishmaniasis range from self-healing skin lesions to lethal visceral infection with chances of relapse. Although treatments are available, secondary effects limit their use outside the clinic and negatively impact the quality of life of patients in endemic areas. Other non-medicinal treatments, such as thermotherapies, are limited to use in patients with cutaneous leishmaniasis but not with visceral infection. Recent studies shed light to mechanisms through which Leishmania can persist by hiding in cellular safe havens, even after chemotherapies. This review focuses on exploring the cellular niches that Leishmania parasites may be leveraging to persist within the host. Also, the cellular, metabolic, and molecular implications of Leishmania infection and how those could be targeted for therapeutic purposes are discussed. Other therapies, such as those developed against cancer or for manipulation of the ferroptosis pathway, are proposed as possible treatments against leishmaniasis due to their mechanisms of action. In particular, treatments that target hematopoietic stem cells and monocytes, which have recently been found to be necessary components to sustain the infection and provide a safe niche for the parasites are discussed in this review as potential field-deployable treatments against leishmaniasis.
Keywords: leishmaniasis, field-deployable therapies, hematopoietic stem cells, monocytes, ferroptosis, repurposing drugs
Introduction
Leishmaniases are chronic, neglected tropical diseases with three major clinical manifestations: cutaneous, mucocutaneous, and visceral leishmaniasis. These diseases are all caused by the Leishmania parasite which is endemic to 88 countries spanning tropical and subtropical regions.1 Leishmania parasites are spread by the bite of an infected sandfly, and the species of parasite transmitted to the host largely determines clinical outcomes.2 Cutaneous leishmaniasis (CL) is characterized by papules which progress to ulcerative lesions that may result in permanent disfigurement or scarring.2–6 In the Old World, CL is largely caused by L. major and L. tropica, while New World cases typically arise from L. mexicana infections.7 Visceral leishmaniasis (VL), the chronic, fatal form of disease primarily manifests through fevers, spleen and liver enlargement, weight loss, and multiple hematological changes as parasites spread systemically through the host.4–6,8,9 VL is caused by L. donovani and L. infantum in the Old and New World, respectively.7 Individuals with visceral infections may later present with post-kala-azar dermal leishmaniasis (PKDL) even after apparent cure. PKDL is characterized by a widespread maculopapular rash due to immune responses against parasites persisting in the skin.4,9 A rare form of disease, mucocutaneous leishmaniasis (MCL), presents in an estimated 1–10% of individuals with prior CL infection, and is primarily caused by L. braziliensis infection in the New World.2,7 This particular manifestation progresses as destructive lesions or ulcers of the mucosa, particularly surrounding the nose and mouth and, like CL, may lead to permanent disfigurement.2 While cutaneous versus visceral pathologies are largely species-specific, hybrid Leishmania parasite infections and their associated clinical manifestations are more arbitrary with complicated clinical portraits and heightened disease severity. In Sri Lanka, a hybrid L. donovani parasite is responsible for causing thousands of cases of cutaneous leishmaniasis, although L. donovani is typically a visceralizing parasite.10 In Peru and Bolivia, hybrid L. braziliensis strains harboring endosymbiotic Leishmania RNA viruses are associated with an increased risk of treatment failure.11 The need to inhibit both Leishmania and its associated endosymbionts through antiviral therapies has been considered.12,13 The complexities surrounding atypical pathologies and worsened symptomology of hybrid Leishmania parasites raises concerns for current field-deployable treatments and emphasizes the importance of therapeutic advancement in the realm of leishmaniases.
Given the severity of the diseases and the barriers for treatment of impoverished patients, especially in rural endemic areas of the world, developing and advancing field-deployable treatments to combat leishmaniases is imperative. In the present review, field-deployable therapies are defined as those that are largely accessible, easy to administer, and able to be deployed in areas of endemicity without the requirement of high costs and recurrent clinic visits. While the spread of Leishmania parasites is global, the greatest burden lies in low- and middle-income countries, where both quality of life and economic growth are negatively impacted as a result of disease. Therefore, the use of field-deployable therapies, novel treatments targeting parasite persistence, and repurposed drugs such as those developed against other diseases, where there is more funding and economic interest, is warranted in populations affected by Leishmania. Here, such current treatments against leishmaniases are summarized (Figure 1) and possible future perspectives and cellular targets for new treatments which have potential as improved field-deployable therapies are explored.
 |
Figure 1 Current field deployable treatments for leishmaniasis. Current field deployable therapies include chemotherapies which are the first-line treatment. Other mechanical therapies such as cryotherapy and thermotherapy can be operated in the field to treat skin lesions. Finally, future treatments include immunotherapies, nanocarriers and repurposing drugs to target the specific cellular niches of the parasite. |
Current Therapies and Field-Deployable Therapies
Chemotherapies
Current treatments for leishmaniasis include various modes of chemotherapy, thermotherapy, and other localized treatments (summarized in Table 1). Pentavalent antimonial drugs are the first line of treatment with other chemotherapies available including liposomal amphotericin B (L-AmB), pentamidine, miltefosine, paromomycin, and antifungal azole drugs.3,5,6,14–20 Antimonials currently in use against CL include sodium stibogluconate (SSG) and meglumine antimonate (MA).17 Cure rates of up to 100% have been documented with this class of drugs but vary based on geographical location, treatment duration, and the species of parasite responsible for infection.17,21 Liposomal amphotericin B is an alternative treatment for VL that has demonstrated cure rates ranging from 50% to 85% and is primarily used when antimonial-resistant parasites are the causative agents of infection.17,22 Pentamidine, while recommended for only a minimal number of Leishmania species, has led to CL cure rates of up to 96%.17,23 Paromomycin is an antibiotic typically used against intestinal parasitic infections but when administered to treat CL has reportedly resulted in cure rates of 50–85%.17,24,25 Miltefosine, originally classified as an anti-cancer drug, is a safe and effective alternative CL treatment, yet many patients experience disease relapse following treatment due to the drug’s inability to achieve complete parasite clearance, or sterile immunity.17,26 Azoles are also used to treat CL, but varying treatment outcomes and rates of parasite clearance have been documented given their primary activity as an antifungal agent.17
 |
Table 1 Current Treatments Against Leishmaniasis. Administration routes, and therapeutic characteristics including use against which manifestation of leishmaniasis and other diseases, if repurposed |
In general, chemotherapies are largely outdated with high levels of toxicity, and many are non-specific for Leishmania parasites.3,5,14,15,17,27 Additional barriers related to chemotherapeutics include the emergence of drug-resistant parasites. Drug resistance further complicates treatment efficacy, with cases in some regions being completely unresponsive to first-line treatments.16,17 Other challenges related to chemotherapeutics include drug costs and accessibility, treatment length and adverse effects leading to noncompliance, and variability in host genetics and immune responses.3,5,15,17,20 Additionally, most drugs like pentavalent antimonials and L-AmB may require numerous injections over the course of days to weeks, further hindering treatment feasibility and their implication as field-deployable treatments.28,38 To lower toxicity, improve drug tolerability, and reduce drug resistance, combination therapies may be administered. By delivering drugs in tandem, the dosage of each individual drug can be decreased, and treatment time may be shortened, which are both desirable factors with regard to field-deployable therapies.5 Current combination treatments include amphotericin B with miltefosine or paromomycin, and other drugs or immunomodulators in supplement with pentavalent antimonials.5,29,30 Further, to overcome negative drug effects, new drugs have been recently developed or repurposed against leishmaniasis.39 This includes drugs entering Phase 1 clinical trials such as DNDi-0690, an antitubercular nitroimidazole-class drug with potential use against VL.40 Other drug candidates in pre-clinical trials phase are DNDi-6174 and DNDi 6148, with the interesting characteristic of being effective against drug-resistant strains of Leishmania.41 Other promising treatments on the trajectory to clinical trials include oligonucleotides, proteasome inhibitors of the triazolopyrimidine class, and drugs of the imadozopyrimidine and pyrazolopyrimidine class.39 While these novel and repurposed therapies are currently not approved and largely experimental, they are promising options for the future of Leishmania treatments.
Therapeutic Vaccines and Immunotherapies
Therapeutic vaccines are a promising treatment option administered post-exposure to Leishmania parasites. These vaccines work to boost the host immune system in an attempt to reduce parasite burden and resolve infection after it has already been established; therefore, therapeutic vaccines are a promising form of immunotherapy against leishmaniasis. Historically, therapeutic vaccines have generated higher CL cure rates in trials and has been favored over chemo-monotherapies with regard to duration, overall treatment outcomes, and adverse effects, as side effects are primarily local to the lesion site.28,42–47 Immunotherapy has been shown to be an effective tool in efficacy trials against PKDL, which is known to be difficult to treat with chemotherapies.28,45 A barrier associated with therapeutic vaccines in field-related settings, however, is the need for boosters or multiple doses.28 In addition to therapeutic vaccines, immunomodulators, like toll-like receptor (TLR) agonists and cytokines have been experimentally tested against leishmaniases, administered both alone and in combination with other drugs, yet efficacy must be further explored and cost remains an issue.28,30 In general, first- and second-generation therapeutic vaccines are currently undergoing clinical trials, yet they have room for advancement and will likely be a vital tool in the future of field-deployable treatments.28
Thermotherapies and Cryotherapies
A variety of forms of thermotherapies and cryotherapies are also used in the field to treat leishmaniasis. Thermotherapies can be administered in the field with battery-operated machines delivering heat in the form of infrared light, lasers, hot water soaks, or radiofrequencies directly applied to lesions to slow growth and eradicate parasites.27,31–34 These therapies have been used for decades in various settings and in some cases, only require one session for cure.32 Carbon dioxide lasers are another form of heat therapy administered directly at the site of infection requiring minimal treatment sessions, emphasizing cost-effectiveness and favorability in the field.27 Thermotherapies are safe with minimal off-target effects and are favored for use in patients with contraindications to other drugs and in regions where healthcare infrastructure is not easily accessible.27 Cryotherapy is also used to treat cutaneous leishmaniasis lesions by direct application of liquid nitrogen or, in resource-limited settings, carbon dioxide solids at temperatures lethal to parasites.31,35,36,48 While these therapies have been deemed safe and effective with few side effects, cure rates vary based on a multitude of factors such as parasite species and lesion size. With these limitations in mind, combination therapies with chemotherapeutics may still be recommended, but these methods alone are especially favorable for patients who cannot receive traditional chemotherapies such as pregnant or immunosuppressed patients.31,35,37 In summary, the lack of sterile immunity and complete parasite clearance achieved following administration of current treatments emphasizes the need for new developments in the realm of field-based therapies.
Parasite clearance or sterile immunity may be desirable outcomes following treatment for leishmaniasis, yet current therapies are incapable of achieving this aim due to inadequate effectiveness or penetration. Future therapies should aim to reach this threshold because latent infections may lead to later reactivation and further clinical complications, as in the case of PKDL and MCL. These cases are responsible for continued parasite transmission and therefore promote disease reemergence and are considered a threat to leishmaniasis elimination in many regions.49 Likewise, the lack of parasite clearance following treatment increases the risk of parasite transmission in endemic and nonendemic regions via other modes such as blood transfusions and organ transplants.50–52 Barriers in achieving sterile immunity and mechanisms in which Leishmania may evade existing treatments are therefore important to discuss when considering future directions of Leishmania therapies.
Roadblocks to Sterile Immunity – Leishmania Refuges in the Body with Potential for Latent Infection
One challenge field-deployable therapies must face is the array of bodily sites and cell types which Leishmania spp. can reside in. This flexibility may contribute to treatment failure, latent infection, and relapse infections in some patients as some sites are immune-permissive or may have less drug accessibility. Furthermore, application of subcurative drug doses and drug resistance of the parasite may raise the likelihood of latent Leishmania infection. While Leishmania has been reported to asymptomatically colonize several unusual areas of the body - including skeletal muscle, female urogenital tract, and mammary glands,53–56 atypical symptomatic leishmaniasis has also been reported in the eye,57 central nervous system,58–60 cartilage,61,62 testes,56,63 and placenta.64–67 Most notably, VL has been well characterized for its ability to infect hematopoietic stem cells (HSCs) of the bone marrow.68,69 Evidence of Leishmania colonization at these sites is significant as it supports the possibility of these sites as potential refuges for latent infection. Therefore, current and future field-deployable treatments against leishmaniases should aim to clear asymptomatic reservoirs of Leishmania throughout the entire body to guarantee sterile immunity and prevent reinfection and disease transmission.
