Back to Journals » Cancer Management and Research » Volume 18

Ferroptosis: A Double-Edged Sword in Cisplatin-Based Cancer Therapy and Acute Kidney Injury
Authors Zhou S, Xiao Y, Fang L
Received 15 December 2025
Accepted for publication 13 February 2026
Published 4 March 2026 Volume 2026:18 589114
DOI https://doi.org/10.2147/CMAR.S589114
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Dr Alessandro Rizzo
Suansuan Zhou,1 Yue Xiao,2 Li Fang3
1Intensive Care Unit, The Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, 130021, People’s Republic of China; 2Department of Pulmonology, The Third Affiliated Clinical Hospital to Changchun University of Chinese Medicine, Changchun, 130117, People’s Republic of China; 3Department of Emergency, The Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, 130021, People’s Republic of China
Correspondence: Li Fang, Department of Emergency, The Affiliated Hospital to Changchun University of Chinese Medicine, No. 1643, Jingyue Street, Nanguan District, Changchun, Jilin, 130021, People’s Republic of China, Email [email protected]
Abstract: Ferroptosis, an emerging type of regulated cell death, continues to attract significant focus over recent years, especially within the areas of cancer therapy and acute kidney injury (AKI) research. In therapies that use platinum-based drugs, particularly cisplatin (Cisplatin), ferroptosis exhibits complex, dual role. In a certain context, ferroptosis enhances clinical effects in platinum-based chemotherapy through promoting tumor cell death, while in a different context, it induces excessive damage to kidney cells, potentially leading to onset as well as worsening of AKI. Consequently, gaining a more comprehensive grasp regarding ferroptosis in this context of platinum-based chemotherapy treatment proves essential, as it could improve the efficacy of cancer therapies while also reducing kidney damage. This knowledge forms the theoretical foundation for developing novel treatment strategies, which can enable precision therapies, reduce side effects, as well as eventually enhance well-being in individuals. This narrative review systematically outlines the role and mechanisms of ferroptosis in the anticancer effects of cisplatin and cisplatin-induced acute kidney injury, and discusses targeting ferroptosis as an important strategy for balancing cancer therapy and preventing kidney damage.
Keywords: ferroptosis, cancer therapeutics, AKI, drug side effects, cisplatin, tumor resistance
Introduction
Ferroptosis represents one type among regulated cellular destruction triggered by iron-induced lipid peroxidation.1 In acute kidney injury, both regulated cell death (such as ferroptosis and apoptosis) and non-regulated cell death (such as necrosis) can coexist. However, because regulated cell death can be precisely regulated through molecular mechanisms and serves as a signal for recovery and repair, while non-regulated cell death typically indicates irreversible cellular damage, regulated cell death, especially ferroptosis, has become a research hotspot. Unlike other well-known forms of cell death, including programmed tissue elimination as well as uncontrolled biological destruction, ferroptosis differs in its distinction due to a buildup with metallic ions inside the cell and the generation resulting from oxidized fats.2 Specifically, excessive Fe2⁺ within the cell can react with hydrogen peroxide (H2O2) through the Fenton reaction (Fe2++H2O2→Fe3++OH−+OH⋅), generating free radicals that further induce lipid peroxidation, damage the cell membrane, and ultimately lead to ferroptosis.3 The process of ferroptosis is driven by critical mechanisms, including the excessive buildup of intracellular iron, the dysfunction of the antioxidant system (particularly through the inhibition of glutathione peroxidase 4, GPX4), and the amplification of lipid peroxidation.4 While ferroptosis’ involvement across both normal as well as disease-related conditions remains under investigation, it has already demonstrated significant biological relevance, especially in cancer and acute organ injury.5–7
Cisplatin, a platinum-containing chemotherapy drug, remains extensively utilized within clinical practice to treat various neoplastic growths, for example, lung, ovarian, and head and neck carcinomas.8–12 The classical DNA damage mechanism of cisplatin primarily involves the formation of adducts with purine bases in DNA, particularly guanine, leading to DNA crosslinking and inhibiting DNA replication and transcription. This triggers a DNA damage response, ultimately resulting in cell apoptosis or arrest.13 However, the use of cisplatin is linked to significant complications, with AKI being particularly prevalent. A retrospective study found that thirty percent among individuals receiving platinum-based treatment develop AKI, with those surviving five or more years experiencing lasting reductions to kidney function rate.14 Cisplatin’s accumulation in the kidneys triggers a series of damaging mechanisms, including oxidative stress and inflammation, which contribute to death in kidney proximal tissues along with onset in AKI.15,16
Recent investigations emphasize a crucial contribution from ferroptosis toward enhancing cisplatin’s anticancer effects,17 while also exacerbating its side effects, particularly AKI, through the induction of ferroptosis.18 Cisplatin initiates iron-dependent lipid oxidation within renal as well as cancer tissues, driving onset ferroptosis.19 Such a mechanism entails a buildup within intracellular iron along with disruption of the antioxidant system, which intensifies cellular damage. As a result, while cisplatin enhances its anticancer efficacy by promoting ferroptosis, it also potentially worsens kidney damage, thereby increasing the risk of AKI.
Basic Processes Involved in Ferroptosis
Ferroptosis was first recognized by Dolma S in 2003 after tumor cells were treated with the chemical agent Erastin, which led to a form of cell death distinct from apoptosis, though it had not yet been named at that time.20 In 2012, S. Dixon et al formally introduced the concept of ferroptosis, defining it as a form of regulated cell death regulated through genetic inhibition or pharmacological chelation of iron via cellular iron uptake, distinguishing this process from alternative types of cell death, such as apoptosis, pyroptosis, as well as necroptosis. Over time, this concept was refined, with ferroptosis ultimately being defined like a type of regulated cell death resulting from mechanisms that rely on metal ions, wherein fatty acid oxidation damages a cellular structure, leading to demise within an organism. Such a phenomenon differs compared to traditional types in cell death such as apoptosis and necrosis.1,21–23 Initially, ferroptosis was thought to be primarily associated with cancer therapy, as studies demonstrated that inhibition of the intracellular antioxidant system could result in iron buildup along with fatty acid oxidation, effectively inducing tumor cell death.24–26 As research on the mechanisms of ferroptosis advanced, it became clear that it is vital to tumor tissues, as well as strongly associated with progression in several conditions, such as cardiovascular disorders, degenerative brain conditions (like Alzheimer’s or Parkinson’s disorders), along with drug-induced damage, like cisplatin-induced renal injury.27–32 Throughout this period, regulatory mechanisms of ferroptosis—such as iron transport, the antioxidant system (particularly GPX4), and their interaction with lipid peroxidation—have been increasingly elucidated, with efforts to explore targeting these mechanisms for therapeutic intervention. Research on ferroptosis continues to evolve, offering fresh perspectives regarding variety in cell death while offering potential avenues for innovative clinical therapies (Figure 1).
