Back to Journals » Drug Design, Development and Therapy » Volume 13
Fenofibrate modified-release pellets with lag phase and high oral bioavailability
Authors Li F, Zheng X, Bao YC, Chen T, Zeng J, Xu XL, Yan C ![]() , Feng LL
, Feng LL
Received 5 July 2018
Accepted for publication 15 October 2018
Published 24 December 2018 Volume 2019:13 Pages 141—151
DOI https://doi.org/10.2147/DDDT.S179266
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Fang Li,1,* Xin Zheng,2,* YongChu Bao,3 Ting Chen,3 Jia Zeng,1 XiaoLi Xu,4 Chao Yan,5 LingLin Feng1
1NHC Key Laboratory of Reproduction Regulation (Shanghai Institute of Planned Parenthood Research), Fudan University, and Shanghai Engineer and Technology Research Center of Reproductive Health Drug and Devices, Shanghai, China; 2Harro Hoefliger Shanghai Representative Office, Shanghai, China; 3Zhitong Laboratories Co., Ltd, Shanghai, China; 4Traditional Chinese Medicine Testing Department, Chongqing Institute for Food and Drug Control, Chongqing, China; 5Schoolof Pharmacy, Shanghai Jiao Tong University, Shanghai, China
*These authors contributed equally to this work
Purpose: Fenofibrate and statin combination therapy is highly recommended by the current clinical guidelines for treatment of mixed dyslipidemia. In this study, an innovative delayed-release preparation of fenofibrate was designed to reduce the risk of muscle toxicity, caused by simultaneous administration of this combination therapy, by altering the pharmacokinetic profile of fenofibrate, as well as to improve the oral bioavailability of the modified-release formulation.
Methods: Micronized fenofibrate was used to prepare drug-loaded cores via a powder layering process before multiparticulate pellet coating. Different coating formulations (Eudragit® RS PO/E100, Eudragit® RS PO/RL PO, Eudragit® NE30D/HPMC, and EC/HPMC) were screened, and their in vitro release was compared with the commercial sustained-release pellets Lipilfen®. Two optimized formulations were evaluated in beagle dogs using two commercial preparations of fenofibrate (the immediate-release preparation Lipanthyl® and the sustained-release pellets Lipilfen®) as references.
Results: The in vivo release of fenofibrate from R1 and R2 selected from in vitro tests exhibited a lag phase, and then rapid and complete drug release. The relative bioavailabilities of R1 and R2 were 100.4% and 201.1%, respectively, which were higher than that of Lipilfen® (67.2%).
Conclusion: The modified fenofibrate pellets developed showed enhanced bioavailability and delayed-release properties. They have the potential to improve safety and compliance when co-administrated with statins. This is the first report of a delayed-release fenofibrate preparation.
Keywords: fenofibrate, modified-release pellets, coated multiparticulate pellet, pharmacokinetics, in vivo studies
Introduction
Cardiovascular diseases (CVDs) still rank as the number one cause of death globally and are responsible for 17.5 million or 31% of all global deaths per year according to the WHO.1 Since dyslipidemias are recognized as a primary major risk factor for CVDs, aggressive lipid modulation has a positive and profound impact on the prevention and treatment of CVDs.2,3 Over the past three decades, statin therapy aimed at lowering low-density lipoprotein cholesterol (LDL-C) has become the cornerstone of reducing CVDs. The high effectiveness of statins for lowering LDL-C levels and the benefit of this approach have been clearly demonstrated by many studies.4,5 However, it has been reported that 14.1% of statin-treated patients experienced vascular events compared with 17.8% of control subjects, indicating a high residual relative risk of 79% based on the fact that statin therapy alone has less substantial effect on other lipid fractions.6
The residual risk is largely attributed to other lipid abnormalities including high levels of triglycerides (TG) and low concentration of high-density lipoprotein cholesterol (HDL-C). This abnormal lipid profile is a characteristic of mixed dyslipidemia, which is an increasingly common type of dyslipidemia and is more prevalent in the growing population of patients with type 2 diabetes mellitus and metabolic syndrome.6,7 Owing to the limitations of statin monotherapy, some more appropriate and effective therapeutic strategies have been explored for the treatment of mixed hyperlipidemia. For example, combination of fenofibrate and statins is highly recommended by the current clinical guidelines.7
Fenofibrate is another widely used treatment for dyslipidemia. Unlike statins, fenofibrate monotherapy tends to significantly improve TG and HDL-C levels.8,9 Furthermore, it has nonlipid, pleiotropic effects (eg, reducing levels of fibrinogen, improving flow-mediated dilatation, improving insulin sensitivity, and reducing urine albumin concentrations) that may contribute to reducing coexisting symptoms in the same patients.10,11 Therefore, the combination of fenofibrate and statin for the treatment of dyslipidemia could produce not only complementary but also synergistic effects and could be particularly favorable for patients with type 2 diabetes mellitus or metabolic syndrome. In the lipid component of the Action to Control Cardiovascular Risk in Diabetes trial, the only complete outcome of the study on combination therapy showed that, compared with simvastatin monotherapy, fenofibrate–simvastatin combination therapy reduced the CVD risk by 31% in a subgroup of diabetic patients with increased TG levels and reduced HDL-C levels. However, the trial was not specifically designed for mixed dyslipidemia with HDL-C and TG below or above a specific cutoff value.12,13
Based on these encouraging benefits, fenofibrate and statins are frequently co-prescribed in clinical practice. However, both fenofibrate and statin monotherapy are associated with increased myopathy risk, indicating potentially greater side effects when these drugs are co-administered, particularly if the doses are high. Therefore, patients who require combination therapy should be advised on the increased risk of muscle toxicity. In order to reduce the potential side effects, instead of simultaneous combination, fenofibrate and statins should preferably be taken in a staggered manner (eg, statins given in the evening and fenofibrate in the morning) to minimize peak dose.14,15 However, a prolonged staggered regimen increases the possibility of mistaken or missed drug doses, leading to poor patient compliance. Fenofibrate modified-release preparations, which delay or sustain release of fenofibrate for more than 8 hours, are expected to overcome this problem by preventing the two drugs reaching their maximum plasma concentrations at the same time after simultaneous administration. Good patient compliance and low risk of side effects therefore make modified-release fenofibrate formulations an attractive approach to achieving overall lipid control.