Re-Purposing Therapies for Use Against Cellular Hosts of Leishmania
While many therapies have been investigated for treatment of leishmaniases, future treatments could be improved by targeting specific cell types which host Leishmania infection. Therefore, it is necessary to understand the characteristics of these cellular permissive hosts and explore the phenotype and microenvironment of such susceptible cells to identify mechanisms to directly target them. Here, the possibility of repurposing drugs for cellular targeting against leishmaniasis is discussed.
As discussed previously, first-line treatments against leishmaniasis are chemotherapeutic agents originally developed to treat different pathologies and later found to possess anti-parasitic properties. This approach is particularly beneficial in the field of the neglected tropical diseases, where drug repurposing can improve the treatment’s availability, despite the lack of financial incentive for new drug discovery.70 Examples of such repurposed drugs include both amphotericin B and azole drugs, discovered as antifungals with the ability to generate instability in cell membranes containing ergosterols.71,72 Decades later, both drugs were observed to have leishmanicidal effects due to the ergosterol content in Leishmania membranes.73–75 Similarly, miltefosine, a drug advanced to clinical trials targeting breast cancer and other malignancies,76 was found to have leishmanicidal potential which led to the approval of a topical miltefosine formulation.77 While the use of miltefosine against breast cancer did not prosper due to its secondary effects, it was approved in 2013 for the treatment of both VL and CL.77,78 Further, drug repurposing has been used by Bustamante et al to perform pharmacokinetic simulations between approved drugs and parasite proteins with homology to the known targets of such drugs.79 The group succeeded in finding 33 drugs that could potentially be used against leishmaniasis, with two drugs, rifabutin and pherphenazine, even displaying anti-leishmanial activity in vitro.79 Further in silico analysis may continue yielding older drugs for use against leishmaniasis. With greater understanding of the mechanisms aiding Leishmania survival and persistence, currently studied therapies can be matched to specific mechanisms and repurposed for use in leishmaniasis cases. However, due to high costs of advanced treatments, some alternatives proposed here are far from clinical and especially field-based use against leishmaniasis. Nevertheless, it is relevant to incorporate these experimental approaches in the pursuit of treating leishmaniases, with a particular interest going forward on targeting safe niches for Leishmania that facilitate persistence of the disease.
Hematopoietic Stem Cells (HSCs)
Immune Mechanisms in the BM and HSCs
The interplay between HSCs and the parasite is a constant fight in VL. On the one hand, HSCs can phagocytose L. infantum and produce pro-inflammatory cytokines such as TNF-α in response to infection.80 On the other hand, hematopoiesis-sustaining HSCs can serve as safe niches for L. donovani, protecting the parasite from chemotherapy.81 The relationship between hematopoiesis and VL has been long accepted, with a number of hematological issues arising in human VL patients caused by L. donovani infection.81 Experimental results show that L. donovani modulates host hematopoietic activity to allow its own survival and replication.82,83 In healthy mice, most long term HSCs (LT-HSCs) are quiescent, but after L. donovani infection there is an accumulation of CD4+ T-cells which, in response to TNF-α present in the BM environment, induces their production of IFN-γ.82 As recently described, a BM immune environment with a higher proportion of IFN-γ- and IL-17-producing effector T-cells correlates with low parasitic load, while greater FoxP3+ IL-10-producing cells and TGF-β levels are related to higher parasitic loads.84 Such immune balance is delicate as, despite IFN- γ being necessary for parasitic control in VL,85 it also induces LT-HSCs to enter the cell cycle, losing quiescence and causing the accumulation of hematological progenitor cells which undergo exhaustion after chronic stimulation.82 On the other hand, this excessive regulatory response can also undermine the health of the patient. As observed in VL patients, the accumulation of IL-10 producing T-regs in the BM inhibits T-cell activity,86 and has been related to PKDL development and resistance to sodium antimony gluconate (SAG) treatment.87,88 Furthermore, chemotherapy itself generates changes in the immune environment. CD8+CD25+Foxp3+ T cells frequency augments in all tissues, including the BM, of L. infantum-infected dogs relative to healthy ones, yet following chemotherapeutic treatments, cell populations returned to baseline levels.89 Although in the absence of treatment, presence of IL-10, TGF- β-producing CD4+ Tregs have been shown to exacerbate L. donovani susceptibility.89,90 Therefore, while immunotherapy against VL has been explored before,91 future therapies targeting the elimination of the parasites hiding in the HSCs will require fine modulation of the immune response in the BM specifically to promote parasitic control without risking hematopoietic balance.
Several reports of the presence of L. donovani in the BM of VL patients show that the BM is a frequent parasite niche.92–96 In VL infection models, both L. infantum and L. donovani show preferential infection of LT-HSCs over short term HSCs (ST-HSCs).97 Further, in the Frizzled6 (Fzd6) deficient VL mouse model in which HSC expansion is dampened, a reduction in parasitic burden during the chronic phase of the L. donovani infection has been seen.98 Additionally, Leishmania parasites may leverage LT-HSCs as niches of protection against chemotherapies. An experiment showed that after treatment with paromomycin, L. infantum disappeared from almost all tissues of the body except HSCs, where it hid in very low numbers. Three weeks after treatment cessation, the hiding parasites were then able to reinitiate HSC infection.97 Moreover, LT-HSCs were shown to be safer niches for both L. infantum and L. donovani than BM-derived macrophages after ex vivo treatment with paromomycin, miltefosine and SSG.97 Therefore, these results demonstrate how LT-HSCs niches are responsible for infection persistence in VL. Interestingly, the authors characterized a specific transcriptional profile in the LT-HSCs and found a reduction of Nos2 expression meaning that, although healthy LT-HSCs balance expression of nitric oxide (NO) and reactive oxygen species (ROS), infected LT-HSCs downregulate this mechanism to permit a safer space for the parasite.97 Although more research must be completed with regard to the specific biology and interaction of Leishmania with LT-HSCs, knowing the phenotype of this specific niche may facilitate the development of future therapies.
Leishmania infection also increases the generation of progenitor cells of the myeloid lineage which further serve as cellular hosts for the parasites.99 In a murine model of chronic VL, L. donovani infection caused the expansion of HSC and myeloid progenitor cells. The progeny of such cells were Ly6Chi/int monocytes with a regulatory phenotype, characterized by IL-10+ and arginase1+, which allow the persistence and proliferation of the parasite.83 Accordingly, in murine CL, it was observed that infection with the persistent strain L. major Seidman led to the recruitment of myeloid cells with a regulatory profile with lower levels of Type I and Type II interferons into the lesion site and the spleen. Moreover, the enhanced cell influx was sustained by the increase of HSCs with a myeloid-biased profile in the BM, compared to the infection with a self-healing inducing parasite.98 Therefore, the mechanism of susceptibility and persistence of VL in the BM include inflammatory immune response disruption, generation of immune-privileged cellular hosts, and generation of monocytic cellular hosts. Future leishmaniasis therapies that aim to cease reactivation of infection should therefore target the LT-HSCs.
Perspectives in Therapies Against Leishmania Infection in HSCs
The potential chemotherapeutic intervention in the BM immune ecosystem may help to eliminate parasites, although there is a remaining risk of disrupting immune balance and causing BM-HSC exhaustion with detrimental patient outcomes. Previously described findings could suggest that inducing ROS and NO by HSCs may favor Leishmania elimination.97 Nevertheless, studies of other diseases including leukemia and diabetes show that ROS dysregulation in the BM plays a role in both disease pathogenesis and the dysfunction of BM cells and hematopoiesis.100–103 Therefore, directly increasing the levels of NO and other oxygen species could be detrimental for the leishmaniasis patient, rather than therapeutic. Nevertheless, REDOX-reactive materials have been used to generate nanocarriers that deliver therapeutic drugs to cells depending on their ROS production, particularly against cancer.104,105 In settings with VL cases, nanocarrier therapies could be used to deliver drugs directly to infected HSCs, also reducing the rates of drug resistance and preventing parasites from reinitiating the infection.
Drug delivery systems are becoming desirable in the treatment of multiple diseases of the bone and BM including multiple myeloma, hematopoietic dysfunction, rheumatoid arthritis, bone metastases, and osteoporosis.106,107 Conventional drug therapy requires the administration of high doses of drugs in which only a small portion reaches the BM, yet these doses are associated with adverse side effects.108 Therefore, the design of drug delivery systems has been widely explored and reviewed.106,108,109 Liposomes and chimeric nanoparticles have been shown to be taken up by the BM.108 Therefore, in Leishmania infection, such technologies could be applied to reduce dosages and increase organ specificity.14,110 In that sense, liposome carriers, whose pharmacokinetics can be controlled through carrier surface modifications, have been designed to target the BM, examples have been listed in Table 2. Furthermore, the liposomal formulation of amphotericin B (Ambisome®) has significantly reduced the toxic effects of the conventional non-liposomal formulation (amphotericin B deoxycholate).111 Hence, the use of liposomal nanocarrier formulations is a plausible next step in the Leishmania drug development.
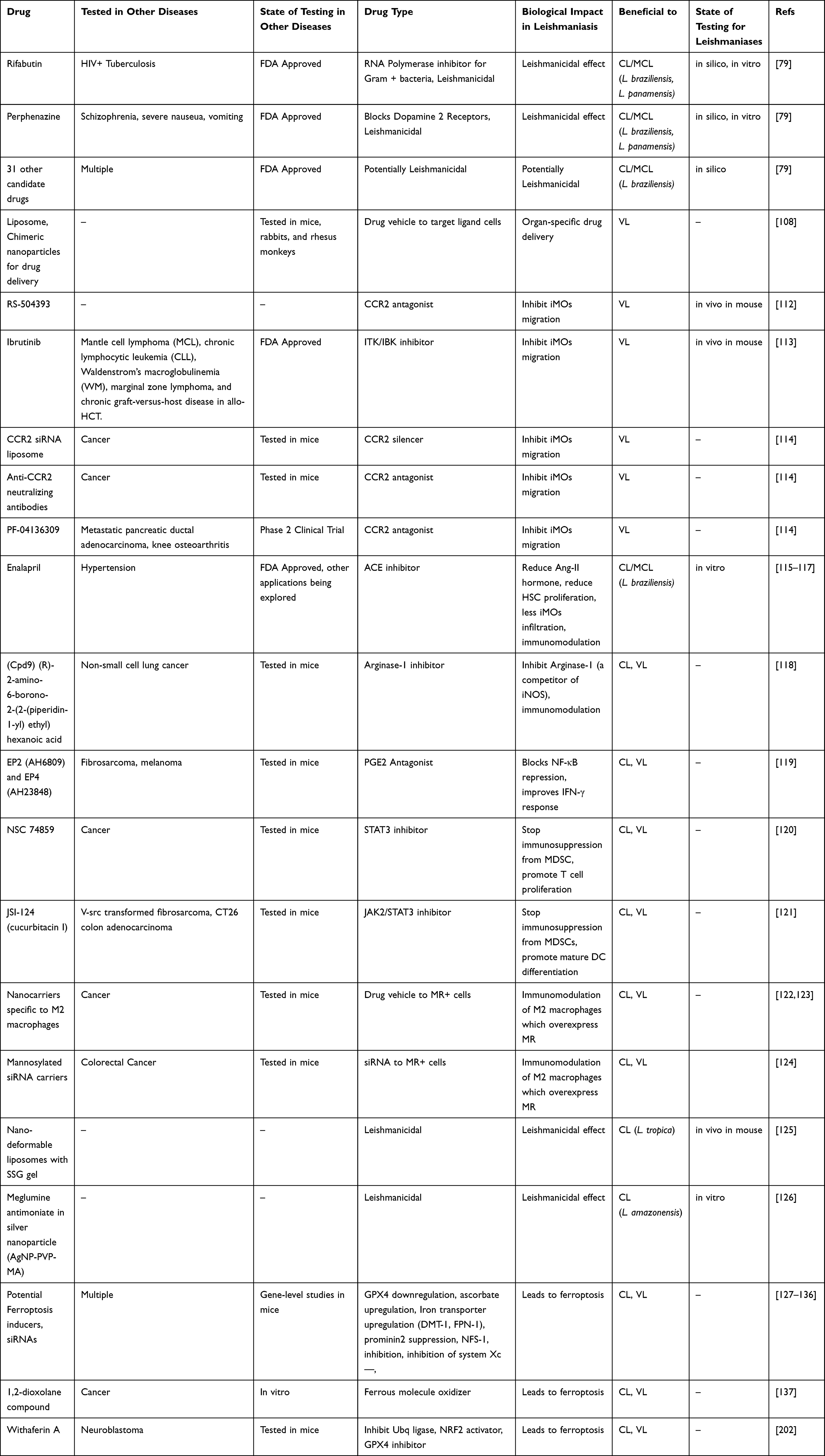 |
Table 2 Future Directions of Drugs for Leishmaniasis. Summary of Future Therapies That Could Be Repurposed for Their Use Against Leishmaniasis, the Disease for Which It Was Designed, Their State of Development and Testing Against Such Disease and Against Leishmaniasis, Mechanism of Action and Their Potential to Be Beneficial to Different Types of Leishmaniasis |
Monocytes
The proliferation of HSCs in response to infection generates more host cells for the parasite in the form of monocytes. Monocytes are circulating myeloid cells derived from HSCs and represent an intermediate state before becoming macrophages or certain types of DCs.139 Although both macrophages and DCs have been widely reported to be infected by Leishmania, macrophages are considered to be the main cells housing Leishmania replication.140–142 Nevertheless, much remains to be explored for their common ancestor. In this section, the characteristics of monocyte and macrophage hosts, as well as possible therapies targeting such niches are explored.
