Back to Journals » Lung Cancer: Targets and Therapy » Volume 11
Favorable Immune Microenvironment in Patients with EGFR and MAPK Co-Mutations
Authors Yang W, Chen N, Li L, Chen X, Liu X, Zhang Y, Cui J
Received 3 July 2020
Accepted for publication 13 August 2020
Published 7 September 2020 Volume 2020:11 Pages 59—71
DOI https://doi.org/10.2147/LCTT.S262822
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Wang Yang, Naifei Chen, Lingyu Li, Xiao Chen, Xiangliang Liu, Yongfei Zhang, Jiuwei Cui
The Cancer Center of the First Hospital of Jilin University, Changchun, Jilin 130021, People’s Republic of China
Correspondence: Jiuwei Cui Tel +86 431-8878-2178
Fax +86-0431-88786134
Email [email protected]
Purpose: Although EGFR-mutated patients generally do not benefit from checkpoint inhibitors (ICIs), some patients in the KEYNOTE-001 study consistently benefited from this treatment. This study investigated immune microenvironment characteristics to identify the subgroup of patients that may benefit from ICIs.
Materials and Methods: Using data from The Cancer Genome Atlas Program (TCGA) and Cancer Proteome Atlas, TMB and protein level of PD-L1 were explored in the patients with EGFR mutations and wild-type patients. Different patterns of EGFR mutations (according to EGFR co-mutation with different downstream pathway genesets) were used to group EGFR mutation population. Estimated infiltration analyses were used to explore changes in the immune microenvironment.
Results: This study analyzed somatic mutation data from 1287 patients from five cohorts (TCGA, Broad, The Tumour Sequencing Project, Memorial Sloan Kettering Cancer Center, Catalogue Of Somatic Mutations In Cancer database). The probability of EGFR mutation was approximately 14.30% (184/1287) and the co-mutation rate was 11.41% (21/184) in patients with EGFR mutations. Glycosaminoglycan-related pathways were significantly upregulated in the EGFR mutant group. EGFR-mutated patients had lower TMB and PD-L1 protein levels than those in wild-type patients. Increase immature DCs infiltration and decreased NK CD56dim, T gamma delta, cytotoxic, and Th2 cell infiltration were the main immune changes in EGFR-mutated patients. Patients with EGFR-MAPK co-mutations had higher levels of TMB and PD-L1 protein expression. Meanwhile, the co-mutated patients had a similar immune microenvironment as that in wild-type patients.
Conclusion: In this study, we defined a subgroup of patients with EGFR-MAPK co-mutations. These co-mutated patients may benefit from ICI treatment.
Keywords: epidermal growth factor receptor, EGFR mutation, tumor mutation burden, TMB, immune checkpoint inhibitor, ICI, lung adenocarcinoma, LUAD
Introduction
Lung cancer is the leading cause of cancer-related morbidity and mortality.1 Non-small cell lung cancer (NSCLC) accounts for approximately 80% of all lung cancers and mainly comprises lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC). Epidermal growth factor receptor (EGFR) mutations are common in LUAD. The incidence of EGFR mutations in LUAD is as low as 12%, while in the Asia-Pacific region, the prevalence is approximately 50%.2 The first-generation EGFR tyrosine kinase inhibitor (TKI) erlotinib has shown remarkable efficacy in the treatment of NSCLC, opening the door to a generation of driver gene-positive targeted therapies.3 However, drug resistance is the primary challenge of EGFR-TKIs, with acquired resistance occurring in almost all patients within approximately one year.4 While immune checkpoint inhibitors (ICIs) significantly improved the survival of patients with NSCLC, they did not provide benefits to patients with EGFR mutations.5
Thus, EGFR-mutated LUAD faces two dilemmas: targeted therapy resistance and ineffective immunotherapy. EGFR-TKI cannot prevent downstream signal-mediated resistance mechanisms.6 EGFR mutations combined with downstream pathway mutations downregulated the efficacy of EGFR-TKI,7 suggesting the vital role of EGFR downstream signal transduction in the therapeutic effects in EGFR-mutated LUAD. Although NSCLC patients with EGFR mutations do not generally benefit from ICIs, some EGFR-mutated patients in the KEYNOTE-001 study benefited from immunotherapy.8 Multiple EGFR mutations subtypes have been reported, which may affect TKI treatment.9 Thus, the EGFR mutation masked a group of people who benefited from ICI treatment due to EGFR mutation heterogeneity.
