Back to Journals » Journal of Inflammation Research » Volume 15
Ferroptosis and Its Potential Role in the Nervous System Diseases
Authors Zhou Y ![]() , Lin W, Rao T, Zheng J, Zhang T, Zhang M, Lin Z
, Lin W, Rao T, Zheng J, Zhang T, Zhang M, Lin Z ![]()
Received 30 November 2021
Accepted for publication 9 February 2022
Published 3 March 2022 Volume 2022:15 Pages 1555—1574
DOI https://doi.org/10.2147/JIR.S351799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yiyang Zhou,1,* Wei Lin,1,* Tian Rao,1 Jinyu Zheng,2 Tianlei Zhang,1 Min Zhang,1 Zhenlang Lin1
1Department of Pediatrics, The Second School of Medicine, Taizhou Women and Children’s Hospital of Wenzhou Medical University, The Second Affiliated Hospital and Yuying Children’s Hospital of Wenzhou Medical University, Wenzhou, 325000, Zhejiang, People’s Republic of China; 2Department of Clinical Medicine, Wenzhou Medical University, Wenzhou, 325000, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhenlang Lin, Email [email protected]
Abstract: Ferroptosis is a novel regulated cell death characterized by metabolic disorders and iron-dependent oxidative destruction of the lipid bilayer. It is primarily caused by the imbalance of oxidation and anti-oxidation in the body and is precisely regulated by numerous factors and pathways inside and outside the cell. Recent studies have indicated that ferroptosis plays a vital role in the pathophysiological process of multiple systems of the body including the nervous system. Ferroptosis may be closely linked to the occurrence and development of neurodegenerative diseases, strokes, and brain tumors. It may also be involved in the development, maturation, and aging of the nervous system. Therefore, this study aims to investigate ferroptosis’s occurrence and regulatory mechanism and summarize its research progress in the pathogenesis and treatment of neurological diseases. This would allow for novel ideas for basic and clinical research of neurological diseases.
Keywords: ferroptosis, iron, lipid peroxidation, molecular mechanism, neurodevelopment, neurological diseases
Introduction
Cell death is an irreversible cessation of life phenomena and a basic biological phenomenon necessary to maintain the typical morphology and functionality of tissues. Based on the difference in morphology and function, the nomenclature committee on cell death (NCCD) divides cell death into accidental cell death (ACD) and regulated cell death (RCD).1 Unlike ACD, RCD primarily relies on specific molecular mechanisms and signaling pathways that can mediate the involvement of various effector molecules in related pathophysiological processes.2,3 Without intervention by an adventitious agent, RCD can act as a physiological effector and promote the development and metabolism in the body.4 In addition, RCD can be induced, and pathological damage occurs when the intracellular and extracellular microenvironment changes drastically and lasts for a lengthy duration, resulting in the inability of the body to adapt.5
Ferroptosis is a novel iron-dependent RCD discovered in recent years. Unlike the previously discovered RCD, it has certain morphology, biochemistry, and genetics characteristics. Morphologically, it did not exhibit chromatin condensation and marginalization typical of apoptosis, cytoplasmic, organelle swelling and plasma membrane rupture typical of necrosis, and double-membrane-enclosed vesicle formation typical of autophagy.6 At the ultrastructural level, ferroptotic cells are typically realized as mitochondrial abnormalities, including mitochondrial shrinkage, increased bilayer membrane density, reduced or even removed mitochondrial cristae, membrane potential changes, and mitochondrial outer membrane rupture.7 As an oxidative cell death, ferroptosis has two biochemical characteristics: iron accumulation and lipid peroxidation. Excess intracellular iron can directly produce large amounts of reactive oxygen species (ROS) through the Fenton reaction, increasing oxidative damage. It can also disrupt oxygen homeostasis by increasing the activity of oxidases such as lipoxygenases (LOXs).6,8 Ferroptosis-induced lipid peroxidation mainly affects polyunsaturated fatty acid (PUFA) on the cell membrane and produces lipid hydroperoxides (LOOHs). The functional defects in the LOOHs scavenging system allow these LOOHs to accumulate continuously and reach lethal levels, where cellular ferroptosis occurs.9 Overexpression of oxidative/antioxidant genes such as cytochrome c oxidase subunit 2 (COX2), acyl-CoA synthetase long-chain family member 4 (ACSL4), and nuclear factor E2 related factor 2 (NFE2L2, also known as Nrf2), may be a genetic feature and biomarker of ferroptosis.10–12
Abnormal regulation of ferroptosis may be closely linked to the pathogenesis of various neurological diseases such as neurodegenerative diseases, strokes, and glioma.13–15 In recent studies, iron toxicity phenomena (such as iron overload) have been observed in neurological diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), epilepsy, and stroke and traumatic brain injury (TBI).16–21 In this study, the primary mechanism of ferroptosis and its research progress in the development and metabolism of the nervous system and neurological diseases are summarized to provide a novel perspective for the functional evaluation of the nervous system and the pathogenesis research and clinical treatment of neurological diseases.
Major Induced Mechanisms of Ferroptosis
Ferroptosis is an RCD triggered by oxidative stress in the intracellular microenvironment, which typically involves iron metabolism and lipid metabolism, ultimately causing iron-dependent peroxidation and oxidative cell death. Most classical ferroptosis inducers such as Erastin and RSL3 are also inhibitors of the cellular antioxidant system,22 suggesting that the antioxidant network is extensively involved in the cellular ferroptosis process. Three main ferroptotic protection systems have recently been found in the body to combat lipid peroxidation. The first is the widespread intracellular glutathione peroxidase 4 (GPX4) system, which catalyzes the reduction of lipid peroxides (LPO) in a glutathione (GSH)-dependent manner.6 The second is the ferroptosis suppressor protein 1 (FSP1) system present on the plasma membrane, which catalyzes the regeneration of reduced coenzyme Q10 (CoQ10), which captures LPO.23,24 The third is the DHODH system in mitochondria, which promotes CoQ reduction to CoQH2, thereby removing LPO and inhibiting ferroptosis.25 We summarize the major induced mechanisms of ferroptosis hereafter (Figure 1).
 |
Figure 1 The major induced pathways of ferroptosis. The induced pathways of ferroptosis, including iron metabolism disorder, lipid peroxidation, and failure of antioxidant systems, are summarized. Iron metabolism and lipid metabolism disorders lead to LPO accumulation, the failure of antioxidant systems leads to LPO not being removed in time, and the above process ultimately induces ferroptosis. Abbreviations: Tf, transferrin; TfR1, transferrin receptor 1; Fe2+, ferrous iron; Fe3+, ferric iron; LIP, labile iron pool; FPN, ferroportin; FT, ferritin; NCOA4, nuclear receptor coactivator 4; HO•, hydroxyl radicals; ROS, reactive oxygen species; LOXs, lipoxygenases; PL, phospholipid; PL•, phospholipid radical; PLOO•, phospholipid peroxyl radical; PLOOH, phospholipid hydroperoxide; PUFA, polyunsaturated fatty acid; GPX4, glutathione peroxidase 4; GSH, glutathione; GS-SG, oxidized glutathione; GCL, glutamate-cysteine ligase; Glu, glutamic acid; CMA, chaperone-mediated autophagy; FSP1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10; DHODH, dihydroorotate dehydrogenase; DHO, dihydroorotate; OA, orotate; iFSP1, inhibitor of ferroptosis suppressor protein 1. |
Iron Homeostasis Imbalance
Iron is a metal element crucial for numerous biological processes in mammals, but excess free iron readily accepts or provides electrons to participate in redox reactions, resulting in cytotoxicity.26 Therefore, a precise regulatory network is required to regulate a range of iron metabolic processes, including absorption, utilization, storage, and recycling, thereby maintaining iron homeostasis.27 The process is primarily regulated by hepatogenic hepcidin and a series of iron regulatory proteins (IRP).28 Dietary iron taken up by enterocytes and circulating iron phagocytosed by macrophages are released into the blood by ferroportin (FPN) on the cell membrane surface.28 FPN is an important intracellular iron efflux protein and is negatively regulated by hepcidin.29,30 Transferrin (TF) binds Fe3+ (Fe2+ is mostly oxidized to Fe3+ by ceruloplasmin or hephaestin) in blood with high affinity, preserving it in a redox-inert state. TF-Fe3+ complexes bind to transferrin receptor 1 (TFR1) on the cell membrane surface and can be endocytosed to form endosomes. Fe3+ in endosomes is reduced to Fe2+ by the ferric reductase STEAP331 and then transported by DMT1 into the labile iron pool (LIP)32 for body metabolism. Similarly, lactotransferrin can mediate the process of iron transfer into cells.33 Excess intracellular iron can be stored in ferritin or transported out of the cell by the hepcidin-FPN axis. Lysosomes can recycle iron from mitochondrial and cytosolic ferritin by selective autophagy.34 Additionally, lipocalin-2 (LCN2) was found to be involved in the regulation of intracellular iron homeostasis. After binding to the LCN2 receptor expressed on the cell surface, iron-loaded LCN2 can be internalized into cells to release iron, thereby increasing intracellular iron content.35 This non-Tf-bound iron (NTBI) uptake may be more significant in the brain (where interstitial NTBI content is higher than Tf-bound iron) and in response to inflammatory stimuli.36 On the other hand, iron-lacking LCN2, which internalizes into cells, can chelate intracellular iron and may transfer it to the extracellular environment, thereby reducing intracellular free iron content.35
The abnormal regulation of iron metabolism leads to imbalance of intracellular iron homeostasis and accumulation of redox-active iron, thereby inducing ferroptosis. Pseudolaric acid B (PAB) can induce the expression of transferrin receptors (TFRC) on glioma cell membranes and enhance the intake of iron, which in turn triggers ferroptosis.37 Nuclear receptor coactivator 4 (NCOA4)-mediated ferritin selective autophagy can increase the free iron content and promote ferroptosis.38 Mitochondrial ferritin (FTMT) overexpression inhibits Erastin-induced ferroptosis in neuroblastoma cells by taking up accumulated labile iron.39 Enhancing FPN expression reduces intracellular iron accumulation and attenuates neuronal ferroptotic damage.40 On the one hand, excessive intracellular accumulation of reactive iron can produce many hydroxyl radicals (HO•, an important ROS acting on lipid peroxidation) through the redox cycle of Fenton and Haber-Weiss reactions, thus causing oxidative damage.41 On the other hand, it can increase the activity of peroxidases such as LOXs and prolyl hydroxylase domain (PHD), thereby improving the sensitivity of cells to ferroptosis.8 The increase of intracellular-free iron caused by iron metabolism disorder is among the characteristics of ferroptosis, but it is unclear whether other metals such as Cu, Ni, and Co, which can produce ROS through Fenton reaction,41 can also induce ferroptosis. If not, this warrants further investigation into whether iron accumulation activates specific downstream effectors necessary to promote ferroptosis after ROS production.