Migration of Monocytes in Anti-Leishmania Therapies
Monocytes are more than just transition cells; they delineate the interactions between host responses and the parasite by shaping immunity against disease.143 In leishmaniasis, inflammatory monocytes (iMOs) have been shown to provide safe environments for parasite replication in both L. donovani and L. major infection models.144,145 Particularly, the influx of iMOs to the liver and spleen seems to be detrimental during experimental VL. In murine infection with L. donovani, migration of iMOs into the liver and spleen can be reduced with the use of the drugs RS-504393, or ibrutinib (Table 2). This resulted in a reduced parasitic burden in the liver and spleen.112,113 The activity of such agents also impacted immune regulation. The use of RS-504393 resulted in no granuloma formation and the decrease of IFN-γ IL-10 double-producer CD4+ T cells and total CD4+ T cells.112 In the case of ibrutinib administration, there were increases in granuloma formation, IFN-γ and IL-4 producing NK cells, as well as the levels of protective cytokines such as IFN-γ, TNF-α, IL-4, and IL-13.113
Interestingly, CL studies show that monocyte migration is beneficial for the host, as monocytes tend to migrate to the lesion site rather than to the visceral organs. In L. major murine infection, CCR2 and platelet activation are responsible for Ly6C+ monocytes reaching the lesion site where they can efficiently participate in parasite elimination.146 Moreover, CCR2 favors DC migration to the draining lymph nodes where the lack of such chemokine receptor leads to a Th2-biased immune phenotype, providing a permissive environment for L. major.147 However, such responses differ depending on the infection route and mouse strain.148 Therefore, targeting monocytic migration through CCR2 inhibition treatments may be beneficial against L. donovani infection, but not against L. major infection. Multiple approaches to inhibit monocytic migration by targeting CCR2 have been tested against different cancer types (Table 2), as the monocytes can be detrimental to the patients of different malignancies.114 These already existing anti-cancer experimental therapies have not been tested against VL, but could lead to favorable results as observed in the cancer studies and have potential in the realm of field-deployable leishmaniasis treatments.
Monocyte Immune Balance
Besides iMOs migration, the phenotype resulting from the interaction between iMOs and the parasite highlights the role of iMOs during infection. For example, among other immune-modulatory mechanisms,149–151 L. donovani is capable of inhibiting acidification and superoxide production in the parasitophorous vacuole of iMOs, which then provides a permissive environment for the parasite.144 In L. donovani murine infection, the balance of inducible nitric oxide synthase (iNOS) and arginase-1 expression by iMOs correlates with the increase or decrease of parasitic burden in visceral organs.152,153 In the spleen, the arginase-1/iNOS ratio increases at 30 days post-infection (d.p.i.) while the infection becomes progressive. At the same time, the arginase-1/iNOS ratio diminishes in the liver as the infection starts resolving112 These findings correspond to observations in human patients diagnosed with VL, in which monocytes obtained from peripheral blood display an anti-inflammatory phenotype characterized by low levels of IL-1β and IL-6, as well as a reduction in phagocytosis capacity and production of ROS.154
Monocyte responses are dependent on the Leishmania strain infecting them, and this in turn seems to influence or relate to disease manifestation. Ex vivo infection of human monocytes with an L. infantum, strain causative of severe VL, induced the downmodulation of the immune response characterized by reduced TLR2, TLR9, and HLA-DR expression, and a lower TNF-α/IL-10 ratio. On the other hand, infection with L. braziliensis, causative of mild CL, has not only shown increased expression of the same receptor and the TNF-α/IL-10 ratio, but also better controls infection by reducing parasite counts by 100-fold.155 In the case of murine L. major infection, monocytes displayed altered maturation and became safe hosts for the parasite; yet, after a second inoculation, monocytes exhibited a stronger anti-parasitic response. While L. major inhibited monocyte maturation into a DC phenotype in secondary infections, monocytes were still able to eliminate parasites in an IFN-γ- and iNOS-dependent manner.145 Similarly, monocytes could eliminate the parasites more efficiently when they were not differentiated into macrophages. Murine infections with L. major generate monocytes with myeloid-derived suppressor cells (MDSCs) (Gr1hiCD11bhiF4/80int). When MDSCs were exposed to IFN-γ and IL-4, MDSCs increased their NO production, hence ramping up their killing capacity. Interestingly, when differentiation from MDSCs into macrophages was chemically induced, there was an increase in lesion sites and parasitic burden, showing that monocytes can eliminate the parasite, even when not fully matured, if they can produce NO.156 Therefore, immunomodulatory therapies could improve the monocytic capacity of eliminating parasites in both VL and CL manifestations.
The Interplay of Monocytes and HSCs: Therapeutic Potential
NO not only has a potent anti-parasitic effect, but also shapes the microenvironment of the infection site by preventing monocyte recruitment from the bloodstream and therefore inhibiting Leishmania from reaching potential host cells.157,158 As discussed previously, HSCs favor Leishmania infection by generating more monocytes that serve as host cells. Hence, HSCs and monocytes play an interconnecting role in sustaining Leishmania survival and proliferation. The interplay between these two cell types is similar to cancer scenarios in which HSCs support the generation and consequent accumulation of monocytes in the tumor, and when exposed to the microenvironment, gain a regulatory phenotype.114 In a murine model of lung cancer, the drug enalapril reduced the levels of the hormone responsible for amplifying HSCs and HSC-derived myeloid progenitors and increasing monocytes and macrophages in the lung.115 Enalapril (Table 2) is an FDA-approved drug commonly used for the treatment of hypertension.116 These results have been extrapolated to the treatment of leishmaniasis. The in vitro administration of enalapril to murine macrophages infected with L. braziliensis increased NO, IFN-γ, IL-12 and TNF-α production by macrophages while reducing the levels of IL-10.117 The hypothetical scenario where enalapril favors both reducing the permissive environment and limiting the expansion of the permissive niches would be beneficial in the treatment of leishmaniasis and the prevention of relapse. More importantly, the use of a drug available in more than 100 countries worldwide such as enalapril159 would be an ideal and affordable treatment to be deployed in the field in Leishmania-endemic areas. It is important to keep in mind that the profile and activation of iMOs differ among Leishmania species and mouse strain,160 so further therapeutic target studies must be specifically designed against different clinical manifestations of leishmaniasis. Still, immunomodulatory therapies directly targeting iMOs could facilitate the immune phenotype necessary to eliminate intracellular parasites.
Reprogramming Monocytes: Lessons from Cancer
In leishmaniasis, the parasite can reshape the immune activation of monocytes toward a regulatory profile, similar to that of M2 macrophages.161 In cancer, the tumor microenvironment reprograms monocytes toward regulatory phenotypes, preventing T-cell cytotoxicity and the recruitment of T-regs that allow tumor growth, metastasis, and immune evasion. However, exposing monocytes in vitro to IFN-γ or IFN-α activates tumoricidal activity by phagocytosis, cytokine-mediated killing, and antibody-dependent cytotoxicity.114 In summary, both cancer and Leishmania infections share the need for monocytes with an effector profile to favor the host and control disease. In a mouse model of non-small cell lung cancer, suppression of arginase-1 activity, an enzymatic competitor of iNOS, restored IFN-γ and IL-2 production by splenocytes and the cytotoxic phenotype of CD8+ T cells.118 This immune profile is beneficial to fight Leishmania infections,162 so it is possible to hypothesize that arginase-1 suppression could be a therapeutic target. An additional mechanism to reprogram monocyte phenotypes is by targeting prostaglandin E2 (PGE2).119 Downstream, PGE2 controls p50 NF-KB accumulation and translocation, and diverts signaling in response to IFN-γ. The administration of a PGE2 agonist to MDSCs with monocytic profiles reshaped this cascade and restored IFN-γ-mediated NO production of monocytes in tumor bearing mice.119 Similarly, the pharmacological inhibition of STAT3 activation reduced the immunosuppressive characteristics of suppressor monocytes.120,121,156 We speculate that the use of immune modulator compounds, such as the examples presented above (Table 2), could skew monocytic profiles in leishmaniasis towards a less permissive phenotype.
Macrophages
Macrophages compound a heterogeneous population of cells derived from monocyte differentiation and have been widely accepted as the main permissive hosts for Leishmania.140,141,163 As for monocytes, the phenotype of macrophages is determinant on their role as effector anti-leishmanial cells or as permissive hosts.164–166 Therefore, the use of immunotherapies is a promising approach to tilt immune balance towards the macrophage effector phenotype, some of which are shared with the monocyte-targeting therapies, a topic which has been recently reviewed in more depth.167 Next, examples are provided of how macrophages could be targeted with and without immunomodulation.
Macrophages can be targeted by loading drugs into specific delivery systems (Table 2), such as the previously mentioned nano-carriers that have shown promising results in the development of therapies against leishmaniasis.122 In the persistent CL model, it was demonstrated that L. major Siedman preferably infects M2 dermal macrophages with high expression of the mannose receptor (MR). Such strategies of immune evasion can keep the parasite protected inside the M2-macrophage from an environment rich in IFN-γ and TNF-α.141 In cancer research specifically, MR is commonly overexpressed; therefore, many nano-sized carriers have been developed to target MR+ cells.123 MR nano-carriers were designed to be covered by multiple MR-ligands on their surface to allow binding to MR+ cells. Once captured by their final target, nano-carriers can work both as drug and antigen delivery systems to antigen presenting cells.123 The clinical applications of MR-directed carriers could be expanded to Leishmania treatment to deliver drugs already in use to improve solubility and specificity, therefore reducing adverse effects of chemotherapies.123,168 Mannosylated carriers can also guide plasmids and siRNA for delivering gene therapy, which could be used to manipulate immune response within the lesion site.124 Daret al have recently developed a nano-deformable liposomes (NDLs) system to deliver SSG in a gel-form administration. The advantages of NDLs include 10-fold better retention of SSG in the deeper layers of the skin in an ex vivo model, compared to regular SSG. NDLs also showed effective anti-leishmanial activity in an in vitro model of L. infantum-infected macrophages.125
Meglumine antimoniate (Glucantime®) has also been successfully loaded to a silver nanoparticle (AgNP-PVP-MA) and its effect was tested in an in vitro model of macrophages infected with L. amazonensis. The administration of AgNP-PVP-MA led to a higher production of NO, TNF-α and IL-6 than that of infected macrophages treated with amphotericin B.126 More importantly, these silver nanoparticles reduced the viability of promastigotes, achieving the two objectives of directly targeting the parasite while modulating the immune response to benefit the patient. Therefore, the development of nano-delivery systems could improve efficacy and safety of current anti-leishmanial drugs.