The heterogeneity of EGFR mutations has gradually revealed in recent years. The tumor mutation burden (TMB) was first used as a valid predictor of ICI benefit.10 Studies have shown a lower TMB11 and the programmed death-ligand 1 (PD-L1) tumor proportion score (TPS) in NSCLC patients with EGFR mutations, suggesting that EGFR-mutated patients are not suitable for ICI therapy.12 Furthermore, the tumor immune microenvironment is closely related to the response to ICI therapy.13 An immunosuppressive microenvironment could partially explain ICI ineffectiveness. The proportion of PD-L1+/CD8+ tumor-infiltrating lymphocytes (TILs) in EGFR-mutated NSCLC was reportedly lower than that in wild-type NSCLC; therefore, patients with EGFR mutations could not benefit from ICI.14 However, patients with L858R had a higher TMB than those with exon 19 deletions; thus, patients with L858R might benefit more from ICI.15,16 Furthermore, Wu et al reported disease control in a patient with EGFR and KRAS mutations after treatment with erlotinib, bevacizumab, and nivolumab,17 suggesting that EGFR mutations could benefit from ICI therapy.
The massive data resources of The Cancer Genome Atlas Program (TCGA), the Cancer Proteome Atlas (TCPA),18 cBioPortal,19,20 and Catalogue Of Somatic Mutations In Cancer (COSMIC) database21 were used to explore whether patients with EGFR mutations could benefit from ICI treatment. Furthermore, the Reactome22 or Kyoto Encyclopedia of Genes and Genomes (KEGG)23 database provided a detailed overview of disease pathways. This study sought to identify potential beneficiaries of ICI therapy and elucidate possible mechanisms based on an exploration of mutation patterns between EGFR and its downstream pathway genesets. This study investigated the characteristics of the immune microenvironment in EGFR-mutant patients by analyzing PD-L1 protein level, TMB, and tumor immune infiltrate. Based on the analysis above, we investigated whether patients with EGFR mutations might benefit from immunotherapy.
Materials and Methods
Lung Adenocarcinoma Somatic Variants Data Acquisition
In the TCGA database (https://portal.gdc.cancer.gov/), somatic mutation data of 567 lung adenocarcinoma (TCGA-LUAD) were downloaded in the “mutect2” pipeline by TCGAbiolinks (October 2019). It was worth mentation that mutations classification with “silent”, “3ʹUTR”, “3ʹFlank”, “5ʹUTR”, “5ʹFlank”, “intron”, “IGR (intergenic region)”, “RNA”, and “Target_region” were not described as mutant type (https://bioconductor.org/packages/release/bioc/vignettes/ELMER/inst/doc/pipe.html). Since “Splice_Region” can occur in an exon region, “Splice_Region” was treated as a mutant type. TCGA somatic variants were identified by whole-exome sequencing (WES) and whole-genome sequencing (WGS) data.
In cBioPortal,19,20 Broad-Lung Adenocarcinoma (Broad, Cell 2012)24 somatic mutation data were obtained from 183 patients with a combination of whole-exome sequencing (WES) or whole-genome sequencing (WGS). TSP-Lung Adenocarcinoma (The Tumour Sequencing Project (TSP), Nature 2008)25 somatic mutation data were obtained from 163 lung adenocarcinoma using targeted sequencing. MSKCC-Non-Small Cell Lung Cancer (Memorial Sloan Kettering Cancer Center (MSKCC), J Clin Oncol 2018)26 somatic mutation data were obtained from 240 NSCLC patients using IMPACT sequencing, and 186 lung adenocarcinoma patients were selected for follow-up analysis.