Lipid Peroxidation
Lipid peroxidation is a prominent manifestation of ferroptotic damage that mainly involves PUFAs on the cell membrane. Having an intact diallyl structure is a vital prerequisite for PUFA peroxidation. The C-H bond at the diallyl group is less stable, making PUFAs on the membrane easily oxidized and thus becoming a major target for intracellular ROS attack.42 MUFAs are less susceptible to peroxidation since they do not possess diallyl groups;43 deuterated PUFAs reduce oxidative stress and attenuate Erastin- or RSL3-induced ferroptotic damage.44 Lipid peroxidation eventually produces LOOHs and consequent reactive aldehydes such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE).41 Among them, MDA is the most mutagenic product in lipid peroxidation, while 4-HNE is the most toxic product.41,45 The above LPO causes oxidative damage to the lipid bilayer and may eventually causes cellular ferroptosis.
Arachidonic acid (AA) and adrenic acid (AdA) are the primary substrates for the peroxidation reaction.10 Cells with up-regulated expression of these PUFAs are more sensitive to ferroptosis.44 Free PUFAs are sequentially synthesized into lipids and bound to membrane phospholipids (PL) to generate PUFA-PL complexes under the action of two enzymes, namely, ACSL4 and LPCAT3. PUFA-PL complex can further generate PUFA-PLOOHs catalyzed by peroxidase LOXs.10,11 Although various cell membrane lipids can be oxidized, the peroxidation of PUFAs in phospholipids by LOXs tends to be particularly important for ferroptosis.46 At the molecular level, HO• accumulated in cells can abstract an allylic hydrogen atom from lipid molecules, form the carbon-centered phospholipid radicals (PL•), and rapidly react with oxygen to form phospholipid peroxyl radicals (PLOO•). PLOO• can extract hydrogen from neighboring lipids molecules, thus generating phospholipid hydroperoxides (PLOOH) and triggering ferroptosis. Simultaneously, this process generates new PL•, forming a free radical chain reaction that drives ferroptosis to continue to progress.42 The body’s antioxidant systems inhibit ferroptosis by providing electrons that reduce PLOOH to PLOH.
Failure of GPX4 Antioxidant System
GPX4 is a selenocysteine-containing and glutathione-dependent enzyme that can convert lipid hydroperoxides to lipid alcohols10,47 and is a key inhibitory protein for the body’s resistance to ferroptosis. The loss of activity or inhibition of expression of GPX4 leads to the accumulation of fatal lipid peroxides, followed by cellular ferroptosis.48 Differing from other members of the glutathione peroxidase family, GPX4 is mostly in monomeric form and can reduce both free present LPO and membrane-bound or protein-bound LPO.49 The enzymatic reaction catalyzed by GPX4 is a recyclable antioxidant pathway, and its enzymatic active site switches between the oxidized and reduced states to continuously reduce LPO. First, selenol (-SeH) in the active site of GPX4 is oxidized to selenic acid (-SeOH) by LPO. Then, -SeOH is reduced by the first GSH to intermediate selenide (-Se-SG) and is further reduced by the second GSH to generate -SeH, completing a full cycle.50 Simultaneously, two GSH molecules produce one molecule of glutathione disulfide (GS-SG) during this process. Therefore, the antioxidant protective effect of GPX4 against ferroptosis depends on the activity of selenium at the catalytic site and a certain amount of GSH substrate. As among the important triggering mechanisms of ferroptosis, failure of GPX4 antioxidant system can be accomplished by directly inhibiting GPX4 through some small molecules (eg, RSL3, FIN56, and FINO2)48,51,52 and indirectly inhibiting GPX4 by inhibiting GSH production, depleting GSH and inhibiting selenium metabolism.
The synthesis of GSH is limited by the content of cysteine (mainly produced by cystine) and the glutamate-cysteine ligase (GCL) activity. Cystine/glutamate antiporter (system Xc-/xCT) on the cell membrane surface, composed of the light chain subunit SLC7A11 and the heavy chain subunit SLC3A2,53 can transfer extracellular cystine into the cell while also transferring the same amount of intracellular glutamate out of the cell. Cystine transferred intracellularly can be reduced to cysteine by GSH or thioredoxin reductase 1 (TXNRD1).54 Cysteine, glutamate, and glycine ultimately generate GSH through a series of enzymatic reactions, of which GCL is the rate-limiting enzyme.55 Erastin, sorafenib, sulfasalazine, etc., can act as SLC7A11 inhibitors that reduce cysteine transfer, thereby inhibiting the production of GSH and triggering ferroptosis.6,53 Interestingly, cysteine can also be produced from methionine via the transsulfuration pathway, thus avoiding the necessity of cystine uptake via system Xc-.56 Therefore, some cells are insensitive to system Xc- inhibitors. In addition, buthionine sulfoximine (BSO), an inhibitor of GCL, can limit GSH production, directly trigger cellular ferroptosis, or enhance cellular sensitivity to ferroptosis.57 The biosynthesis of selenoproteins is mainly regulated by the mevalonate pathway. GPX4 contains a selenocysteine (Sec) at its active site. The genetic code for Sec is UGA, but UGA typically acts as a stop signal in mRNA translation, resulting in selenoproteins from being mistranslated.58 The body requires specific selenocysteine-tRNA (Sec-tRNA) to incorporate Sec into the UGA codon of GPX4. In contrast, prenylation modification of specific adenine sites (using the product of mevalonate pathway as a donor) is a guarantee of efficient binding of Sec-tRNA to Sec.58 Inhibitors of the mevalonate pathway such as statins have been demonstrated to interfere with Sec-tRNA maturation, inhibiting GPX4 biosynthesis.59 This warrants further investigation on ferroptosis regulation by the mevalonate pathway.
Failure of FSP1 Antioxidant System
FSP1, as the first GPX4 replenishing enzyme, is important for ferroptosis development when GPX4 is inhibited. FSP1 was originally discovered as a pro-apoptotic gene mediated by p5360 and had a protective effect against ferroptosis caused by GPX4 loss. Due to its similarity to the amino acid sequence of apoptosis-inducing factor (AIF), it is called mitochondria-associated apoptosis-inducing factor 2 (AIFM2).61 Subsequently, it was found that N-terminus of FSP1 lacks a mitochondrial targeting sequence and does not locate in mitochondria. Ferroptosis inhibition is primarily mediated by ubiquinone (ie, coenzyme Q10, CoQ10).24 FSP1 can catalyze CoQ10 regeneration through NAD (P) H, while reduced CoQ10 inhibits the proliferation of lipid peroxidation and ferroptosis by capturing LPO. In GPX4-knockdown cells, FSP1 effectively inhibits ferroptosis, while iFSP1 (inhibitor of ferroptosis suppressor protein 1), an effective inhibitor of FSP1, selectively induces ferroptosis in GPX4 knockdown cells overexpressing FSP1.23 Therefore, FSP1-NAD (P) H-CoQ10 axis protects against cellular ferroptosis in a GPX4- and ACSL4-independent manner, and myristoylation modification of the amino terminus of FSP1 is essential for its anti-ferroptosis activity.24 This suggests that failure of FSP1 system would greatly increase the sensitivity of cells to ferroptosis, and regulating the activity of FSP1 may have a potential neuroprotective effect in patients with neurological diseases such as PD by inhibiting ferroptosis.62
Failure of DHODH Antioxidant System
Mitochondria is the primary site of aerobic respiration and energy generation in cells and an important source of intracellular ROS.63 The electron transport chain at the inner mitochondrial membrane also generates a large volume of superoxide anion (O2•-) during ATP production. On the one hand, O2•- in mitochondria increases ROS content; on the other hand, it can also be converted into H2O2 under superoxide dismutase (SOD) and diffuse from mitochondria to the cytosol. Consequently, the increased H2O2 in the cytosol can produce highly reactive HO• and other ROS through the Fenton reaction in the high iron environment of the cytosol. Mitochondrial ROS are significant for inducing lipid peroxidation and cellular ferroptosis within mitochondria and cell membranes.
DHODH is an enzyme located on the outer surface of the inner mitochondrial membrane64 and can catalyze the oxidation of dihydroorotate (DHO) to orotate (OA) and the reduction of CoQ to CoQH2. It plays an important role in the development of ferroptosis when the GPX4 system is inhibited. Mao et al25 found that inhibiting DHODH induced ferroptosis in low-expressing GPX4 cells and increased the sensitivity of high-expressing GPX4 cells to ferroptosis, and that treatment with ferroptosis inhibitor liproxstatin-1 largely rescued this cellular ferroptosis exacerbated by DHODH knockdown. This highly suggests that DHODH is involved in the ferroptotic defense mechanism of mitochondria. Further studies have revealed that inhibiting DHODH increased mitochondria’s CoQ/CoQH2 ratio. Overexpression of ciona intestinalis AOX (CiAOX) oxidized CoQH2 to CoQ in mitochondria65 and enhanced RSL3-induced mitochondrial peroxidation and ferroptosis in HT-1080 cells, but DHODH inhibition on this basis did not have an additional ferroptosis-sensitizing effect. In addition, the down-regulation of DHODH did not affect the expression levels of GSH, GPX4, SLC7A11, or ACSL4, among others. Therefore, DHODH-CoQ pathway is critical as an independent defense mechanism in mitochondrial ferroptotic defense, and the inhibition of this pathway will increase the sensitivity of ferroptosis.
Potential Regulatory Mechanisms of Ferroptosis
Ferroptosis is primarily characterized by iron-dependent oxidative and antioxidant imbalances. Numerous factors in the oxidative/antioxidant network of the body may be involved in ferroptosis regulation, producing or scavenging accumulated ROS and LPO. The occurrence of ferroptosis may be regulated by miRNAs, histones, and numerous signaling pathway networks (Table 1).