Overall, the identification of cellular hiding niches, such as HSCs and the main cellular host, monocytes/macrophages, allowed the characterization of mechanisms and phenotypes related to susceptibility. Therefore, we propose that future therapies target such pathways (Figure 2) for therapies that completely eradicate the parasite from the host.
 |
Figure 2 Interplay of Leishmania and its cellular hosts: HSCs and monocytes. Hematopoietic stem cells (HSCs) respond to Leishmania infection. During this process, the microenvironment of the bone marrow (BM) changes, an inflammatory Th1 response usually prevents parasite expansion although infection can lead to local T-cell exhaustion, whereas an abundance of a regulatory environment, leaded by regulatory T cells (T regs) result in an elevated parasitic burden (PB) and is related to drug resistance. The nitric oxide (NO) and reactive oxygen species (ROS) production is also reduced and allows the generation of a permissive environment for the parasite. Although HSCs expand and differentiate to fight the infection, the resulting monocytes become the main host for the parasite, therefore resulting detrimental for the host. These newly differentiated monocytes express IL-10 and arginase (Arg)-1, and a reduction in iNOS and TNF-α, the characteristic phenotype that allows parasite survival. Finally, monocytes play a role by migrating to either the visceral organs in visceral leishmaniasis (VL) or to the skin lesion in cutaneous leishmaniasis (CL) with different roles for each manifestation. All this process can be targeted by the use of different therapies (bold), such as immunotherapies to reinstall the immune balance and favor an inflammatory response which allows parasite killing, also nanocarriers could be used to develop drugs directly to the infected cells. Other drugs limiting the expansion of HSCs could stop the generation of more cellular hosts as the monocytes, whereas migration of monocytes can also be driven into a favorable result for the patient. |
Ferroptosis – a Novel Target for Future Therapies Affecting Multiple Cell Types
While discussing the function of cell death in infectious disease settings, we are often reminded of our decades-old understanding that hosts effectively eliminate pathogenic niches through mechanisms such as apoptosis, necroptosis, pyroptosis, and autophagy. Alternatively, intracellular pathogens attempt to delay or avoid cell death by eliminating host immune cells, thus destabilizing the immune response. Broadly speaking, researchers are making significant efforts to control cell death procedures that may aid in the eradication of diseases, and certainly many pharmacological/natural compounds have been identified to achieve this. Similarly, over the past few years, an important mechanism, ferroptosis, has been characterized as being equally engaged in cell death regulation for multiple cell types.
Ferroptosis is a newly discovered programmed cell death, and it plays a vital regulatory role in the occurrence and development of many diseases. The ferroptosis process (Figure 3) is defined by the accumulation of lethal lipid species derived from lipid peroxidation. The process involves three major metabolisms: lipids, thiols, and iron-dependent lipid peroxidation. It can be precluded through the enzymatic reaction of glutathione peroxidase 4 (GPX4), that catalyzes the reduction of lipid peroxidation and ferroptosis suppressor protein 1 (FSP-1) which then catalyzes the regeneration of ubiquinone (Co-enzyme Q10).169,170 Through this unique pathway, ferroptosis undergoes a completely different course of action from apoptosis, necroptosis, autophagy, and all other forms of cell death.
 |
Figure 3 Regulatory pathways involved in ferroptosis. In ferroptosis, PUFAs are oxidized by free radicals due to lipid peroxidation. Hydroxyl facilitates lipid peroxidation, which causes stress-induced cell damage. Ferrous iron catalyzes these lipid hydroperoxides (LOOHs) into ROS molecules, followed by impairing cell membrane integrity. By turning damaging lipid hydroperoxides into lipid alcohols with the help of glutathione (GSH), GPX4 controls ferroptosis. Cystine is crucial for controlling lipid peroxidation by keeping GSH levels stable. SLC7A11 is a key regulator of SystemXc and is responsible for facilitating cystine/glutamate transport. Lipoperoxidation is inhibited by FSP-1, which is a NADPH-dependent coenzyme Q10 oxidoreductase located in cell membranes. Lipophilic radicals are contained within them, stopping the dissemination of lipid peroxide. This figure was created using Servier Medical Art Commons Attribution 3.0 Unported Licence (https://smart.servier.com/). Abbreviations: GSH, glutathione; GPX4, Glutathione peroxidase 4; PUFA, polyunsaturated fatty acids with carbon-carbon double bonds; AscH-, ascorbate autoxidation; TFR1, transferrin receptor-like; GLS, glutamate by glutaminase; DMT1, divalent metal transporter 1; LOXs, lipoxygenases; SLC7A11, Solute Carrier Family 7 Member 11; LOOH, lipid hydroperoxides; FSP-1, ferroptosis suppressor protein; Ubiquinone-10, NADPH-dependent coenzyme Q10. |
Unrestricted lipid peroxidation causes ferroptosis. Lipid peroxidation can be defined as a series of events where various oxidants (free radicals) strike on lipid containing carbon-carbon double bond(s). In this oxidation process, free radicals such as hydroxyl (-OH) are created by excess of ferrous iron (Fe2+) through the Fenton reaction. Subsequently, hydroxyls then attack polyunsaturated fatty acids (PUFAs).171 Hydroxyls are important for lipid peroxidation and in excess cause oxidative cell damage that can affect cellular DNA and lipid membrane damage or can alter the functional protein under stress conditions. Ferrous iron catalyzes lipid hydroperoxide into ROS molecules.171,172 Eventually, lipid peroxidation impairs cell membrane integrity and fluidity, causing cell damage or death. GPX4 is a key enzyme in ferroptosis regulation as it reacts with unstable intercellular lipid peroxides to form stable nontoxic lipid alcohols with the help of glutathione (GSH).173,174 Abridged GSH inhibits GPX4 and restrains the host’s ability to repair peroxidized lipids, triggering ferroptosis. Gao et al explained that cystine and cysteine are important for the synthesis of intercellular GSH, and their absence encourages ROS production.175 SLC7A11 (Solute Carrier Family 7 Member 11) is another critical component of system Xc’s cystine/glutamate transporter system. SLC7A11 controls cystine absorption and glutamine metabolism.176 Besides GPX4 and GSH, ferroptosis suppressor protein (FSP-1) is a NADPH-dependent coenzyme Q10 (ubiquinone-10) oxidoreductase on the cell membrane that inhibits lipid peroxidation. FSP-1, also known as apoptosis-inducing factor mitochondrial 2 (AIFM2), captures lipophilic radicals to stop lipid peroxide dissemination.170
Ferroptosis in Parasitic and Other Infectious Diseases
Moderating the above-mentioned pathways leads to ferroptosis-related cell death in a range of infectious diseases, including parasitic infections. Matsushita et al, showed that L. major infection in mice with GPX4-deficient T cells resulted in a decreased CD4+ T cell count, which play a major role in the persistence of Leishmania in vivo.127 Both CD8+ and CD4+ T cells deficient of GPX4 were unable to expand and protect the immune cell population from L. major infection. Also, in an ex vivo experiment, T cells lacking GPX4 rapidly accumulated membrane lipid peroxidase, which led to ferroptotic cell death instead of necroptosis.127 Leishmania can also express ascorbate peroxidase (APX), a gene associated with the trypanothione pathway that converts host hydrogen peroxide into water molecules and alters GXP expression, as well.177,178 It has been observed that L. braziliensis and L. infantum strains which highly express APX are more likely to develop an antimony-resistant phenotype.177,179 Similarly, elevated APX was found to regulate amphotericin-B resistance in clinical isolates of L. donovani.180,181 APX is also an essential factor to control metacyclogenesis in L. major promastigotes and causes antimonial resistance.182,183 Ascorbate-induced cell death and ferroptosis processes share common features including ROS generation, iron dependency, caspase independency and lipid peroxidation.128 Lower concentrations of ascorbate could prevent ferroptosis, but ascorbate overexpression causes cellular cytotoxicity and ferroptosis.128 Interestingly, anemia and abnormal erythrophagocytosis and hemophagocytosis have been implicated in L. donovani infections.184 Leishmania infection also stimulates CD163 expression, boosts cytosolic heme oxygenase-1 (HMOX1) levels, and reduces ROS generation to promote parasite survival and replication.185 Corroborating this with macrophage function for their important role in iron storage and recycling of senescent red blood cells (RBCs), macrophages ingest senescent RBCs, harvest a larger amount of iron, and facilitate iron breakdown into cytosolic heme which is then used by HMOX1 and binds to hemopexin to be internalized in macrophages through the CD91 receptors.186–189
The exploration of the role of ferroptosis during Leishmania infections has only recently begun, however observations of ferroptosis have been implied in other infections. Thus, these studies can help extrapolate possible therapeutic targets for leishmaniasis. For example, some studies suggested that Trypanosoma cruzi extract contained ascorbate-dependent peroxidase activity.190–192 GPX-like glutathione peroxidases (Px I–III) are found in trypanosomes, however, these peroxidases are type II tryparedoxin peroxidases that reduce tryparedoxin at significantly greater folds than GSH.193 Similarly, a large amount of ROS is generated in host cells infected with malaria parasites through activation of PAMPs and DAMPs, leading to cell death due to lipid peroxidation and ferroptosis.194 Moreover, an instance of mutations in human TP53 caused defective p53 which led to accumulation of excessive iron in macrophages, and promoted M2 polarization, but inhibited Plasmodium infection and supported Listeria bacteria.195 Like parasitic infection, GPX4-deficient T cells in mice infected with choriomeningitis virus also showed impaired T cell development due to abnormal lipid peroxidation, leading to T cell ferroptosis.127 Additionally, when injecting ovalbumin into mice and humans, Yao et al, observed follicular helper T cells underwent ferroptosis, with higher lipid ROS and more of the final products of polyunsaturated fatty acid peroxidation, MDA (malondialdehyde) and 4-HNE (4-hydroxynonenal), in comparison to non-follicular helper T cells.196 In the same vein, Mycobacterium tuberculosis (Mtb) can use host-stored iron for its own growth.197,198 However, Mtb-infected macrophages have shown unregulated lipid peroxidation and decreased GPX4, which in turn significantly increased cell death.199 Iron accumulation and lipid peroxidation also occurs in patients with cryptococcal meningitis.200,201 Particularly, Cryptococcus infections in AIDS patients have been shown to activate macrophages in CSF and promote pro-inflammatory cytokines and chemokines that lead to abundance of iron transporters, like divalent metal transporter 1 (DMT-1) and ferroportin 1 (FPN-1), that promote increased extracellular iron uptake, which could be associated with ferroptotic conditions.129–131
In addition to conditions seen in infectious diseases, cancer cells also accumulate high levels of iron; therefore, ferroptosis has been more deeply studied in cancer models.132 Although parasitic diseases and cancer are quite different, the basic knowledge, pharmacological targets and approaches in cancer studies can be leveraged for the investigation of leishmaniasis. Imbalanced iron metabolism is correlated with numerous cancer types such as breast, ovarian, brain, renal, and lung cancer.138 For example, ferritin dissociation induces reduction of ferric (Fe3+) to ferrous (Fe2+) iron, thus upregulating lipid peroxidation and mitochondrial superoxide generation which selectively kills cancer cells.133 Similarly, a 1,2-dioxolane compound can oxidize ferrous molecules, leading to lipid peroxidation that eliminates cancer cells through ferroptotic mechanisms.137 Suppressing or silencing the prominin2 gene in breast cancer cells has been shown to decrease intercellular iron export through formation of ferritin-associated multivesicular bodies.134 In lung cancer cells, inhibition of nitrogen fixation-1 (NFS-1), an iron-sulfur cluster biosynthetic enzyme, stimulates transferrin receptors and downmodulates ferritin that leads to ferroptosis.135 In high-risk neuroblastoma, obstruction of a ubiquitin ligase complex leads to the ability to reduce nuclear factor erythroid 2-related factor 2 (NRF2) inhibition, upsurging heme oxygenase-1 activity and suppressing GPX4 to kill cancer cells through a natural ferroptosis inducer compound withaferin A (WA).202 Furthermore, ferroptosis may contribute to tumor immunity beyond direct killing, particularly in association with dendritic cells that play an important role against tumor cells. Studies have shown that gene ALox15-derived lipid peroxidation regulates dendritic cells and modulates adaptive immune responses, while in contrast, oxidizing phosphatidylcholine inhibits dendritic cell maturation and decreases differentiation of Th17 cells.203 Additionally, IFN-γ plays an indispensable role in antitumor activity. Surprisingly IFN-γ can suppress ferroptosis-suppressor genes that aid with GSH production in cancer.136 Thus, through both the ability of ferroptosis to induce direct killing of cancer cells and infected cells, and through the immunomodulation that can follow lipid peroxidation, these studies demonstrate the potential for new directions of research in developing leishmaniasis treatments where similar mechanisms may exist.