In the COSMIC database,21 EGFR, GRB2, SOS1, SOS2, HRAS, KRAS, NRAS, ARAF, RAF1, BRAF, MAP2K1, MAP2K2, MAPK1, MAPK3, and CCND1 somatic mutation data of lung cancer, including negative and positive mutation results, were downloaded (COSMIC v90, released 05-SEP-19) (https://cancer.sanger.ac.uk/cosmic). And 188 patients with mutation data of these fifteen genes were used to follow-up analysis.
The Cancer Genome Atlas Lung Adenocarcinoma Data Acquisition
The Cancer Genome Atlas Lung Adenocarcinoma (TCGA-LUAD) RNA-seq data (including read counts and fragments per kilobase of exon model per million reads mapped (FPKM)) of 533 primary tumors were downloaded from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) by TCGAbiolinks27 in R (October 2019). Moreover, RNA-seq data were annotated by GENCODE v22 (https://www.gencodegenes.org/human/release_22.html). Lung adenocarcinoma (LUAD) Level 4 Reverse phase protein array (RPPA) data of 362 primary tumors were downloaded from the Cancer Proteome Atlas (TCPA)18 (https://www.tcpaportal.org/tcpa/download.html). Only 529 LUAD RNA-seq data and 358 LUAD RPPA data with mutation data were used to follow-up analysis. The corresponding LUAD clinical data were also downloaded by TCGAbiolinks27 in R (October 2019).
Estimation of Tumor Mutation Burden
For this study, TCGA-LUAD mutation data were then processed by “maftools”28 in R. By using “variants.per.sample” function in “maftools” R package, the total number of nonsynonymous mutations for each sample can be roughly calculated. TCGA used the Genome Reference Consortium (GRCh38) reference genome, so we used 35 Mb as the estimate of the exome size. Lung Adenocarcinoma (Broad, Cell 2012)24 somatic data was used 36.6 Mb as the estimate of the exome size. TMB = variants.per.sample/exome size (mut/MB). Data from the MSKCC cohort included a calculated “mutation rate” that was used to measure the TMB levels of each sample. Via “maftools” R package, the TSP cohort data used the number of variants per sample to represent the TMB level of each sample.
Estimation of Tumor Immune Infiltrates
Tumor IMmune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/)29,30 was a comprehensive database of immune infiltrates. Via TIMER, we obtained a relationship between EGFR mutation and the abundance of immune infiltrates (B Cell, CD8+ T Cell, CD4+ T Cell, Macrophage, Neutrophil, Dendritic Cell).
Single sample gene set enrichment analysis (ssGSEA) could also estimate tumor immune infiltrates. Via the “GSVA” R package,31 we estimated the tumor immune infiltrates of TCGA-LUAD using the markers of 24 kinds of immune cells (Dendritic cell [DC], activated DCs [aDC], plasmacytoid DCs [pDC], immature DCs [iDC], B cells, CD8 T cells, cytotoxic cells, Eosinophils, Macrophages, Mast cells, Neutrophils, NK CD56bright cells, NK CD56dim cells, NK cells, T cells, T helper [Th] cells, Th1 cells, Th17 cells, Th2 cells, T central memory cell (Tcm), T effector memory cell [Tem], T follicular helper cell [TFH], T gamma delta [Tgd], regulatory T cell [TReg]) in Bindea et al study.32 For ssGSEA analysis, we converted the TCGA-LUAD counts data to transcripts per million (TPM) data.
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) could determine whether the two physiological states showed statistically consistent differences.33,34 GSEA analysis was performed using 529 LUAD-FPKM data in GSEA_4.0.2. Pathways in Reactome22 or Kyoto Encyclopedia of Genes and Genomes (KEGG)23 database with p-value<0.05, the false discovery rate (FDR)<0.25, and the normalized enrichment score (NES)>1.5 were considered as the critical pathways. In the EGFR mutation group, the key genes of the significant pathways were defined as the contribution genes before the enrichment score (ES) reached the peak.