 |
Figure 1 This diagram illustrates the interplay between iron homeostasis, lipid metabolism, and their roles in cellular oxidative stress and ferroptosis. Iron is transported across membranes via SLC40A1 and TFRC, stored in ferritin, and can be released into the labile iron pool through NCOA4-mediated ferrophagy. Lipids and fatty acids undergo processes such as lipophagy, conversion to PUFA (polyunsaturated fatty acids), and enzymatic activity of LPCAT3 and ACSL4 to generate PL-PUFA (phospholipid-polyunsaturated fatty acids). In ferroptosis, GPX4 plays a central role in reducing lipid peroxides and protecting cells from oxidative damage, with AJFM2 involved in regulating lipid peroxidation. The Xc-system transports cysteine and glutamate, supporting the synthesis of glutathione (GSH), a critical antioxidant for cellular protection. CoQ10 (Coenzyme Q10) modulates ROS levels in mitochondria and supports energy metabolism. The TCA cycle (Tricarboxylic acid cycle) and ETC (electron transport chain) generate energy while contributing to ROS production. Enzymes such as GPX4, GCH1, and the reduction of GSSG (oxidized glutathione) maintain redox balance, preventing cellular damage from oxidative stress. Abbreviations: SLC40A1, Solute Carrier Family 40 Member 1; TFRC, Transferrin Receptor; NCOA4, Nuclear Receptor Coactivator 4; LPCAT3, Lysophosphatidylcholine Acyltransferase 3; ACSL4, Acyl-CoA Synthetase Long-Chain Family Member 4; PL-PUFA, Phospholipid-Polyunsaturated Fatty Acids; GPX4, Glutathione Peroxidase 4; AJFM2, Atypical Protein Family Member 2; Xc-system, Cystine/Glutamate Antiporter; GSH, Glutathione; CoQ10, Coenzyme Q10; TCA, Tricarboxylic Acid; ETC, Electron Transport Chain; GSSG, Oxidized Glutathione; GCH1, GTP Cyclohydrolase 1. |
Ferroptosis first emerged within the realm of basic medical sciences, accompanied by discussions on its primary regulatory mechanisms. However, subsequent clinical investigations have mainly concentrated on understanding how various proteins or drugs influence the ferroptosis process in disease states by targeting essential mechanisms, such as iron transport, fat oxidative damage, and the antioxidant system. To better understand ferroptosis’ involvement in disease, the mechanisms governing ferroptosis will be detailed in the following section.
Iron Transport
Iron accumulation is a defining feature of ferroptosis, with the Fenton reaction playing a central role in driving ferroptosis due to the buildup of iron.33 As a catalyst in the Fenton reaction, iron interacts with peroxides to produce free radicals, such as reactive hydroxide species, that initiate lipid peroxidation, ultimately leading to impairment of cell membrane. Consequently, a number of key molecules that regulate iron transport, storage, metabolism, and other related processes also have an impact on ferroptosis. For example, transferrin receptor 1 (TfR1) facilitates iron uptake by cells, and modulating TfR1 can influence ferroptosis in those cells.34–37 Iron serves as an essential component in numerous essential physiological functions, contributing significantly to enzyme activity, such as arachidonic acid 5-lipoxygenase (ALOX), which are involved in redox reactions and lipid peroxidation.38 When iron metabolism becomes unbalanced, ferritinophagy, a vital mechanism in controlling iron concentrations, gets triggered. By the action of nuclear receptor coactivator 4 (NCOA4), ferritin degradation promotes breakdown associated with iron storage proteins.39 Ferroportin 1 (FPN1) promotes iron export, while Hepcidin can inhibit FPN1, leading to iron accumulation and subsequently triggering ferroptosis.
Antioxidant Enzyme System
Lipid oxidation serves as a key hallmark in ferroptosis, and antioxidant system involved are essential for preventing ferroptosis initiation. GPX4 serves as a central component in an antioxidant system and functions as a key indicator for assessing damage associated with ferroptosis. As a result, the antioxidant system can be divided into two types: the GPX4-dependent as well as the non-GPX4-dependent pathways. In the GPX4-dependent pathway, selenium along with glutathione (GSH) contribute to regulating GPX4 synthesis as well as its function.40–43 GPX4 converts harmful fatty acid hydroperoxides into their respective hydroxyl compounds, with GSH undergoing a redox reaction in this process. Within the cell, GSH acts as a protective molecule, with synthesis primarily reliant upon cysteine absorption, along with its transformation into homocysteine, a process facilitated via peptide uptake mechanisms (SLC7A11). The SLC7A11/GPX4 axis plays an essential role in the GPX4-dependent antioxidant system.44 In the GPX4-independent antioxidant system, its primary function is carried out through the reduction of coenzyme Q10 (CoQ). Within the mitochondrial inner membrane, dihydroorotate dehydrogenase (DHODH) transforms CoQ into its reduced form, CoQH2. The increased activity of DHODH enhances CoQH2 synthesis, which efficiently neutralizes fat oxidative damage while reducing ferroptosis within the mitochondria.45 Additionally, the FSP1-CoQ10 axis is also an important component of the GPX4-independent antioxidant system. FSP1 enhances its antioxidant activity by catalyzing the reduction of CoQ10, thereby protecting cells from ferroptosis-induced damage. The FSP1-CoQ10 axis and the DHODH-CoQH2 axis work together, providing two independent antioxidant pathways that regulate the occurrence of ferroptosis.
Lipid Peroxidation
Lipid peroxidation represents a key feature in ferroptosis. Unsaturated fatty acids are crucial in driving free radical-induced lipid peroxidation across cellular membranes, while their by-products accumulate during ferroptosis. While various membrane lipids can be oxidized, peroxidation of Polyunsaturated Fatty Acids (PUFAs) is particularly crucial in the development of ferroptosis. Enzymes like Acyl-CoA Synthetase Long-Chain Family Member 4 (ACSL4) as well as Lysophosphatidylcholine Acyltransferase 3 (LPCAT3) are involved in embedding PUFAs within cellular structures. Specifically, ACSL4 catalyzes CoA attachment to arachidonic acid (AA), forming CoA-AA intermediates, which are subsequently esterified by LPCAT3 to produce phosphatidylethanolamine, culminating in production arachidonic acid-phosphatidylethanolamine (PE-AA). Both forms of PE-AA undergo oxidation—enzymatically (via lipoxygenase, such as ALOX12 or ALOX15) and non-enzymatically (via auto-oxidation to PE-AA-OOH)—which ultimately contributes to cell death. In this process, ALOX family members (such as ALOX12, ALOX15, etc.) catalyze the oxidation of PE-AA, generating lipid peroxides (such as PE-AA-OOH), which damage the cell membrane, leading to membrane disruption and cell death. Particularly, ALOX12 and ALOX15 promote the accumulation of lipid peroxidation by oxidizing PE-AA, playing a key role in the mechanism of ferroptosis. The enhanced activity of ALOX enzymes promotes lipid peroxidation, further driving the occurrence of ferroptosis. Therefore, LPCAT3, ACSL4, and ALOX family members are important molecules in regulating lipid peroxidation and ferroptosis.46–49
The interactions between these three pathways collectively regulate the occurrence of ferroptosis. The iron metabolism pathway influences the extent of lipid peroxidation by regulating the intracellular iron concentration, while the antioxidant system prevents the accumulation of lipid peroxides by clearing excess oxidative molecules, thus inhibiting the occurrence of ferroptosis. The intricate regulation among these pathways highlights the complexity of ferroptosis and provides new therapeutic targets for related diseases.
Advances in Ferroptosis Research and Its Role in Cisplatin’s Anticancer Effects
Ferroptosis was first identified in tumor cells treated with inducers for ferroptosis, and subsequent research established enhancing ferroptosis may act as a potent approach to inhibit tumor growth. Numerous studies have demonstrated that cisplatin triggers ferroptosis within cancerous tissues.50 For instance, Wang et al reported that cisplatin treatment significantly enhanced lipid peroxidation and reduced GPX4 protein levels in gastric cancer cells.51 Similarly, Yang JY’s team found platinum-based treatment led to a decrease in xCT protein levels in esophageal cancer epithelial cells.52 While numerous studies support the conclusion that cisplatin promotes ferroptosis to inhibit tumor progression, the specific mechanisms by which cisplatin regulates ferroptosis in tumor cells remain largely unexplored. This is likely due to the prevailing belief that cisplatin exerts its effects by interacting with DNA, creating DNA-platinum complexes, and disrupting DNA synthesis as well as gene expression, eventually leading to apoptosis within cancerous tissues. This widely accepted view has limited the investigation of cisplatin’s role in modulating ferroptosis in tumors. However, recent studies have placed greater emphasis regarding ferroptosis to enhancing cisplatin’s antitumor effects. Several studies suggest that modulating ferroptosis can reduce tumor cell resistance to cisplatin, and that inducing ferroptosis in tumor cells can enhance cisplatin’s inhibitory actions. Certain molecules have been found to regulate ferroptosis, thereby reducing tumor cell resistance to cisplatin.53–55 For example, Xiang L along with colleagues found inhibition targeting long-chain non-coding RNA (lncRNA) MAFG antisense RNA 1 (MAFG-AS1) enhances responsiveness toward cisplatin within BUC tissue cultures through triggering ferroptosis. Mechanistically, the binding of MAFG-AS1 with poly(rC)-binding protein 2 (PCBP2) facilitates its recruitment by the deubiquitinase UCHL5, which stabilizes the PCBP2 protein. PCBP2 then interacts with iron transporter ferroportin 1 (FPN1), thereby inhibiting ferroptosis. In conclusion, suppression in MAFG-AS1 and MAFG enhances cisplatin responsiveness within BUC tissues through induction ferroptosis.56 A separate body of research has explored the use of natural or herbal components to promote ferroptosis, thereby enhancing cisplatin’s antitumor effects57–72 (Table 1). These herbal formulations or components facilitate ferroptosis within cancerous tissues through modulation key proteins associated with iron transport, lipid peroxidation, as well as antioxidant mechanisms. However, many of these studies lack depth, focusing primarily on the regulation of ferroptosis by these drugs without fully exploring the specific mechanisms involved, such as how they modulate ferroptosis-related proteins.