In adaptive immunity, GPX4 knockout mice showed the presence of antigen specific CD8+ and CD4+ cells, yet failed to expand T cell counts during chronic infection.127 Mechanically, IFN-γ released from CD8+ T cells triggers lipid peroxidation and ferroptosis through the inhibition of system Xc—.136 Similarly, cysteine and PD-L1 strongly inhibit ID8 cell derived tumors along with increasing lipid peroxidation in tumor cells and elevating proportions of IFN-γ+ and TNF-α+ CD8+ and CD4+ T cells in the tumor microenvironment.136,204,205 Remarkably, expression of system Xc− was negatively associated with CD8+ T cells and IFN-γ in cancer treatments and their outcome. To summarize, ferroptosis consists of a series of events involving redox imbalance, iron release ferritin, and lipid peroxidation, all which lead to the occurrence of cell death in various diseases and stress conditions. Ferroptotic cell death has been demonstrated to occur in tumor cells and in multiple infectious diseases with beneficial impacts for the host and reduction of disease burden. These findings support the further investigation of the several compounds identified to regulate ferroptotic pathways. More work in stimulation of ferroptosis in infected cells could further improve field-deployable therapies against Leishmania and other infectious diseases.
Conclusion
Overall, the realm of field-deployable therapies has room for improvement and advancement, and faces discrepancies in which treatments are truly and universally field-deployable. With current treatments being variable with regard to their accessibility, feasibility to be administered in the field, achieval of sterile immunity and risk of relapse, and more, a number of new prospects should be considered for Leishmania treatment going forward. Leishmania parasites have been shown to infect multiple cell types including monocytes, macrophages, dendritic cells, and neutrophils in multiple areas of the body. Consideration of this fact will allow for the development of directed therapies to eliminate the parasites from its cellular hosts for generation of sterile immunity. This is particularly appealing for targeting monocytes/macrophages, which are considered to be the main cellular host of the parasite in the body. Also, targeting HSCs is desirable, since recent discoveries reveal how they provide a safe-heaven for Leishmania to avoid the immune system and a lair to the parasite to allow reinfection. By taking advantage of drugs and therapies developed to be used against other diseases, leishmaniasis patients can be treated with more advanced and specific therapies. The advantages of repurposing drugs impact a multitude of factors. First, patients will benefit from therapies with higher efficacy and less adverse effects. Second, since leishmaniasis is a neglected tropical disease causing economic burden to low- and middle-income countries, repurposing therapies may become a more affordable pathway to alleviate disease burden and eventually achieve eradication of the disease. Third, some therapies have already been approved for their use against other diseases, therefore the time to reach and treat Leishmania patients could be reduced. Lastly, by targeting unique pathways utilized by the parasites, future treatments may be enhanced. One prominent pathway which is being explored relatively recently is the ferroptosis cell death pathway. Although drugs targeting this pathway have not yet been universally approved, there are promising results in the case of other intracellular infections and cancer settings that indicate stimulation of ferroptosis or inactivation of ferroptosis-inhibiting genes have shown beneficial effects in these disease settings and thus merit investigation for leishmaniasis. Overall, current and potential treatments for leishmaniases, both field-deployable and experimental, provide promising candidates for the mitigation of disease and promotion of host outcomes to Leishmania infection.
Acknowledgments
Our contributions are an informal communication and represent our own best judgment. These comments do not bind or obligate FDA.
Disclosure
The US Food and Drug Administration and Ohio State Innovation Foundation report a patent 17-786,129 pending to Gennova Biopharmaceuticals. The authors report no other conflict of interest in this work.
References
1. CDC. DPDx – Leishmaniasis:23. Available from: https://www.cdc.gov/dpdx/leishmaniasis/index.html.
2. David CV, Craft N. Cutaneous and mucocutaneous leishmaniasis. Dermatol Ther. 2009;22(6):491. doi:10.1111/j.1529-8019.2009.01272.x
3. Reithinger R, Dujardin JC, Louzir H, Pirmez C, Alexander B, Brooker S. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7(9):581–596. doi:10.1016/S1473-3099(07)70209-8
4. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet. 2018;392(10151):951–970. doi:10.1016/S0140-6736(18)31204-2
5. Alcantara LM, Ferreira TCS, Gadelha FR, Miguel DC. Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. Int J Parasitol Drugs Drug Resist. 2018;8(3):430–439. doi:10.1016/j.ijpddr.2018.09.006
6. Tiuman TS, Santos AO, Ueda-Nakamura T, Filho BP, Nakamura CV. Recent advances in leishmaniasis treatment. Int J Infect Dis. 2011;15(8):e525–32. doi:10.1016/j.ijid.2011.03.021
7. McGwire BS, Satoskar AR. Leishmaniasis: clinical syndromes and treatment. QJM. 2014;107(1):7–14. doi:10.1093/qjmed/hct116
8. Varma N, Naseem S. Hematologic changes in visceral leishmaniasis/kala azar. Indian J Hematol Blood Transfus. 2010;26(3):78. doi:10.1007/s12288-010-0027-1
9. Zijlstra EE. The immunology of post-kala-azar dermal leishmaniasis (PKDL). Parasit Vectors. 2016;9:464. doi:10.1186/s13071-016-1721-0
10. Lypaczewski P, Matlashewski G. Leishmania donovani hybridisation and introgression in nature: a comparative genomic investigation. Lancet Microbe. 2021;2(6):e250–e258. doi:10.1016/S2666-5247(21)00028-8
11. Adaui V, Lye LF, Akopyants NS, et al. Association of the endobiont double-stranded RNA virus LRV1 with treatment failure for human leishmaniasis caused by leishmania braziliensis in Peru and Bolivia. J Infect Dis. 2016;213(1):112–121. doi:10.1093/infdis/jiv354
12. Kuhlmann FM, Robinson JI, Bluemling GR, Ronet C, Fasel N, Beverley SM. Antiviral screening identifies adenosine analogs targeting the endogenous dsRNA Leishmania RNA virus 1 (LRV1) pathogenicity factor. Proc Natl Acad Sci U S A. 2017;114(5):E811–E819. doi:10.1073/pnas.1619114114
13. Hartley MA, Ronet C, Zangger H, Beverley SM, Fasel N. Leishmania RNA virus: when the host pays the toll. Front Cell Infect Microbiol. 2012;2:99. doi:10.3389/fcimb.2012.00099
14. Uliana SRB, Trinconi CT, Coelho AC. Chemotherapy of leishmaniasis: present challenges. Parasitology. 2018;145(4):464–480. doi:10.1017/S0031182016002523
15. Moore EM, Lockwood DN. Treatment of visceral leishmaniasis. J Glob Infect Dis. 2010;2(2):151–158. doi:10.4103/0974-777X.62883
16. Ponte-Sucre A, Gamarro F, Dujardin JC, et al. Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl Trop Dis. 2017;11(12):e0006052. doi:10.1371/journal.pntd.0006052
17. Madusanka RK, Silva H, Karunaweera ND. Treatment of cutaneous leishmaniasis and insights into species-specific responses: a narrative review. Infect Dis Ther. 2022;11(2):695–711. doi:10.1007/s40121-022-00602-2
18. Alrajhi AA, Ibrahim EA, De Vol EB, Khairat M, Faris RM, Maguire JH. Fluconazole for the treatment of cutaneous leishmaniasis caused by Leishmania major. N Engl J Med. 2002;346(12):891–895. doi:10.1056/NEJMoa011882
19. Galvao EL, Rabello A, Cota GF. Efficacy of azole therapy for tegumentary leishmaniasis: a systematic review and meta-analysis. PLoS One. 2017;12(10):e0186117. doi:10.1371/journal.pone.0186117
20. Saenz RE, Paz H, Berman JD. Efficacy of ketoconazole against Leishmania braziliensis panamensis cutaneous leishmaniasis. Am J Med. 1990;89(2):147–155. doi:10.1016/0002-9343(90)90292-l
21. Ballou WR, McClain JB, Gordon DM, et al. Safety and efficacy of high-dose sodium stibogluconate therapy of American cutaneous leishmaniasis. Lancet. 1987;2(8549):13–16. doi:10.1016/s0140-6736(87)93053-4
22. Sundar S, Chakravarty J. Liposomal amphotericin B and leishmaniasis: dose and response. J Glob Infect Dis. 2010;2(2):159–166. doi:10.4103/0974-777X.62886
23. Gadelha EPN, Ramasawmy R, da Costa Oliveira B, et al. An open label randomized clinical trial comparing the safety and effectiveness of one, two or three weekly pentamidine isethionate doses (seven milligrams per kilogram) in the treatment of cutaneous leishmaniasis in the Amazon Region. PLoS Negl Trop Dis. 2018;12(10):e0006850. doi:10.1371/journal.pntd.0006850
24. Krause G, Kroeger A. Topical treatment of American cutaneous leishmaniasis with paramomycin and methylbenzethonium chloride: a clinical study under field conditions in Ecuador. Trans R Soc Trop Med Hyg. 1994;88(1):92–94. doi:10.1016/0035-9203(94)90517-7
25. Soto J, Grogl M, Berman J, Olliaro P. Limited efficacy of injectable aminosidine as single-agent therapy for Colombian cutaneous leishmaniasis. Trans R Soc Trop Med Hyg. 1994;88(6):695–698. doi:10.1016/0035-9203(94)90235-6
26. Zerpa O, Ulrich M, Blanco B, et al. Diffuse cutaneous leishmaniasis responds to miltefosine but then relapses. Br J Dermatol. 2007;156(6):1328–1335. doi:10.1111/j.1365-2133.2007.07872.x
27. Wolf Nassif P, De Mello TF, Navasconi TR, et al. Safety and efficacy of current alternatives in the topical treatment of cutaneous leishmaniasis: a systematic review. Parasitology. 2017;144(8):995–1004. doi:10.1017/S0031182017000385
28. Khamesipour A. Therapeutic vaccines for leishmaniasis. Expert Opin Biol Ther. 2014;14(11):1641–1649. doi:10.1517/14712598.2014.945415
29. Husein-ElAhmed H, Gieler U, Steinhoff M. Evidence supporting the enhanced efficacy of pentavalent antimonials with adjuvant therapy for cutaneous leishmaniasis: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2020;34(10):2216–2228. doi:10.1111/jdv.16333
30. Berbert TRN, de Mello TFP, Wolf Nassif P, et al. Pentavalent antimonials combined with other therapeutic alternatives for the treatment of cutaneous and mucocutaneous leishmaniasis: a systematic review. Dermatol Res Pract. 2018;2018:1–21. doi:10.1155/2018/9014726
31. Garza-Tovar TF, Sacriste-Hernandez MI, Juarez-Duran ER, Arenas R. An overview of the treatment of cutaneous leishmaniasis. Fac Rev. 2020;9:28. doi:10.12703/r/9-28
32. Velasco-Castrejon O, Walton BC, Rivas-Sanchez B, et al. Treatment of cutaneous leishmaniasis with localized current field (radio frequency) in Tabasco, Mexico. Am J Trop Med Hyg. 1997;57(3):309–312. doi:10.4269/ajtmh.1997.57.309
33. Aronson NE, Wortmann GW, Byrne WR, et al. A randomized controlled trial of local heat therapy versus intravenous sodium stibogluconate for the treatment of cutaneous Leishmania major infection. PLoS Negl Trop Dis. 2010;4(3):e628. doi:10.1371/journal.pntd.0000628
34. Volpedo G, Huston RH, Holcomb EA, et al. From infection to vaccination: reviewing the global burden, history of vaccine development, and recurring challenges in global leishmaniasis protection. Expert Rev Vaccines. 2021;20(11):1431. doi:10.1080/14760584.2021.1969231
35. Panagiotopoulos A, Stavropoulos PG, Hasapi V, Papakonstantinou AM, Petridis A, Katsambas A. Treatment of cutaneous leishmaniasis with cryosurgery. Int J Dermatol. 2005;44(9):749–752. doi:10.1111/j.1365-4632.2005.02628.x
36. Al-Qubati Y, Janniger EJ, Schwartz RA. Cutaneous leishmaniasis: cryosurgery using carbon dioxide slush in a resource-poor country. Int J Dermatol. 2012;51(10):1217–1220. doi:10.1111/j.1365-4632.2011.04958.x
37. Lopez-Carvajal L, Cardona-Arias JA, Zapata-Cardona MI, Sanchez-Giraldo V, Velez ID. Efficacy of cryotherapy for the treatment of cutaneous leishmaniasis: meta-analyses of clinical trials. BMC Infect Dis. 2016;16:360. doi:10.1186/s12879-016-1663-3
38. Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the infectious diseases society of America (IDSA) and the American society of tropical medicine and hygiene (ASTMH). Clin Infect Dis. 2016;63(12):1539. doi:10.1093/cid/ciw742
39. Pinheiro AC, de Souza MVN. Current leishmaniasis drug discovery. RSC Med Chem. 2022;13(9):1029. doi:10.1039/d1md00362c
40. Thompson AM, O’Connor PD, Marshall AJ, et al. 7-substituted 2-Nitro-5,6-dihydroimidazo 2,1-b 1,3 oxazines: novel antitubercular agents lead to a new preclinical candidate for visceral leishmaniasis. J Med Chem. 2017;60(10):4212. doi:10.1021/acs.jmedchem.7b00034