EGFR Mutation Subgroup Analysis
Based on the KEGG database, the NSCLC pathway (hsa05223 Non-small cell lung cancer) was used to identify EGFR-related downstream pathways (https://www.genome.jp/dbget-bin/www_bget?hsa05223). There were three pathways (29 genes): 1) MAPK signaling pathway (14 genes), 2) Calcium signaling pathway (5 genes), 3) the PI3K-Akt signaling pathway (10 genes). In this study, co-mutation group was defined as an EGFR mutation with at least one gene mutation of EGFR-related downstream pathways. Patients with EGFR mutations not accompanied by gene mutations of the downstream pathway were assigned to the EGFR-mutated group, and other patients were assigned to the wild-type group (WT). According to the above grouping, both three EGFR-related downstream genesets and all genesets were analyzed. Then, the best grouping criteria were selected for follow-up analysis. Both EGFR mutation traits and co-mutation distributions were plotted as snake graphs by “ggalluvial” R package.
Upset Plot for Five Cohorts
To demonstrate the distribution of 15 gene mutations in the EGFR and downstream MAPK signaling pathways, “UpSetR” was used to plot upset diagrams.
Progression-Free Survival Analysis in Checkpoint Inhibitor Treatment
Clinical data from the MSKCC cohort26 treated with anti-PD-(L)1-based therapy was analyzed to investigate whether co-mutated patients could benefit from ICI treatment. To reduce the effect of combination therapy as well as a history of multiple lines of therapy, only patients (108/186) who were in the first- and second-line monotherapy group were included in this analysis. Furthermore, the Kaplan-Meier plot of progression-free survival was plotted by “survival” and “survminer” R packages.
Statistical Analysis
The violin diagrams were plotted by “ggstatsplot” in R. For all statistical analysis, the p-value<0.05 was determined to be significant. Mann–Whitney U-test was used when the number of groups compared = 2. Kruskal–Wallis one-way ANOVA was used when the number of groups compared > 2, and the Dwass-Steel-Crichtlow-Fligner test was used for pairwise comparisons. The holm test was used to correct for p-values of multiple comparisons.
Results
Generally Lower TMB and PD-L1 Protein Expression in Patients with EGFR Mutations
The TCGA-LUAD patients included 67 EGFR-mutated patients, corresponding to a total EGFR mutation site frequency of 79. The most common sites of EGFR mutation were on exons 18–21 (64/79), mainly L858R mutation and exon 19 deletions (Figure S1). The results showed that TMB (EGFR-mutated vs wild-type [WT] [mean] [mut/Mb]: 5.1 vs 6.88, p=<0.001) and PD-L1 protein (EGFR-mutated vs WT [mean]: 0.01 vs 0.17, p=0.009) levels in EGFR-mutated patients were significantly lower than those in EGFR-WT patients (Figure 1A and B). The Broad’s dataset also showed lower TMB levels in patients with EGFR mutations (EGFR-mutated vs WT [mean] [mut/Mb]: 3.44 vs 7.7, p=0.001) (Figure S2A). Although the result of two large panel next-generation sequencing (NGS) (MSK and TSP cohort) did not show significant differences, the median TMB was lower in patients with EGFR mutations (Figure S2B, S2C).