And yet, few studies have reported the development of modified-release fenofibrate preparations. Low bioavailability is one of the significant challenges in the development of modified-release fenofibrate preparations. Fenofibrate, which belongs to Biopharmaceutical Classification System class II, is poorly water soluble but highly permeable, and dissolution is the rate-limiting step in drug absorption. Fenofibrate was initially approved as a standard formulation of 100 mg capsules, which was administrated three times daily. Owing to poor compliance and bioavailability, this standard formulation is rarely used and the bioavailability of fenofibrate has subsequently been improved by other formulations such as a 250 mg sustained-release capsule, Lipilfen®, a 200 mg micronized fenofibrate capsule, Lipanthyl®, and a 145 mg nanonized tablet, Tricor®. Many strategies such as micronization and nanoparticle technology, solid dispersion technology, and emulsifying technology have been used to enhance the bioavailability of fenofibrate.16–19 However, these methods were generally used for the development of immediate-release preparations, and their high bioavailability relied heavily on a rapid release rate. Conversely, the delayed or sustained release of fenofibrate generally prevents enhancement of bioavailability. Therefore, the development of modified-release fenofibrate pellets with high bioavailability is considered more difficult than development of immediate-release pellets.
The objective of the present research was to develop a fenofibrate modified-release formulation using coated multiparticulate pellet technology, which could achieve 8 hours delayed release. To increase the bioavailability, micronized fenofibrate and a powder layering process were employed to prepare drug-loaded cores. To achieve delayed and complete release, different coating formulations (Eudragit® RS PO/E100, Eudragit® RS PO/RL PO, Eudragit® NE30D/HPMC, and EC/HPMC) were screened and their in vitro release was compared with that of the sustained-release pellets Lipilfen®. Furthermore, two optimized coating formulations were evaluated in an in vivo pharmacokinetic study using two commercial preparations (the immediate-release product Lipanthyl® and sustained-release pellets Lipilfen®) as controls.
Materials and methods
Materials
Fenofibrate was purchased from Jiangsu Nhwa Pharmaceutical Co. (Xuzhou, China). Fenofibric acid and 4-chlorophenyl-4′-hydroxyphenyl methanone (internal standard) were acquired from Zhejiang Yidu Bio-tech Co. (Jiangshan, China). Commercial capsules of Lipilfen® and Lipanthyl® were purchased from Shanghai Ethypharm Pharmaceuticals Co. (Shanghai, China) and Laboratories Fournier (Dijon, France), respectively. Sucrose/starch nonpareil seeds (0.4–0.6 mm) were purchased from Hangzhou Gaocheng Biological Nutrient Technology Co. (Hangzhou, China). Aerosil® 200, Eudragit® RS PO, Eudragit® E100 (Evonik, Essen, Germany), hydroxypropyl methylcellulose (HPMC) E5, ethyl cellulose (EC) 10 cPs (Dow Chemical Co., Midland, MI, USA), HPMC 6 cPs (Shin-Etsu Chemical Co., Tokyo, Japan), triethyl citrate (TEC [A Johnson Matthey, London, UK]), talc (passed through 200 mesh; Sichuan Serpentine Mineral Factory, Sichuan, China), polyethylene glycol (PEG) 4000, and lactose monohydrate (SLS, chemical grade) (Sinopharm Chemical Reagent Co., Shanghai, China) were used. Ethyl acetate, n-hexane, acetonitrile, and methanol were of chromatographic grade. All other reagents were of analytical grade or better.
