Back to Journals » International Journal of Nanomedicine » Volume 14
Ferumoxytol and CpG oligodeoxynucleotide 2395 synergistically enhance antitumor activity of macrophages against NSCLC with EGFRL858R/T790M mutation
Authors Wang G, Zhao J, Zhang M, Wang Q, Chen B, Hou Y ![]() , Lu K
, Lu K
Received 6 November 2018
Accepted for publication 22 April 2019
Published 24 June 2019 Volume 2019:14 Pages 4503—4515
DOI https://doi.org/10.2147/IJN.S193583
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Guoqun Wang,1 Jiaojiao Zhao,2 Meiling Zhang,1 Qian Wang,1 Bo Chen,3 Yayi Hou,2,4 Kaihua Lu1
1Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, People’s Republic of China; 2The State Key Laboratory of Pharmaceutical Biotechnology, Division of Immunology, Medical School, Nanjing University, Nanjing 210093, People’s Republic of China; 3Institute of Materials Science and Devices, Suzhou University of Science and Technology, Suzhou 215009, People’s Republic of China; 4Jiangsu Key Laboratory of Molecular Medicine, Nanjing University, Nanjing 210093, People’s Republic of China
Purpose: Drug resistance is a major challenge for epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs) treatment of lung cancer. Ferumoxytol (FMT) drives macrophage (MΦ) transformation towards a M1-like phenotype and thereby inhibits tumor growth. CpG oligodeoxynucleotide 2395 (CpG), a toll-like receptor 9 (TLR9) agonist, is an effective therapeutic agent to induce anticancer immune responses. Herein, the effect of co-administered FMT and CpG on MΦ activation for treating non-small cell lung cancer (NSCLC) was explored.
Methods: The mRNA expression levels of M1-like genes in RAW 264.7 MΦ cells stimulated by FMT, CpG and FMT and CpG (FMT/CpG) were evaluated by quantitative reverse transcription PCR (qRT-PCR). Then, the effects of FMT/CpG-pretreated MΦ supernatant on apoptosis and proliferation of H1975 cells were detected by flow cytometry, and the expression of EGFR and its downstream signaling pathway in H1975 cells were explored by western blotting. Finally, a H1975 cell xenograft mouse model was used to study the anti-tumor effect of the combination of FMT and CpG in vivo.
Results: FMT and CpG synergistically enhanced M1-like gene expression in MΦ, including tumor necrosis factor-α, interleukin (IL)-12, IL-1α, IL-1β, IL-6 and inducible nitric oxide synthase (iNOS). FMT/CpG-pretreated MΦ supernatant inhibited proliferation and induced apoptosis of H1975 cells, accompanied by down-regulation of cell cycle-associated proteins and up-regulation of apoptosis-related proteins. Further studies indicated that the FMT/CpG-pretreated MΦ supernatant suppressed p-EGFR and its downstream AKT/mammalian target of rapamycin signaling pathway in H1975 cells. Furthermore, FMT/CpG suppressed tumor growth in mice accompanied by a decline in the EGFR-positive tumor cell fraction and increased M1 phenotype macrophage infiltration.
Conclusion: FMT acted synergistically with CpG to activate MΦ for suppressed proliferation and promoted apoptosis of NSCLC cells via EGFR signaling. Thus, combining FMT and CpG is an effective strategy for the treatment of NSCLC with EGFRL858R/T790M, mutation.
Keywords: ferumoxytol, CpG oligodeoxynucleotide, macrophages, non-small cell lung cancer, epidermal growth factor receptor
Introduction
The incidence of lung cancer has been increasing yearly with a mortality rate ranking first among malignant tumors.1 EGFR-tyrosine kinase inhibitors (TKIs) are effective for the treatment of lung cancer patients with EGFR mutation,2–4 however, drug resistance develops in most cases after 9–13 months.5 Although molecular targeted therapy improves patient survival as compared to conventional treatment approaches such as radiotherapy, chemotherapy, and surgery, the 5-year overall survival rate is still <15%.6 Therefore, there is an urgent need to develop alternative treatment strategies for non-small cell lung cancer (NSCLC) harboring resistance mutations.
Immunotherapy has shown great potential for cancer treatment.7–9 Macrophages (MΦ) are heterogeneous immunocytes accounting for a significant proportion in the tumor microenvironment (TME).10 Activated MΦ can be divided into two subtypes according to function:11,12 M1, which mediates inflammation and the anti-tumor immune response by producing pro-inflammatory factors such as tumor necrosis factor (TNF)-α and IL-12; and M2, which secretes high levels of immunosuppressive mediators to promote tumor growth, invasion, and metastasis. NSCLC is associated with greater infiltration of MΦ than other cancers13 and the density of M2 MΦ was negatively correlated with the survival of lung cancer patients who had undergone surgery.14 In addition, M2 MΦ infiltration reduces the sensitivity of EGFR-TKIs in lung adenocarcinoma.15,16 Whereas gefitinib inhibits M2-like polarization of MΦ in Lewis lung cancer,17 which represents a promising approach in modulating the TME for lung cancer treatment. However, although restoring the immunological activity of MΦ has substantial antitumor effects, treatment of NSCLC with EGFR mutation—particularly T790M—by steering MΦ to M1 phenotype has not been reported.
Ferumoxytol (FMT) is a versatile nanoparticle that has been widely used as a drug carrier.18–21 However, its potential as an immune-activator to treat tumors has been overlooked. A recent study has showed that iron released by lysed red blood cells induces the conversion of MΦ into a pro-inflammatory phenotype capable of directly killing tumor cells; besides, an abundant of iron-loaded MΦ is correlated with reduced tumor size in NSCLC patients.13 In addition, FMT synergized with TLR3 agonist poly (I:C) significantly suppressed the growth of melanoma by shifting macrophages to a tumoricidal phenotype in our previous work.22 This suggests that activation of MΦ by FMT may be an effective strategy for the treatment of lung cancer, although it has not been investigated in NSCLC with EGFRT790M mutation.
CpG oligodeoxynucleotide 2395 (CpG) is an artificially synthesized oligodeoxynucleotide containing unmethylated CpG motifs that triggers a pro-inflammatory immune response by interacting with Toll-like receptor (TLR) 9 in MΦ.23,24 The antitumor effect of CpG has been demonstrated in several malignancies including stage I/II melanoma,25 glioblastoma,26,27 and non-Hodgkin’s lymphoma.28 However, not all clinical studies on CpG reported the improved patient outcomes. For instance, CpG did not enhance the therapeutic effect of erlotinib in patients with advanced recurrent EGFR-positive NSCLC.29 Whereas, on the other hand, CpG in combination with the first-line drug taxane and platinum chemotherapy prolonged survival in patients with advanced NSCLC30 indicating that CpG may potentially maximize the benefits of immunotherapy in patients with EGFRT790M mutation by combined therapy.