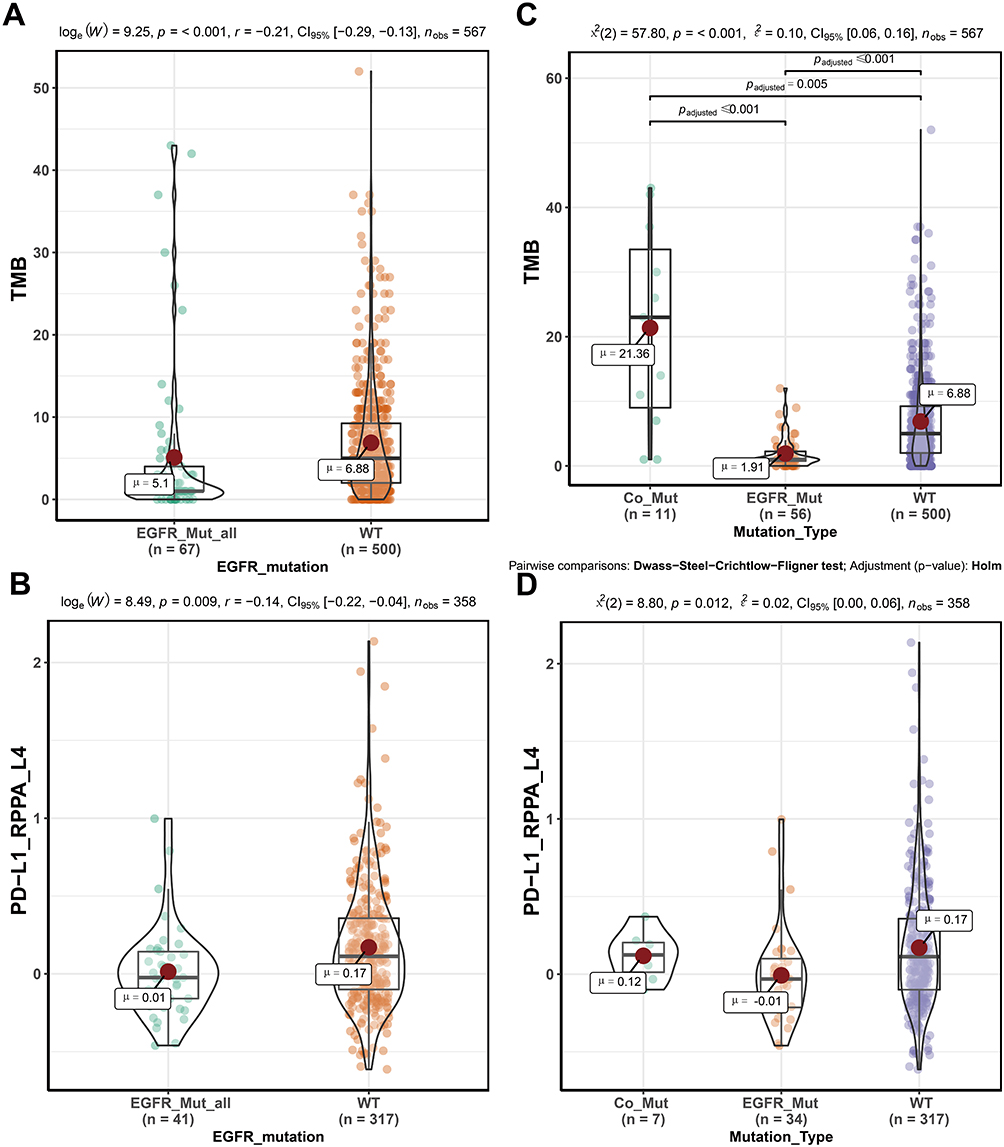 |
Figure 1 TMB and PD-L1 Protein Levels in Different Subtypes of Lung Adenocarcinoma. (A) showed TMB levels in patients with EGFR-mutated and wild-type patients. (B) showed PD-L1 protein expression levels in patients with EGFR-mutated and wild-type patients. (C) showed TMB levels in different subtypes. (D) showed PD-L1 protein expression levels in different subtypes. Abbreviations: TMB, tumor mutation burden; PD-L1, programmed death-ligand 1; LUAD, lung adenocarcinoma; EGFR_Mut_all, all EGFR-mutated patients; Co_Mut, EGFR-MAPK co-mutated patients; EGFR_Mut, EGFR-mutated patients; WT, wild-type patients. |
The Immunosuppressive Microenvironment of LUAD Patients with EGFR Mutation
To investigate the effect of EGFR mutations on the immune microenvironment of LUAD, we used TIMER and ssGSEA to estimate the tissue’s immune infiltrates. A higher degree of dendritic cells (DC) (0.001 ≤ p-value < 0.01) and B cells (0.01 ≤ p-value < 0.05) infiltrating in EGFR-mutated patients were identified in the TIMER database (Figure S3). ssGSEA analysis showed that DC (p-value = 0.026), mast cells (p-value = 0.048), and T follicular helper (TFH) cells (p-value = 0.026) were significantly upregulated in patients with EGFR mutations. In particular, immature dendritic cells (iDC) (p-value = 0.004) showed significantly infiltrated in the EGFR-mutated population whereas NK CD56dim (p-value = 0.009), T helper 2 (Th2) (p-value = 0.002), T gamma delta (Tgd) (p-value = 0.001), and cytotoxic cells (p-value = 0.017) were downregulated in patients with EGFR mutations (Figure S4).