Solubility determination
An excess of fenofibrate was added to test tubes containing 10 mL of different solvents, including distilled water, aqueous solution containing 0.5% (w/v) and 0.1% (w/v) Tween 80, and aqueous solution containing 0.5%, 1%, 2%, and 2.88% (w/v) (ie, 0.1 mol/L) SLS, and then shaken in a water bath at 25°C for 24 hours. The suspensions were filtered through a 0.45 μm membrane filter and diluted with methanol. The fenofibrate content was analyzed using HPLC-UV system.
Micronized fenofibrate preparation
Micronized fenofibrate was prepared using a JGM-H100 jet mill (Shanghai HLSF Technology Co. Ltd, Shanghai, China) at 8 MPa air pressure and a feed rate of 3 kg/h. Jet milling was conducted twice sequentially, and then the particle size distribution of the milled and unmilled drug powder was measured using Mastersizer MS 2000 (Malvern Instruments, Malvern, UK).
Angle of repose measurement
The angle of repose was determined with a Granulate Flow Tester (GT; ERWEKA GmbH, Heusenstamm, Germany) according to ISO4324-1977. About 150 mL of each powder was poured into a funnel and then allowed to pass through a nozzle onto a balance-plate. Angle of repose was calculated as the angle of the powder’s cone to the plate. The measurements were performed in triplicate.
Drug-loaded pellet core preparation
The drug-loaded pellet cores were prepared by powder layering using a tangential-spray GPCG1.1 rotary fluidized-bed granulator (Glatt, Binzen, Germany). The mixture of fenofibrate and Aerosil® 200 (96/4, w/w) powders was applied to sucrose/starch nonpareil seeds (0.4–0.6 mm in diameter) through a powder feeder at a rate of 25 g/min until 70% drug loading was achieved (ie, the drug loading is as same as that of Lipilfen®). The rotor speed was maintained at 518 rpm. The inlet air and product temperatures were controlled at 45°C–48°C and 28°C–30°C, respectively. The binder solution containing 5% HPMC was applied through a 1.0 mm nozzle at a rate of 15 g/min. After layering, the pellets were passed through a 40-mesh sieve to remove undersized material.
Film-coated pellet preparation
The drug-loaded cores were transferred into a Mini-Glatt fluidized-bed coater (Glatt, Binzen, Germany). Four different coating systems (R1–R4 in Table 1) were applied to the same drug-loaded cores to achieve the desired cumulative release profile. The polymer dispersions (R1–R3) were prepared by adding the polymers to 80% (v/v) ethanol solution, followed by stirring overnight. R4 was used as its aqueous dispersion. After coating with polymer dispersion, the pellets were removed and dried at 40°C for 2 hours in an air-circulated oven for further study. Where necessary, Aerosil® 200 was added to prevent the sticking of pellets during storage.
 | Table 1 Formulations and coating procedures for different coating systems |
In vitro characterization
Differential scanning calorimetry (DSC)
The drug-loaded cores were milled into powders using a mortar before DSC analysis. The powders were in sealed aluminum pans fitted with perforated lids. The thermal properties of unmilled fenofibrate powder and milled drug-loaded core powder were analyzed using a Thermal Analyzer-60WS, DSC-60 (Shimadzu Corp., Kyoto, Japan). The temperature range for the measurements was 40°C–90°C, at a heating rate of 5°C/min.
Scanning electron microscopy
The morphological characteristics of the drug-loaded cores and Lipilfen® were evaluated by environmental scanning electron microscopy (SEM; FEI Quanta 250 FEG; FEI Inc., Hillsboro, OR, USA). The samples were secured to a brass specimen stub using double-side adhesive tape. A Leica ion sputter coater (EM SCD050; Leica Microsystems, Wetzlar, Germany) was used to make the pellets electrically conductive. The stub was coated with platinum at 40 mA in vacuum (<10−1 mbar) for 60 seconds.
Drug content analysis
The drug content of the samples was analyzed using a Shimadzu HPLC-UV system (Shimadzu Corp., Kyoto, Japan) with a Shim-pack VP-ODS (4.6×250 mm, 5 μm) column at a detection wavelength of 286 nm. The mobile phase consisted of methanol–water (80:20, v/v). The flow rate was 1 mL/min, and the injection volume was 20 μL.
Approximately 600 mg of pellets was ground to fine powder using a mortar and pestle before quantification. Approximately 30 mg of powder was then accurately weighed and added to a 100 mL volumetric flask containing 80 mL of mobile phase. After ultrasonic extraction for 10 minutes, the volume was adjusted to 100 mL with mobile phase. The sample solution was prepared by diluting 1 mL of the above solution with 9 mL of mobile phase and analyzed by HPLC.