41. Van den Kerkhof M, Leprohon P, Mabille D, et al. Identification of resistance determinants for a promising antileishmanial oxaborole series. Microorganisms. 2021;9(7):1408. doi:10.3390/microorganisms9071408
42. Machado-Pinto J, Pinto J, da Costa CA, et al. Immunochemotherapy for cutaneous leishmaniasis: a controlled trial using killed Leishmania (Leishmania) amazonensis vaccine plus antimonial. Int J Dermatol. 2002;41(2):73–78. doi:10.1046/j.1365-4362.2002.01336.x
43. Mayrink W, Botelho AC, Magalhaes PA, et al. Immunotherapy, immunochemotherapy and chemotherapy for American cutaneous leishmaniasis treatment. Rev Soc Bras Med Trop. 2006;39(1):14–21. doi:10.1590/s0037-86822006000100003
44. Convit J, Ulrich M, Zerpa O, et al. Immunotherapy of American cutaneous leishmaniasis in Venezuela during the period 1990–99. Trans R Soc Trop Med Hyg. 2003;97(4):469–472. doi:10.1016/s0035-9203(03)90093-9
45. Datta A, Podder I, Das A, Sil A, Das NK. Therapeutic modalities in post kala-azar dermal leishmaniasis: a systematic review of the effectiveness and safety of the treatment options. Indian J Dermatol. 2021;66(1):34–43. doi:10.4103/ijd.IJD_264_20
46. Taslimi Y, Zahedifard F, Rafati S. Leishmaniasis and various immunotherapeutic approaches. Parasitology. 2018;145(4):497–507. doi:10.1017/S003118201600216X
47. Moafi M, Rezvan H, Sherkat R, Taleban R. Leishmania vaccines entered in clinical trials: a review of literature. Int J Prev Med. 2019;10:95. doi:10.4103/ijpvm.IJPVM_116_18
48. Shaddel M, Sharifi I, Karvar M, Keyhani A, Baziar Z. Cryotherapy of cutaneous leishmaniasis caused by Leishmania major in BALB/c mice: a comparative experimental study. J Vector Borne Dis. 2018;55(1):42–46. doi:10.4103/0972-9062.234625
49. Gedda MR, Singh B, Kumar D, et al. Post kala-azar dermal leishmaniasis: a threat to elimination program. PLoS Negl Trop Dis. 2020;14(7):e0008221. doi:10.1371/journal.pntd.0008221
50. Mansueto P, Seidita A, Vitale G, Cascio A. Transfusion transmitted leishmaniasis. What to do with blood donors from endemic areas? Travel Med Infect Dis. 2014;12(6 Pt A):617–627. doi:10.1016/j.tmaid.2014.10.011
51. Mirzabeigi M, Farooq U, Baraniak S, Dowdy L, Ciancio G, Vincek V. Reactivation of dormant cutaneous Leishmania infection in a kidney transplant patient. J Cutan Pathol. 2006;33(10):701–704. doi:10.1111/j.1600-0560.2006.00532.x
52. Antinori S, Cascio A, Parravicini C, Bianchi R, Corbellino M. Leishmaniasis among organ transplant recipients. Lancet Infect Dis. 2008;8(3):191–199. doi:10.1016/S1473-3099(08)70043-4
53. Peris MP, Esteban-Gil A, Ares-Gómez S, Morales M, Castillo JA, Moreno B. Characterization of lesions in the temporal muscle and the male reproductive system (epididymis and testicle) of dogs experimentally infected with Leishmania infantum with different clinical stages. Vet Parasitol. 2022;305. doi:10.1016/j.vetpar.2022.109700
54. Boechat VC, Pereira SA, Júnior AAVM, et al. Frequency, active infection and load of Leishmania infantum and associated histological alterations in the genital tract of male and female dogs. PLoS One. 2020;15(9):e0238188. doi:10.1371/journal.pone.0238188
55. Boechat VC, Mendes Junior AAV, Madeira M, et al. Occurrence of Leishmania infantum and associated histological alterations in the genital tract and mammary glands of naturally infected dogs. Parasitol Res. 2016;115(6):2371. doi:10.1007/s00436-016-4987-4
56. Turchetti AP, Souza TD, Paixão TA, Santos RL. Sexual and vertical transmission of visceral leishmaniasis. J Infect Dev Ctries. 2014;8(4):403. doi:10.3855/jidc.4108
57. Mignot G, Bhattacharya Y, Reddy A. Ocular Leishmaniasis - A systematic review. Indian J Ophthalmol. 2021;69(5):1052. doi:10.4103/ijo.IJO_2232_20
58. Maia CSF, Monteiro MC, Gavioli EC, Oliveira FR, Oliveira GB, Romão PRT. Neurological disease in human and canine leishmaniasis--clinical features and immunopathogenesis. Parasite Immunol. 2015;37(8):385. doi:10.1111/pim.12203
59. Petersen CA, Greenlee MHW. Neurologic manifestations of Leishmania spp. infection. J Neuroparasitology. 2011;2:1–5. doi:10.4303/jnp/N110401
60. Llanos-Cuentas A, Valencia BM, Petersen CA. Neurological manifestations of human leishmaniasis. Handb Clin Neurol. 2013;114:193. doi:10.1016/b978-0-444-53490-3.00013-3
61. Sbrana S, Marchetti V, Mancianti F, Guidi G, Bennett D. Retrospective study of 14 cases of canine arthritis secondary to Leishmania infection. J Small Anim Pract. 2014;55(6):309. doi:10.1111/jsap.12204
62. Guidelli GM, De Stefano R, Galeazzi M, Selvi E. Synovial Leishmaniasis. Arthritis Rheumatol. 2016;68(4):931. doi:10.1002/art.39541
63. Mehrotra R, Choudhry VP, Saxena R, Kapila K, Saraya AK. Asymptomatic visceral leishmaniasis in a child with acute lymphoblastic leukaemia. J Infect. 1995;30(2):157. doi:10.1016/s0163-4453(95)80012-3
64. Grinnage-Pulley T, Scott B, Petersen CA. A mother’s gift: congenital transmission of trypanosoma and leishmania species. PLoS Pathog. 2016;12(1):e1005302. doi:10.1371/journal.ppat.1005302
65. Berger BA, Bartlett AH, Saravia NG, Galindo Sevilla N. Pathophysiology of leishmania infection during pregnancy. Trends Parasitol. 2017;33(12):935. doi:10.1016/j.pt.2017.08.012
66. Figueiró-Filho EA, Duarte G, El-Beitune P, Quintana SM, Maia TL. Visceral leishmaniasis (kala-azar) and pregnancy. Infect Dis Obstet Gynecol. 2004;12(1):31. doi:10.1080/1064744042000210384
67. Pagliano P, Carannante N, Rossi M, et al. Visceral leishmaniasis in pregnancy: a case series and a systematic review of the literature. J Antimicrob Chemother. 2005;55(2):229. doi:10.1093/jac/dkh538
68. Gajurel K, Dhakal R, Deresinski S. Leishmaniasis in solid organ and hematopoietic stem cell transplant recipients. Clin Transplant. 2017;31(1):e12867. doi:10.1111/ctr.12867
69. Tatarelli P, Fornaro G, Del Bono V, et al. Visceral leishmaniasis in hematopoietic cell transplantation: case report and review of the literature. J Infect Chemother. 2018;24(12):990. doi:10.1016/j.jiac.2018.05.008
70. Makoni M. New threats to visceral leishmaniasis control. Lancet Microbe. 2021;2(11):e574. doi:10.1016/S2666-5247(21)00285-8
71. Mesa-Arango AC, Scorzoni L, Zaragoza O. It only takes one to do many jobs: amphotericin B as antifungal and immunomodulatory drug. Front Microbiol. 2012;3:286. doi:10.3389/fmicb.2012.00286
72. Shafiei M, Peyton L, Hashemzadeh M, Foroumadi A. History of the development of antifungal azoles: a review on structures, SAR, and mechanism of action. Bioorg Chem. 2020;104:104240. doi:10.1016/j.bioorg.2020.104240
73. Cohen BE, Gamargo M. Concentration and time dependence of amphotericin B-induced permeability changes across plasma membrane vesicles from Leishmania sp. Drugs Exp Clin Res. 1987;13(9):539–546.
74. Ramos H, Valdivieso E, Gamargo M, Dagger F, Cohen BE. Amphotericin B kills unicellular leishmanias by forming aqueous pores permeable to small cations and anions. J Membr Biol. 1996;152(1):65–75. doi:10.1007/s002329900086
75. Keighobadi M, Emami S, Fakhar M, Shokri A, Mirzaei H, Hosseini Teshnizi S. Repurposing azole antifungals into antileishmanials: novel 3-triazolylflavanones with promising in vitro antileishmanial activity against Leishmania major. Parasitol Int. 2019;69:103–109. doi:10.1016/j.parint.2018.12.006
76. Leonard R, Hardy J, van Tienhoven G, et al. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J Clin Oncol. 2001;19(21):4150–4159. doi:10.1200/JCO.2001.19.21.4150
77. Dorlo TP, Balasegaram M, Beijnen JH, de Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67(11):2576–2597. doi:10.1093/jac/dks275
78. Van Bocxlaer K, Yardley V, Murdan S, Croft SL. Topical formulations of miltefosine for cutaneous leishmaniasis in a BALB/c mouse model. J Pharm Pharmacol. 2016;68(7):862–872. doi:10.1111/jphp.12548
79. Bustamante C, Ochoa R, Asela C, Muskus C. Repurposing of known drugs for leishmaniasis treatment using bioinformatic predictions, in vitro validations and pharmacokinetic simulations. J Comput Aided Mol Des. 2019;33(9):845–854. doi:10.1007/s10822-019-00230-y
80. Carvalho-Gontijo R, Moreira DR, Resende M, et al. Infection of hematopoietic stem cells by Leishmania infantum increases erythropoiesis and alters the phenotypic and functional profiles of progeny. Cell Immunol. 2018;326:77. doi:10.1016/j.cellimm.2017.10.016
81. Challen GA, Boles N, Lin KK, Goodell MA. Mouse hematopoietic stem cell identification and analysis. Cytometry A. 2009;75(1):14–24. doi:10.1002/cyto.a.20674
82. Pinto AI, Brown N, Preham O, Doehl JSP, Ashwin H, Kaye PM. TNF signalling drives expansion of bone marrow CD4+ T cells responsible for HSC exhaustion in experimental visceral leishmaniasis. PLoS Pathog. 2017;13(7):e1006465. doi:10.1371/journal.ppat.1006465
83. Abidin BM, Hammami A, Stager S, Heinonen KM. Infection-adapted emergency hematopoiesis promotes visceral leishmaniasis. PLoS Pathog. 2017;13(8):e1006422. doi:10.1371/journal.ppat.1006422
84. Kumar P, Misra P, Thakur CP, Saurabh A, Rishi N, Mitra DK. T cell suppression in the bone marrow of visceral leishmaniasis patients: impact of parasite load. Clin Exp Immunol. 2018;191(3):318–327. doi:10.1111/cei.13074
85. Kima PE, Soong L. Interferon gamma in leishmaniasis. Front Immunol. 2013;4:156. doi:10.3389/fimmu.2013.00156
86. Rai AK, Thakur CP, Singh A, et al. Regulatory T cells suppress T cell activation at the pathologic site of human visceral leishmaniasis. PLoS One. 2012;7(2):e31551. doi:10.1371/journal.pone.0031551
87. Murray HW. Clinical and experimental advances in treatment of visceral leishmaniasis. Antimicrob Agents Chemother. 2001;45(8):2185–2197. doi:10.1128/AAC.45.8.2185-2197.2001
88. Saha S, Mondal S, Ravindran R, et al. IL-10- and TGF-beta-mediated susceptibility in kala-azar and post-kala-azar dermal leishmaniasis: the significance of amphotericin B in the control of Leishmania donovani infection in India. J Immunol. 2007;179(8):5592–5603. doi:10.4049/jimmunol.179.8.5592
89. Santos MF, Alexandre-Pires G, Pereira MA, et al. Immunophenotyping of peripheral blood, lymph node, and bone marrow t lymphocytes during canine leishmaniosis and the impact of antileishmanial chemotherapy. Front Vet Sci. 2020;7. doi:10.3389/fvets.2020.00375
90. Tiwananthagorn S, Iwabuchi K, Ato M, Sakurai T, Kato H, Katakura K. Involvement of CD4+ Foxp3+ regulatory T cells in persistence of leishmania donovani in the liver of alymphoplastic aly/aly mice. PLoS Negl Trop Dis. 2012;6(8):e1798. doi:10.1371/journal.pntd.0001798
91. Roatt BM, Aguiar-Soares RD, Coura-Vital W, et al. Immunotherapy and immunochemotherapy in visceral leishmaniasis: promising treatments for this neglected disease. Front Immunol. 2014;5:272. doi:10.3389/fimmu.2014.00272
92. Ali N, Hussain S. Leishmania donovani bodies in bone marrow. Clin Case Rep. 2014;2(5):238–239. doi:10.1002/ccr3.97
93. Dantas Brito M, Campilho F, Branca R, et al. Visceral leishmaniasis: a differential diagnosis to remember after bone marrow transplantation. Case Rep Hematol. 2014;2014:587912. doi:10.1155/2014/587912
94. Gawade S, Nanaware M, Gokhale R, Adhav P. Visceral leishmaniasis: a case report. Australas Med J. 2012;5(2):130–134. doi:10.4066/AMJ.2012997
95. Dittus C, Semmel D. Leishmania amastigotes visualized on bone marrow aspirate in a leishmaniasis and HIV coinfected patient presenting with pancytopenia. Blood. 2013;122(26):4162. doi:10.1182/blood-2013-08-519306
96. Zhang G, Zhong J, Wang T, Zhong L. Erratum: a case of visceral leishmaniasis found by left oblique hernia: a case report. Exp Ther Med. 2021;21(1):43. doi:10.3892/etm.2020.9477
97. Dirkx L, Hendrickx S, Merlot M, et al. Long-term hematopoietic stem cells as a parasite niche during treatment failure in visceral leishmaniasis. Commun Biol. 2022;5(1):626. doi:10.1038/s42003-022-03591-7
98. Bandeira Ferreira FL, Séguin O, Descoteaux A, Heinonen KM. Persistent cutaneous Leishmania major infection promotes infection-adapted myelopoiesis. Microorganisms. 2022;10(3):535. doi:10.3390/microorganisms10030535
99. Mirkovich AM, Galelli A, Allison AC, Modabber FZ. Increased myelopoiesis during Leishmania major infection in mice: generation of ‘safe targets’, a possible way to evade the effector immune mechanism. Clin Exp Immunol. 1986;64(1):1–7.