Functional Enrichment of LUAD Patients with EGFR Mutation
We performed GSEA enrichment analysis to investigate the effect of EGFR mutations on signaling pathway. The results showed that the genes related to glycosaminoglycan metabolism (“REACTOME HS GAG DEGRADATION”, “REACTOME HEPARAN SULFATE HEPARIN HS GAG METABOLISM”, “REACTOME A TETRASACCHARIDE LINKER SEQUENCE IS REQUIRED FOR GAG SYNTHESIS”, “REACTOME CHONDROITIN SULFATE DERMATAN SULFATE METABOLISM”, “REACTOME GLYCOSAMINOGLYCAN METABOLISM”), diseases of glycosylation (“REACTOME DISEASES ASSOCIATED WITH GLYCOSAMINOGLYCAN METABOLISM”, “REACTOME DEFECTIVE B4GALT7 CAUSES EDS PROGEROID TYPE”), and receptor tyrosine kinases signaling (“REACTOME PHOSPHOLIPASE C MEDIATED CASCADE FGFR2”) were generally over-expressed in patients with EGFR-mutated LUAD (Figure 2A). Seven genes were identified by intersecting key genes in seven GAG-related pathways, with syndecan-2 (SDC2) showing significant downregulation in the EGFR-MAPK co-mutated patients (Figure S5A–G)
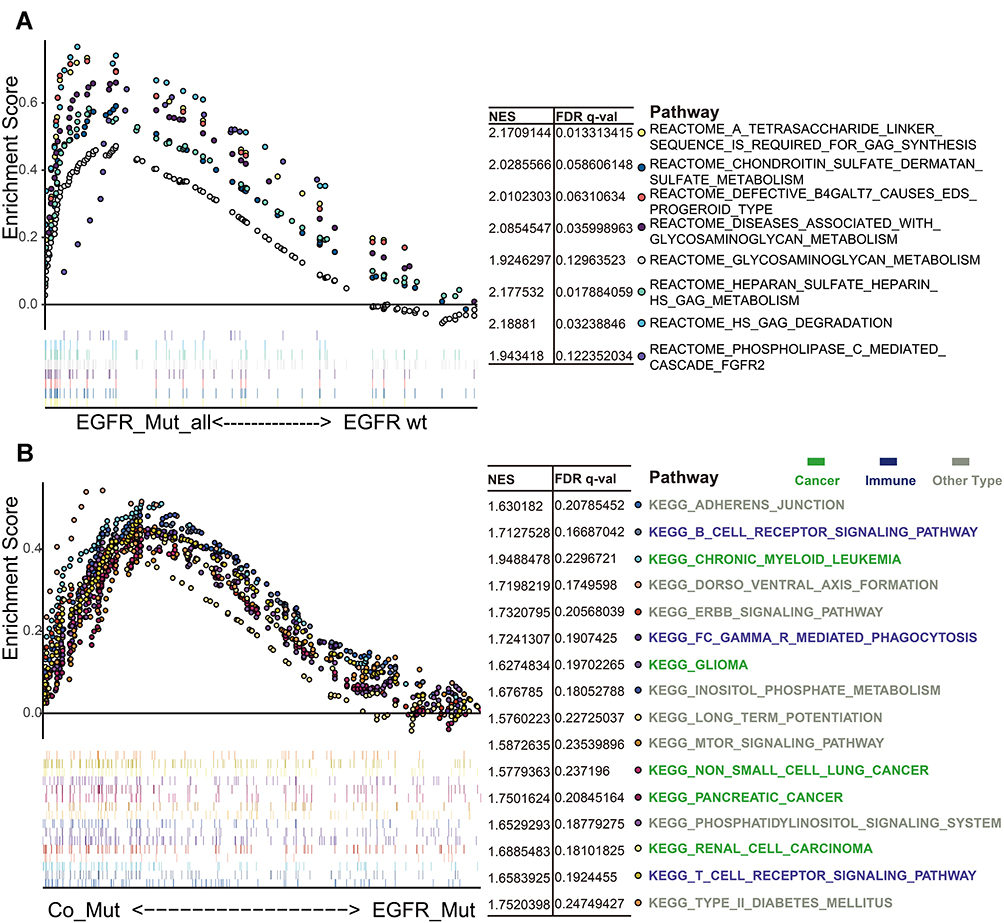 |
Figure 2 Enrichment Analysis of Different EGFR Mutation Types. (A) showed Reactome pathways that are significantly related to EGFR mutations. (B) showed KEGG pathways that are significantly associated with EGFR-MAPK co-mutation. Pathways with p-value<0.05, FDR<0.25, and NES>1.5 were shown. Different colors represented different signal pathways. Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes; FDR, the false discovery rate; NES, the normalized enrichment score. |
Heterogeneity in Patients with EGFR Mutations
While the TMB level of EGFR-mutated patients was generally low, there was still heterogeneity in this population. To investigate the heterogeneity, we first chose the most common mutation sites (exon 18–21), which did not significantly differ from each other (TCGA: p=0.623; Broad: p=0.528) (Figure S6A and B); secondly, EGFR was involved in the development of NSCLC through its downstream pathway (Figure S7). The TMB and PD-L1 protein levels of patients with MAPK signaling geneset mutations were similar to those of wild-type patients (Figure S8A and B). The results showed that patients with EGFR-MAPK co-mutations have high TMB and high PD-L1 protein levels compared to other EGFR co-mutated patterns (Figure 1C and D, Figure S9A – F). Meanwhile, data from the Broad (p=<0.001), MSKCC (p=0.043), and TSP (p=0.006) cohorts confirmed higher TMB levels in patients with EGFR-MAPK co-mutations (Figure S2D – F). Furthermore, patients with L858R mutation were more likely to have co-mutation compared to those with exon 19 deletions (Exon 19 deletion vs L858R: 4.76% [1/21] vs 19.05% [4/21]) (Figure 3).