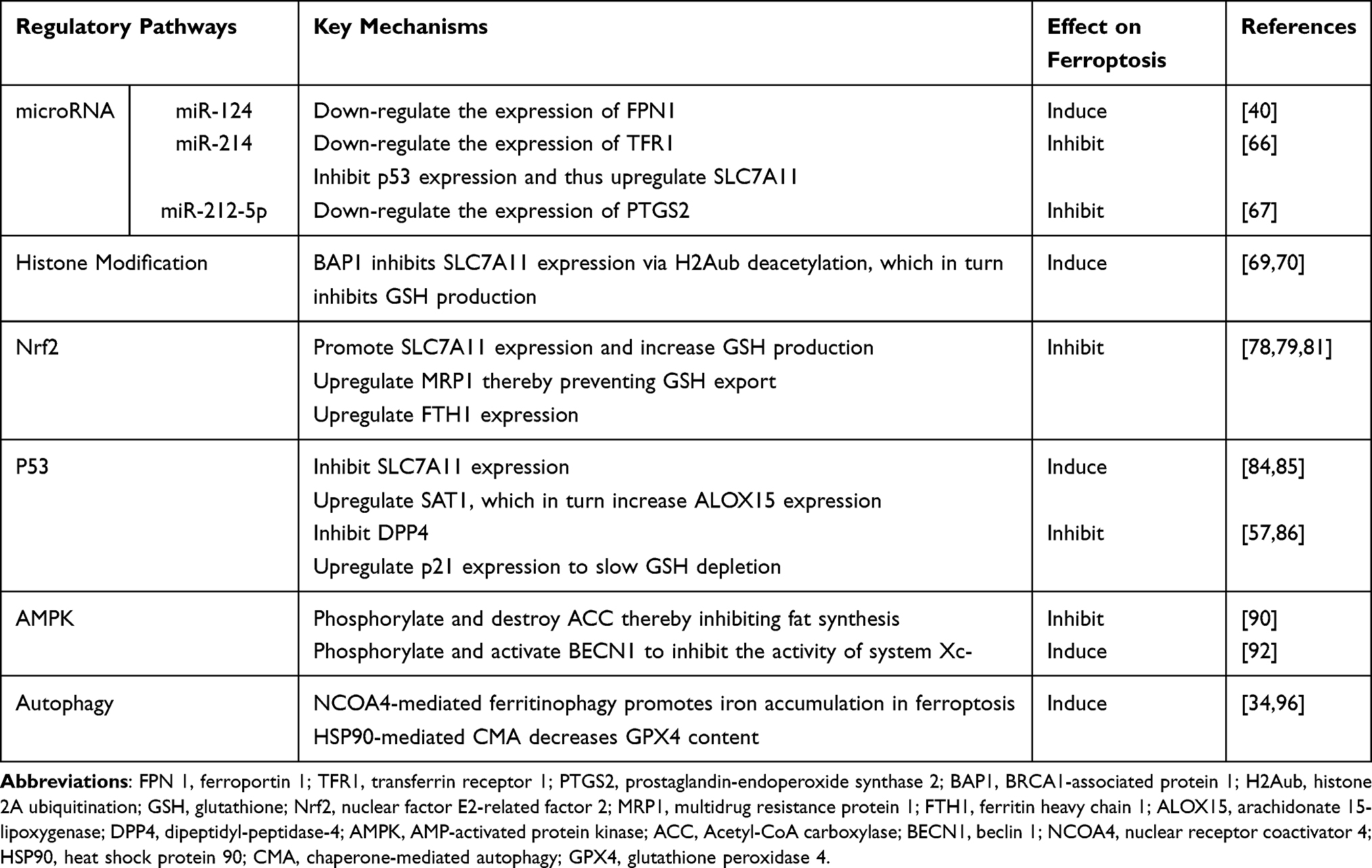 |
Table 1 Potential Regulatory Pathways of Ferroptosis |
MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNAs that bind to the 3' untranslated region of mRNAs and induce rapid degradation of mRNAs, thereby achieving gene transcription regulation. MiRNAs inhibit or induce ferroptosis by negatively regulating the expression of ferroptosis-related genes. MiR-214 can reduce TFR1 expression levels and elevate SLC7A11 levels by decreasing p53 levels, thereby reducing lipid peroxidation and iron deposition in vitro and in vivo and significantly inhibiting ferroptosis.66 Bao et al40 found that miR-124 levels in the perihematomal tissue of ICH model mice were lower than those of sham mice, and inhibition of miR-124 could up-regulate the expression of FPN and reduce iron accumulation and ferroptosis in neurons, thereby alleviating ICH symptoms. In addition, miR-212-5p may play an ferroptosis protective role by down-regulating prostaglandin-endoperoxide synthase-2 (Ptgs2) levels.67 In summary, perfecting the miRNA regulatory network associated with the ferroptosis pathway may be significant for the mechanistic study and targeted therapy of ferroptosis.
Histone Modification
Histone modification can affect the affinity of histones to DNA duplexes, thereby changing the loose and condensed state of chromatin. This, in turn, affects the binding of regulatory proteins such as transcription factors to chromatin and gene expression. The interaction between BRCA1-associated protein 1 (BAP1) and ASXL1 produces a deubiquitinase that removes monoubiquitin from histone 2A ubiquitination (H2Aub)68 and plays a vital role in the epigenetic regulation of gene transcription. Zhang et al69 found that BAP1 inhibited SLC7A11 expression at a certain degree through H2Aub deubiquitination, thereby inhibiting cystine uptake and upregulating cells’ sensitivity ferroptosis. Additionally, the transcription factors hepatocyte nuclear factor 4 alpha (HNF4A) and hypermethylated in cancer 1 (HIC1) can competitively bind the histone acetyltransferase KAT2B to affect GSH production and resist and promote ferroptosis in HCC cells, respectively.70
Nrf2
The transcription factor Nrf2 protects cells from oxidative stress by regulating various endogenous antioxidant responses. Downstream targets of Nrf2 include various ferroptosis-related genes such as HO-1, TFRC, FPN1, SLC7A11, glutathione synthetase (GSS), and GPX4.71–75 Nrf2 is an essential transcriptional regulator of ferroptosis, and its activity is tightly regulated by Kelch-like ECH-associated protein 1 (Keap1). Under normoxic conditions, Nrf2 binds to Keap1 and is inactivated by ubiquitination and proteasomal degradation, so it is expressed at low levels.76 Upon oxidative stress, Nrf2 is released from the Keap1 binding site and rapidly translocates into the nucleus, followed by binding to antioxidant response element (ARE) in the promoter region. The activation of transcription of downstream antioxidant gene to maintain cellular redox homeostasis.77 Nrf2 can directly bind to the ARE sequence in the promoter region of SLC7A11, promoting SLC7A11 expression78 and increasing GSH production. In addition, Keap1 inhibition to increase the activity of Nrf2 leads to up-regulation of multidrug resistance protein 1 (MRP1), thereby preventing GSH efflux and inhibiting cellular ferroptosis.79 Therefore, we speculate that Nrf2 can regulate ferroptosis by targeting GPX4 antioxidant system. Moreover, GMF gene knockdown using CRISPR/Cas9 method activates Nrf2-HO-1 pathway in BV2 microglia, thereby upregulating ferritin and reducing intracellular free iron to improve microglia hyperplasia.80 In addition, Nrf2 activation can increase ferritin heavy chain 1 (FTH1) expression in PC12 cells to enhance iron storage capacity and thus play an ferroptosis regulatory role.81 These suggest that Nrf2 may also be involved in ferroptosis regulation by regulating intracellular iron metabolism.
p53
The tumor suppressor protein p53 plays a core role in various cellular stress events, including DNA damage, hypoxia, nutrient starvation, and oncogene activation.82 The outcome of p53 activation depends on the level of stress events, and low levels of stress or damage-triggered p53 activation can induce cell cycle arrest, DNA repair, and survival, which can protect against oxidative stress damage by down-regulating ROS production in cells. In contrast, high-stress levels or damage-induced p53 activation can induce apoptosis.83 Similarly, p53 plays a dual role in regulating cellular ferroptosis. On the one hand, p53 can enhance ferroptosis by inhibiting SLC7A11 expression or promoting SAT1 expression. Inhibition of p53 can increase SLC7A11 expression at the transcriptional level, thereby exerting an anti-ferroptosis effect and ameliorating nerve injury after intracerebral hemorrhage.84 SAT1, upregulated by p53, induces cellular ferroptosis by increasing ALOX15 expression, a lipoxygenase that catalyzes AA peroxidation.85 On the other hand, p53 can inhibit ferroptosis by inhibiting dipeptidyl-peptidase-4 (DPP4) activity or p21 expression. P53 can block DPP4 activity to weaken plasma membrane-associated DPP4-dependent peroxidation, thereby inhibiting Erastin-induced ferroptosis.57 In addition, p53-mediated p21 expression slows down intracellular GSH depletion and ROS accumulation, thereby delaying ferroptosis induced by system Xc- inhibition or direct cystine deprivation in many cancer cells.86 Although the bidirectional regulatory role of p53 on ferroptosis is widely recognized, it remains unknown whether there exists a factor or mechanism that controls the differential response of p53 to ferroptosis.
AMPK
The consumption of nutrients and energy causes energy stress and leads to cell death,87 as evidenced by intracellular ATP depletion and a corresponding increase in AMP levels. AMP-activated protein kinase (AMPK), as a key sensor of intracellular energy status, can be activated by increased AMP and the phosphorylation of upstream kinases. This promotes body catabolism and inhibits anabolism to increase ATP levels, restoring energy balance and maintaining overall cell survival.88,89 Prior studies have demonstrated that AMPK regulates ferroptosis to maintain cell survival to a certain degree through phosphorylation and inactivation of acetyl-CoA carboxylase (ACC).90 ACC1 and ACC2 are two related enzymes that catalyze the synthesis of malonyl-CoA from acetyl-CoA, promote fatty acid synthesis, and inhibit fatty acid oxidation. AMPK phosphorylates and kills ACC, resulting in the inhibition of biosynthesis of PUFAs and other fatty acids, thereby inhibiting ferroptosis. LKB1 was found to be one of the major upstream kinases that activate AMPK after energy stress.91 Additionally, AMPK directly activated by Erastin can phosphorylate and activate downstream beclin1 (BECN1, an autophagy key protein), which selectively binds SLC7A11 to inhibit the activity of system Xc- in a transcription-independent form, thereby depleting GSH and inducing ferroptosis.92 However, this ferroptosis-inducing effect of AMPK, which was only found when ferroptosis was induced by erastin, needs to be further confirmed. Collectively, AMPK plays an important role in the regulation of ferroptosis, especially in the context of energy stress, and the upstream and downstream factors of AMPK regulating ferroptosis deserve more in-depth study.
Autophagy
Recent studies have proven that autophagy is closely linked to ferroptosis. Oxidative stress and ROS can induce autophagy, and autophagy-related signaling pathways regulate ferroptosis and jointly affect cell death.38,93 Selective autophagy is significant in ferroptosis regulation. Ferritinophagy is a process that selectively degrades ferritin through autophagy, thereby increasing intracellular iron levels and inducing ferroptosis. NCOA4 is a selective transport receptor that selectively degrades ferritin through lysosomes.94 Knockdown of coatomer protein complex subunit zeta 1 (COPZ1) can induce ferroptosis in glioblastoma cells by upregulating NCOA4 and promoting ferritinophagy.34 Ferritinophagy to promote cellular ferroptosis has also been found to be involved in subarachnoid hemorrhage (SAH) in rats95. Chaperone-mediated autophagy (CMA) is an autophagic process in which substrate proteins in the cytosol are selectively bound by molecular chaperones and transported into lysosomes for digestion and degradation by lysosomal enzymes. Lamp-2a acts as a receptor to transport the CMA substrate GPX4 to the lysosomal matrix for degradation, thereby reducing cellular antioxidant capacity and promoting ferroptosis in mouse neural cell line 661W cells.96 In summary, autophagy plays a crucial role in ferroptosis development, and regulating ferroptosis by adjusting autophagic activity provides a novel perspective for treating diseases such as tumors.
Effects of Ferroptosis on the Development, Functional Maintenance, and Aging of the Nervous System
The nervous system is the most important system in the human body and plays a dominant role in regulating physiological functional activities. Therefore, it is vital to ensure the normal development and maturation, functional maintenance, and normal aging of the nervous system. High-throughput sequencing technologies enable deep gene expression profiling on a genome-wide scale and have been used for gene transcriptome data analysis in the human brain.97,98 The data demonstrate that the expression of ferroptosis-related genes such as GPX4, Nrf2, SLC7A11, TFRC, TF, and YAP have different characteristics during various developmental stages and in different brain regions.99 This suggests that ferroptosis and its related genes may be involved in a series of processes such as development, functional maintenance, and aging of the nervous system. Therefore, exploring the role of ferroptosis-related genes in the nervous system and investigating the association between the nervous system and ferroptosis are necessary for assessing nervous system functionality.