100. Richardson C, Yan S, Vestal CG. Oxidative stress, bone marrow failure, and genome instability in hematopoietic stem cells. Int J Mol Sci. 2015;16(2):2366–2385. doi:10.3390/ijms16022366
101. Mangialardi G, Spinetti G, Reni C, Madeddu P. Reactive oxygen species adversely impacts bone marrow microenvironment in diabetes. Antioxid Redox Signal. 2014;21(11):1620–1633. doi:10.1089/ars.2014.5944
102. Porto ML, Rodrigues BP, Menezes TN, et al. Reactive oxygen species contribute to dysfunction of bone marrow hematopoietic stem cells in aged C57BL/6 J mice. J Biomed Sci. 2015;22:97. doi:10.1186/s12929-015-0201-8
103. Zhu H, Kwak HJ, Liu P, et al. Reactive Oxygen species-producing myeloid cells act as a bone marrow niche for sterile inflammation-induced reactive granulopoiesis. J Immunol. 2017;198(7):2854–2864. doi:10.4049/jimmunol.1602006
104. Liu J, Li Y, Chen S, et al. Biomedical application of reactive oxygen species-responsive nanocarriers in cancer, inflammation, and neurodegenerative diseases. Front Chem. 2020;8:838. doi:10.3389/fchem.2020.00838
105. Kulbacka J, Wilk KA, Bazylińska U, Dubińska-Magiera M, Potoczek S, Saczko J. Curcumin loaded nanocarriers with varying charges augmented with electroporation designed for colon cancer therapy. Int J Mol Sci. 2022;23(3):1377. doi:10.3390/ijms23031377
106. Chen Y, Wu X, Li J, Jiang Y, Xu K, Su J. Bone-targeted nanoparticle drug delivery system: an emerging strategy for bone-related disease. Front Pharmacol. 2022;13:909408. doi:10.3389/fphar.2022.909408
107. Mu CF, Shen J, Liang J, et al. Targeted drug delivery for tumor therapy inside the bone marrow. Biomaterials. 2018;155:191–202. doi:10.1016/j.biomaterials.2017.11.029
108. Sou K, Goins B, Oyajobi BO, Travi BL, Phillips WT. Bone marrow-targeted liposomal carriers. Expert Opin Drug Deliv. 2011;8(3):317–328. doi:10.1517/17425247.2011.553218
109. Jiang Y, Lin W, Zhu L. Targeted drug delivery for the treatment of blood cancers. Molecules. 2022;27(4). doi:10.3390/molecules27041310
110. Oliveira LF, Schubach AO, Martins MM, et al. Systematic review of the adverse effects of cutaneous leishmaniasis treatment in the New World. Acta Trop. 2011;118(2):87–96. doi:10.1016/j.actatropica.2011.02.007
111. Stone NR, Bicanic T, Salim R, Hope W. Liposomal Amphotericin B (AmBisome(®)): a review of the pharmacokinetics, pharmacodynamics, clinical experience and future directions. Drugs. 2016;76(4):485–500. doi:10.1007/s40265-016-0538-7
112. Terrazas C, Varikuti S, Oghumu S, et al. Ly6Chi inflammatory monocytes promote susceptibility to Leishmaia donovani infection. Sci Rep. 2017;7(1):14693. doi:10.1038/s41598-017-14935-3
113. Varikuti S, Volpedo G, Saljoughian N, et al. The potent ITK/BTK inhibitor ibrutinib is effective for the treatment of experimental visceral leishmaniasis caused by leishmania donovani. J Infect Dis. 2019;219(4):599–608. doi:10.1093/infdis/jiy552
114. Olingy CE, Dinh HQ, Hedrick CC. Monocyte heterogeneity and functions in cancer. J Leukoc Biol. 2019;106(2):309–322. doi:10.1002/JLB.4RI0818-311R
115. Cortez-Retamozo V, Etzrodt M, Newton A, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38(2):296–308. doi:10.1016/j.immuni.2012.10.015
116. (FDA) USFada. VASOTEC®, Enalapril maleate tablets. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/pre96/018998_Vasotec.cfm.
117. Ferreira Lopes de Araújo C, Ribeiro da Silva Nunes J, Alexandre Nunes Dos Santos R, Rios Santos F. Leishmaniasis: effect of enalaprilat on the production of (ON) and cytokines in vitro. Res Soc Dev. 2021;10(3). doi:10.33448/rsd-v10i3.12949
118. Miret JJ, Kirschmeier P, Koyama S, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. 2019;7(1):32. doi:10.1186/s40425-019-0504-5
119. Porta C, Consonni FM, Morlacchi S, et al. Tumor-derived prostaglandin E2 promotes p50 NF-κB-dependent differentiation of monocytic MDSCs. Cancer Res. 2020;80(13):2874–2888. doi:10.1158/0008-5472.CAN-19-2843
120. Holtzhausen A, Harris W, Ubil E, et al. TAM family receptor kinase inhibition reverses MDSC-mediated suppression and augments anti-PD-1 therapy in melanoma. Cancer Immunol Res. 2019;7(10):1672–1686. doi:10.1158/2326-6066.CIR-19-0008
121. Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005;65(20):9525–9535. doi:10.1158/0008-5472.CAN-05-0529
122. de Souza A, Marins DSS, Mathias SL, et al. Promising nanotherapy in treating leishmaniasis. Int J Pharm. 2018;547(1–2):421–431. doi:10.1016/j.ijpharm.2018.06.018
123. Dalle Vedove E, Costabile G, Merkel OM. Mannose and mannose-6-phosphate receptor-targeted drug delivery systems and their application in cancer therapy. Adv Healthc Mater. 2018;7(14):e1701398. doi:10.1002/adhm.201701398
124. Pan Z, Kang X, Zeng Y, et al. A mannosylated PEI–CPP hybrid for TRAIL gene targeting delivery for colorectal cancer therapy. Polym Chem. 2017;8(35):5275–5285. doi:10.1039/C7PY00882A
125. Dar MJ, Din FU, Khan GM. Sodium stibogluconate loaded nano-deformable liposomes for topical treatment of leishmaniasis: macrophage as a target cell. Drug Deliv. 2018;25(1):1595–1606. doi:10.1080/10717544.2018.1494222
126. Gélvez APC, Diniz Junior JAP, Brígida RTSS, Rodrigues APD. AgNP-PVP-meglumine antimoniate nanocomposite reduces Leishmania amazonensis infection in macrophages. BMC Microbiol. 2021;21(1):211. doi:10.1186/s12866-021-02267-2
127. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555–568. doi:10.1084/jem.20140857
128. Lőrincz T, Holczer M, Kapuy O, Szarka A. The interrelationship of pharmacologic ascorbate induced cell death and ferroptosis. Pathol Oncol Res. 2019;25(2):669–679. doi:10.1007/s12253-018-0539-9
129. Urrutia P, Aguirre P, Esparza A, et al. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem. 2013;126(4):541–549. doi:10.1111/jnc.12244
130. Jarvis JN, Meintjes G, Bicanic T, et al. Cerebrospinal fluid cytokine profiles predict risk of early mortality and immune reconstitution inflammatory syndrome in HIV-associated cryptococcal meningitis. PLoS Pathog. 2015;11(4):e1004754. doi:10.1371/journal.ppat.1004754
131. Xiao L, Huang H, Fan S, et al. Ferroptosis: a mixed blessing for infectious diseases. Front Pharmacol. 2022;13:992734. doi:10.3389/fphar.2022.992734
132. Spangler B, Morgan CW, Fontaine SD, et al. A reactivity-based probe of the intracellular labile ferrous iron pool. Nat Chem Biol. 2016;12(9):680–685. doi:10.1038/nchembio.2116
133. Sato K, Shi L, Ito F, et al. Non-thermal plasma specifically kills oral squamous cell carcinoma cells in a catalytic Fe(II)-dependent manner. J Clin Biochem Nutr. 2019;65(1):8–15. doi:10.3164/jcbn.18-91
134. Brown CW, Amante JJ, Chhoy P, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575–586.e4. doi:10.1016/j.devcel.2019.10.007
135. Alvarez SW, Sviderskiy VO, Terzi EM, et al. Author Correction: NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2022;609(7929):E12. doi:10.1038/s41586-022-05323-7
136. Wang W, Green M, Choi JE, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. doi:10.1038/s41586-019-1170-y
137. Gaschler MM, Andia AA, Liu H, et al. FINO2 initiates ferroptosis though GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14(5):507–515. doi:10.1038/s41589-018-0031-6
138. Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC. PCBP1 and NCOA4 regulate erythroid iron storage and heme biosynthesis. J Clin Invest. 2017;127(5):1786–1797. doi:10.1172/JCI90519
139. McGrath KE, Frame JM, Palis J. Early hematopoiesis and macrophage development. Semin Immunol. 2015;27(6):379–387. doi:10.1016/j.smim.2016.03.013
140. Liu D, Uzonna JE. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front Cell Infect Microbiol. 2012;2:83. doi:10.3389/fcimb.2012.00083
141. Lee SH, Charmoy M, Romano A, et al. Mannose receptor high, M2 dermal macrophages mediate nonhealing Leishmania major infection in a Th1 immune environment. J Exp Med. 2018;215(1):357–375. doi:10.1084/jem.20171389
142. Woelbing F, Kostka SL, Moelle K, et al. Uptake of Leishmania major by dendritic cells is mediated by Fcgamma receptors and facilitates acquisition of protective immunity. J Exp Med. 2006;203(1):177–188. doi:10.1084/jem.20052288
143. Raybarman C, Bhattacharjee S. Central and local controls of monocytopoiesis influence the outcome of Leishmania infection. Cytokine. 2021;147:155325. doi:10.1016/j.cyto.2020.155325
144. Matte C, Arango Duque G, Descoteaux A. Leishmania donovani metacyclic promastigotes impair phagosome properties in inflammatory monocytes. Infect Immun. 2021;89(7):e0000921. doi:10.1128/IAI.00009-21
145. Romano A, Carneiro MBH, Doria NA, et al. Divergent roles for Ly6C+CCR2+CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PLoS Pathog. 2017;13(6):e1006479. doi:10.1371/journal.ppat.1006479
146. Goncalves R, Zhang X, Cohen H, Debrabant A, Mosser DM. Platelet activation attracts a subpopulation of effector monocytes to sites of Leishmania major infection. J Exp Med. 2011;208(6):1253–1265. doi:10.1084/jem.20101751
147. Sato N, Ahuja SK, Quinones M, et al. CC chemokine receptor (CCR)2 is required for Langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med. 2000;192(2):205–218. doi:10.1084/jem.192.2.205
148. Quinones MP, Estrada CA, Jimenez F, et al. CCL2-independent role of CCR2 in immune responses against Leishmania major. Parasite Immunol. 2007;29(4):211–217. doi:10.1111/j.1365-3024.2006.00935.x
149. de Freitas EO, Leoratti FM, Freire-de-lima CG, Morrot A, Feijó DF. The contribution of immune evasive mechanisms to parasite persistence in visceral leishmaniasis. Front Immunol. 2016;7:153. doi:10.3389/fimmu.2016.00153