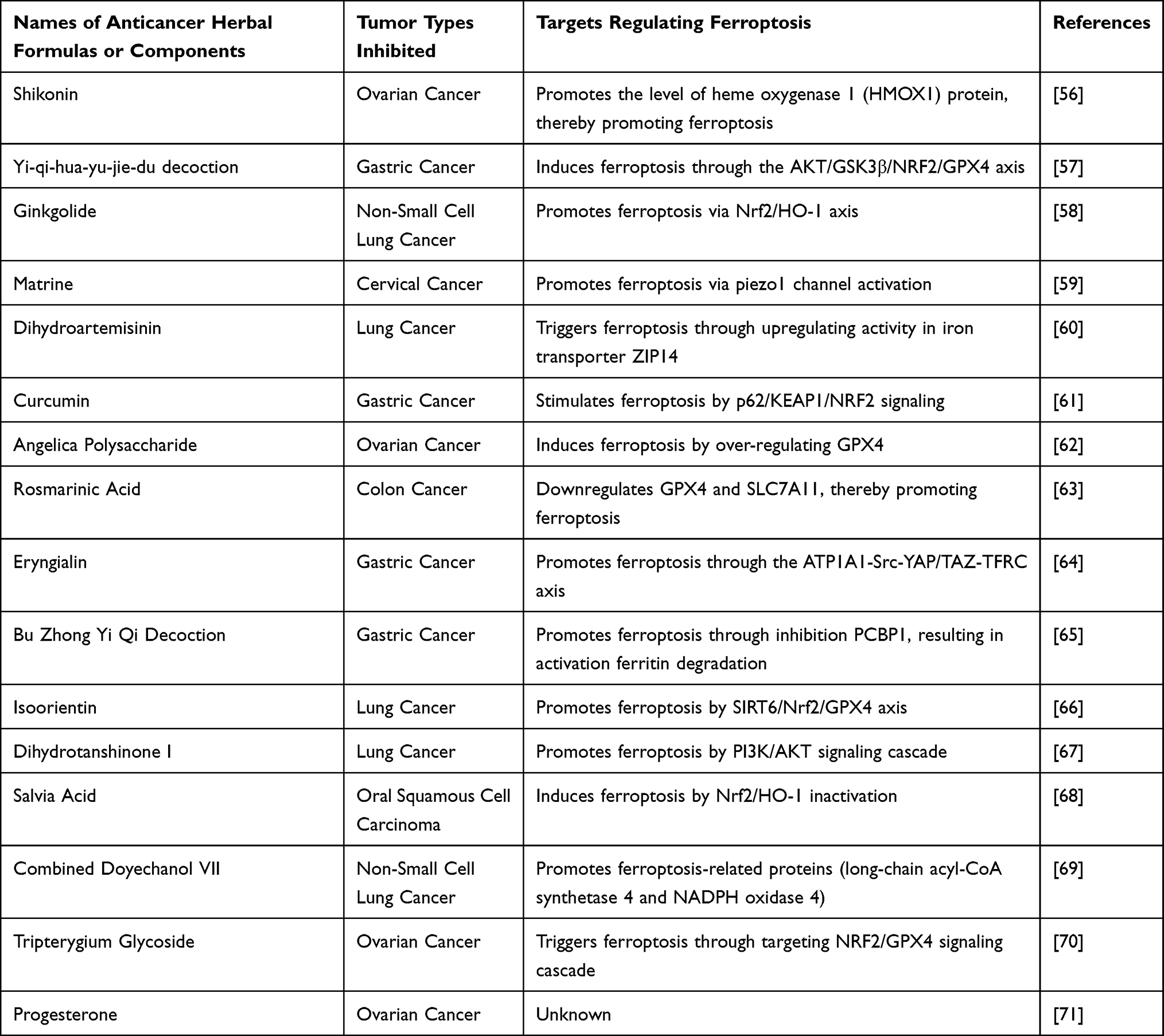 |
Table 1 Anticancer Herbal Formulations or Components That Regulate Ferroptosis to Inhibit Tumor Growth |
In conclusion, recent research indicates that cisplatin may inhibit tumor progression through the induction of ferroptosis; nonetheless, precise processes involved remain to be fully elucidated. Furthermore, certain genes and proteins are believed to regulate ferroptosis-related pathways, which could either amplify cisplatin’s antitumor effects or mitigate the malignant progression of cisplatin-resistant tumor cells. Additionally, various natural compounds and traditional Chinese medicine formulations demonstrated ability in inducing ferroptosis in malignant tissue, thus improving cisplatin efficacy in targeting these cells.
Advances on Ferroptosis Study Within Cisplatin-Triggered AKI
Since ferroptosis was first identified, its significant involvement within cisplatin-triggered AKI has been recognized among numerous researchers. A following section will review various studies, highlighting their strengths and limitations (Figure 2).
 |
Figure 2 Demonstrates that cisplatin induces ferroptosis through the activation of intracellular signaling pathways. Platinum-based drugs are transported into cells via Copper Transporter-1 (CTR1), where they activate several molecular mechanisms, including Akt, NF-κB, and Wnt. These pathways subsequently trigger inflammation, leading to increased levels of TNF-α and IL-6. Additionally, they initiate DNA damage, cell cycle arrest, and apoptosis. Key contributors to ferroptosis include the accumulation of iron ions (Fe2⁺) and the process of polyunsaturated fatty acid oxidation. Furthermore, elevation within reactive oxygen species (ROS) and cellular membrane oxidation further accelerates ferroptosis progression within cells, ultimately resulting in cellular impairment as well as apoptosis. |
Deng F’s team was the first to establish a connection between ferroptosis and cisplatin-induced AKI. Their research demonstrated that renal tubular epithelial cell death induced by cisplatin could be reversed using iron inhibitors, marking the first evidence of ferroptosis’s role in this condition.73 However, their study was relatively superficial and did not investigate specific ferroptosis biomarkers. Similarly, Mishima et al found that several FDA-approved drugs and hormones, including rifampin, propranolol, carvedilol, and thyroid hormones, exhibit anti-ferroptosis properties, helping to mitigate cisplatin-triggered AKI within scavenging free radicals related to lipid peroxidation.74 These findings provide additional evidence for crucial involvement with ferroptosis during development associated with cisplatin-triggered AKI.