In this study, we investigated the above possibility in the present study using the human NSCLC cell line H1975 with EGFRL858R/T790M mutations and a xenograft mouse model. Although treatment with FMT and CpG alone or in combination did not have an inhibitory effect on tumor cells, the supernatant of MΦ pretreated with both FMT and CpG (FMT/CpG) promoted apoptosis and inhibited proliferation in tumor cells by suppressing the expression of EGFR signaling pathway components. These results provide the first demonstration that in addition to traditional chemotherapy and molecular targeted therapy, MΦ activation by FMT/CpG is an effective strategy for the treatment of NSCLC with EGFRL858R/T790M mutations.
Materials and methods
Cell culture and reagents
The TLR9 agonist CpG (class C ODN 2395) was purchased from InvivoGen (San Diego, CA, USA; #tlrl-2395–5). FMT was a gift from Prof. Ning Gu.31,32 The RAW 264.7 MΦ cell line was obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). H1975 cells with EGFRL858R/T790M mutation were provided by Prof. Yongqian Shu (The First Affiliated Hospital of Nanjing Medical University, Nanjing, China) and the use of H1975 cells are approved by the Ethics Committee of the First Affiliated Hospital of Nanjing Medical University for the experimental study. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin (Thermo Fisher Scientific) at 37°C in a humidified atmosphere of 5% CO2.
The cells were passaged at 70–80% confluence. RAW 264.7 cells were seeded in 24-well plates and FMT (100 μg/mL) and CpG (2.5 μg/mL) were added to the medium; the MΦ supernatant was collected after stimulation for 12 h. H1975 cells were seeded in a 24-well plate. When the cells were attached, the supernatant was discarded and the cells were treated with FMT (100 μg/mL), CpG (2.5 μg/mL), or the supernatant of MΦ grown for 12 hrs with or without FMT/CpG. H1975 cells were cultured for 48 hrs at 37°C in a 5% CO2 incubator and used for cell proliferation, apoptosis, and Western blotting experiments.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR)
Total RNA was extracted from harvested cells using TRIzol reagent (Thermo Fisher Scientific; #15596018). The mRNA was reverse transcribed into cDNA using HiScript II 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co., Ltd, Nanjing, China; #R211) according to the instructions. QRT-PCR was performed in SYBR Green PCR Master Mix (Thermo Fisher Scientific; #4309155) on a Step One Plus system (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. The relative expression levels of target genes were calculated with the 2−ΔΔCt method relative to the endogenous control glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Primer sequences are shown in Table 1.
 | Table 1 Sequences of forward and reverse primers used for qRT-PCR |
Cell viability assay
The effect of FMT and CpG on NSCLC progression was examined using Cell Counting Kit (CCK)-8 (Dojindo Laboratories, Kumamoto, Japan; #CK04). The cells were seeded at a density of 5×103/well in 96-well plates and incubated overnight at 37°C in an atmosphere of 5% CO2. The cells were then treated with fresh medium containing different concentrations of FMT, CpG, and FMT/CpG for 48 h. The supernatant was discarded and replaced with fresh medium containing 10% CCK-8 solution, followed by incubation for 1–4 h. The absorbance at 450 nm was measured using a multi-well spectrophotometer (BioTek, Winooski, VT, USA). There were six replicate wells for each sample and the experiment was repeated three times.
Flow cytometry (FCM) analysis of cell apoptosis
Cell apoptosis was detected by FCM using the Annexin V-Alexa Fluor 488/PI Apoptosis Assay kit (FcMACS, Nanjing, China; #FMSAV488-100). H1975 cells were collected, washed twice in phosphate-buffered saline at 4°C, and resuspended in binding buffer. A 100-μL of the cell suspension was transferred to a flow tube, and 5 μL Annexin V-Alexa Fluor 488 and 10 μL propidium iodide (PI) were added, followed by mixing and incubation at 4°C for 15 mins in the dark. The cell suspension was sorted on a FACS Calibur instrument (BD Biosciences, San Jose, CA, USA). Early and late apoptotic cell fractions (Annexin V-positive and Annexin V/PI double-positive, respectively) were quantified.
FCM analysis of cell proliferation
H1975 cells were labeled with 10 μM 5(6)-carboxyfluorescein diacetate N-hydroxysuccinimidyl ester (CFSE; Thermo Fisher Scientific; #65-0850-84) and incubated in CFSE staining solution for 15 mins at 37°C. An equal volume of culture medium (containing serum) was added to the cells along with CFSE staining solution, followed by incubation for 5 mins. The CFSE-containing solution was removed and the cells were washed twice with an equal volume of culture medium. The fluorescently labeled cells were used for subsequent experiments. Cell proliferation was detected by FCM. The proliferation index was calculated using ModFit LT software (Verity Software House, Topsham, ME, USA) as the total number of divisions divided by the number of proliferating parent cells.33,34
Colony formation assay
H1975 cells were seeded at a density of 1×103/well in 12-well plates and incubated overnight at 37°C in an atmosphere of 5% CO2. The supernatant was discarded, and the cells were cultured for 2 weeks under different treatment conditions. The medium was refreshed at appropriate intervals, which was determined according to the pH of the culture supernatant. The experiment was terminated when macroscopic clones appeared in the culture plates, which were then washed twice with PBS, fixed in 4% formalin for 10–15 mins, dried, and stained with 0.1% crystal violet for 15 mins. The number of colonies was counted under a microscope and those with >50 cells were defined as positive.
Western blotting
Cells subjected to different treatments were lysed in ice-cold radioimmunoprecipitation assay buffer (Beyotime, Shanghai, China; #P0013K), and protein concentration was determined using a bicinchoninic acid protein quantitation kit (Vazyme Biotech Co., Ltd; #E112-01). Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Bio-Rad) that was blocked in 5% bovine serum albumin for 1 hrs at room temperature and then incubated overnight at 4°C with primary antibodies. After washing three times with Tris-buffered saline with 0.1% Tween 20, the membrane was incubated with secondary antibody (Thermo Fisher Scientific; #A16110SAMPLE; #A16078SAMPLE) for 1 hrs at room temperature. Protein expression was visualized by enhanced chemiluminescence Western blotting detection reagent (Millipore, Billerica, MA, USA; #WBULS0500), and protein band intensity was quantified using ImageJ software. Primary antibodies against B-cell lymphoma-2-associated X protein (Bax; #5023), Cleaved Caspase-3 (Asp175; #9661), Cleaved poly(ADP ribose) polymerase (PARP,Asp214; #5625), Cyclin A2 (#4656), Cyclin B1 (#12231), EGFR (#4267), phospho-EGFR (p-EGFR,Tyr1068; #3777), AKT (#4691), p-AKT (Ser473; #4060), mammalian target of rapamycin (mTOR; #2983), p-mTOR (Ser2448; #5536) and GAPDH (#5174) were purchased from Cell Signaling Technology (Beverly, MA, USA).