150. Lambertz U, Silverman JM, Nandan D, et al. Secreted virulence factors and immune evasion in visceral leishmaniasis. J Leukoc Biol. 2012;91(6):887–899. doi:10.1189/jlb.0611326
151. Cecílio P, Pérez-Cabezas B, Santarém N, Maciel J, Rodrigues V, Cordeiro da Silva A. Deception and manipulation: the arms of leishmania, a successful parasite. Front Immunol. 2014;5:480. doi:10.3389/fimmu.2014.00480
152. Murray HW, Nathan CF. Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J Exp Med. 1999;189(4):741–746. doi:10.1084/jem.189.4.741
153. Boitz JM, Gilroy CA, Olenyik TD, et al. Arginase is essential for survival of leishmania donovani promastigotes but not intracellular amastigotes. Infect Immun. 2017;85(1). doi:10.1128/IAI.00554-16
154. Singh N, Kumar R, Chauhan SB, Engwerda C, Sundar S. Peripheral blood monocytes with an antiinflammatory phenotype display limited phagocytosis and oxidative burst in patients with visceral leishmaniasis. J Infect Dis. 2018;218(7):1130–1141. doi:10.1093/infdis/jiy228
155. Viana AG, Magalhães LMD, Giunchetti RC, Dutra WO, Gollob KJ. Infection of human monocytes with Leishmania infantum strains induces a downmodulated response when compared with infection with Leishmania brazilensis. Front Immunol. 2017;8:1896. doi:10.3389/fimmu.2017.01896
156. Pereira WF, Ribeiro-Gomes FL, Guillermo LV, et al. Myeloid-derived suppressor cells help protective immunity to Leishmania major infection despite suppressed T cell responses. J Leukoc Biol. 2011;90(6):1191–1197. doi:10.1189/jlb.1110608
157. Formaglio P, Alabdullah M, Siokis A, et al. Nitric oxide controls proliferation of Leishmania major by inhibiting the recruitment of permissive host cells. Immunity. 2021;54(12):2724–2739.e10. doi:10.1016/j.immuni.2021.09.021
158. Postat J, Olekhnovitch R, Lemaître F, Bousso P. A metabolism-based quorum sensing mechanism contributes to termination of inflammatory responses. Immunity. 2018;49(4):654–665.e5. doi:10.1016/j.immuni.2018.07.014
159. Husain MJ, Datta BK, Kostova D, et al. Access to cardiovascular disease and hypertension medicines in developing countries: an analysis of essential medicine lists, price, availability, and affordability. J Am Heart Assoc. 2020;9(9):e015302. doi:10.1161/JAHA.119.015302
160. Vellozo NS, Rigoni TS, Lopes MF. New Therapeutic Tools to Shape Monocyte Functional Phenotypes in Leishmaniasis. Front Immunol. 2021;12:704429. doi:10.3389/fimmu.2021.704429
161. Roy S, Mukhopadhyay D, Mukherjee S, et al. An IL-10 dominant polarization of monocytes is a feature of Indian Visceral Leishmaniasis. Parasite Immunol. 2018;40(7):e12535. doi:10.1111/pim.12535
162. Pacheco-Fernandez T, Volpedo G, Verma C, Satoskar AR. Understanding the immune responses involved in mediating protection or immunopathology during leishmaniasis. Biochem Soc Trans. 2021;49(1):297–311. doi:10.1042/BST20200606
163. Hume DA, Irvine KM, Pridans C. The Mononuclear Phagocyte System: the Relationship between Monocytes and Macrophages. Trends Immunol. 2019;40(2):98–112. doi:10.1016/j.it.2018.11.007
164. Oliveira WN, Ribeiro LE, Schrieffer A, Machado P, Carvalho EM, Bacellar O. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of human tegumentary leishmaniasis. Cytokine. 2014;66(2):127–132. doi:10.1016/j.cyto.2013.12.016
165. Bogdan C. Macrophages as host, effector and immunoregulatory cells in leishmaniasis: impact of tissue micro-environment and metabolism. Cytokine X. 2020;2(4):100041. doi:10.1016/j.cytox.2020.100041
166. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol. 2016;16(9):581–592. doi:10.1038/nri.2016.72
167. Vellozo NS, Ribiero-Gomes FL, Lopes MF. Shifting macrophage phenotypes in leishmaniasis. In: Kumar V, editor. Macrophages Celebrating 140 Years of Discovery. Intech Open; 2022:392.
168. Du H, Levine M, Ganesa C, Witte DP, Cole ES, Grabowski GA. The role of mannosylated enzyme and the mannose receptor in enzyme replacement therapy. Am J Hum Genet. 2005;77(6):1061–1074. doi:10.1086/498652
169. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
170. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
171. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev. 2007;107(3):748–766. doi:10.1021/cr040077w
172. Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, and significance. Am J Clin Nutr. 1993;57(5 Suppl):715S–724S. doi:10.1093/ajcn/57.5.715S
173. Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289–3303. doi:10.1016/j.bbagen.2012.11.020
174. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
175. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
176. Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589. doi:10.1038/s41467-021-21841-w
177. Moreira DS, Xavier MV, Murta SMF. Ascorbate peroxidase overexpression protects Leishmania braziliensis against trivalent antimony effects. Mem Inst Oswaldo Cruz. 2018;113(12):e180377. doi:10.1590/0074-02760180377
178. Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019;25:101047. doi:10.1016/j.redox.2018.11.005
179. Andrade JM, Murta SM. Functional analysis of cytosolic tryparedoxin peroxidase in antimony-resistant and -susceptible Leishmania braziliensis and Leishmania infantum lines. Parasit Vectors. 2014;7:406. doi:10.1186/1756-3305-7-406
180. Kumar A, Das S, Purkait B, et al. Ascorbate peroxidase, a key molecule regulating amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother. 2014;58(10):6172–6184. doi:10.1128/AAC.02834-14
181. Das S, Giri S, Sundar S, Shaha C. Functional involvement of leishmania donovani tryparedoxin peroxidases during infection and drug treatment. Antimicrob Agents Chemother. 2018;62(1). doi:10.1128/AAC.00806-17
182. Pal S, Dolai S, Yadav RK, Adak S. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS One. 2010;5(6):e11271. doi:10.1371/journal.pone.0011271
183. Mukherjee A, Boisvert S, Monte-Neto RL, et al. Telomeric gene deletion and intrachromosomal amplification in antimony-resistant Leishmania. Mol Microbiol. 2013;88(1):189–202. doi:10.1111/mmi.12178
184. Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. 2016;44(3):492–504. doi:10.1016/j.immuni.2016.02.016
185. Laranjeira-Silva MF, Hamza I, Pérez-Victoria JM. Iron and heme metabolism at the leishmania-host interface. Trends Parasitol. 2020;36(3):279–289. doi:10.1016/j.pt.2019.12.010
186. Kristiansen M, Graversen JH, Jacobsen C, et al. Identification of the haemoglobin scavenger receptor. Nature. 2001;409(6817):198–201. doi:10.1038/35051594
187. Morimoto A, Omachi S, Osada Y, et al. Hemophagocytosis in experimental visceral leishmaniasis by leishmania donovani. PLoS Negl Trop Dis. 2016;10(3):e0004505. doi:10.1371/journal.pntd.0004505
188. Pham NK, Mouriz J, Kima PE. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect Immun. 2005;73(12):8322–8333. doi:10.1128/IAI.73.12.8322-8333.2005
189. Silva RL, Santos MB, Almeida PL, et al. sCD163 levels as a biomarker of disease severity in leprosy and visceral leishmaniasis. PLoS Negl Trop Dis. 2017;11(3):e0005486. doi:10.1371/journal.pntd.0005486
190. Docampo R, de Boiso JF, Boveris A, Stoppani AO. Localization of peroxidase activity in Trypanosoma cruzi microbodies. Experientia. 1976;32(8):972–975. doi:10.1007/BF01933918
191. Boveris A, Sies H, Martino EE, Docampo R, Turrens JF, Stoppani AO. Deficient metabolic utilization of hydrogen peroxide in Trypanosoma cruzi. Biochem J. 1980;188(3):643–648. doi:10.1042/bj1880643
192. Clark D, Albrecht M, Arévalo J. Ascorbate variations and dehydroascorbate reductase activity in Trypanosoma cruzi epimastigotes and trypomastigotes. Mol Biochem Parasitol. 1994;66(1):143–145. doi:10.1016/0166-6851(94)90045-0
193. Bogacz M, Krauth-Siegel RL. Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. Elife. 2018;7. doi:10.7554/eLife.37503
194. Cotter C, Sturrock HJ, Hsiang MS, et al. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet. 2013;382(9895):900–911. doi:10.1016/S0140-6736(13)60310-4
195. Singh KS, Leu JI, Barnoud T, et al. Author Correction: African-centric TP53 variant increases iron accumulation and bacterial pathogenesis but improves response to malaria toxin. Nat Commun. 2020;11(1):1541. doi:10.1038/s41467-020-15366-x
196. Yao Y, Chen Z, Zhang H, et al. Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol. 2021;22(9):1127–1139. doi:10.1038/s41590-021-00996-0
197. Imlay JA, Chin SM, Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240(4852):640–642. doi:10.1126/science.2834821
198. Rodriguez GM. Control of iron metabolism in Mycobacterium tuberculosis. Trends Microbiol. 2006;14(7):320–327. doi:10.1016/j.tim.2006.05.006
199. Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell dead in tissue necrosis. J Exp Med. 2019;216(3):556–570. doi:10.1084/jem.20181776
200. Xu X, Lin D, Tu S, Gao S, Shao A, Sheng J. Is ferroptosis a future direction in exploring cryptococcal meningitis? Front Immunol. 2021;12:598601. doi:10.3389/fimmu.2021.598601
201. Barluzzi R, Saleppico S, Nocentini A, et al. Iron overload exacerbates experimental meningoencephalitis by Cryptococcus neoformans. J Neuroimmunol. 2002;132(1–2):140–146. doi:10.1016/s0165-5728(02)00324-7
202. Hassannia B, Wiernicki B, Ingold I, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341–3355. doi:10.1172/JCI99032
203. Rothe T, Gruber F, Uderhardt S, et al. 12/15-Lipoxygenase-mediated enzymatic lipid oxidation regulates DC maturation and function. J Clin Invest. 2015;125(5):1944–1954. doi:10.1172/JCI78490
204. Cramer SL, Saha A, Liu J, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23(1):120–127. doi:10.1038/nm.4232
205. Yan HF, Zou T, Tuo QZ, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6(1):49. doi:10.1038/s41392-020-00428-9
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.