Upon discovering ferroptosis involvement in cisplatin-triggered AKI, subsequent research has concentrated on uncovering molecules or drugs that regulate ferroptosis, which in turn can influence AKI. For instance, Hu Z’s team demonstrated that paricalcitol, a Vitamin D Receptor (VDR) agonist, alleviates cisplatin-triggered AKI through lowering oxidative damage in cellular membranes, along with reducing malondialdehyde (MDA) levels, improving both functional and histological outcomes. VDR activation inhibits ferroptosis through the transregulation of GPX4, thus offering protection against cisplatin-induced renal damage.75 Meanwhile, Lu Q’s team identified that the small GTPase Ras homolog enriched in brain 1 (Rheb1) is upregulated within the nephron structures in mice kidneys suffering from cisplatin-triggered AKI. Rheb1-deficient mice showed exacerbated renal impairment, with increased nephron damage due to apoptosis, necroptosis, as well as ferroptosis. Their findings imply Rheb1 might inhibit cisplatin-triggered renal epithelial injury by preserving cellular energy balance.76 Despite these findings, the studies primarily demonstrate that certain molecules influence ferroptosis markers, but they fail to clarify exact processes through which these molecules govern onset as well as development associated with ferroptosis.
As research into mechanisms underlying ferroptosis advances, more comprehensive understanding of its role has facilitated deeper exploration into its contribution toward cisplatin-triggered AKI. Researchers have begun to delve into how specific molecules or target proteins regulate ferroptosis by influencing key components within the ferroptosis pathway. For instance, Fan et al concentrated on heme, a heme-scavenging protein that accumulates in the kidneys during AKI. In their cisplatin-induced AKI model, they observed significant accumulation of hemoglobin and heme, particularly in the proximal tubules of the kidneys. Furthermore, they found that levels related to oxygenase-1, a marker for oxidative damage, were notably elevated in wild-type mice compared to heme knockout mice. Additional studies revealed that heme exacerbates oxidative stress by binding to heme oxygenase-1, thereby worsening kidney damage in AKI.77
The research team led by Kim DH examined the role played by activated farnesoid X receptor (FXR), discovering that FXR could mitigate cisplatin-induced AKI by modulating ferroptosis. They also observed that FXR deficiency resulted in an enhanced ferroptosis response, with increased lipid peroxidation, iron accumulation, alongside elevated oxygenase-1 protein levels within both HK-2 cells and mice. Additionally, they found a reduced ratio of glutathione to glutathione disulfide, alongside lower levels of glutathione peroxidase 4. Chromatin immunoprecipitation assays using kidney tissue from mice demonstrated FXR activation by a ligand, which bound with several well-established regulatory elements, including Slc51a, Slc51b, Osgin1, Mafg, as well as ferroptosis-associated factors such as Aifm2, Ggt6, Gsta4. Moreover, the FXR-regulated transcriptional repressor MAFG was shown to interact with Hmox1, Nqo1, as well as Tf within kidney tissue from mice treated with FXR agonists. These findings suggest that activated FXR contributes to protection against cisplatin-mediated AKI by regulating transcription among genes associated with ferroptosis.78
Additionally, the Wang S research team discovered that Dioscin, a natural compound, alleviates ferroptosis as well as apoptosis by stimulating Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) / Heme Oxygenase 1 (HO-1) mechanism, thus preventing cisplatin-induced AKI.79 Zhou et al found that MicroRNA-214-3p aggravates ferroptosis as well as renal injury during cisplatin-mediated AKI by targeting GPX4.80 The Deng Z team demonstrated that WW domain-binding protein 2 inhibits ferroptosis during cisplatin-triggered AKI by preventing lysosomal degradation within GPX4.81 Additionally, Pan M and his team found that Celastrol, another natural compound, suppresses ferroptosis via the Nrf2/GPX4 pathway, thus mitigating acute kidney injury.82 Collectively, these studies emphasize how specific drugs or molecules regulate ferroptosis by targeting key molecules involved in this pathway.
Post-translational modifications of proteins play a crucial role in regulating protein function or enhancing protein expression through processes such as methylation and acetylation, affecting both histones and non-histone proteins. As research continues to advance, the concept of post-translational modifications has garnered increasing attention, especially within ferroptosis research. More recently, research has concentrated on how post-translational modifications impact ferroptosis-related pathway proteins, influencing the progression of this cellular process. For instance, in the context of acetylation, Yang et al identified that Sirt6, a histone deacetylase, modulates the expression of BRCA1 associated protein 1 (BAP1) through the deacetylation of histones, thus influencing the initiation of ferroptosis.83 Additionally, Liang NN and his team observed that using a murine system for cisplatin-mediated AKI, DHODH acetylation, an essential enzyme involved in pyrimidine synthesis, had increased in the mitochondria, while levels of Sirtuin 3 (SIRT3) were reduced in both in renal tissue from treated mice as well as within HK-2 cellular model. Their laboratory studies further demonstrated how overexpressing SIRT3 alleviated acetylation on mitochondrial DHODH and ferroptosis events within renal tissues under cisplatin treatment. Based on these findings, the researchers concluded that mitochondrial dysfunction drives DHODH acetylation, which subsequently leads to ferroptosis in renal cells during cisplatin-induced AKI.84
Although the concept of ferroptosis has been known for over a decade and research on this process has become more advanced, drugs specifically targeting ferroptosis are still in preclinical stages, with none yet available for clinical application. Despite this, several natural compounds and drugs have demonstrated potential in targeting ferroptosis and mitigating cisplatin-mediated AKI during preclinical investigations. For example, a research by Tu Y and his team revealed that complex carbohydrates isolated out of Flammulina velutipes (FVPs) suppress ferroptosis by modifying the gut microbiome and boosting concentrations in short-chain fatty acids (SCFAs).85 Another study by Hu J and his colleagues investigated natural products and focused on Bergenin, a compound derived from the plant Bougainvillea spectabilis. Bergenin, long utilized in herbal tea, is known for its ability to activate Nrf2 and its anti-inflammatory and antioxidant properties. The researchers found that treatment with Bergenin modulated biomarkers of ferroptosis and ferritin autophagy. Specifically, Bergenin binds to and phosphorylates Glycogen Synthase Kinase 3 Beta (GSK3β), inhibiting its activity, which promotes movement from Nrf2 into a cell’s core. This consequently facilitates binding by Nrf2 with a Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) regulatory region, activating PPARγ. Nevertheless, when either Nrf2 as well as PPARγ became suppressed, Bergenin’s protective effects against ferroptosis, along with ferritin autophagy in cisplatin-treated HK-2 cultures became weakened. In vivo, Bergenin was found to significantly reduce cisplatin-induced kidney damage. Furthermore, Bergenin was shown to decrease ferroptosis mediated by iron autophagy induced by cisplatin. These findings suggest that Bergenin is capable of effectively inhibiting ferroptosis caused by cisplatin-driven AKI, largely via stimulation within p-GSK3β/Nrf2/PPARγ signaling cascade.86 Moreover, Guo S and his team focused on components from traditional Chinese medicine and discovered that Baicalein alleviates cisplatin-triggered kidney impairment as well as tissue injury, while also improving damage within cisplatin-administered HK2 cultures. Mechanistically, baicalein has been found to reduce activity in 12-lipoxygenase (ALOX12), a key enzyme inhibiting cellular structure oxidation, thus preventing ferroptosis during AKI. Structural modeling, along with SPR analysis confirmed that baicalein directly interacts with ALOX12. Furthermore, silencing ALOX12 exhibited regulatory effects similar to those of baicalein. The use of the ALOX12 inhibitor ML355 yielded similar results. Such results emphasize baicalein’s involvement with controlling ALOX12-mediated ferroptosis within nephritic epithelial cultures. Baicalein has shown promise as a viable treatment against AKI, whereas ALOX12 may represent promising therapeutic potential.87 In another study, Abdel-Rahman N and his team found that roflumilast, a selective phosphodiesterase 4 (PDE4) inhibitor used primarily for chronic obstructive pulmonary disease (COPD), lowers MDA levels and increases GSH levels within kidneys from cisplatin-administered AKI mice, demonstrating compound antioxidant effects. Roflumilast also elevated kidney concentrations for GPX4 as well as xCT subunits, as well as the levels of ferritin and Nrf2 in the kidneys. These results suggest that roflumilast protects kidney function by reducing oxidative stress and inhibiting ferroptosis.88
Although a significant portion of current research on targeting ferroptosis to treat cisplatin-associated AKI is still at preclinical stages, most studies aim to confirm whether specific natural compounds or drugs can protect the kidneys by inhibiting ferroptosis. The multitarget properties inherent in classical Chinese medicine and other natural compounds, combined with their ability to address multiple diseases with a single drug, make natural agents aimed at ferroptosis an effective strategy in managing cisplatin-associated AKI. Additionally, development in drugs targeting ferroptosis aimed at treating or preventing cisplatin-associated AKI holds great potential for the future. Therefore, it is essential for researchers to continue exploring this field through further studies.