Tumor xenograft studies
H1975 cells were resuspended in 100 μL sterile PBS, and 2×106 cells were subcutaneously injected into the right flank of 5–6 weeks old female BALB/c nude mice (Shanghai Laboratory Animal Research Center, Shanghai, China). When the tumors reached a volume of 100 mm3, the animals were randomly divided into two groups (n=5 each) that were intratumorally injected with PBS or FMT (10 mg/kg) and CpG (2.5 mg/kg) every 2 days for 10 days. Body weight, maximum tumor length (L), and minimum tumor width (W) were recorded every 3 days. The tumor volume was calculated with the following formula: V = (L × W2)/2. At the end of the experiment, the mice were euthanized and the tumors were excised, washed with PBS, and fixed in formalin for immunohistochemistry. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Nanjing medical University and conformed to the Guidelines for the Care and Use of Laboratory Animals.
Immunohistochemistry
Tumor tissue specimens were fixed with 4% paraformaldehyde, embedded in paraffin, and cut into 4 μm-thick sections that were deparaffinized with xylene, rehydrated in a graded series of ethanol for 5 mins, washed three times with PBS, and then blocked with serum for 30 mins. The sections were incubated overnight at 4°C with primary antibody, washed three times with PBS, incubated with biotinylated secondary antibody for 30 mins at 37°C, and then washed with PBS, followed by staining with 3,3′-diaminobenzidine at room temperature for 10 mins in the dark. After staining with hematoxylin for 2 mins, the sections were subjected to hydrochloric acid/alcohol differentiation, dehydrated with ethanol and xylene, dried, and photographed under a microscope.
Statistical analysis
Statistical analyses were performed using SPSS 19.0 software (SPSS Inc, Chicago, IL, USA). Data were compared by one-way ANOVA or Student’s t-test. All statistical analyses were conducted at the significant level of α=0.05 and the Least Significance Difference or Dunnett’s test were used for post hoc of ANOVA analysis.
Results
Characterization of FMT
The polymer coating the outer layer of FMT was synthesized by terminal aldehyde group reduction and hydroxycarboxymethylation of dextran T10. As indicated in Figure 1A, the average diameter of synthesized dextran T10-coated FMT was about 7 nm and dynamic light scattering showed that the hydration particle size of FMT was 35 nm (Figure 1B). The molecular formula of the FMT external material is shown in Figure 1C, with H or COOH as the R group.31,32
 | Figure 1 Characterization of FMT. (A) Transmission electron micrograph of FMT. (B) Hydration particle size of FMT was shown by dynamic light scattering. (C) Molecular formula of polymer compound coating the outer layer of FMT, where the R group is H or COOH. Abbreviations: COOH, carboxyl; FMT, ferumoxytol. |
FMT and CpG synergistically promote M1-like gene expression in MΦ
To investigate the effects of FMT, CpG, and FMT/CpG on MΦ activation, the mRNA expression levels of M1-like genes in RAW 264.7 cells stimulated for 12 hrs were examined by qRT-PCR. FMT/CpG synergistically enhanced the expression of the M1-like genes of TNF-α, IL-12, IL-1α, IL-1β and IL-6 compared to either agent alone or the lipopolysaccharide (LPS)-stimulated positive control group (Figure 2). Among these altered genes, IL-12 was upregulated to the greatest degree following co-stimulation (Figure 2B), with a transcript level that was 109 times higher than that in control MΦ. FMT or CpG treatment alone had a similar albeit less potent effect (15- and 36-fold higher expression, respectively, as compared to the control. In contrast, in MΦ stimulated with LPS as a positive control, IL-12 mRNA expression was increased by only 20 fold relative to the control. In addition, the mRNA level of the M1-related co-stimulatory molecule cluster of differentiation 86 (CD86) and inducible nitric oxide synthase (iNOS) was also enhanced by FMT/CpG as compared to treatment with each agent alone. Thus, FMT and CpG synergistically promote MΦ activation towards a tumoricidal phenotype, with upregulation of M1-like genes.
 | Figure 2 FMT synergizes with CpG ODN 2395 to promote M1-like gene expression in MΦ. Relative mRNA expression was analyzed by qRT-PCR in RAW 264.7 cells following treatment with FMT (100 µg/mL), CpG (2.5 µg/mL), both FMT and CpG (FMT/CpG), or LPS (100 ng/mL) for 12 hrs. Results are expressed as mean ± SDof three independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control. Abbreviations: FMT, ferumoxytol; LPS, lipopolysaccharide; MΦ, macrophages; ODN, oligodeoxynucleotide; qRT-PCR, quantitative reverse transcription PCR. |
FMT/CpG-pretreated MΦ supernatant reduce NSCLC cell viability
To determine whether FMT and CpG directly suppress NSCLC growth, H1975 cells were treated with FMT, CpG, and a combination of both for 48 hrs. FMT showed no obvious toxicity to H1975 cells (Figure 3A). CpG reduced cell viability in a dose-dependent manner, although the effect was non-significant at low doses (Figure 3B). In addition, the rate of inhibition upon treatment with FMT combined with CpG was only 13% (Figure 3C). These results demonstrate that FMT/CpG shows negligible cytotoxicity towards H1975 cells.
 | Figure 3 FMT/CpG-pretreated MΦ supernatant reduces H1975 cell viability. (A and B) H1975 cells were incubated with different concentrations of FMT (0–400 µg/mL) or CpG (0–20 µg/mL) for 48 hrs and cell viability was evaluated with the CCK-8 assay. (C) Effect of FMT (100 µg/mL), CpG (2.5 µg/mL), FMT/CpG, the supernatant of MΦ grown for 12 hrs without stimulation (−FMT/CpG MΦS), or supernatant of MΦ pretreated with FMT/CpG for 12 hrs (+FMT/CpG MΦS) on the viability of H1975 cells after 48 hrs. Results are expressed as mean ± SDof three independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control. Abbreviations: CCK-8, Cell Counting Kit-8; FMT, ferumoxytol; MΦ, macrophages. |
As demonstrated previously,co-treatment with FMT and CpG synergistically induced MΦ activation towards a tumoricidal phenotype. To investigate whether this phenotypic switch can lead to inhibition of tumor cell growth, H1975 cells were treated with FMT/CpG, the supernatant of MΦ grown for 12 hrs without any stimulation (−FMT/CpG MΦS), or the supernatant of MΦ pretreated with FMT/CpG for 12 hrs (+FMT/CpG MΦS). After 48 hrs, cell viability was detected. As shown in the Figure 3C, the cell viability of H1975 exposed to +FMT/CpG MΦS was only 58.9% compared with the control group, while that in the −FMT/CpG MΦS group was 80.48%. Altogether, FMT/CpG-pretreated MΦ supernatant has a significant inhibitory effect on H1975 cell viability.