In vitro release study
Dissolution testing of fenofibrate pellets was conducted using the USP 37 Apparatus II dissolution method (paddle method, RCZ-8B; Tianjin Tianda Tianfa Technology Co., Ltd., Tianjin, China). Briefly, ~360 mg of pellets (equivalent to 250 mg of fenofibrate) was added to 1,000 mL of dissolution medium containing 0.1 mol/L SLS at 37°C, with a paddle rotation speed of 120 rpm. At predetermined time points, 2 mL samples were withdrawn and centrifuged at 16,000 rpm for 8 minutes before HPLC analysis.
The similarity factor (F2) was used to compare release profiles using the following equation:
|
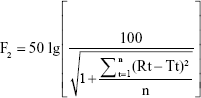
where:
Rt = percentage of drug dissolved at each time point for the reference;
Tt = percentage of drug dissolved at each time point for the test; and
n = number of dissolution sampling time points.
If F2 is ≥50, then the dissolution profiles are considered similar. The larger the F2 value, the greater the similarity.
In vivo pharmacokinetics
Animals and dosing
A single-dose, randomized, four-way crossover study was performed. Four male beagle dogs weighing 15±1 kg were fasted for at least 12 hours before dosing and 4 hours post-dosing and allowed free access to water throughout the experiment. A washout period of 10 days was used for consecutive dosing. The study protocol was approved by the ethics committee of Shanghai Institute of Pharmaceutical Industry (No 2011-009) and followed the National Institutes of Health guide for the care and use of laboratory animals.
Capsules of R1, R2, Lipilfen®, and Lipanthyl® (250 mg, expressed as fenofibrate equivalents) were orally administered. Blood samples (4 mL) were collected from a forearm vein with heparinized syringes at 0 hour (pre-dose) and 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, and 72 hours post-dose. Plasma samples were obtained by centrifugation of the blood samples at 4,000 rpm for 5 minutes and stored at −18°C until use.
Preparation of plasma samples
Plasma sample (800 μL), 20 μL of internal standard solution (25 μg/mL 4-chlorophenyl-4′-hydroxyphenyl methanone in methanol), and 200 μL of hydrochloric acid solution (5 mol/L in distilled water) were added to a 5 mL polyethylene centrifuge tube, vortexed for 5 minutes, and then mixed with 3 mL of solvent (n-hexane and ethyl acetate, 9:1, v/v) for 5 minutes. The mixture was then centrifuged at 13,000 rpm for 5 minutes. Then, 2.5 mL of supernatant was transferred to a 5 mL polyethylene centrifuge tube and evaporated to dryness at 40°C under a gentle stream of oxygen-free nitrogen. The residue was then reconstituted with 200 μL of mobile phase, and an aliquot of 100 μL was injected into the HPLC-UV system.
Chromatography and method validation
The concentration of fenofibrate acid in plasma was determined by HPLC. A Shim-pack VP-ODS column (4.6 mm×150 mm, 5 μm), protected by a guard column (Shim-pack Column Holder, 4.6 mm×10 mm, 5 μm) was used. The mobile phase consisted of acetonitrile–phosphoric acid solution (pH 2.5) (40:60, v/v). The flow rate was 1.2 mL/min. The detection wavelength was 295 nm. To support the pharmacokinetic studies of fenofibrate acid in beagle dog, method validation was performed in terms of determining specificity, linearity, precision, and accuracy. Calibration curves were generated by five standards. Precision and accuracy were assessed by determination of quality control samples using five replicate preparations of rat plasma samples at three concentration levels (low, 0.02 μg/mL; medium, 0.25 μg/mL; and high, 5 μg/mL for fenofibrate acid) in 3 validation days each with independently prepared calibration curves.
Data analysis
The maximum peak concentration (Cmax) and the time to reach maximum plasma concentration (Tmax) were obtained directly from the plasma concentration–time profile. Other pharmacokinetic parameters were computed using the DAS 2.0 software program. The relative bioavailability (FX) was calculated using the equation FX = (AUC(0–t)T/AUC(0–t)R)×100%, where AUC(0–t)T and AUC(0–t)R are the area under the curve after oral administration for the test and reference, respectively.
Results and discussion
Micronized drugs
Fenofibrate raw material is a crystalline powder with a mean particle diameter of 169.6 μm (Figure 1A). This large particle size is unfavorable for the chosen powder layering process, and using smaller drug particles could improve the layering efficiency.20 In this study, the jet milling micronization technique was selected to reduce the particle size of fenofibrate to smaller than 10 μm. The processed fenofibrate particles (with a mean particle diameter of 7.7 μm) were suitable for the powder layering process (with a layering efficiency of 90%) (Figure 1A). It should be noted that although fenofibrate was milled to micrometer size, the DSC results showed that it retained crystallinity (Figure 1B). There are numerous other studies that use smaller drug particles, even in the nanosize range, to improve the bioavailability of fenofibrate preparations.21–24 However, small drug particle sizes can lead to increased agglomeration during the powder layering process. In addition, further reducing the size of drug particles requires more complex processing and advanced equipment. A drug particle size <10 μm rather than on the nanoscale was therefore used for this study.