Targeting Ferroptosis: A Strategy to Balance Cisplatin-Triggered AKI and the Tumor-Inhibiting Activity
Within cancer treatment, while promoting ferroptosis can inhibit tumor growth, it simultaneously elevates the risk of kidney damage. This inherent conflict greatly limits the clinical application of cisplatin, a widely used anti-cancer drug. Consequently, this suggests that we cannot exclusively focus on tumor treatment (eg, using ferroptosis activators to enhance anti-cancer effects), as seen in preclinical studies, nor can we solely concentrate on preventing or managing AKI (eg, using ferroptosis inhibitors to protect the kidneys). As a result, there is a need to identify drugs capable of triggering ferroptosis within malignant tissues, as well as inhibiting it within kidney tissue, thereby excluding drugs that specifically target ferroptosis-related proteins. Traditional Chinese medicine and natural compounds may offer a viable solution.
For instance, research conducted by Guo S and his team demonstrated that Baicalein can alleviate cisplatin-associated AKI through suppressing ALOX12-mediated ferroptosis.87 Similarly, the Lai JQ team found that Baicalein can induce ferroptosis within colon tumor tissues by suppressing JAK2/STAT3/GPX4 molecular mechanisms.89 Furthermore, the Jin Y team showed that Baicalein increases cervical tumor growth’s susceptibility to cisplatin.90 Although current studies on baicalin do not directly confirm that it enhances cisplatin’s anti-cancer effects through ferroptosis, existing evidence suggests that baicalin both inhibits tumor growth and mitigates cisplatin-induced kidney damage.
Several other natural compounds align with this mechanism. For example, Celastrol inhibits ferroptosis via the Nrf2/GPX4 pathway, reducing acute kidney injury82 while also significantly hindering the progression of various cancers, including gastric cancer,91 liver cancer,92–97 clear cell renal cancer,98 breast carcinoma,99 nasopharyngeal carcinoma.100 Another compound, Gastrodin, not only mitigates cisplatin-associated AKI through ferroptosis suppression101 but also promotes ferroptosis in gliomas and colorectal cancer, thereby suppressing tumor progression.102,103
Therefore, natural compounds that target ferroptosis hold great potential in preventing or alleviating cisplatin-associated AKI while simultaneously inhibiting cancer progression, representing a promising area for future research.
Summary
Ferroptosis, one recently identified type in regulated cell death, is increasingly being acknowledged for its critical biological functions in both cancer treatment as well as AKI research. Ferroptosis’s dual nature in cisplatin therapy has attracted significant attention. While cisplatin boosts its anti-cancer efficacy by inducing ferroptosis in tumor cells, it concurrently worsens cisplatin-associated AKI, which presents major obstacle to its broader clinical application. Consequently, understanding and regulating the role of ferroptosis in cisplatin therapy could enhance the therapeutic effectiveness against cancer, while simultaneously minimizing cisplatin’s side effects, especially in the context of preventing and managing AKI.
Current research indicates how ferroptosis induced by cisplatin not only enhances the anti-cancer effects via iron-driven membrane oxidative damage but simultaneously leads to kidney cell damage. Targeting key processes associated with ferroptosis, including the regulation of iron metabolism alongside antioxidant systems, provides new possibilities for precision therapy. For example, research has demonstrated that natural compounds, including baicalin and Celastrol, can modulate ferroptosis, thereby increasing the anti-cancer effects of cisplatin. At the same time, it also demonstrates a protective effect on the kidneys in the cisplatin-induced AKI model. These natural compounds, with their multitarget properties, offer substantial potential for balancing the anti-tumor effects of cisplatin and reducing the impact of AKI.
Regulating ferroptosis offers promising new therapeutic strategies for overcoming the dual challenges of cancer treatment and AKI. However, current preclinical research predominantly centers on laboratory validation, with limited exploration of the underlying mechanisms. For example, while it is well established that certain natural compounds or molecules can regulate ferroptosis-related proteins, the ability to precisely control ferroptosis pathways, especially through selective regulation in kidney and tumor cells, remains a significant unresolved challenge.
In conclusion, regulating ferroptosis presents considerable potential in cisplatin therapy by enhancing its anti-cancer effects and reducing kidney toxicity. However, targeting ferroptosis to improve efficacy and reduce toxicity remains a challenging goal. Firstly, achieving selective targeting between tumors and normal tissues (especially the kidneys) and preventing excessive ferroptosis activation that could damage normal tissues is a significant challenge. Secondly, the issue of drug resistance in tumor cells also hinders ferroptosis-targeted therapies, and overcoming resistance to ensure sustained therapeutic effects is a problem that needs to be addressed. Therefore, future research directions should become more specific. First, the development of tissue-specific ferroptosis inducers or inhibitors is necessary to enhance the selectivity and precision of treatments. Secondly, identifying and validating biomarkers that predict ferroptosis sensitivity will aid in the development of personalized treatment strategies. Finally, exploring the synergistic effects of ferroptosis and immunotherapy is also worthy of further investigation, as this could provide new insights and strategies for cancer treatment. In conclusion, traditional Chinese medicine and natural compounds, with their multitarget effects and fewer side effects, could prove to be invaluable tools for achieving this goal. Subsequent studies ought to focus efforts toward uncovering underlying processes associated with ferroptosis, along with its dual involvement in both tumor suppression and kidney protection, with a particular focus on how these natural substances could be effectively integrated into healthcare to offer safer and more effective treatment options. In addition, further research is needed on tissue-specific targeting, biomarkers, and other related issues.
Funding
The author(s) reported there is no funding associated with the work featured in this article.
Disclosure
No potential conflict of interest was reported by the author(s).