FMT/CpG-pretreated MΦ supernatant induces apoptosis of NSCLC cells
The decrease in cell viability of H1975 may be a combined effect of tumor cell apoptosis and inhibited proliferation. First, H1975 cells were treated with FMT/CpG, −FMT/CpG MΦS or +FMT/CpG MΦS for 48 hrs and cell apoptosis was analyzed by FCM. The results of Figure 4A and B showed that FMT/CpG and −FMT/CpG MΦS induced a low level of apoptosis in H1975 cells (9.18% and 10.6%, respectively). In comparison, the rate of apoptosis was significantly higher in cells exposed to FMT/CpG-pretreated MΦ supernatant than in control cells (46.2% vs 5.01%). To investigate the mechanism underlying this effect, we examined the expression of several apoptosis-related proteins by Western blotting and found that FMT/CpG-pretreated MΦ supernatant increased the expression levels of the apoptosis-related proteins Bax, Cleaved Caspase-3, and Cleaved PARP in H1975 cells (Figure 4C and D).
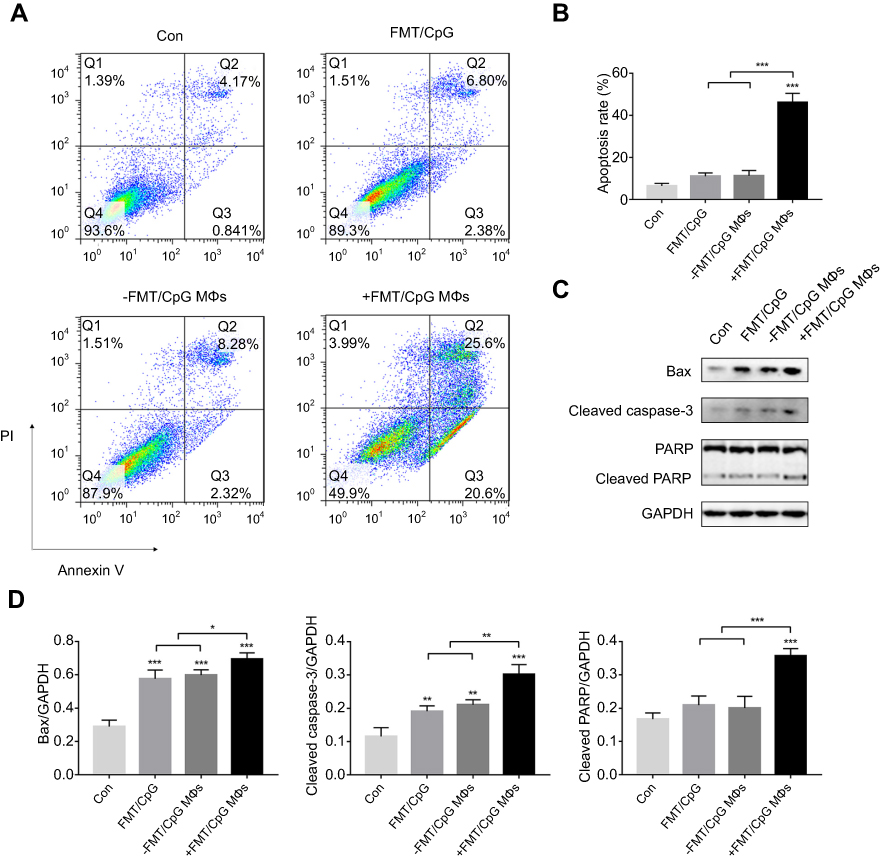 | Figure 4 FMT/CpG-pretreated MΦ supernatant induces apoptosis of H1975 cells. (A) H1975 cells were treated with FMT/CpG, −FMT/CpG MΦS or +FMT/CpG MΦS. After 48 hrs, cells were stained with Annexin V/PI and cell apoptosis was analyzed by FCM. (B) Quantitative analysis of apoptotic H1975 cells. (C) Protein levels of Bax, Cleaved Caspase-3, and Cleaved PARP were evaluated by Western blotting in H1975 cells treated as indicated for 48 hrs. (D) Protein levels were quantified with ImageJ software and normalized to that of GAPDH. Results are expressed as mean ± SDof three independent experiments. *P<0.05, ** P<0.01, ***P<0.001 vs control. Abbreviations: Bax, B-cell lymphoma-2-associated X protein; FCM, flow cytometry; FMT, ferumoxytol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MΦ, macrophages; PARP, poly(ADP-ribose) polymerase; PI, propidium iodide. |
FMT/CpG-pretreated MΦ supernatant suppresses NSCLC cell proliferation
Next, the proliferation of H1975 cells pre-labeled with CFSE and subjected to different treatments was analyzed by FCM. As indicated in Figure 5A and B, the proliferation rate of H1975 cells in the control group was retarded in the fourth generation, with an average proliferation index of 7.2. There was no change in the index of H1975 cells treated with FMT/CpG. The proportion of cells in the fourth generation decreased upon treatment with −FMT/CpG MΦS, yielding an average proliferation index of 5.62 while the proliferation of H1975 cells in the +FMT/CpG MΦS group was markedly blocked in the third generation, with a proliferation index of 4.5, indicating that FMT/CpG-pretreated MΦ supernatant suppressed NSCLC cell growth. In addition, we verified the effect of +FMT/CpG MΦS on H1975 cell proliferation and clump development by colony formation assay. The results showed that FMT/CpG-pretreated MΦ supernatant significantly inhibited tumor growth and interfered development of tumor-like cell clumps (Figure 5C and D). A Western blotting analysis further revealed that the cell cycle-associated S/G2 phase regulatory protein Cyclin A2 and the G2/M phase regulatory protein Cyclin B1 were both downregulated to a greater extent in H1975 cells treated with +FMT/CpG MΦS as compared to the other three groups (Figure 5E and F), suggesting that the cells were arrested in the S35,36 and G2/M37,38 phases of the cell cycle. Collectively, these results confirm that FMT/CpG-pretreated MΦ supernatant inhibits the proliferation of H1975 cells.