Ferroptosis-related genes are highly expressed in the embryonic and postnatal human brain and may be related to brain development. At 37 weeks post-conception, genes such as YAP1, HMOX1, and SLC7A11 were observed to be up-regulated, promote neuronal and non-neuronal cell proliferation, and are critical in cognitive development.100 As an essential nutrient for neurodevelopment, adequate iron is essential for infant growth and development, but excessive iron accumulation can negatively affect brain development.101 Thus, the precise regulation of iron homeostasis is required during brain development. Postnatally, TF and TFRC are highly expressed in the brain100 and are critical in delivering iron. Iron is transported by the blood after binding to TF and crosses the blood-brain barrier by binding to TFRC on the surface of microvascular endothelial cells.102 In addition, YAP expression is increased after birth, especially in non-neuronal cells. YAP is an effector of the Hippo pathway that integrates chemical and mechanical signals within the cell. Highly expressed YAP is activated in Schwann cells and regulates Schwann cells proliferation and the transcription of basal layer receptor genes through Hippo pathway, thus promoting correct radial sorting and myelination of axons, which induce proper brain development.103 Nrf2, as a ferroptosis-suppressor gene, is highly expressed in astrocytes and plays a neuroprotective role in the brain. It is largely activated in abnormal states such as diseases.104
Ferroptosis is also closely related to the functional maintenance of the nervous system. In the adult mammalian brain, inherent neural progenitor cells in the dentate gyrus of the hippocampus produce new functional neurons that are integrated into existing neural circuits to maintain the integrity of hippocampal learning, memory, emotion, pattern separation, and other functions.105,106 Therefore, it is crucial to understand the factors that control maintenance, differentiation, and integration of adult neural stem cells (NSCs). Studies have demonstrated that loss of LCN2, an iron transporter of NSCs, may lead to intracellular iron accumulation and induce iron-dependent endogenous oxidative stress. This leads to cell cycle arrest and defects in the proliferation, differentiation, and maturation of NSCs, after which affects hippocampus-dependent contextual fear discriminative tasks in mice, with manifestations of behavioral and spatial learning disorders similar to anxiety and depression.107
Ferroptosis is also involved in aging and neurodegeneration.108–110 Iron is excessive with aging.108 The reasons for this are the following: (i) the body’s metabolic rate decreases with aging, resulting in a decrease in iron requirements and an increase in iron accumulation; (ii) the amount of hemoglobin that stores 60% of total iron decreases with aging; and (iii) menopause causes relative iron excess in women. It is conceivable that excess iron can adversely affect cell functionality, ultimately leading to cell death. Brain iron accumulation in the caudate nucleus, putamen, globus pallidus, and hippocampus promotes oxidative damage and protein oligomerization, inducing age-related neurological diseases such as AD, PD, and dementia with Lewy bodies.111 Reduced GSH expression in an aging brain induces oxidative stress and cellular ferroptosis. This leads to cognitive dysfunction, which is accelerated in many central nervous system diseases, including AD.112 Currently, the functional study of ferroptosis and its related genes in the nervous system at various stages is imperfect and warrants further investigation.
Ferroptosis and Neurological Diseases
Brain tissue has more oxidative metabolism, fewer antioxidant enzymes, and high lipid content. PUFAs such as AA and docosahexaenoic acid (DHA) are also predominant.113,114 Therefore, brain tissue is more sensitive to lipid peroxidation and ferroptosis. Existing studies have demonstrated that ferroptosis may cause neurological dysfunction and cell death and driver neurological diseases such as stroke, PD, AD, and HD.115 Therefore, ferroptosis modulators may have significant therapeutic potential for neurological disorders. Hereafter, we elaborate on the relationship between ferroptosis and these neurological disorders (Table 2).
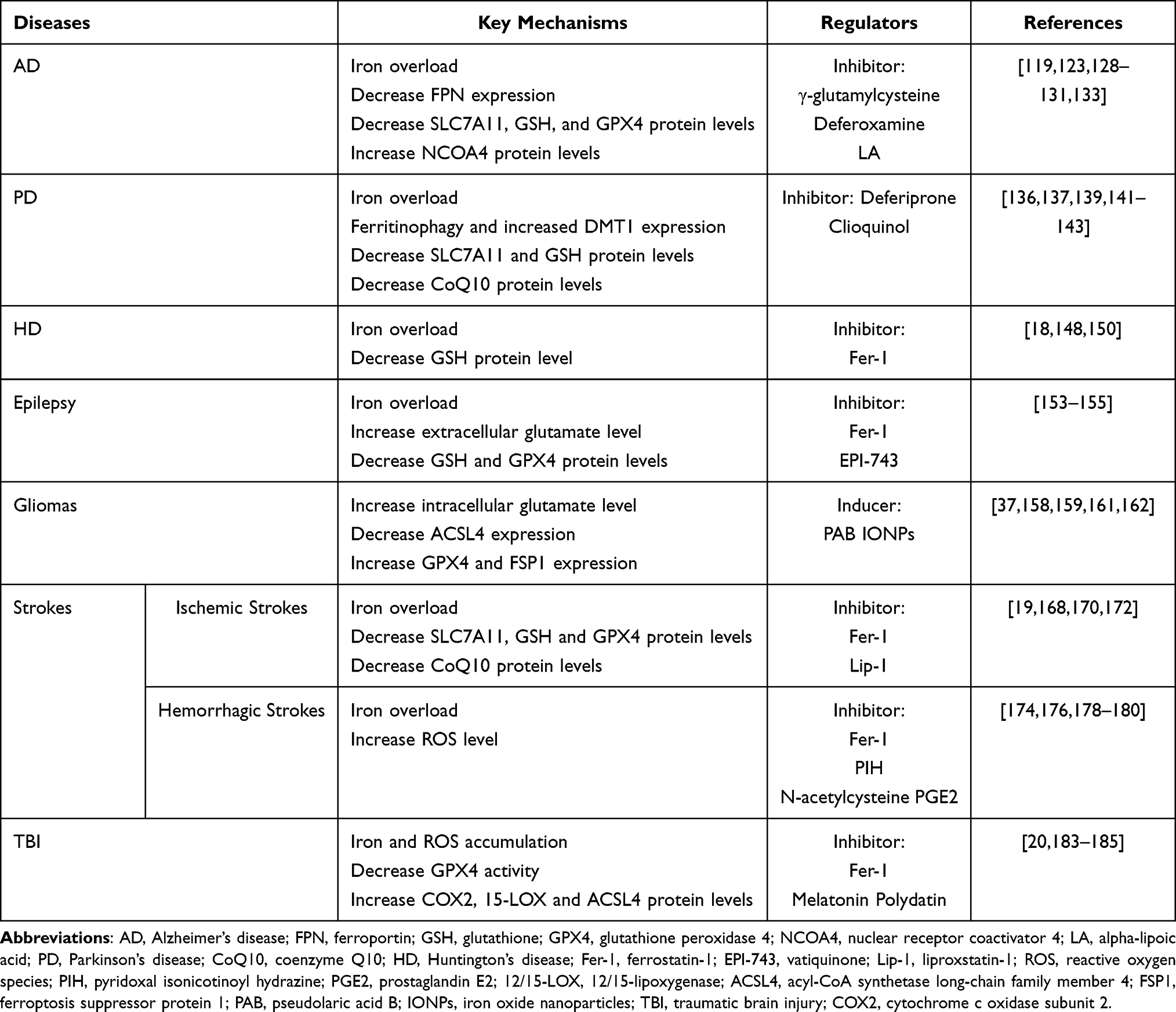 |
Table 2 Key Mechanisms and Regulators of Ferroptosis in Nervous System Diseases |
Alzheimer’s Disease
AD is the most common form of dementia and is characterized by a progressive decline in memory and cognitive function, affecting tens of millions of people on a global scale. The most typical histopathological features of AD are amyloid plaques formed by extracellular amyloid-β (Aβ) deposition and intracellular neurofibrillary tangles (NFTs) formed by tau protein hyperphosphorylation.116 However, Aβ-based treatments have not achieved the desired results, and to date, there are still no drugs that can slow down AD progression.117 Neuronal ferroptosis provides a new direction for future studies on AD.
Iron deposition is among the first reported brain chemical changes in AD patients (first discovered in 1953118). Pathological neuronal iron accumulation in the brain of an AD patient, which produces large amounts of free radicals through the Fenton reaction, is an important factor that leads to oxidative damage in AD.119 Some AD patients similarly induce oxidative stress and experience lipid peroxidation and ferroptosis after the dysregulation of iron homeostasis caused by elevated ferritin levels in the absence of increased iron.120 In fact, ferritin in AD brain differs from physiological ferritin, and its catalytic site can be used in Fenton reaction to enhance oxidative stress.121 It has been found that a relatively high content of iron (the iron content remains within the normal range) may change the processing of the amyloid-β precursor protein (APP) through iron-dependent ferroptosis, accelerate the formation of plaques and NFTs, and play an upstream role in AD pathogenesis.122 The down-regulation of FPN and brain atrophy in the neocortex and hippocampus were observed in the hippocampus of model mice and brain tissue of AD patients, so the down-regulation of FPN may be greatly involved in progressive brain atrophy of AD by promoting ferroptosis.123 In addition, there is increasing evidence that Aβ accumulates in neurons in the AD brain. This can occur in various subcellular regions, producing synaptic disruption, inhibiting the ubiquitin-proteasome system, and inducing toxic effects such as mitochondrial dysfunction and proinflammatory responses.124,125 The cytotoxicity of Aβ was demonstrated to be mediated by ferroptotic cell death, and the slow accumulation of Aβ in neurons may lead to prolonged cellular ferroptosis, thus resulting in additional toxic responses.126 During the exploration of targeting ferroptosis, it was demonstrated that supplementation with tetrahydroxy stilbene glycoside, γ-glutamylcysteine, etc., can restore the GSH-GPX4 antioxidant system and reduce ROS levels in AD patients, thereby reducing Aβ-induced brain injury.127,128 The metal ion chelator deferoxamine (DFO) can reduce the accumulation of iron in the brain region of experimental animals, inhibit Aβ lamellae or plaque formation, and dissolve the already prepared lamellae.129 DFO treatment has been found in the clinical treatment of AD that may slow down the related dementia symptoms.130 Alpha-lipoic acid (LA) has antioxidant and iron chelating properties. LA administration significantly blocked tau-induced iron overload and lipid peroxidation, reducing neuronal loss in AD mice.131 Activation of the Nrf2 pathway can reduce oxidative stress, neuroinflammation, and ferroptosis in AD, thereby improving cognitive deficits.132 In addition, in elderly AD patients with cardiac insufficiency, mitochondrial aldehyde dehydrogenase (ALDH2) was found to eliminate the manifestations of increased NCOA4, decrease GPX4 and SLC7A11 in APP/PS1 mice by inhibiting lipid peroxidase ACSL4-dependent ferroptosis, and improve their cognitive deficits and cardiac insufficiency.133 Targeting ferroptosis key pathways such as iron metabolism, lipid metabolism, and the GPX4 antioxidant system can potentially become a major breakthrough in AD treatment.