References
1. Zhou Q, Meng Y, Li D, et al. Ferroptosis in cancer: from molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9(1):55. doi:10.1038/s41392-024-01769-5
2. Li J, Jia YC, Ding YX, Bai J, Cao F, Li F. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Biol Sci. 2023;19(9):2756–14. doi:10.7150/ijbs.83348
3. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424–442. doi:10.1038/s41580-024-00703-5
4. Zheng J, Conrad M. Ferroptosis: when metabolism meets cell death. Physiol Rev. 2025;105(2):651–706. doi:10.1152/physrev.00031.2024
5. Hao M, Jiang Y, Zhang Y, Yang X, Han J. Ferroptosis regulation by methylation in cancer. Biochim Biophys Acta Rev Cancer. 2023;1878(6):188972. doi:10.1016/j.bbcan.2023.188972
6. Qiao O, Wang X, Wang Y, Li N, Gong Y. Ferroptosis in acute kidney injury following crush syndrome: a novel target for treatment. J Adv Res. 2023;54:211–222. doi:10.1016/j.jare.2023.01.016
7. Zhang L, Chen F, Dong J, et al. HDAC3 aberration-incurred GPX4 suppression drives renal ferroptosis and AKI-CKD progression. Redox Biol. 2023;68:102939. doi:10.1016/j.redox.2023.102939
8. Zoń A, Bednarek I. Cisplatin in ovarian cancer treatment-known limitations in therapy force new solutions. Int J Mol Sci. 2023;24(8). doi:10.3390/ijms24087585
9. Hamaya S, Oura K, Morishita A, Masaki T. Cisplatin in liver cancer therapy. Int J Mol Sci. 2023;24(13). doi:10.3390/ijms241310858
10. Gandin V, Hoeschele JD, Margiotta N. Special issue “Cisplatin in cancer therapy: molecular mechanisms of action 3.0”. Int J Mol Sci. 2023;24(9). doi:10.3390/ijms24097917
11. Li Q, Chen S, Wang X, et al. Cisplatin-Based combination therapy for enhanced cancer treatment. Curr Drug Targets. 2024;25(7):473–491. doi:10.2174/0113894501294182240401060343
12. Bhat A, Verma S, Chander G, et al. Cisplatin-based combination therapy for cancer. J Cancer Res Ther. 2023;19(3):530–536. doi:10.4103/jcrt.jcrt_792_22
13. Galluzzi L, Senovilla L, Vitale I, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–1883. doi:10.1038/onc.2011.384
14. Yu B, Jin L, Yao X, et al. TRPM2 protects against cisplatin-induced acute kidney injury and mitochondrial dysfunction via modulating autophagy. Theranostics. 2023;13(13):4356–4375. doi:10.7150/thno.84655
15. Deng F, Zhang H, Zhou W, et al. TRPA1 promotes cisplatin-induced acute kidney injury via regulating the endoplasmic reticulum stress-mitochondrial damage. J Transl Med. 2023;21(1):695. doi:10.1186/s12967-023-04351-9
16. Li Y, Shi L, Zhao F, et al. PIM1 attenuates cisplatin-induced AKI by inhibiting Drp1 activation. Cell Signal. 2024;113:110969. doi:10.1016/j.cellsig.2023.110969
17. Elmorsy EA, Saber S, Hamad RS, et al. Advances in understanding cisplatin-induced toxicity: molecular mechanisms and protective strategies. Eur J Pharm Sci. 2024;203:106939. doi:10.1016/j.ejps.2024.106939
18. Han Z, Luo Y, Chen H, et al. A deep insight into ferroptosis in renal disease: facts and perspectives. Kidney Dis. 2024;10(3):224–236. doi:10.1159/000538106
19. Su Y, Zhao B, Zhou L, et al. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020;483:127–136. doi:10.1016/j.canlet.2020.02.015
20. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296. doi:10.1016/s1535-6108(03)00050-3
21. Gao W, Wang X, Zhou Y, Wang X, Yu Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct Target Ther. 2022;7(1):196. doi:10.1038/s41392-022-01046-3
22. Dixon SJ, Pratt DA. Ferroptosis: a flexible constellation of related biochemical mechanisms. Mol Cell. 2023;83(7):1030–1042. doi:10.1016/j.molcel.2023.03.005
23. Chen X, Kang R, Kroemer G, Tang D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021;28(10):2843–2856. doi:10.1038/s41418-021-00859-z
24. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–849. doi:10.1016/j.ccell.2019.04.002
25. Zhang C, Liu X, Jin S, Chen Y, Guo R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol Cancer. 2022;21(1):47. doi:10.1186/s12943-022-01530-y
26. Yang F, Xiao Y, Ding JH, et al. Ferroptosis heterogeneity in triple-negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab. 2023;35(1):84–100.e108. doi:10.1016/j.cmet.2022.09.021
27. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. 2023;20(1):7–23. doi:10.1038/s41569-022-00735-4
28. Feng Q, Yang Y, Ren K, et al. Broadening horizons: the multifaceted functions of ferroptosis in kidney diseases. Int J Biol Sci. 2023;19(12):3726–3743. doi:10.7150/ijbs.85674
29. Berndt C, Alborzinia H, Amen VS, et al. Ferroptosis in health and disease. Redox Biol. 2024;75:103211. doi:10.1016/j.redox.2024.103211
30. Lee S, Hwang N, Seok BG, Lee S, Lee S,J, Chung SW. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis. 2023;14(7):464. doi:10.1038/s41419-023-05978-8
31. Zhang Y, Xin L, Xiang M, et al. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother. 2022;145:112423. doi:10.1016/j.biopha.2021.112423
32. Xu S, He Y, Lin L, Chen P, Chen M, Zhang S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021;12(4):289. doi:10.1038/s41419-021-03559-1
33. He YJ, Liu XY, Xing L, Wan X, Chang X, Jiang HL. Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials. 2020;241:119911. doi:10.1016/j.biomaterials.2020.119911
34. Wang D, Liang W, Huo D, et al. SPY1 inhibits neuronal ferroptosis in amyotrophic lateral sclerosis by reducing lipid peroxidation through regulation of GCH1 and TFR1. Cell Death Differ. 2023;30(2):369–382. doi:10.1038/s41418-022-01089-7
35. Wang H, Yu X, Liu D, et al. VDR activation attenuates renal tubular epithelial cell ferroptosis by regulating Nrf2/HO-1 signaling pathway in diabetic nephropathy. Adv Sci. 2024;11(10):e2305563. doi:10.1002/advs.202305563
36. Chen L, Ma Y, Ma X, et al. TFEB regulates cellular labile iron and prevents ferroptosis in a TfR1-dependent manner. Free Radic Biol Med. 2023;208:445–457. doi:10.1016/j.freeradbiomed.2023.09.004
37. Lan T, Hu L, Sun T, et al. H3K9 trimethylation dictates neuronal ferroptosis through repressing Tfr1. J Cereb Blood Flow Metab. 2023;43(8):1365–1381. doi:10.1177/0271678x231165653
38. García-Montero C, Fraile-Martinez O, Cobo-Prieto D, et al. Abnormal histopathological expression of klotho, ferroptosis, and circadian clock regulators in pancreatic ductal adenocarcinoma: prognostic implications and correlation analyses. Biomolecules. 2024;14(8). doi:10.3390/biom14080947
39. Wu H, Liu Q, Shan X, Gao W, Chen Q. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy. 2023;19(7):2062–2077. doi:10.1080/15548627.2023.2170960
40. Liu J, Tang D, Kang R. Targeting GPX4 in ferroptosis and cancer: chemical strategies and challenges. Trends Pharmacol Sci. 2024;45(8):666–670. doi:10.1016/j.tips.2024.05.006
41. Liang D, Feng Y, Zandkarimi F, et al. Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell. 2023;186(13):2748–2764.e2722. doi:10.1016/j.cell.2023.05.003