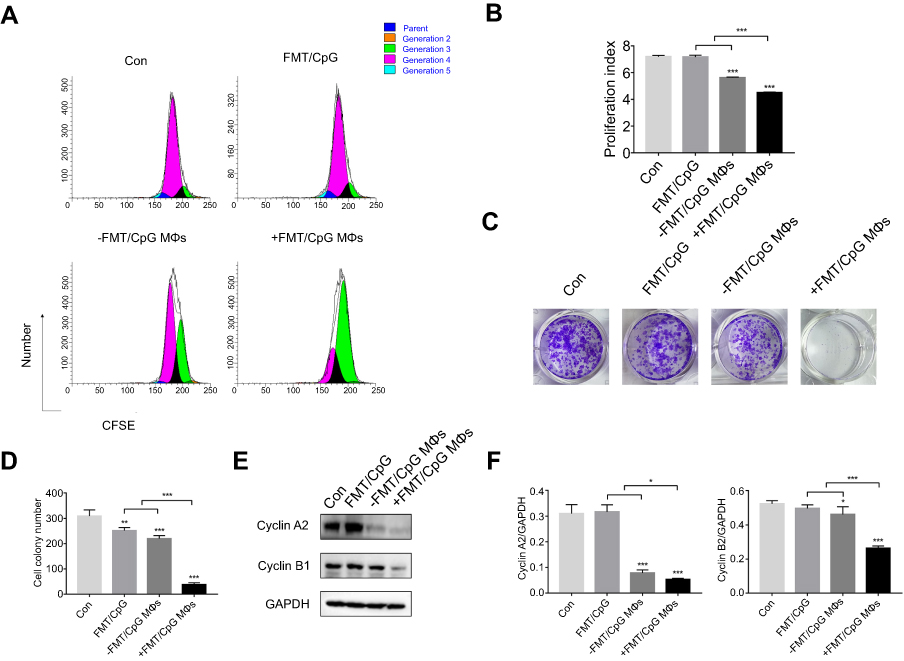 | Figure 5 FMT/CpG-pretreated MΦ supernatant inhibits the proliferation of H1975 cells. (A) H1975 cells labeled with CFSE were subjected to indicated treatments for 48 hrs, and the proliferative fraction was analyzed by FCM. (B) Proliferation index of H1975 cells treated as indicated was determined with ModFit LT software. (C and D) Colony formation of H1975 cells following indicated treatments was quantified and the colonies of >50 cells were defined as positive. (E) Protein levels of Cyclin A2 and Cyclin B1 were detected by Western blotting analysis of H1975 cells following indicated treatments for 48 hrs. (F) Protein levels were quantified with ImageJ software and normalized to that of GAPDH. Results are expressed as mean ± SDof three independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control. Abbreviations: CFSE, 5(6)-carboxyfluorescein diacetate N-hydroxysuccinimidyl ester; FCM, flow cytometry; FMT, ferumoxytol; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MΦ, macrophages. |
FMT/CpG-pretreated MΦ supernatant downregulates p-EGFR and its downstream AKT/mTOR signaling in H1975 cells
Activation of EGFR and its downstream AKT-mTOR signaling pathway leads to increased proliferation and decreased apoptosis of tumor cells.39 Next, Western blotting was used to detect whether the FMT/CpG pre-treated macrophage supernatant affects the expression of EGFR and its downstream signaling pathway proteins in H1975 cells. As shown in Figure 6A and B, in contrast to the control group, the expression levels of p-EGFR, p-AKT, and p-mTOR proteins in H1975 cells treated with −FMT/CpG MΦS were partially inhibited, but not as potently as levels of the +FMT/CpG MΦS group. Compared to the FMT/CpG group, incubated with FMT/CpG pre-treated macrophage supernatant more considerably downregulated the levels of above proteins. Collectively, the results indicated that FMT/CpG pre-treated macrophage supernatant significantly promoted cell apoptosis and inhibited cell proliferation by downregulating phosphorylation of EGFR and its downstream AKT/mTOR protein in H1975 cells.
 | Figure 6 FMT/CpG pretreated MΦ supernatant suppresses the expression of EGFR and downstream signaling in H1975 cells. (A) H1975 cells were treated as indicated for 48 hrs, and protein levels of p-EGFR and its down p-AKT and p-mTOR were analyzed by Western blotting. (B) Protein levels were quantified with ImageJ software and phosphorylated protein levels were normalized to total protein levels. Results are expressed as mean ± SDof three independent experiments. *P<0.05, **P<0.01, ***P<0.001 vs control. Abbreviations: AKT, protein kinase B; EGFR, epidermal growth factor receptor; FMT, ferumoxytol; MΦ, macrophages; mTOR, mammalian target of rapamycin. |
FMT and CpG synergistically inhibit tumor growth in a xenograft mouse model
We next investigated whether the combination of FMT and CpG has anti-tumorigenic effects in vivo using an H1975 cell xenograft mouse model. As showed in Figure 7A and B, co-administration of FMT and CpG significantly suppressed the tumor growth in tumor-bearing mice. Ki-67, an antigen present in the nuclei of proliferating cells, is widely used to evaluate the proliferative activity of tumors.40 The immunohistochemical analysis revealed that the percentage of Ki-67- and EGFR-positive tumor cells in the FMT/CpG group was markedly lower than that in the control group (Figure 7D and E), whereas the percentage of M1 macrophages stained with F4/80 and iNOS in tumor tissues was significantly up-regulated (Figure 7F). In addition, the mice showed no obvious weight loss or treatment-related death, indicating that the combined treatment was non-toxic (Figure 7C). Thus, FMT acts synergistically with CpG to suppress tumor growth in vivo.