Parkinson’s Disease
PD is a common progressive neurodegenerative disorder characterized by dyskinesias such as resting tremor and myotonia and non-motor symptoms, including anosmia, constipation, anxiety, depression, and cognitive impairment. Dopamine-based therapy has a significant impact on symptoms during the early stages of the disease, with no disease-modifying effect. PD continues to deteriorate after medication and cannot be cured.134 Studies have found that, in addition to progressively worsening dopaminergic neuron degeneration and Lewy bodies formed by α-synuclein accumulation, many key features, and triggers of ferroptosis pathways are also important pathophysiological features of PD.135 This suggests that ferroptosis has a broad prospect in the research and development of neuroprotective drugs for PD.
An important pathological feature of PD is the imbalance of iron homeostasis and accumulation in the substantia nigra caused by alterations in iron regulatory proteins. In a rat model of PD, ferritin heavy chain FTH1 expressed at low levels recruits LC3 and selectively binds to NCOA4 in the autophagosome, inducing ferritin autophagy to further degrade FTH1 and form a loop.136 This eventually leads to free ferrous ions and ROS accumulation, thus inducing cellular ferroptosis. Elevated levels of DMT1 were found to contribute to intracellular iron accumulation and promote neurodegeneration in PD models.137 Subjects with TF gene variants tend to have a significantly reduced risk of PD.138 This suggests that ferroptosis caused by iron accumulation is critical in neuronal degeneration in PD. A clinical randomized controlled trial also reported a reduction in iron accumulation in the substantia nigra and a slowing in the progression of motor dysfunction in PD patients after treatment with the iron chelator deferiprone.139 The decrease in SLC7A11 expression and GSH depletion increase ROS content, induce cell ferroptosis, and promote the degradation of dopaminergic neurons in PD.140,141 Meanwhile, GSH supplementation or activation of Nrf2 pathway is expected to be an effective therapeutic procedure for neuroprotection in PD. There exists a deficiency of CoQ10 in PD patients,142 which may induce ferroptosis by reducing the body’s antioxidant capacity, thereby promoting PD progression. It was shown that Clioquinol treatment significantly improved both motor and non-motor symptoms in a monkey model of Parkinson’s disease, which may act by decreasing iron content in the substantia nigra and inhibiting ferroptosis.143 Targeting ferroptosis may have an important role in slowing or even inhibiting the sustained progression of PD.
Huntington’s Disease
HD is an autosomal dominant neurodegenerative disease caused by the repeated amplification of dominant CAG trinucleotide of Huntington gene on chromosome 4, primarily manifested in dance-like involuntary movement, cognitive impairment, and mental symptoms.144 Although HD has an explicit genetic origin and causes the accumulation of mutant huntingtin protein (mHTT) in neural cells, the specific molecular mechanism causing neuronal death is unknown. Therefore, there is no effective strategy to prevent or delay the process of HD, and only supportive and symptomatic treatments are clinically available. Prior studies have reported that mHTT is expressed in neurons and glial cells and can trigger oxidative stress and increase intracellular ROS levels.145 Ferroptosis manifestations such as iron accumulation and oxidative stress have also been observed in HD patients and model animals.18,146 This suggests that ferroptosis may potentially be involved in neuronal cell death in HD. Decreased GSH expression has been detected in the striatum, cortex, and hippocampus of HD patients and model animals,147,148 and insufficient GSH reduces the body’s antioxidant capacity, causing LPO accumulation and inducing ferroptosis. Quinti et al149 detected an Nrf2 activation signal in neural stem cells of HD patients and demonstrated that this signal inhibited the release of inflammatory factors. It is well-known that Nrf2 activates downstream pathways to play a role in inhibiting ferroptosis, so it is important to further explore Nrf2 downstream ferroptosis-related genes. In addition, Skouta et al150 found that treatment with ferrostatin-1 (Fer-1), a ferroptosis inhibitor, inhibited lipid peroxidation in HD cells and had a significant neuroprotective effect. In summary, an in-depth investigation of the specific mechanism of ferroptosis involved in HD pathogenesis and nerve cell death, and targeting HD with ferroptosis modulators, is a worthy direction of basic and clinical research of HD.
Epilepsy
Epilepsy is a central nervous system dysfunction caused by highly synchronized abnormal discharges of neurons in the brain, which is clinically characterized by repetitive and refractory manifestations due to its diverse etiologies and complex pathogenesis. Recent studies have revealed that ferroptosis is highly associated with epilepsy. On the one hand, stroke and traumatic brain injury are common causes of seizures that may be linked to the breakdown of hemoglobin and abnormal accumulation of iron.151,152 In addition, the up-regulation of 4-HNE and ferritin expression and intracellular iron accumulation have been found in tissues of tubular sclerosis (TS), which is a common cause of drug resistance in epilepsy. These all suggest that abnormal iron metabolism and ferroptosis it causes may play a role in seizures. On the other hand, the dysfunctional GPX4 antioxidant system may also be an important pathogenesis of epilepsy. During seizures, the brain often has a high expression of extracellular glutamate, which is also among the causes of recurrent seizures. High extracellular glutamate levels can inhibit system Xc- and reduce GSH synthesis, thereby inducing ferroptosis.153 GPX4 expression was decreased, and lipid peroxidation appeared in the hippocampus of epileptic model rats, which could be reversed by Fer-1.154 Alternatively, related studies suggest that vatiquinone can modulate ferroptosis by targeting GPX4 and 15-lipoxygenase. Vatiquinone effectively prevented cellular ferroptosis from patients with epilepsy.155 In summary, ferroptosis inhibition may be an effective treatment for patients with epilepsy and warrants further investigation.
Glioma
Glioma, especially glioblastoma (GBM), is the most common primary malignancy in the central nervous system, with complex drug resistance and survival mechanisms and high patient mortality.156 Cancer cells require more iron than normal tissue to support the strong energy demands imposed by unlimited proliferation. This cannot avoid significantly expanding the LIP of cancer cells and allows cancer cells to become more dependent on cellular defense mechanisms to protect against ROS and ferroptosis.157 Prior studies have reported that the reason for poor prognosis and strong drug resistance of gliomas may be that these tumors can effectively avoid ferroptosis. Gliomas exhibit metabolically-adaptive glutamine addiction158 that produces large amounts of glutamate and promotes system Xc- to transport cystine, thereby improving ferroptosis resistance. ACSL4 is down-regulated in human glioma tissues and cells, reducing lipid peroxidation and ferroptosis.159 MiR-670-3p can then inhibit human glioblastoma ferroptosis by downregulating ACSL4 expression.160 Additionally, GPX4 and FSP1, key peroxidation inhibitors of ferroptosis, are highly expressed in gliomas, inhibiting cellular ferroptosis and promoting glioma survival.161
Targeting and regulating key proteins of ferroptosis to induce ferroptosis would potentially inhibit glioma cell proliferation. Knockdown of COPZ1 can up-regulate NCOA4, induce ferritin degradation, increase intracellular ferrous ion levels and lipid ROS accumulation, and ultimately lead to ferroptosis.34 PAB increases intracellular iron content by up-regulating TFRC, leading to the overproduction of H2O2 and lipid peroxides, which induces ferroptosis. Meanwhile, PAB can reduce intracellular GSH levels through the p53-mediated system Xc- pathway and further exacerbate ferroptosis in glioma cells.37 Iron oxide nanoparticles (IONPs) target glioblastoma and significantly increase ionic iron and ROS content, followed by the initiation of ferroptosis for local chemotherapy. The co-released si-GPX4 in nanoparticles can also play a synergistic therapeutic effect by inhibiting the expression of GPX4 and triggering ferroptosis.162 Inducing ferroptosis in early glioma cells can stimulate the release of DAMPs such as ATP and HMGB1 to promote the activation and maturation of bone marrow-derived dendritic cells (BMDCs), initiating adaptive immune responses, and injecting early ferroptosis cells induced in vitro into model mice, can have a similar vaccine effect.163 This provides us with new ideas for immunotherapy of tumors. In addition, ferroptosis-inducing agents are expected to synergize with anti-PD-1 antibodies for the immunotherapy of GBM and increase the sensitivity of tumor cells to immunotherapy to counteract the powerful immunosuppressive and immune evasion properties of GBM.164 The induction of ferroptosis can also reduce drug resistance in gliomas. Protein inhibitors such as GPX4 and SLC7A11 can reverse the resistance of Temozolomide (TMZ, the current first-line treatment for glioblastoma) by inducing ferroptosis and synergistically be used to treat glioma patients.165
Interestingly, recent studies have revealed that neutrophils can regulate the expression of genes such as GPX4, ACSL4 by transferring myeloperoxidase (MPO) into tumor cells, which in turn induces iron-dependent accumulation of lipid peroxides and ferroptosis in tumor cells. This pro-tumor cellular ferroptosis effect may be involved in and promote late GBM necrosis and enhance tumor aggressiveness causing advanced GBM necrosis and enhancing tumor invasiveness.15 Therefore, ferroptosis modulators have a positive therapeutic effect on gliomas during various stages, and targeting ferroptosis may provide a new direction for glioma therapy.
Strokes
Strokes are among the leading cause of death and disability worldwide, including ischemic strokes and hemorrhagic strokes.166 To date, the exact mechanism that leads to neuronal cell death in strokes remains unclear. There are great challenges in preventing and rescuing neuronal cell death in strokes. Therefore, elucidating the role of ferroptosis in neuronal cell death in strokes is vital for etiological research and drug development.
Ischemic stroke is a clinical syndrome in which hypoxic-ischemic damage of brain tissue and a series of neurological deficits occur due to various cerebrovascular lesions and cerebral blood supply disorder, accounting for approximately 85% of all stroke cases.167 Ischemic brain tissue can be functionally divided into the central necrotic areas and the surrounding ischemic penumbra. Saving the ischemic penumbra and avoiding or alleviating primary brain injury are the treatment priorities for ischemic strokes. However, the exact mechanism of neuronal death in ischemic strokes remains unclear. The acidic environment of brain tissue after cerebral ischemia has been found to reduce the pH-dependent affinity of TF for iron, leading to iron release from TF. Unbound iron can be easily absorbed by neurons, causing iron accumulation, leading to ferroptosis.19 Multiple iron chelates or iron export compounds have indicated neuroprotective effects in ischemic strokes models,168,169 supporting iron accumulation as a therapeutic target in ischemic strokes. In addition, GSH, SLC7A11, and GPX4 levels were significantly reduced in ischemic brain tissue of ischemic stroke model rats, while cysteine supplementation to restore depleted GSH levels or administration of ferroptosis inhibitors (ferrostatin-1 or liproxstatin-1) significantly reduced neuronal injuries after ischemic strokes.168,170,171 CoQ10 deficiency is widespread in stroke patients taking statins for a lengthy duration, while CoQ10 supplementation was effective in improving functional deficits and reducing cerebral infarction volume in rats after ischemic strokes.172 This may be associated with regulating inflammation, degrading ROS, and inhibiting ferroptosis. The above studies indicate that ferroptosis plays a vital role in neuronal death after ischemic strokes, and it is important to identify appropriate ferroptosis inhibitors to protect ischemic penumbra neurons.