42. Lei G, Mao C, Horbath AD, et al. BRCA1-Mediated dual regulation of ferroptosis exposes a vulnerability to GPX4 and PARP Co-Inhibition in BRCA1-Deficient cancers. Cancer Discov. 2024;14(8):1476–1495. doi:10.1158/2159-8290.Cd-23-1220
43. Zhou L, Lian G, Zhou T, et al. Palmitoylation of GPX4 via the targetable ZDHHC8 determines ferroptosis sensitivity and antitumor immunity. Nat Cancer. 2025;6(5):768–785. doi:10.1038/s43018-025-00937-y
44. Wang Y, Zheng L, Shang W, et al. Wnt/beta-catenin signaling confers ferroptosis resistance by targeting GPX4 in gastric cancer. Cell Death Differ. 2022;29(11):2190–2202. doi:10.1038/s41418-022-01008-w
45. Lin X, Zhang Q, Li Q, et al. Upregulation of CoQ shifts ferroptosis dependence from GPX4 to FSP1 in acquired radioresistance. Drug Resist Updat. 2024;73:101032. doi:10.1016/j.drup.2023.101032
46. Iqbal S, Jabeen F, Kahwa I, Omara T. Suberosin alleviates thiazolidinedione-induced cardiomyopathy in diabetic rats by inhibiting ferroptosis via modulation of ACSL4-LPCAT3 and PI3K-AKT signaling pathways. Cardiovasc Toxicol. 2023;23(9–10):295–304. doi:10.1007/s12012-023-09804-7
47. Cui J, Wang Y, Tian X, et al. LPCAT3 is transcriptionally regulated by YAP/ZEB/EP300 and Collaborates with ACSL4 and YAP to determine ferroptosis sensitivity. Antioxid Redox Signal. 2023;39(7–9):491–511. doi:10.1089/ars.2023.0237
48. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biology. 2021;10(3). doi:10.3390/biology10030184
49. Gupta G, Bhat AA, Goyal A, et al. Exploring ACSL4/LPCAT3/ALOX15 and SLC7A11/GPX4/NFE2L2 as potential targets in ferroptosis-based cancer therapy. Future Med Chem. 2023;15(14):1209–1212. doi:10.4155/fmc-2023-0125
50. Du J, Wang X, Li Y, et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 2021;12(7):705. doi:10.1038/s41419-021-03996-y
51. Wang H, Lu C, Zhou H, et al. Synergistic effects of dihydroartemisinin and cisplatin on inducing ferroptosis in gastric cancer through GPX4 inhibition. Gastric Cancer. 2025;28(2):187–210. doi:10.1007/s10120-024-01574-7
52. Yang JY, Lei XY, He KY, et al. HMGA1 drives chemoresistance in esophageal squamous cell carcinoma by suppressing ferroptosis. Cell Death Dis. 2024;15(2):158. doi:10.1038/s41419-024-06467-2
53. Valashedi MR, Roushandeh AM, Tomita K, et al. CRISPR/Cas9-mediated knockout of Lcn2 in human breast cancer cell line MDA-MB-231 ameliorates erastin-mediated ferroptosis and increases cisplatin vulnerability. Life Sci. 2022;304:120704. doi:10.1016/j.lfs.2022.120704
54. Luo Y, Liu X, Chen Y, et al. Targeting PAX8 sensitizes ovarian cancer cells to ferroptosis by inhibiting glutathione synthesis. Apoptosis. 2024;29(9–10):1499–1514. doi:10.1007/s10495-024-01985-y
55. Wang Y, Wang C, Guan X, et al. PRMT3-Mediated arginine methylation of METTL14 promotes malignant progression and treatment resistance in endometrial carcinoma. Adv Sci. 2023;10(36):e2303812. doi:10.1002/advs.202303812
56. Xiang L, Zeng Q, Liu J, et al. MAFG-AS1/MAFG positive feedback loop contributes to cisplatin resistance in bladder urothelial carcinoma through antagonistic ferroptosis. Sci Bull. 2021;66(17):1773–1788. doi:10.1016/j.scib.2021.01.027
57. Ni M, Zhou J, Zhu Z, et al. Shikonin and cisplatin synergistically overcome cisplatin resistance of ovarian cancer by inducing ferroptosis via upregulation of HMOX1 to promote Fe(2+) accumulation. Phytomedicine. 2023;112:154701. doi:10.1016/j.phymed.2023.154701
58. Huang W, Wen F, Yang P, Li Y, Li Q, Shu P. Yi-qi-hua-yu-jie-du decoction induces ferroptosis in cisplatin-resistant gastric cancer via the AKT/GSK3β/NRF2/GPX4 axis. Phytomedicine. 2024;123:155220. doi:10.1016/j.phymed.2023.155220
59. Lou JS, Zhao LP, Huang ZH, et al. Ginkgetin derived from Ginkgo biloba leaves enhances the therapeutic effect of cisplatin via ferroptosis-mediated disruption of the Nrf2/HO-1 axis in EGFR wild-type non-small-cell lung cancer. Phytomedicine. 2021;80:153370. doi:10.1016/j.phymed.2020.153370
60. Jin J, Fan Z, Long Y, et al. Matrine induces ferroptosis in cervical cancer through activation of piezo1 channel. Phytomedicine. 2024;122:155165. doi:10.1016/j.phymed.2023.155165
61. Yang Z, Zhou Z, Meng Q, et al. Dihydroartemisinin sensitizes lung cancer cells to cisplatin treatment by upregulating ZIP14 expression and inducing ferroptosis. Cancer Med. 2024;13(19):e70271. doi:10.1002/cam4.70271
62. Feng T, Zhou Y, Mao X, Rui X, Cai L. Curcumol enhances the sensitivity of gastric cancer to cisplatin resistance by inducing ferroptosis through the P62/KEAP1/NRF2 pathway. Integr Cancer Ther. 2024;23:15347354241294043. doi:10.1177/15347354241294043
63. Guo W, Wang W, Lei F, et al. Angelica sinensis polysaccharide combined with cisplatin reverses cisplatin resistance of ovarian cancer by inducing ferroptosis via regulating GPX4. Biomed Pharmacother. 2024;175:116680. doi:10.1016/j.biopha.2024.116680
64. Huang JY, Hsu TW, Chen YR, Kao SH. Rosmarinic acid potentiates cytotoxicity of cisplatin against colorectal cancer cells by enhancing apoptotic and ferroptosis. Life. 2024;14(8). doi:10.3390/life14081017
65. Ke A, Yang W, Zhang W, et al. The cardiac glycoside periplocymarin sensitizes gastric cancer to ferroptosis via the ATP1A1-Src-YAP/TAZ-TFRC axis. Phytomedicine. 2025;142:156804. doi:10.1016/j.phymed.2025.156804
66. Liu YT, Cai HR, Li H, Mu QR, Huang JY, Gao Y. Buzhong Yiqi decoction improves cisplatin resistance in non-small cell lung cancer by inhibiting PCBP1 to activate the ferritinophagy-mediated ferroptosis pathway. J Ethnopharmacol. 2025;120317. doi:10.1016/j.jep.2025.120317
67. Feng S, Li Y, Huang H, et al. Isoorientin reverses lung cancer drug resistance by promoting ferroptosis via the SIRT6/Nrf2/GPX4 signaling pathway. Eur J Pharmacol. 2023;954:175853. doi:10.1016/j.ejphar.2023.175853
68. Li FJ, Gao LC, Long HZ, et al. Dihydroisotanshinone I regulates ferroptosis via PI3K/AKT pathway to enhance cisplatin sensitivity in lung adenocarcinoma. J Pharm Pharmacol. 2025;77(6):752–767. doi:10.1093/jpp/rgae085
69. Han L, Li L, Wu G. Induction of ferroptosis by carnosic acid-mediated inactivation of Nrf2/HO-1 potentiates cisplatin responsiveness in OSCC cells. Mol Cell Probes. 2022;64:101821. doi:10.1016/j.mcp.2022.101821
70. Wu Y, Yang L, Li Z, Chen Q, Hu J. Polyphyllin VII enhances the antitumor activity of cisplatin in non-small cell lung cancer cells by inducing ferroptosis and enhancing apoptosis. J Biochem Mol Toxicol. 2025;39(4):e70186. doi:10.1002/jbt.70186
71. Koyanagi T, Saga Y, Takahashi Y, et al. Progesterone enhances sensitivity of ovarian cancer cells to SN38 through inhibition of topoisomerase I and inducing ferroptosis. Cancer Rep. 2025;8(4):e70202. doi:10.1002/cnr2.70202
72. Ma B, Zhong Y, Chen R, et al. Tripterygium glycosides reverse chemotherapy resistance in ovarian cancer by targeting the NRF2/GPX4 signal axis to induce ferroptosis of drug-resistant human epithelial ovarian cancer cells. Biochem Biophys Res Commun. 2023;665:178–186. doi:10.1016/j.bbrc.2023.04.111