 | Figure 1 Comparison of unprocessed and milled fenofibrate powders. |
Drug loading of cores
There are two approaches to drug loading of cores in a fluidized-bed, solution/suspension layering and powder layering. The latter is suitable for all kinds of drugs with different solubility properties and is particularly suitable for drugs required in high doses. In this study, the oral dose of fenofibrate (250 mg/day) was high, while the solubility of fenofibrate in distilled water at 25°C (<0.02 μg/mL, Table 2) was low. Powder layering was therefore chosen for the preparation of fenofibrate pellet cores.
 | Table 2 Solubility of fenofibrate in different solvents (n=3) |
Although using micronized drug powder can improve layering efficiency, as previously mentioned, drug powders in the micrometer size range usually have poor flowability. Therefore, addition of glidant into micronized drug powders is usually necessary. The angle of repose of micronized drug powder was 47.1°±0.4°, indicating poor flowability. After it was mixed with 2% and 4% Aerosil® 200, the angle of repose decreased to 40.3°±0.3° and 37.7°±1.3°, respectively. Agglomeration was not observed when using 4% Aerosil® 200, but was noted twice during the powder laying process when using only 2% Aerosil® 200. In addition to its glidant effect, Aerosil® 200 can also be used as an anticaking agent to prevent powders from clumping during storage. Therefore, 4% Aerosil® 200 was used. The powder laying process was stable and reproducible, producing homogeneous products with a layering efficiency of 90%, drug loading of 70%, and bulk density of 0.6 g/mL. The prepared drug-loaded cores were spherical, and the drug layer was found to be loose relative to the core as observed by SEM (Figure 2).
 | Figure 2 SEM micrographs of drug-loaded cores. |
Effect of pellet coating film system
The in vitro release of fenofibrate from the uncoated pellets was very fast, similar to that of the immediate-release preparation Lipanthyl® (Figure 3). Coating the pellets with a film was therefore necessary to modify the release of fenofibrate. In this study, four different types of film systems were investigated, including: 1) Eudragit® RS PO/E100, 2) Eudragit® RS PO/RL PO, 3) Eudragit® NE30D/HPMC, and 4) EC/HPMC. In addition, Lipilfen®, the only marketed fenofibrate modified-release product, was used as a positive control. When Eudragit® NE30D was used as the coating film, the fenofibrate formulation exhibited an inappropriate “too slow to release completely” profile (Figure 4, R4). In the late phase, the cumulative amount of the formulation released was far less than for Lipilfen® (7 hours: 61.9% vs 90.4%), indicating poor bioavailability even though delayed or sustained drug release was achieved. By comparison, formulations prepared using the other three coating film systems showed better release profiles (Figure 4, R1, R2, and R3). The F2 factors of the R1 and R2 formulations compared with Lipilfen® were 72 and 62, respectively, higher than that of R3 (F2 factor 57), indicating that the release of R1 and R2 was more similar to Lipilfen®. These two formulations were therefore selected for the subsequent in vivo pharmacokinetic studies. The effects of coating weight and the weight ratio of the polymers in the coating film systems on the release of R1 and R2 were investigated. As shown in Figure 5, the release rate of fenofibrate increased with decreasing coating weight and increasing ratio of the water-insoluble component.
 | Figure 3 In vitro release of two immediate-release preparations (n=3). |
 | Figure 4 In vitro release of fenofibrate pellets with four different film coating systems and Lipilfen® (n=3 or 12). |