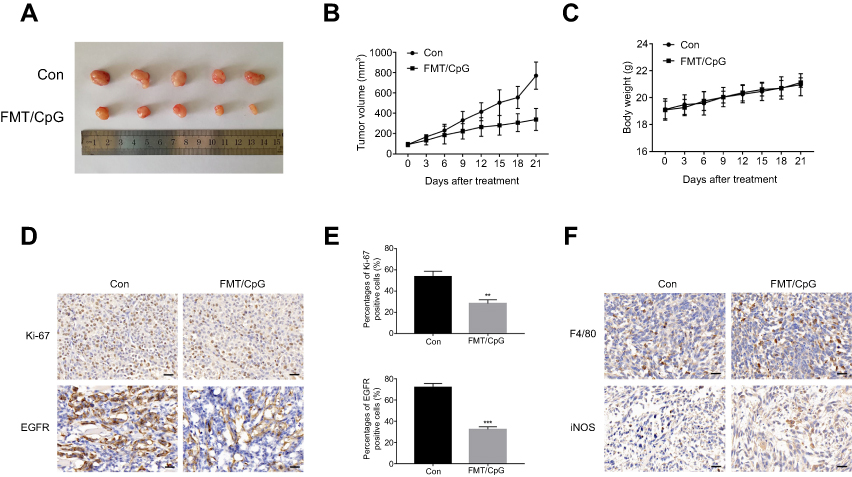 | Figure 7 FMT and CpG synergistically inhibit tumor growth in a nude mouse xenograft model. (A) Image of tumors harvested at the end of treatment. (B) Tumor growth curves of tumor-bearing mice. (C) Body weight of tumor-bearing mice during the experiment. (D) Ki-67 and EGFR expression of tumor cells detected by immunohistochemistry. Scale bar: 50 μm. (E) Quantitative analysis of (D). (F) Macrophages stained with F4/80 and iNOS in tumor tissues detected by immunohistochemistry. Scale bar: 50 μm. Results are expressed as mean ± SDof three independent experiments. **P<0.01, ***P<0.001 vs control. Abbreviation: EGFR, epidermal growth factor receptor; FMT, ferumoxytol. |
Discussion
Osimertinib, a third-generation EGFR-TKI that has been approved for the treatment of EGFRT790M-positive NSCLC patients, acts by inhibiting p-EGFR and downstream signaling.41 However, with changes in the TME and the continuous transformation of cancer cells, the emergence of new drug-resistant mutations is inevitable.42 Various strategies have been developed to overcome resistance to EGFR-TKI treatment including combination chemotherapy,43 antitumor vascular therapy,44 and immunotherapy based on inhibitors of programmed cell death-1 and its ligand.45 However, the toxicity of standard drugs and the high cost of the latter two approaches limit their universal application. As such, there is an urgent need for safer, more effective, and affordable therapeutic strategies to improve the outcome of NSCLC patients with EGFR mutation.
NSCLC is characterized by a large number of MΦ in the TME.13 Given their diversity and plasticity, treatment approaches that induce a phenotype switch in tumor-associated MΦ can be effective against NSCLC. Although MΦ are known to be affected by EGFR-TKIs,15,16 it is unclear whether inducing their transformation can inhibit the progression of NSCLC with EGFRT790M mutation. FMT is a nanomaterial with good biocompatibility and low toxicity that has been approved by the US Food and Drug Administration for the treatment of anemia.46,47 CpG has been used for cancer therapy in clinical trials.30 Many studies have demonstrated the advantages of combined immunotherapy over monotherapy in cancer treatment,48,49and our previous studies also showed that the combined treatment of FMT and TLR3 agonist poly (I:C) induced synergistically induces macrophage activation for melanoma regression.22 Since they are both activators of MΦ, in this study we investigated whether FMT and CpG used in combination can suppress NSCLC by inducing MΦ activation.
IL-12 blocked the migration and invasion of lung adenocarcinoma cells50 and suppressed lung tumor growth, thereby prolonging the survival of lung cancer-bearing mice51 while TNF-α is a cytotoxic protein produced by MΦ that can directly induce apoptosis in NSCLC cells.52,53 In the present study, we found that FMT/CpG was relatively non-toxic to H1975 cells, whereas FMT/CpG-pretreated MΦ supernatant inhibited H1975 cell proliferation and induced apoptosis, which would be related to the anti-tumor effects of IL-12 and TNF-α as well as other cytotoxic cytokines generated by MΦ induced by FMT/CpG. The caspase family plays critical regulatory roles in cell apoptosis51,54 In our study, FMT/CpG-pretreated MΦ supernatant significantly upregulated the protein levels of apoptosis-related Cleaved Caspase-3 and its substrate Cleaved PARP in H1975 cells, indicating that the cell apoptosis of H1975 was through activating caspase-3 pathway. As abnormal activation of EGFR signaling pathway leads to sustained growth of lung cancer cells.39 We also observed that p-EGFR and the downstream factors p-AKT and p-mTOR were downregulated in H1975 cells exposed to FMT/CpG-pretreated MΦ supernatant. In addition, the growth of subcutaneous xenograft tumors in mice was inhibited by treatment with FMT/CpG, which was accompanied by a decline in the EGFR-positive tumor cell fraction and increased M1 type macrophage infiltration. Thus, the combination of the two agents synergistically activated MΦ through upregulation of tumoricidal genes and induction of apoptosis, which was achieved by inhibiting the phosphorylation of EGFR and its downstream signaling in NSCLC cells (Figure 8).
 | Figure 8 Schematic illustration of synergistic MΦ activation by FMT and CpG for the treatment of NSCLC with EGFRT790M mutation. Abbreviations: EGFR, epidermal growth factor receptor; FMT, ferumoxytol; MΦ, macrophages; NSCLC, non-small cell lung cancer. |
Although systemic administration of TLR-9 agonists has been unsuccessful,55,56 local intratumoral injection of CpG ODN is still an effective strategy for tumor treatment by activation of innate responses.28 Gallotta et al found that delivery of a TLR9 agonist through the airways could effectively render lung tumors permissive to PD-1 blockade by promoting optimal CD4+ and CD8+ T-cell Interplay,57 which characterizes a strategy to apply localized TLR9 stimulation to a tumor type not accessible for direct injection. Our previous studies have showed that, compared with combined treatment with FMT/PIC, systemic administration of FP-NPs surface functionalized with poly (I:C) showed a better inhibition of lung metastasis.22 Based on the above findings, we are here to initially explore the effects of FMT combined with CpG on NSCLC with EGFR T790M mutation. Interestingly, similar to the previous results, we found that the combination of FMT and CpG significantly inhibited tumor growth in mice by synergistically activating macrophages. Although this route of administration has some limitations in clinical application it provides a good reference for us to explore novel methods for CpG delivery in the future.