Endothelial cells, astrocytes, pericytes, and neurons constitute the neurovascular unit (NVU) and play an important role in maintaining the structural and functional integrity of the blood-brain barrier (BBB).173 BBB dysfunction is among the important mechanisms in the development of hemorrhagic strokes. After this type of stroke, various cells constituting NVU are associated with iron metabolism disorders and ROS accumulation.174 This suggests a significant association between ferroptosis and hemorrhagic strokes. Further studies revealed that vascular rupture causes blood spillage and damage to red blood cells and hemoglobin in hemorrhagic strokes, releasing large amounts of iron and ROS and inducing neuronal ferroptosis. After Fer-1 treatment, hemoglobin-induced iron accumulation and ROS production were significantly reduced, and neural cells were protected.175,176 Targeting GPX4 also showed significant neuroprotection. The upregulation of GPX4 and the inhibition of ferroptosis can significantly alleviate BBB injury, oxidative stress, and inflammation after intracerebral hemorrhage (ICH) and restore its neuronal function.177 Treatment with the lipophilic iron chelator pyridoxal isonicotinoyl hydrazine (PIH) can up-regulate GPX4 and down-regulate epoxylyase-2, IL-1β, and TNF-α to reduce ROS production and iron accumulation in perihematomal tissues, and inhibit lipid peroxidation and inflammation, thereby reducing cellular ferroptosis and brain tissue damage after ICH.178 In addition, overexpression of miR-137 may reduce hemoglobin-induced apoptosis and mitochondrial dysfunction by inhibiting ferroptosis through the miR-137-COX2/PGE2 signaling pathway.179 On the other hand, N-acetylcysteine can reduce neuronal ferroptosis and neurological dysfunction after ICH by reducing toxic ROS produced catalyzed by ALOX5. This can take effect synergistically with clinically approved prostaglandin E2 (PGE2).180 Recent studies have reported that selenium (Se) adaptively enhances transcription to protect cells from ferroptosis after hemorrhagic strokes. However, selenium supplements can drive many selenoprotein transcriptions and synergistically activate transcription factors TFAP2c and Sp1 to up-regulate GPX4, inhibit cell ferroptosis, and improve hemorrhagic stroke symptoms. Different doses of selenium supplements only show strong or weak effects without evident toxicity, which suggests that selenium supplements are effective and relatively safe. Therefore, targeted ferroptosis therapy is promising for hemorrhagic strokes.
Traumatic Brain Injury
TBI typically originates from primary injury, attributable to brain trauma, which gradually develops into secondary injury. This releases glutamate, ROS, and Fe2+, inducing mitochondrial injury and inflammatory response and resulting in temporary or permanent neurological deficits.181,182 The primary features of ferroptosis are iron overload and ROS accumulation. Ferroptosis may thus play a prominent role in inducing cell death and secondary injury of TBI, which is considered a significant cause of high mortality and poor prognosis of TBI.183 Xie et al20 found that ferroptosis manifestations such as iron accumulation, decreased GPX4 activity, and LPO accumulation occurred in a controlled cortical impact (CCI) mouse TBI model. Transmission electron microscopy confirmed the presence of mitochondrial contraction, a typical feature of ferroptosis. Intraventricular injection of Fer-1 significantly reduced iron accumulation and neuronal degeneration while improving long-term motor and cognitive functionality. As the primary form of intracellular iron storage, ferritin is composed of heavy chain subunit (FTH) and light chain subunit (FTL). FTH comprises ferroxidase activity, converting Fe2+ to Fe3+, while FTL stores excess iron by nucleation uptake. Rui et al184 found that melatonin inhibited iron accumulation and the expression of ferroptosis-related proteins such as COX2 after TBI to rescue neuronal ferroptosis and improve neurological deficits. Melatonin acts in part by inhibiting neuronal ferritin H. In addition, the up-regulation of lipid metabolism-related proteins such as 15-LOX and ACSL4 was also observed in the CCI model. This allows the abundant presence of AA/AdA in the brain to be oxidized, leading to lipid peroxidation injuries and inducing ferroptosis.185 Recent studies have also confirmed that polydatin can exert long-term neuroprotective effects and significantly improve subacute motor dysfunction in TBI by maintaining GPX4 activity in nerve cells and inhibiting neuronal ferroptosis.183 Ferroptosis may be an important therapeutic target for TBI patients; targeting this may significantly improve TBI patient outcomes.
Conclusion and Outlook
Ferroptosis is primarily caused by a disruption in the body’s oxidative balance. Disturbances in iron and lipid metabolism cause excessive accumulation of LPO within the lipid bilayer, causing oxidative destruction of the membrane. Disruption of antioxidant systems such as GPX4, FSP1, and DHODH result in the inability to timely remove LPO continuously produced in the cell, ultimately leading to the massive destruction of the lipid bilayer membrane and cell death. However, the exact cause of cell death in ferroptosis is not clear. With the ongoing progress of ferroptosis research, numerous regulators and pathophysiological phenomena such as Nrf2, p53, and autophagy are closely linked to ferroptosis. The occurrence and regulatory network of ferroptosis are constantly improving, laying the foundation for ferroptosis to serve in clinics.
Ferroptosis and its related genes may be involved in a series of processes of maturation, functional maintenance, and aging of the nervous system. They may also play an important role in neurodegenerative diseases, strokes, epilepsy, brain tumors, as well as other neurological diseases with unclear mechanisms and poor prognosis. Therefore, attempts to detect ferroptosis and its related genes may have a significant role in the early diagnosis and prevention of neurological dysplasia or congenital functional defects. Further studies of the role of ferroptosis in neurological diseases may reveal part of the pathogenesis of complex refractory diseases such as AD, PD, and epilepsy and provide novel ideas for the development of new drugs to slow or even reverse the progression of the mentioned diseases. Exploring the occurrence and development of ferroptosis in brain tumors and attempting to induce tumor cell death with ferroptosis inducers are expected to cooperate with traditional tumor therapy and immunotherapy in the treatment of GBM and other refractory tumors and drug-resistant tumors to prolong the life of tumor patients and improve their overall quality of life. However, in the context of glioma, by promoting ferroptosis to kill tumor cells, whether it is possible to damage nerve cells at the same time, aggravating the original combined neurodegenerative diseases, stroke and other neurological diseases or leading to similar symptoms in glioma patients without original related complications. This conundrum needs further study. In conclusion, exploring the underlying mechanisms of ferroptosis and effective and specific ferroptosis regulators is a worthy direction for future research on neurological diseases.
Consent for Publication
All the authors read the manuscript carefully and gave their consent for publication.
Acknowledgments
Wei Lin and Yiyang Zhou are co-first authors for this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Wenzhou Science and Technology Plan Project (Y20210008).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kane AB. Redefining cell death. Am J Pathol. 1995;146(1):1–2.
2. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
3. Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22(1):58–73. doi:10.1038/cdd.2014.137
4. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742–758. doi:10.1016/j.cell.2011.10.033
5. Galluzzi L, Bravo-San Pedro JM, Kepp O, et al. Regulated cell death and adaptive stress responses. Cell Mol Life Sci. 2016;73(11–12):2405–2410. doi:10.1007/s00018-016-2209-y
6. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
7. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–147. doi:10.1038/nrm3737
8. Chen X, Yu C, Kang R, et al. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226.
9. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30(6):478–490.
10. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi:10.1038/nchembio.2238
11. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
12. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
13. Do Van B, Gouel F, Jonneaux A, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi:10.1016/j.nbd.2016.05.011
14. Alim I, Caulfield JT, Chen Y, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. 2019;177(5):1262–1279 e25. doi:10.1016/j.cell.2019.03.032
15. Yee PP, Wei Y, Kim SY, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun. 2020;11(1):5424. doi:10.1038/s41467-020-19193-y
16. Yan N, Zhang J. Iron metabolism, ferroptosis, and the links with Alzheimer’s disease. Front Neurosci. 2019;13:1443. doi:10.3389/fnins.2019.01443
17. Belaidi AA, Bush AI. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem. 2016;139(Suppl 1):179–197. doi:10.1111/jnc.13425
18. Dominguez JF, Ng AC, Poudel G, et al. Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry. 2016;87(5):545–549. doi:10.1136/jnnp-2014-310183
19. Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. 2022;42(1):259–305.
20. Xie BS, Wang YQ, Lin Y, et al. Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther. 2019;25(4):465–475. doi:10.1111/cns.13069
21. Chen S, Chen Y, Zhang Y, et al. Iron metabolism and ferroptosis in epilepsy. Front Neurosci. 2020;14:601193. doi:10.3389/fnins.2020.601193
22. Kuang F, Liu J, Tang D, et al. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. doi:10.3389/fcell.2020.586578
23. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
24. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
25. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
26. Wang CY, Babitt JL. Liver iron sensing and body iron homeostasis. Blood. 2019;133(1):18–29. doi:10.1182/blood-2018-06-815894
27. Gozzelino R, Arosio P. Iron homeostasis in health and disease. Int J Mol Sci. 2016;17(1):130. doi:10.3390/ijms17010130
28. Muckenthaler MU, Rivella S, Hentze MW, et al. A red carpet for iron metabolism. Cell. 2017;168(3):344–361. doi:10.1016/j.cell.2016.12.034
29. Aschemeyer S, Qiao B, Stefanova D, et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood. 2018;131(8):899–910. doi:10.1182/blood-2017-05-786590
30. Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1(3):155–157. doi:10.1016/j.cmet.2005.02.005
31. Ohgami RS, Campagna DR, Greer EL, et al. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet. 2005;37(11):1264–1269. doi:10.1038/ng1658
32. Fleming MD, Romano MA, Su MA, et al. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proc Natl Acad Sci U S A. 1998;95(3):1148–1153. doi:10.1073/pnas.95.3.1148
33. Wang Y, Liu Y, Liu J, et al. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531(4):581–587. doi:10.1016/j.bbrc.2020.07.032
34. Zhang Y, Kong Y, Ma Y, et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene. 2021;40(8):1425–1439. doi:10.1038/s41388-020-01622-3
35. Devireddy LR, Gazin C, Zhu X, et al. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123(7):1293–1305. doi:10.1016/j.cell.2005.10.027
36. McCarthy RC, Sosa JC, Gardeck AM, et al. Inflammation-induced iron transport and metabolism by brain microglia. J Biol Chem. 2018;293(20):7853–7863. doi:10.1074/jbc.RA118.001949
37. Wang Z, Ding Y, Wang X, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21–33. doi:10.1016/j.canlet.2018.04.021
38. Santana-Codina N, Mancias JD. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals (Basel). 2018;11(4):114. doi:10.3390/ph11040114
39. Wang YQ, Chang SY, Wu Q, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308. doi:10.3389/fnagi.2016.00308
40. Bao WD, Zhou XT, Zhou LT, et al. Targeting miR-124/Ferroportin signaling ameliorated neuronal cell death through inhibiting apoptosis and ferroptosis in aged intracerebral hemorrhage murine model. Aging Cell. 2020;19(11):e13235. doi:10.1111/acel.13235
41. Ayala A, Munoz MF, Arguelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi:10.1155/2014/360438
42. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944–5972. doi:10.1021/cr200084z
43. Magtanong L, Ko PJ, To M, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26(3):420–432 e9. doi:10.1016/j.chembiol.2018.11.016
44. Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–75. doi:10.1073/pnas.1603244113
45. Esterbauer H, Eckl P, Ortner A. Possible mutagens derived from lipids and lipid precursors. Mutat Res. 1990;238(3):223–233. doi:10.1016/0165-1110(90)90014-3
46. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628–641 e26. doi:10.1016/j.cell.2017.09.044
47. Chen JJ, Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol. 2018;28(2):77–78. doi:10.1016/j.tcb.2017.11.007
48. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
49. Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289–3303. doi:10.1016/j.bbagen.2012.11.020
50. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. 2019;19(18):e1800311. doi:10.1002/pmic.201800311
51. Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503. doi:10.1038/nchembio.2079
52. Gaschler MM, Andia AA, Liu H, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14(5):507–515. doi:10.1038/s41589-018-0031-6
53. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi:10.7554/eLife.02523
54. Mandal PK, Seiler A, Perisic T, et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. 2010;285(29):22244–22253. doi:10.1074/jbc.M110.121327
55. Francis Stuart SD, Villalobos AR. GSH and zinc supplementation attenuate cadmium-induced cellular stress and stimulation of choline uptake in cultured neonatal rat choroid plexus epithelia. Int J Mol Sci. 2021;22(16):8857. doi:10.3390/ijms22168857
56. Hayano M, Yang WS, Corn CK, et al. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278. doi:10.1038/cdd.2015.93
57. Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–1704. doi:10.1016/j.celrep.2017.07.055
58. Kryukov GV, Castellano S, Novoselov SV, et al. Characterization of mammalian selenoproteomes. Science. 2003;300(5624):1439–1443. doi:10.1126/science.1083516
59. Warner GJ, Berry MJ, Moustafa ME, et al. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J Biol Chem. 2000;275(36):28110–28119. doi:10.1074/jbc.M001280200
60. Wu M, Xu LG, Li X, et al. AMID, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem. 2002;277(28):25617–25623. doi:10.1074/jbc.M202285200
61. Gong M, Hay S, Marshall KR, et al. DNA binding suppresses human AIF-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J Biol Chem. 2007;282(41):30331–30340. doi:10.1074/jbc.M703713200
62. Chen L, Xie J. Ferroptosis-suppressor-protein 1: a potential neuroprotective target for combating ferroptosis. Mov Disord. 2020;35(3):400. doi:10.1002/mds.27990
63. Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
64. Vasan K, Werner M, Chandel NS. Mitochondrial metabolism as a target for cancer therapy. Cell Metab. 2020;32(3):341–352. doi:10.1016/j.cmet.2020.06.019
65. Hakkaart GA, Dassa EP, Jacobs HT, et al. Allotopic expression of a mitochondrial alternative oxidase confers cyanide resistance to human cell respiration. EMBO Rep. 2006;7(3):341–345. doi:10.1038/sj.embor.7400601
66. Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020;260:118305. doi:10.1016/j.lfs.2020.118305
67. Xiao X, Jiang Y, Liang W, et al. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol Brain. 2019;12(1):78. doi:10.1186/s13041-019-0501-0
68. LaFave LM, Beguelin W, Koche R, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21(11):1344–1349. doi:10.1038/nm.3947
69. Zhang Y, Shi J, Liu X, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–1192. doi:10.1038/s41556-018-0178-0
70. Zhang X, Du L, Qiao Y, et al. Ferroptosis is governed by differential regulation of transcription in liver cancer. Redox Biol. 2019;24:101211. doi:10.1016/j.redox.2019.101211