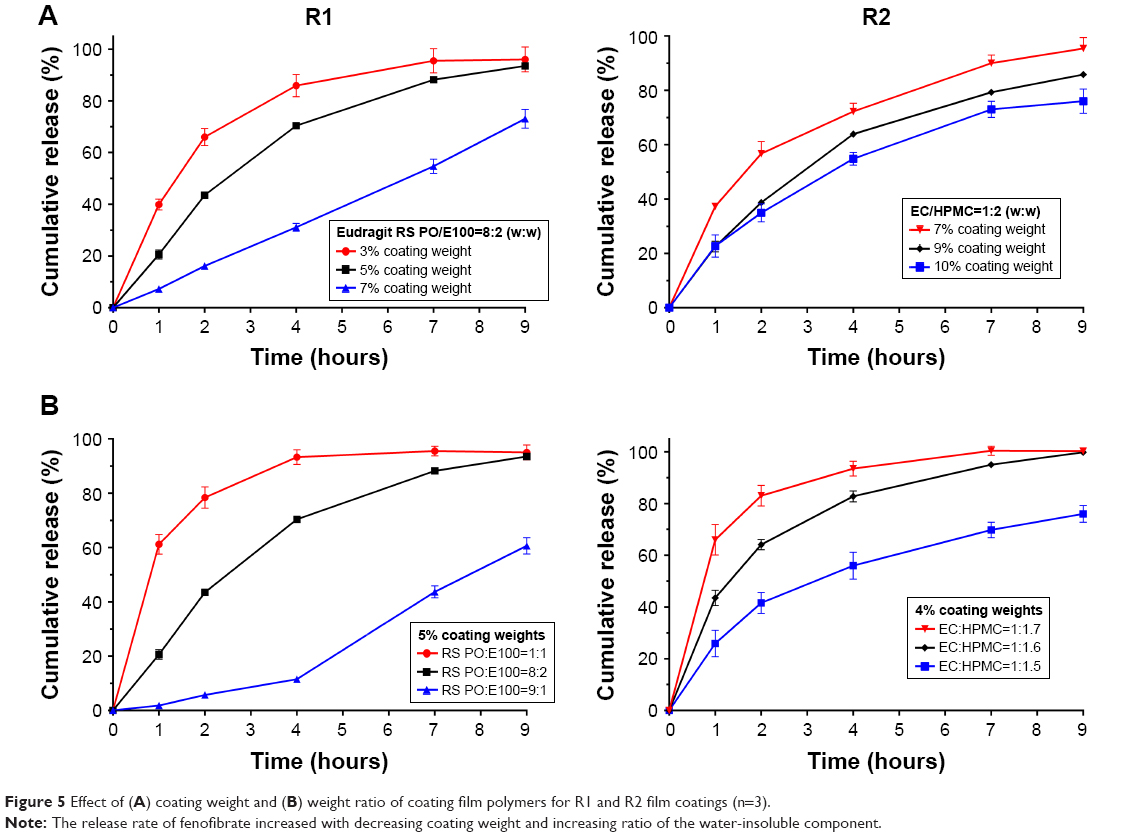 | Figure 5 Effect of (A) coating weight and (B) weight ratio of coating film polymers for R1 and R2 film coatings (n=3). |
Dissolution of fenofibrate pellets
Fenofibrate is a poorly water-soluble drug. To increase the solubility of fenofibrate in the media, the effect of different solubilizers was investigated. As shown in Table 2, compared with Tween 80, addition of SLS in the media resulted in higher fenofibrate solubility. In this study, 1,000 mL of 0.1 mol/L SLS was used as the dissolution media, and these sink conditions for dissolution of the fenofibrate pellet were also applied to Lipilfen®, the commercial sustained preparation of fenofibrate.
Pharmacokinetic study of fenofibrate pellets in beagle dogs
Fenofibrate is in fact a prodrug that is rapidly metabolized after absorption, mainly to its active metabolite, fenofibric acid. It has been reported that almost no unchanged fenofibrate can be detected in blood plasma after administration of an oral dose.25,26 Therefore, the pharmacokinetic study of fenofibrate focused on the plasma concentration–time profiles of fenofibric acid.
Under the chromatographic conditions described previously, optimized separation and detection conditions were achieved in plasma26 and method validation was performed.
- The method was specific and selective with no interference from endogenous material present in dog plasma at the retention times of fenofibric acid and the internal standard 4-chlorophenyl-4′-hydroxyphenyl methanone (Figure 6).
- Results were calculated using the peak area ratios of fenofibric acid and an internal standard. The calibration curve, generated using a weighted (1/Y) linear least-squares regression (Y=0.726X–0.002, r=1.000, n=5), showed linearity from 0.02 μg/mL (the lower limit of quantification) up to 5.00 μg/mL (the upper limit of quantification) for fenofibric acid in rat plasma.
- The precision and accuracy of the quantification method for fenofibrate met the acceptable criteria. The mean intra-assay precision (%CV) for fenofibric acid was 5.3%, 5.7%, and 5.1% at low, medium, and high levels, respectively; inter-assay precision for fenofibric acid was 11.8%, 5.3%, and 5.5% at low, medium, and high levels, respectively. The method recovery was 90.1%, 99.4%, and 97.8% at low, medium, and high levels, respectively.
 | Figure 6 Typical HPLC chromatograms of (A) blank beagle dog plasma, (B) blank beagle dog plasma spiked with the internal standard, (C) blank beagle dog plasma spiked with fenofibric acid, and (D) plasma sample after oral administration of fenofibrate preparation spiked with the internal standard. |
The pharmacokinetics of fenofibric acid after oral administration of R1, R2, Lipilfen® (commercial sustained pellets), and Lipanthyl® (commercial immediate-release preparation) was investigated based on the same dose of fenofibrate (Figure 7; Table 3).
 | Figure 7 Mean plasma concentration–time profiles for fenofibric acid in beagle dogs after oral administration of marketed fenofibrate products and two fenofibrate delayed-release pellets (equivalent to 250 mg of fenofibrate) (n=3 or 4). |
 | Table 3 Pharmacokinetic parameters for fenofibric acid after oral administration of marketed fenofibrate products and two fenofibrate delayed-release pellets in beagle dogs |
Following administration, Lipanthyl® showed a typical immediate-release profile. Specifically, the plasma level of fenofibric acid in beagle dogs increased quickly after oral administration of Lipanthyl® and reached the maximum concentration (1.562 μg/mL) at 1.7 hours, followed by a marked decrease between 1.7 and 4 hours.