73. Deng F, Sharma I, Dai Y, Yang M, Kanwar YS. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest. 2019;129(11):5033–5049. doi:10.1172/jci129903
74. Mishima E, Sato E, Ito J, et al. Drugs repurposed as antiferroptosis agents suppress organ damage, including AKI, by functioning as lipid peroxyl radical scavengers. J Am Soc Nephrol. 2020;31(2):280–296. doi:10.1681/asn.2019060570
75. Hu Z, Zhang H, Yi B, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020;11(1):73. doi:10.1038/s41419-020-2256-z
76. Lu Q, Wang M, Gui Y, et al. Rheb1 protects against cisplatin-induced tubular cell death and acute kidney injury via maintaining mitochondrial homeostasis. Cell Death Dis. 2020;11(5):364. doi:10.1038/s41419-020-2539-4
77. Fan X, Zhang X, Liu LC, et al. Hemopexin accumulates in kidneys and worsens acute kidney injury by causing hemoglobin deposition and exacerbation of iron toxicity in proximal tubules. Kidney Int. 2022;102(6):1320–1330. doi:10.1016/j.kint.2022.07.024
78. Kim DH, Choi HI, Park JS, et al. Farnesoid X receptor protects against cisplatin-induced acute kidney injury by regulating the transcription of ferroptosis-related genes. Redox Biol. 2022;54:102382. doi:10.1016/j.redox.2022.102382
79. Wang S, Zheng Y, Jin S, Fu Y, Liu Y. Dioscin protects against cisplatin-induced acute kidney injury by reducing ferroptosis and apoptosis through activating Nrf2/HO-1 signaling. Antioxidants. 2022;11(12). doi:10.3390/antiox11122443
80. Zhou J, Xiao C, Zheng S, et al. MicroRNA-214-3p aggravates ferroptosis by targeting GPX4 in cisplatin-induced acute kidney injury. Cell Stress Chaperones. 2022;27(4):325–336. doi:10.1007/s12192-022-01271-3
81. Deng Z, Wang Y, Liu J, et al. WBP2 restrains the lysosomal degradation of GPX4 to inhibit ferroptosis in cisplatin-induced acute kidney injury. Redox Biol. 2023;65:102826. doi:10.1016/j.redox.2023.102826
82. Pan M, Wang Z, Wang Y, et al. Celastrol alleviated acute kidney injury by inhibition of ferroptosis through Nrf2/GPX4 pathway. Biomed Pharmacother. 2023;166:115333. doi:10.1016/j.biopha.2023.115333
83. Yang S, Chen L, Din S, et al. The SIRT6/BAP1/xCT signaling axis mediates ferroptosis in cisplatin-induced AKI. Cell Signal. 2025;125:111479. doi:10.1016/j.cellsig.2024.111479
84. Liang NN, Guo YY, Zhang XY, et al. Mitochondrial Dysfunction-Evoked DHODH acetylation is involved in renal cell ferroptosis during cisplatin-induced acute kidney injury. Adv Sci. 2024;11(43):e2404753. doi:10.1002/advs.202404753
85. Tu Y, Tang E, Ye H, et al. Flammulina velutipes polysaccharides ameliorate cisplatin-induced acute kidney injury in mice via regulation of gut microbiota and ferroptosis pathway. Int J Biol Macromol. 2025;290:138526. doi:10.1016/j.ijbiomac.2024.138526
86. Hu J, Zhang Y, Zhang Y, et al. Bergenin inhibits ferritinophagy and ferroptosis in cisplatin-induced acute kidney injury by activating the p-GSK3β/Nrf2/PPARγ pathway. Int Immunopharmacol. 2025;147:114004. doi:10.1016/j.intimp.2024.114004
87. Guo S, Zhou L, Liu X, Gao L, Li Y, Wu Y. Baicalein alleviates cisplatin-induced acute kidney injury by inhibiting ALOX12-dependent ferroptosis. Phytomedicine. 2024;130:155757. doi:10.1016/j.phymed.2024.155757
88. Abdel-Rahman N, Abdel-Rahman AM, Sharawy MH. Regulating ferroptosis by roflumilast attenuates cisplatin-induced kidney injury. Int Immunopharmacol. 2025;151:114331. doi:10.1016/j.intimp.2025.114331
89. Lai JQ, Zhao LL, Hong C, et al. Baicalein triggers ferroptosis in colorectal cancer cells via blocking the JAK2/STAT3/GPX4 axis. Acta Pharmacol Sin. 2024;45(8):1715–1726. doi:10.1038/s41401-024-01258-z
90. Jin Y, Wu Q, Pan S, et al. Baicalein enhances cisplatin sensitivity in cervical cancer cells by promoting cuproptosis through the Akt pathway. Biomed Pharmacother. 2024;179:117415. doi:10.1016/j.biopha.2024.117415
91. Yang C, Xue R, Qin C, et al. Celastrol induces ferroptosis by regulating CERKL to exert anti-gastric cancer effect. Am J Chin Med. 2025;53(3):931–949. doi:10.1142/s0192415x25500351
92. Cai B, Qi M, Zhang X, Zhang D. Integrating network pharmacology with in vitro experiments to validate the efficacy of celastrol against hepatocellular carcinoma through ferroptosis. Drug Des Devel Ther. 2024;18:3121–3141. doi:10.2147/dddt.S450324
93. Zhang X, Qi M, Huo K, et al. Celastrol induces ferroptosis by suppressing RRM2 in hepatocellular carcinoma. Heliyon. 2024;10(13):e33936. doi:10.1016/j.heliyon.2024.e33936
94. Yan J, Xu T. Celastrol targets FANCD2 to induce autophagy-dependent ferroptosis in hepatocellular carcinoma cells. Discov Med. 2025;37(195):669–684. doi:10.24976/Discov.Med.202537195.58
95. Luo P, Zhang Q, Shen S, et al. Mechanistic engineering of celastrol liposomes induces ferroptosis and apoptosis by directly targeting VDAC2 in hepatocellular carcinoma. Asian J Pharm Sci. 2023;18(6):100874. doi:10.1016/j.ajps.2023.100874
96. Qi M, Zhang K, Zhang X, et al. Arginine tagged liposomal carrier for the delivery of celastrol for ferroptosis-induced hepatocellular carcinoma therapy. Colloids Surf B Biointerfaces. 2025;250:114546. doi:10.1016/j.colsurfb.2025.114546
97. Zhang X, Chen Y, Li X, et al. Carrier-free self-assembled nanomedicine based on celastrol and galactose for targeting therapy of hepatocellular carcinoma via inducing ferroptosis. Eur J Med Chem. 2024;267:116183. doi:10.1016/j.ejmech.2024.116183
98. Li H, Deng C, Zhu N, Zhang C, Zeng Q, Qin L. An ultrasensitive GSH-specific fluorescent probe unveils celastrol-induced ccRCC ferroptosis. Bioorg Chem. 2023;134:106454. doi:10.1016/j.bioorg.2023.106454
99. Shi QL, Feng CR, Li HY, et al. Celastrol inhibits the DPYSL2-JAK/STAT pathway by targeting mito-IDHs mediated mitochondrial metabolism to exhaust breast cancer. Acta Pharmacol Sin. 2025. doi:10.1038/s41401-025-01548-0
100. Feng T, Hu G, Luo Y, et al. The combination of celastrol and curcumin enhances the antitumor effect in nasopharyngeal carcinoma by inducing ferroptosis. Biol Pharm Bull. 2025;48(7):1096–1106. doi:10.1248/bpb.b25-00053
101. Qiu CW, Chen B, Zhu HF, Liang YL, Mao LS. Gastrodin alleviates cisplatin nephrotoxicity by inhibiting ferroptosis via the SIRT1/FOXO3A/GPX4 signaling pathway. J Ethnopharmacol. 2024;319(Pt 3):117282. doi:10.1016/j.jep.2023.117282
102. Cao W, Lan J, Zeng Z, Yu W, Lei S. Gastrodin induces ferroptosis of glioma cells via upregulation of homeobox D10. Molecules. 2023;28(24). doi:10.3390/molecules28248062
103. Zhang Q, Xu Z, Liu W, et al. Gastrodin promotes ferroptosis in CRC cells by inhibiting SKP2 to reduce NCOA4 ubiquitination. Tissue Cell. 2025;95:102793. doi:10.1016/j.tice.2025.102793
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Network Pharmacology and Metabolomics Reveal Anti-Ferroptotic Effects of Curcumin in Acute Kidney Injury
Liu X, Zhou Y, Lu Z, Yang F, Wang Y, Zhang S, Zhang J, Zou H, Lin M
Drug Design, Development and Therapy 2024, 18:6223-6241
Published Date: 21 December 2024