71. Hassannia B, Wiernicki B, Ingold I, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest. 2018;128(8):3341–3355.
72. Fillebeen C, Charlebois E, Wagner J, et al. Transferrin receptor 1 controls systemic iron homeostasis by fine-tuning hepcidin expression to hepatocellular iron load. Blood. 2019;133(4):344–355.
73. Brissot P, Pietrangelo A, Adams PC, et al. Haemochromatosis. Nat Rev Dis Primers. 2018;4:18016.
74. Liu T, Jiang L, Tavana O, et al. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79(8):1913–1924.
75. Li F, Cui L, Yu D, et al. Exogenous glutathione improves intracellular glutathione synthesis via the gamma-glutamyl cycle in bovine zygotes and cleavage embryos. J Cell Physiol. 2019;234(5):7384–7394.
76. Lu K, Alcivar AL, Ma J, et al. NRF2 induction supporting breast cancer cell survival is enabled by oxidative stress-induced DPP3-KEAP1 interaction. Cancer Res. 2017;77(11):2881–2892.
77. Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34(6):340–346.
78. Carpi-Santos R, Calaza KC. Alterations in system xc(-) expression in the retina of type 1 diabetic rats and the role of Nrf2. Mol Neurobiol. 2018;55(10):7941–7948.
79. Cao JY, Poddar A, Magtanong L, et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 2019;26(6):1544–1556 e8.
80. Selvakumar GP, Ahmed ME, Raikwar SP, et al. CRISPR/Cas9 editing of Glia maturation factor regulates mitochondrial dynamics by attenuation of the NRF2/HO-1 dependent ferritin activation in Glial cells. J Neuroimmune Pharmacol. 2019;14(4):537–550.
81. Liu Z, Lv X, Song E, et al. Fostered Nrf2 expression antagonizes iron overload and glutathione depletion to promote resistance of neuron-like cells to ferroptosis. Toxicol Appl Pharmacol. 2020;407:115241.
82. Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–1078.
83. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405.
84. Zhao H, Li X, Yang L, et al. Isorhynchophylline relieves ferroptosis-induced nerve damage after intracerebral hemorrhage via miR-122-5p/TP53/SLC7A11 Pathway. Neurochem Res. 2021;46(8):1981–1994.
85. Ou Y, Wang SJ, Li D, et al. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–E6812.
86. Tarangelo A, Magtanong L, Bieging-Rolett KT, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–575.
87. Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345(6203):1250256.
88. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262.
89. Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26(3):190–201.
90. Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234.
91. Li C, Dong X, Du W, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther. 2020;5(1):187.
92. Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28(15):2388–2399 e5.
93. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388.
94. Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109.
95. Liang Y, Deng Y, Zhao J, et al. Ferritinophagy is involved in experimental subarachnoid hemorrhage-induced neuronal ferroptosis. Neurochem Res. 2022;47:692–700.
96. Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996–3005.
97. Kang HJ, Kawasawa YI, Cheng F, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478(7370):483–489.
98. Miller JA, Ding SL, Sunkin SM, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508(7495):199–206.
99. Werling DM, Pochareddy S, Choi J, et al. Whole-genome and RNA sequencing reveal variation and transcriptomic coordination in the developing human prefrontal cortex. Cell Rep. 2020;31(1):107489.
100. Kim SW, Kim Y, Kim SE, et al. Ferroptosis-related genes in neurodevelopment and central nervous system. Biology (Basel). 2021;10(1):35.
101. Wang Y, Wu Y, Li T, et al. Iron metabolism and brain development in premature infants. Front Physiol. 2019;10:463. doi:10.3389/fphys.2019.00463
102. Yu P, Chang YZ. Brain iron metabolism and regulation. Adv Exp Med Biol. 2019;1173:33–44.
103. Poitelon Y, Lopez-Anido C, Catignas K, et al. YAP and TAZ control peripheral myelination and the expression of laminin receptors in Schwann cells. Nat Neurosci. 2016;19(7):879–887. doi:10.1038/nn.4316
104. Liddell JR. Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants (Basel). 2017;6(3):65.
105. Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687–702. doi:10.1016/j.neuron.2011.05.001
106. Clelland CD, Choi M, Romberg C, et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325(5937):210–213. doi:10.1126/science.1173215
107. Ferreira AC, Santos T, Sampaio-Marques B, et al. Lipocalin-2 regulates adult neurogenesis and contextual discriminative behaviours. Mol Psychiatry. 2018;23(4):1031–1039. doi:10.1038/mp.2017.95
108. Toyokuni S, Yanatori I, Kong Y, et al. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci. 2020;111(8):2665–2671. doi:10.1111/cas.14496
109. Mazhar M, Din AU, Ali H, et al. Implication of ferroptosis in aging. Cell Death Discov. 2021;7(1):149. doi:10.1038/s41420-021-00553-6
110. Zhou RP, Chen Y, Wei X, et al. Novel insights into ferroptosis: implications for age-related diseases. Theranostics. 2020;10(26):11976–11997. doi:10.7150/thno.50663
111. Bartzokis G, Tishler TA, Lu PH, et al. Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol Aging. 2007;28(3):414–423. doi:10.1016/j.neurobiolaging.2006.02.005
112. Currais A, Maher P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid Redox Signal. 2013;19(8):813–822. doi:10.1089/ars.2012.4996
113. Olmez I, Ozyurt H. Reactive oxygen species and ischemic cerebrovascular disease. Neurochem Int. 2012;60(2):208–212. doi:10.1016/j.neuint.2011.11.009
114. Larrieu T, Laye S. Food for mood: relevance of nutritional omega-3 fatty acids for depression and anxiety. Front Physiol. 2018;9:1047. doi:10.3389/fphys.2018.01047
115. Weiland A, Wang Y, Wu W, et al. Ferroptosis and its role in diverse brain diseases. Mol Neurobiol. 2019;56(7):4880–4893. doi:10.1007/s12035-018-1403-3
116. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. doi:10.1111/ene.13439
117. Hodson R. Alzheimer’s disease. Nature. 2018;559(7715):S1. doi:10.1038/d41586-018-05717-6
118. Goodman L. Alzheimer’s disease; a clinico-pathologic analysis of twenty-three cases with a theory on pathogenesis. J Nerv Ment Dis. 1953;118(2):97–130. doi:10.1097/00005053-195308000-00001
119. Smith MA, Harris PL, Sayre LM, et al. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94(18):9866–9868. doi:10.1073/pnas.94.18.9866
120. Ashraf A, Jeandriens J, Parkes HG, et al. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: evidence of ferroptosis. Redox Biol. 2020;32:101494. doi:10.1016/j.redox.2020.101494
121. Quintana C, Bellefqih S, Laval JY, et al. Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol. 2006;153(1):42–54. doi:10.1016/j.jsb.2005.11.001
122. Ayton S, Portbury S, Kalinowski P, et al. Regional brain iron associated with deterioration in Alzheimer’s disease: a large cohort study and theoretical significance. Alzheimers Dement. 2021;17(7):1244–1256. doi:10.1002/alz.12282
123. Bao WD, Pang P, Zhou XT, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021;28(5):1548–1562. doi:10.1038/s41418-020-00685-9
124. Takahashi RH, Nagao T, Gouras GK. Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol Int. 2017;67(4):185–193. doi:10.1111/pin.12520
125. Bayer TA, Wirths O. Intracellular accumulation of amyloid-Beta - a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front Aging Neurosci. 2010;2:8. doi:10.3389/fnagi.2010.00008
126. Huang L, McClatchy DB, Maher P, et al. Intracellular amyloid toxicity induces oxytosis/ferroptosis regulated cell death. Cell Death Dis. 2020;11(10):828. doi:10.1038/s41419-020-03020-9
127. Gao Y, Li J, Wu Q, et al. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int Immunopharmacol. 2021;99:108002. doi:10.1016/j.intimp.2021.108002
128. Liu Y, Chen Z, Li B, et al. Supplementation with gamma-glutamylcysteine (gamma-GC) lessens oxidative stress, brain inflammation and amyloid pathology and improves spatial memory in a murine model of AD. Neurochem Int. 2021;144:104931. doi:10.1016/j.neuint.2020.104931
129. Agrawal M, Saraf S, Saraf S, et al. Nose-to-brain drug delivery: an update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J Control Release. 2018;281:139–177. doi:10.1016/j.jconrel.2018.05.011
130. Crapper McLachlan DR, Dalton AJ, Kruck TP, et al. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337(8753):1304–1308. doi:10.1016/0140-6736(91)92978-B
131. Zhang YH, Wang DW, Xu SF, et al. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol. 2018;14:535–548. doi:10.1016/j.redox.2017.11.001
132. Shao L, Dong C, Geng D, et al. Ginkgolide B protects against cognitive impairment in senescence-accelerated P8 mice by mitigating oxidative stress, inflammation and ferroptosis. Biochem Biophys Res Commun. 2021;572:7–14. doi:10.1016/j.bbrc.2021.07.081