By contrast, Lipilfen® exhibited typical sustained-release characteristics. After oral administration, the plasma level of fenofibric acid gradually reached the maximum concentration (0.721 μg/mL) at 10.8 hours, and then decreased at a very slow rate. The plasma drug level was maintained at a relatively constant concentration. However, the bioavailability of Lipilfen® was much lower compared with that of Lipanthyl®, only 67.2%.
In the R1 and R2 dosing groups, a lag release phase was observed (for R1, almost non-release in 8 hours; for R2, sustained release). The plasma drug level then increased quickly to reach maximum concentrations at 11–12 hours (1.175 and 2.486 μg/mL for R1 and R2, respectively). Compared with Lipanthyl®, the relative bioavailabilities of R1 and R2 were 100.4% and 201.1%, respectively. The bioavailability of R1 was equivalent to Lipanthyl®, and the bioavailability of R2 was twice that of Lipanthyl®.
Although the drug release was delayed by almost 8 hours, the modified pellets developed (R1 and R2) had enhanced bioavailability. The improved bioavailability of R1 and R2 could be attributed to the immediate drug release in the late phase, which could be a result of the following factors:
- The high permeability of the coating films did not slow down the influx of water into the particles. In R1, Eudragit® E100, one of the film components, could be easily dissolved below pH 5. In R2, a high ratio of water-soluble polymer HPMC was present in the film. It is therefore speculated that water could infuse into the particles from the beginning of the lag phase leading to concurrent exposure of fenofibrate to the intestinal fluid, although drug release was not observed owing to the hydrophobicity of fenofibrate.
- Both the increased specific surface area of the microsize particles and the loose structure of the drug layer produced by the powder layering process were favorable for improving the wettability of the drug particles in the lag phase. Once the lag phase was over, an immediate release of fenofibrate was possible as the drug particles had been pre-wetted.
- HPMC used in R2 helped to further improve absorption. The release of poorly soluble drugs is often associated with the generation of supersaturation, which implies the risk of drug precipitation and reduced availability for absorption. Polymer HPMC was considered as the precipitation inhibitors to stabilize the supersaturated drug solution in the small intestine, and then increased the availability.27
The results revealed that the fenofibrate preparations (R1 and R2) achieved clear delayed-release properties and high bioavailability.
Compared with the in vivo results, the dissolution method requires further development. Although the in vitro dissolution method could differentiate the immediate-release and modified-release preparations, it was unable to differentiate the delayed-release and sustained-release preparations (ie, R1 and R2). In fact, it is often challenging to determine whether or how a change in dissolution observed may impact in vivo performance in the absence of an in vitro-in vivo correlation (IVIVC), because the relationship between in vitro behaviors and in vivo performance of modified-release drug products is complex and dependent highly on many variables related to drug substance, product design, formulation, test method, and their interplays. Lin et al also showed four possible scenarios linking in vitro drug release to in vivo performance, including the case that formulations passing F2 criteria exhibited different in vivo performance.28 In the current study, other dissolution conditions such as different pH and different paddle rotation speed were optimized (data not shown), but establishing IVIVC for the modified-release fenofibrate pellets remains a challenge.
It should also be pointed out that elimination half-life (t1/2z) of four formulations reported in Table 3 should not differ much, because t1/2z for a given drug in a given person (or animal) is not changed by the route of administration. In our current study, by using the DAS 2.0 software program, the final four data points (24, 36, 48, and 72 hours) were chosen as default to compute Ke. Some of these final four data points may still be in the distribution phase because fenofibrate has a very long half-life. So, the estimate of Ke is not as accurate as the estimate from the four points selected during the elimination phase. To minimize errors in estimates, more data points during the elimination phase should be needed in the pharmacokinetic study.
Conclusion
The modified fenofibrate pellets developed showed enhanced bioavailability and delayed-release properties. They have the potential to improve safety and compliance when co-administrated with statins. This is the first report of a delayed-release fenofibrate preparation.