Conclusion
FMT combined with CpG induced the activation of MΦ towards a tumoricidal phenotype; this was accompanied by increased apoptosis and suppression of cell proliferation and EGFR signaling in NSCLC cells. Our findings for the first time provide evidence of FMT/CpG as a novel and effective treatment for NSCLC with EGFRT790M mutation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81672949) and National Key Research and Development Program of China (2017YFA0104303). We thank Prof. Ning Gu (Southeast University) for providing FMT and Prof. Yongqian Shu (The First Affiliated Hospital of Nanjing Medical University) for providing the H1975 (EGFRL858R/T790M mutation) human NSCLC cell line.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi:10.3322/caac.21492
2. Zhou C, Yao LD. Strategies to improve outcomes of patients with EGRF-mutant non-small cell lung cancer: review of the literature. J Thorac Oncol. 2016;11:174–186. doi:10.1016/j.jtho.2015.10.002
3. Yue D, Xu S, Wang Q, et al. Erlotinib versus vinorelbine plus cisplatin as adjuvant therapy in Chinese patients with stage IIIA EGFR mutation-positive non-small-cell lung cancer (EVAN): a randomised, open-label, phase 2 trial. Lancet Respir Med. 2018;6:863–873. doi:10.1016/S2213-2600(18)30277-7
4. Wu YL, Ahn MJ, Garassino MC, et al. CNS efficacy of osimertinib in patients with T790M-positive advanced non-small-cell lung cancer: data from a randomized phase III trial (AURA3). J Clin Oncol. 2018;36:2702–2709. doi:10.1200/JCO.2018.77.9363
5. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med. 2017;376:629–640. doi:10.1056/NEJMoa1612674
6. Siegel RL, Miller KD. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi:10.3322/caac.21551
7. Conway EM, Pikor LA, Kung SH, et al. Macrophages, inflammation, and lung cancer. Am J Respir Crit Care Med. 2016;193:116–130. doi:10.1164/rccm.201508-1545CI
8. Morgensztern D, Herbst RS. Nivolumab and pembrolizumab for non-small cell lung cancer. Clin Cancer Res. 2016;22:3713–3717. doi:10.1158/1078-0432.CCR-16-0190
9. Sanmamed MF, Chen L, Paradigm A. Shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175:313–326. doi:10.1016/j.cell.2018.07.049
10. Yang M, McKay D, Pollard JW. diverse functions of macrophages in different tumor microenvironments. Cancer Res. 2018;78:5492–5503. doi:10.1158/0008-5472.CAN-18-1367
11. Hume DA. The many alternative faces of macrophage activation. Front Immunol. 2015;6:370. doi:10.3389/fimmu.2015.00370
12. Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. 2015;12:1–4. doi:10.1038/cmi.2014.83
13. Costa da Silva M, Breckwoldt MO, Vinchi F, et al. Iron induces anti-tumor activity in tumor-associated macrophages. Front Immunol. 2017;8:1479.
14. Chen JJ, Yao PL, Yuan A, et al. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin Cancer Res. 2003;9:729–737.
15. Chung FT, Lee KY, Wang CW, et al. Tumor-associated macrophages correlate with response to epidermal growth factor receptor-tyrosine kinase inhibitors in advanced non-small cell lung cancer. Int J Cancer. 2012;131:E227–35. doi:10.1002/ijc.27592
16. Zhang B, Zhang Y, Zhao J, et al. M2-polarized macrophages contribute to the decreased sensitivity of EGFR-TKIs treatment in patients with advanced lung adenocarcinoma. Med Oncol. 2014;31:127. doi:10.1007/s12032-014-0374-0
17. Tariq M, Zhang JQ, Liang GK, et al. Gefitinib inhibits M2-like polarization of tumor-associated macrophages in Lewis lung cancer by targeting the STAT6 signaling pathway. Acta Pharmacol Sin. 2017;38:1501–1511. doi:10.1038/aps.2016.89
18. Sneha M, Sundaram NM. Preparation and characterization of an iron oxide-hydroxyapatite nanocomposite for potential bone cancer therapy. Int J Nanomed. 2015;10:99–106.
19. Johnson B, Toland B, Chokshi R, et al. Magnetically responsive paclitaxel-loaded biodegradable nanoparticles for treatment of vascular disease: preparation, characterization and in vitro evaluation of anti-proliferative potential. Curr Drug Deliv. 2010;7:263–273. doi:10.2174/156720110793360621
20. Klenk C, Gawande R, Uslu L, et al. Ionising radiation-free whole-body MRI versus (18)F-fluorodeoxyglucose PET/CT scans for children and young adults with cancer: a prospective, non-randomised, single-centre study. Lancet Oncol. 2014;15:275–285. doi:10.1016/S1470-2045(13)70510-2
21. Neuwelt EA, Varallyay CG, Manninger S, et al. The potential of ferumoxytol nanoparticle magnetic resonance imaging, perfusion, and angiography in central nervous system malignancy: a pilot study. Neurosurgery. 2007;60:601–611. doi:10.1227/01.NEU.0000255350.71700.37
22. Zhao J, Zhang Z, Xue Y, et al. Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I: C) promote melanoma regression. Theranostics. 2018;8:6307–6321. doi:10.7150/thno.29746
23. Nikoofal-Sahlabadi S, Matbou Riahi M, Sadri K, et al. Liposomal CpG-ODN: an in vitro and in vivo study on macrophage subtypes responses, biodistribution and subsequent therapeutic efficacy in mice models of cancers. Eur J Pharm Sci. 2018;119:159–170. doi:10.1016/j.ejps.2018.04.018
24. He XY, Liu BY, Wu JL, et al. A dual macrophage targeting nanovector for delivery of oligodeoxynucleotides to overcome cancer-associated immunosuppression. ACS Appl Mater Interfaces. 2017;9:42566–42576.
25. Koster BD, van Den Hout M, Sluijter BJR, et al. Local adjuvant treatment with low-dose CpG-B offers durable protection against disease recurrence in clinical stage I-II melanoma: data from two randomized phase II trials. Clin Cancer Res. 2017;23:5679–5686. doi:10.1158/1078-0432.CCR-17-0944
26. Carpentier A, Metellus P, Ursu R, et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 2010;12:401–408. doi:10.1093/neuonc/nop047
27. Ursu R, Carpentier AF. Immunotherapeutic approach with oligodeoxynucleotides containing CpG motifs (CpG-ODN) in malignant glioma. Adv Exp Med Biol. 2012;746:95–108.
28. Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28:4324–4332. doi:10.1200/JCO.2010.28.9793
29. Belani CP, Nemunaitis JJ, Chachoua A, et al. Phase 2 trial of erlotinib with or without PF-3512676 (CPG 7909, a Toll-like receptor 9 agonist) in patients with advanced recurrent EGFR-positive non-small cell lung cancer. Cancer Biol Ther. 2013;14:557–563. doi:10.4161/cbt.24598
30. Manegold C, Gravenor D, Woytowitz D, et al. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3979–3986. doi:10.1200/JCO.2007.15.2777
31. Chen B, Sun J, Fan F, et al. Ferumoxytol of ultrahigh magnetization produced by hydrocooling and magnetically internal heating co-precipitation. Nanoscale. 2018;10:7369–7376. doi:10.1039/C8NR00736E
32. Chen B, Li Y, Zhang X, et al. An efficient synthesis of ferumoxytol induced by alternating-current magnetic field. Mater Lett. 2016;170:93–96. doi:10.1016/j.matlet.2016.02.006
33. Fulcher D, Wong S. Carboxyfluorescein succinimidyl ester-based proliferative assays for assessment of T cell function in the diagnostic laboratory. Immunol Cell Biol. 1999;77:559–564. doi:10.1046/j.1440-1711.1999.00870.x
34. Asquith B, Debacq C, Florins A, et al. Quantifying lymphocyte kinetics in vivo using carboxyfluorescein diacetate succinimidyl ester (CFSE). Proc Biol Sci. 2006;273:1165–1171. doi:10.1098/rspb.2005.3432
35. Vigneron S, Sundermann L, Labbe JC, et al. Cyclin A-cdk1-dependent phosphorylation of bora is the triggering factor promoting mitotic entry. Dev Cell. 2018;45:
36. Zheng G, Yu H. Cyclin a turns on bora to light the path to mitosis. Dev Cell. 2018;45:542–543. doi:10.1016/j.devcel.2018.05.017
37. Kalimutho M, Sinha D, Jeffery J, et al. CEP55 is a determinant of cell fate during perturbed mitosis in breast cancer. EMBO Mol Med. 2018;10:e8566. doi:10.15252/emmm.201708566
38. Mohamed TMA, Ang YS, Radzinsky E, et al. Regulation of cell cycle to stimulate adult cardiomyocyte proliferation and cardiac regeneration. Cell. 2018;173:104–116. doi:10.1016/j.cell.2018.02.014
39. Hu J, Zhang H, Cao M, et al. Auranofin enhances ibrutinib‘s anticancer activity in EGFR-mutant lung adenocarcinoma. Mol Cancer Ther. 2018;17:2156–2163. doi:10.1158/1535-7163.MCT-17-1173
40. Temraz S, Shamseddine A, Mukherji D, et al. Ki67 and P53 in relation to disease progression in metastatic pancreatic cancer: a single institution analysis. Pathol Oncol Res. 2018. doi:10.1007/s12253-018-0464-y
41. Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi:10.1158/2159-8290.CD-13-0646
42. Thress KS, Paweletz CP, Felip E, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med. 2015;21:560–562. doi:10.1038/nm.3854
43. Nie K, Zhang Z, Zhang C, et al. Osimertinib compared docetaxel-bevacizumab as third-line treatment in EGFR T790M mutated non-small-cell lung cancer. Lung Cancer. 2018;121:5–11. doi:10.1016/j.lungcan.2018.04.012
44. Hata A, Katakami N, Kaji R, et al. Afatinib plus bevacizumab combination after acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: multicenter, single-arm, phase 2 trial (ABC Study). Cancer. 2018;124:3830–3838. doi:10.1002/cncr.31678
45. Tozuka T, Seike M, Minegishi Y, et al. Pembrolizumab and salvage chemotherapy in EGFR T790M-positive non-small-cell lung cancer with high PD-L1 expression. Onco Targets Ther. 2018;11:5601–5605. doi:10.2147/OTT.S168598
46. Futterer S, Andrusenko I, Kolb U, et al. Structural characterization of iron oxide/hydroxide nanoparticles in nine different parenteral drugs for the treatment of iron deficiency anaemia by electron diffraction (ED) and X-ray powder diffraction (XRPD). J Pharm Biomed Anal. 2013;86:151–160. doi:10.1016/j.jpba.2013.07.013
47. Lu M, Cohen MH, Rieves D, et al. FDA report: ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85:315–319.
48. Adeegbe DO, Liu S, Hattersley MM, et al. BET bromodomain inhibition cooperates with PD-1 blockade to facilitate antitumor response in Kras-mutant non-small cell lung cancer. Cancer Immunol Res. 2018;6:1234–1245. doi:10.1158/2326-6066.CIR-18-0077
49. Horn L, Mansfield AS, Szczesna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379:2220–2229. doi:10.1056/NEJMoa1809064
50. Li X, Zhang P, Liu X, et al. Expression of interleukin-12 by adipose-derived mesenchymal stem cells for treatment of lung adenocarcinoma. Thorac Cancer. 2015;6:80–84. doi:10.1111/1759-7714.12151
51. Yue T, Zheng X, Dou Y, et al. Interleukin 12 shows a better curative effect on lung cancer than paclitaxel and cisplatin doublet chemotherapy. BMC Cancer. 2016;16:665. doi:10.1186/s12885-016-2701-7
52. Zhu J, Xin Y, Liu X, et al. Nimotuzumab enhances the sensitivity of non-small cell lung cancer cells to tumor necrosis factor-alpha by inhibiting the nuclear factor-small ka, CyrillicB signaling pathway. Exp Ther Med. 2018;15:3345–3351.
53. Harada M, Morimoto K, Kondo T, et al. Quinacrine inhibits ICAM-1 transcription by blocking DNA binding of the NF-kappaB subunit p65 and sensitizes human lung adenocarcinoma A549 cells to TNF-alpha and the fas ligand. Int J Mol Sci. 2017;18(12):e2603. doi:10.3390/ijms18122603
54. Chen MF, Huang SJ, Huang CC, et al. Saikosaponin d induces cell death through caspase-3-dependent, caspase-3-independent and mitochondrial pathways in mammalian hepatic stellate cells. BMC Cancer. 2016;16:532. doi:10.1186/s12885-016-2599-0
55. Li J, Song W, Czerwinski DK, et al. Lymphoma immunotherapy with CpG oligodeoxynucleotides requires TLR9 either in the host or in the tumor itself. J Immunol. 2007;179:2493–2500. doi:10.4049/jimmunol.179.4.2493
56. Manegold C, van Zandwijk N, Szczesna A, et al. A phase III randomized study of gemcitabine and cisplatin with or without PF-3512676 (TLR9 agonist) as first-line treatment of advanced non-small-cell lung cancer. Ann Oncol. 2012;23:72–77. doi:10.1093/annonc/mdr030
57. Gallotta M, Assi H, Degagne E, et al. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4(+) and CD8(+) T-cell interplay. Cancer Res. 2018;78:4943–4956. doi:10.1158/0008-5472.CAN-18-0729
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.