133. Zhu ZY, Liu YD, Gong Y, et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol Sin. 2022;43(1):39–49.
134. Verschuur CVM, Suwijn SR, Boel JA, et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s disease. N Engl J Med. 2019;380(4):315–324. doi:10.1056/NEJMoa1809983
135. Mahoney-Sanchez L, Bouchaoui H, Ayton S, et al. Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog Neurobiol. 2021;196:101890. doi:10.1016/j.pneurobio.2020.101890
136. Tian Y, Lu J, Hao X, et al. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson’s disease. Neurotherapeutics. 2020;17(4):1796–1812. doi:10.1007/s13311-020-00929-z
137. Bi M, Du X, Jiao Q, et al. Alpha-synuclein regulates iron homeostasis via preventing Parkin-mediated DMT1 ubiquitylation in Parkinson’s disease models. ACS Chem Neurosci. 2020;11(11):1682–1691. doi:10.1021/acschemneuro.0c00196
138. Rhodes SL, Buchanan DD, Ahmed I, et al. Pooled analysis of iron-related genes in Parkinson’s disease: association with transferrin. Neurobiol Dis. 2014;62:172–178. doi:10.1016/j.nbd.2013.09.019
139. Devos D, Moreau C, Devedjian JC, et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal. 2014;21(2):195–210. doi:10.1089/ars.2013.5593
140. Vallerga CL, Zhang F, Fowdar J, et al. Analysis of DNA methylation associates the cystine-glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat Commun. 2020;11(1):1238. doi:10.1038/s41467-020-15065-7
141. Asanuma M, Miyazaki I. Glutathione and related molecules in Parkinsonism. Int J Mol Sci. 2021;22(16):8689. doi:10.3390/ijms22168689
142. Mischley LK, Allen J, Bradley R. Coenzyme Q10 deficiency in patients with Parkinson’s disease. J Neurol Sci. 2012;318(1–2):72–75. doi:10.1016/j.jns.2012.03.023
143. Shi L, Huang C, Luo Q, et al. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging (Albany NY). 2020;12(10):9515–9533. doi:10.18632/aging.103225
144. McColgan P, Tabrizi SJ. Huntington’s disease: a clinical review. Eur J Neurol. 2018;25(1):24–34. doi:10.1111/ene.13413
145. Hands S, Sajjad MU, Newton MJ, et al. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J Biol Chem. 2011;286(52):44512–44520. doi:10.1074/jbc.M111.307587
146. Pinho BR, Duarte AI, Canas PM, et al. The interplay between redox signalling and proteostasis in neurodegeneration: in vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic Biol Med. 2020;146:372–382. doi:10.1016/j.freeradbiomed.2019.11.021
147. Klepac N, Relja M, Klepac R, et al. Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: a cross-sectional study. J Neurol. 2007;254(12):1676–1683. doi:10.1007/s00415-007-0611-y
148. Kumar P, Kalonia H, Kumar A. Nitric oxide mechanism in the protective effect of antidepressants against 3-nitropropionic acid-induced cognitive deficit, glutathione and mitochondrial alterations in animal model of Huntington’s disease. Behav Pharmacol. 2010;21(3):217–230. doi:10.1097/FBP.0b013e32833a5bf4
149. Quinti L, Dayalan Naidu S, Trager U, et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc Natl Acad Sci U S A. 2017;114(23):E4676–E4685. doi:10.1073/pnas.1614943114
150. Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–4556. doi:10.1021/ja411006a
151. Zelano J, Holtkamp M, Agarwal N, et al. How to diagnose and treat post-stroke seizures and epilepsy. Epileptic Disord. 2020;22(3):252–263. doi:10.1684/epd.2020.1159
152. van Vliet EA, Ndode-Ekane XE, Lehto LJ, et al. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol Dis. 2020;145:105080. doi:10.1016/j.nbd.2020.105080
153. Fricker M, Tolkovsky AM, Borutaite V, et al. Neuronal cell death. Physiol Rev. 2018;98(2):813–880. doi:10.1152/physrev.00011.2017
154. Mao XY, Zhou HH, Jin WL. Ferroptosis induction in pentylenetetrazole kindling and pilocarpine-induced epileptic seizures in mice. Front Neurosci. 2019;13:721. doi:10.3389/fnins.2019.00721
155. Kahn-Kirby AH, Amagata A, Maeder CI, et al. Targeting ferroptosis: a novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS One. 2019;14(3):e0214250. doi:10.1371/journal.pone.0214250
156. Rasmussen BK, Hansen S, Laursen RJ, et al. Epidemiology of glioma: clinical characteristics, symptoms, and predictors of glioma patients grade I-IV in the the Danish Neuro-Oncology Registry. J Neurooncol. 2017;135(3):571–579. doi:10.1007/s11060-017-2607-5
157. Zhao N, Huang Y, Wang YH, et al. Ferronostics: measuring tumoral ferrous iron with PET to predict sensitivity to iron-targeted cancer therapies. J Nucl Med. 2021;62(7):949–955. doi:10.2967/jnumed.120.252460
158. Obara-Michlewska M, Szeliga M. Targeting glutamine addiction in gliomas. Cancers (Basel). 2020;12(2):310. doi:10.3390/cancers12020310
159. Cheng J, Fan YQ, Liu BH, et al. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. 2020;43(1):147–158. doi:10.3892/or.2019.7419
160. Bao C, Zhang J, Xian SY, et al. MicroRNA-670-3p suppresses ferroptosis of human glioblastoma cells through targeting ACSL4. Free Radic Res. 2021;55(7):853–864. doi:10.1080/10715762.2021.1962009
161. Huang R, Dong R, Wang N, et al. Adaptive changes allow targeting of ferroptosis for glioma treatment. Cell Mol Neurobiol. 2021. doi:10.1007/s10571-021-01092-5
162. Zhang Y, Fu X, Jia J, et al. Glioblastoma therapy using codelivery of cisplatin and glutathione peroxidase targeting siRNA from iron oxide nanoparticles. ACS Appl Mater Interfaces. 2020;12(39):43408–43421. doi:10.1021/acsami.0c12042
163. Efimova I, Catanzaro E, Van der Meeren L, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer. 2020;8(2):e001369. doi:10.1136/jitc-2020-001369
164. Zhuo S, Chen Z, Yang Y, et al. Clinical and biological significances of a ferroptosis-related gene signature in glioma. Front Oncol. 2020;10:590861. doi:10.3389/fonc.2020.590861
165. Hu Z, Mi Y, Qian H, et al. A potential mechanism of temozolomide resistance in glioma-ferroptosis. Front Oncol. 2020;10:897. doi:10.3389/fonc.2020.00897
166. Campbell BCV, Khatri P. Stroke. Lancet. 2020;396(10244):129–142. doi:10.1016/S0140-6736(20)31179-X
167. Feigin VL, Nguyen G, Cercy K, et al.; Collaborators, G.B.D.L.R.o.S. Global, regional, and country-specific lifetime risks of stroke, 1990 and 2016. N Engl J Med. 2018;379(25):2429–2437.
168. Tuo QZ, Lei P, Jackman KA, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017;22(11):1520–1530. doi:10.1038/mp.2017.171
169. Hanafy KA, Gomes JA, Selim M. Rationale and current evidence for testing iron chelators for treating stroke. Curr Cardiol Rep. 2019;21(4):20. doi:10.1007/s11886-019-1106-z
170. Liu Y, Min JW, Feng S, et al. Therapeutic role of a cysteine precursor, OTC, in ischemic stroke is mediated by improved proteostasis in mice. Transl Stroke Res. 2020;11(1):147–160. doi:10.1007/s12975-019-00707-w
171. Lan B, Ge JW, Cheng SW, et al. Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J Integr Med. 2020;18(4):344–350. doi:10.1016/j.joim.2020.01.008
172. Nasoohi S, Simani L, Khodagholi F, et al. Coenzyme Q10 supplementation improves acute outcomes of stroke in rats pretreated with atorvastatin. Nutr Neurosci. 2019;22(4):264–272. doi:10.1080/1028415X.2017.1376928
173. Jiang X, Andjelkovic AV, Zhu L, et al. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog Neurobiol. 2018;163–164:144–171. doi:10.1016/j.pneurobio.2017.10.001
174. Fang Y, Gao S, Wang X, et al. Programmed cell deaths and potential crosstalk with blood-brain barrier dysfunction after hemorrhagic stroke. Front Cell Neurosci. 2020;14:68. doi:10.3389/fncel.2020.00068
175. Zille M, Karuppagounder SS, Chen Y, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48(4):1033–1043. doi:10.1161/STROKEAHA.116.015609
176. Li Q, Han X, Lan X, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2(7):e90777. doi:10.1172/jci.insight.90777
177. Zhang Z, Wu Y, Yuan S, et al. Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 2018;1701:112–125. doi:10.1016/j.brainres.2018.09.012
178. Zhang H, Wen M, Chen J, et al. Pyridoxal isonicotinoyl hydrazone improves neurological recovery by attenuating ferroptosis and inflammation in cerebral hemorrhagic mice. Biomed Res Int. 2021;2021:9916328. doi:10.1155/2021/9916328
179. Li Y, Wang J, Chen S, et al. miR-137 boosts the neuroprotective effect of endothelial progenitor cell-derived exosomes in oxyhemoglobin-treated SH-SY5Y cells partially via COX2/PGE2 pathway. Stem Cell Res Ther. 2020;11(1):330. doi:10.1186/s13287-020-01836-y
180. Karuppagounder SS, Alin L, Chen Y, et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84(6):854–872. doi:10.1002/ana.25356
181. Hu X, Chen H, Xu H, et al. Role of pyroptosis in traumatic brain and spinal cord injuries. Int J Biol Sci. 2020;16(12):2042–2050. doi:10.7150/ijbs.45467
182. Galgano M, Toshkezi G, Qiu X, et al. Traumatic brain injury: current treatment strategies and future endeavors. Cell Transplant. 2017;26(7):1118–1130. doi:10.1177/0963689717714102
183. Huang L, He S, Cai Q, et al. Polydatin alleviates traumatic brain injury: role of inhibiting ferroptosis. Biochem Biophys Res Commun. 2021;556:149–155. doi:10.1016/j.bbrc.2021.03.108
184. Rui T, Wang H, Li Q, et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J Pineal Res. 2021;70(2):e12704. doi:10.1111/jpi.12704
185. Kenny EM, Fidan E, Yang Q, et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit Care Med. 2019;47(3):410–418. doi:10.1097/CCM.0000000000003555
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.