Acknowledgments
This work was supported by the National Key Technologies R&D Program of China during the 13th Five-Year Plan Period (grant No. 2016YFC1000902) and the Scientific Research Project of Shanghai Municipal Health and Family Planning Commission (grant number: 20144Y0049 and 201540357). The authors thank Sarah Dodds, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
WHO. Global status report on noncommunicable diseases. 2014. Available from: http://www.who.int/nmh/publications/ncd-status-report-2014/en/. Accessed October 17, 2017. | ||
Jellinger PS, Handelsman Y, Rosenblit PD, et al. American association of clinical endocrinologists and American college of endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract. 2017;23(Suppl 2):1–87. | ||
Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care. 2013;40(1):195–211. | ||
Ridker PM, Rose L, Cook NR. A proposal to incorporate trial data into a hybrid ACC/AHA algorithm for the allocation of statin therapy in primary prevention. J Am Coll Cardiol. 2015;65(9):942–948. | ||
Stone NJ, Robinson JG, Lichtenstein AH. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American college of cardiology/American heart association task force on practice guidelines. J Am Coll Cardiol. 2014;63(25 part B):2889–2934. | ||
Miller M. Dyslipidemia and cardiovascular risk: the importance of early prevention. QJM. 2009;102(9):657–667. | ||
Koh KK. How to control residual risk during the statin era? J Am Coll Cardiol. 2015;66(16):1848. | ||
Jacobson TA, Ito MK, Maki KC, et al. National lipid association recommendations for patient-centered management of dyslipidemia: part 1 – full report. J Clin Lipidol. 2015;9(2):129–169. | ||
Tsunoda F, Asztalos IB, Horvath KV, Steiner G, Schaefer EJ, Asztalos BF. Fenofibrate, HDL, and cardiovascular disease in Type-2 diabetes: The DAIS trial. Atherosclerosis. 2016;247:35–39. | ||
MKeage K, Keating GM. Fenofibrate: a review of its use in dyslipidaemia. Drugs. 2011;71(14):1917–1946. | ||
Knickelbein JE, Abbott AB, Chew EY. Fenofibrate and diabetic retinopathy. Curr Diab Rep. 2016;16(10):90. | ||
Athyros VG, Papageorgiou AA, Athyrou VV, Demitriadis DS, Kontopoulos AG. Atorvastatin and micronized fenofibrate alone and in combination in type 2 diabetes with combined hyperlipidemia. Diabetes Care. 2002;25(7):1198–1202. | ||
Durrington PN, Tuomilehto J, Hamann A, Kallend D, Smith K. Rosuvastatin and fenofibrate alone and in combination in type 2 diabetes patients with combined hyperlipidaemia. Diabetes Res Clin Pract. 2004;64(2):137–151. | ||
Reiner Ž, Catapano AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias. Revista Española de Cardiología. 2011;64(12):1168–1168.e60. | ||
Shao K, Tang YB, Zhou D. Comparison of the safety of statin monotherapy and coadministration with fenofibrate in patients with mixed hyperlipidemia: a meta-analysis. International Journal of Clinical and Experimental Medicine. 2016;9:5291–5300. | ||
Yousaf AM, Mustapha O, Kim DW, et al. Novel electrosprayed nanospherules for enhanced aqueous solubility and oral bioavailability of poorly water-soluble fenofibrate. Int J Nanomedicine. 2016;11:213–221. | ||
Chaudhary RS, Amankwaa E, Kumar S, Hu T, Chan M, Sanghvi P. Application of a hot-melt granulation process to enhance fenofibrate solid dose manufacturing. Drug Dev Ind Pharm. 2016;42(7):1137–1148. | ||
Bouledjouidja A, Masmoudi Y, van Speybroeck M, Schueller L, Badens E. Impregnation of Fenofibrate on mesoporous silica using supercritical carbon dioxide. Int J Pharm. 2016;499(1–2):1–9. | ||
Pongsamart K, Kleinebudde P, Puttipipatkhachorn S. Preparation of fenofibrate dry emulsion and dry suspension using octenyl succinic anhydride starch as emulsifying agent and solid carrier. Int J Pharm. 2016;498(1–2):347–354. | ||
Suhrenbrock L, Radtke G, Knop K, Kleinebudde P. Pellet layering: scale-up considerations using different kinds of processing equipment. Drug Dev Ind Pharm. 2012;38(12):1494–1503. | ||
Vogt M, Kunath K, Dressman JB. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: comparison with commercial preparations. Eur J Pharm Biopharm. 2008;68(2):283–288. | ||
Weng T, Qi J, Lu Y, et al. The role of lipid-based nano delivery systems on oral bioavailability enhancement of fenofibrate, a BCS II drug: comparison with fast-release formulations. J Nanobiotechnology. 2014;12:39. | ||
Mochalin VN, Sagar A, Gour S, Gogotsi Y. Manufacturing nanosized fenofibrate by salt assisted milling. Pharm Res. 2009;26(6):1365–1370. | ||
Shegokar R, Müller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399(1–2):129–139. | ||
Hanafy A, Spahn-Langguth H, Vergnault G, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–426. | ||
Lovin I, Albu F, Tache F, David V, Medvedovici A. Solvent and salting effects on sample preparation for the determination of fenofibric acid in human plasma by HPLC-DAD. Microchemical Journal. 2003;75(3):179–187. | ||
Van Speybroeck M, Mols R, Mellaerts R, et al. Combined use of ordered mesoporous silica and precipitation inhibitors for improved oral absorption of the poorly soluble weak base itraconazole. Eur J Pharm Biopharm. 2010;75(3):354–365. | ||
Lin Z, Zhou D, Hoag S, Qiu Y. Influence of drug properties and formulation on in vitro drug release and biowaiver regulation of oral extended release dosage forms. AAPS J. 2016;18(2):333–345. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.