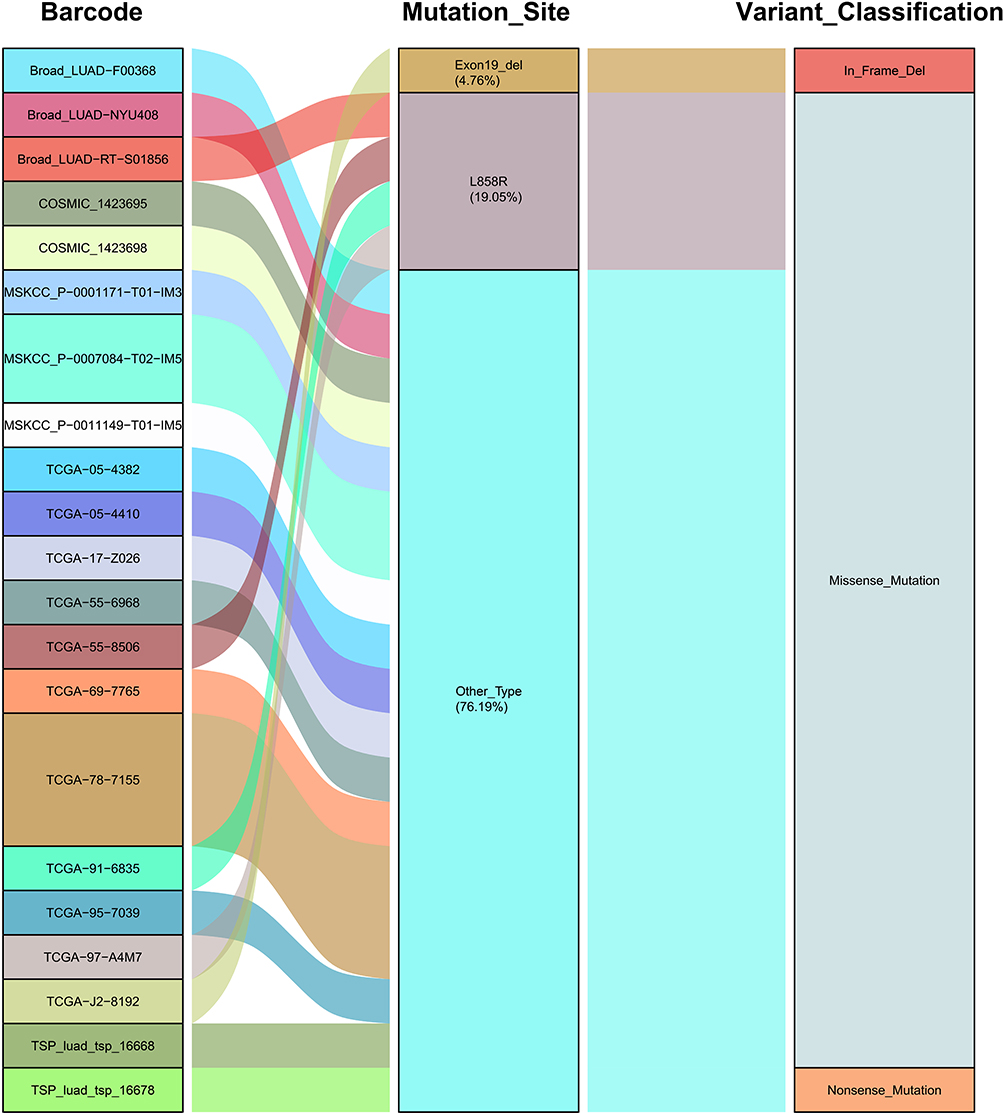 |
Figure 3 Characteristics of EGFR Mutation Sites in Patients with EGFR-MAPK Co-mutation. On the left, barcodes for patients with EGFR-MAPK co-mutation in different cohorts (TCGA, Broad, MSKCC, TSP) was shown; the EGFR mutation sites were shown in the middle and the variant classification on the right. Abbreviations: TCGA, The Cancer Genome Atlas Program; MSKCC, Memorial Sloan Kettering Cancer Center; TSP, The Tumour Sequencing Project. |
Probability of EGFR-MAPK Co-Mutation
Data from 1287 patients with LUAD from the TCGA, Broad, MSKCC, TSP, and COSMIC cohorts were used to assess the probability of EGFR-MAPK co-mutation. The basic characteristics of the patients are shown in Table 1. These five datasets had similar EGFR mutation rates (11.82–18.40%). In the mutation data of TCGA-LUAD, Broad, MSKCC, TSP, and COSMIC, the probability of patients with EGFR-MAPK co-mutation was 16.42% (11/67), 9.38% (3/32), 12.00% (3/25), 6.67% (2/30) and 6.67% (2/30), respectively (Figure 4A–E). The probability of EGFR-mutation was about 14.30% (11.82–18.40%), and EGFR-MAPK co-mutated patients accounted for approximately 11.41% (6.67–16.42%) of EGFR-mutated patients (Table 1).
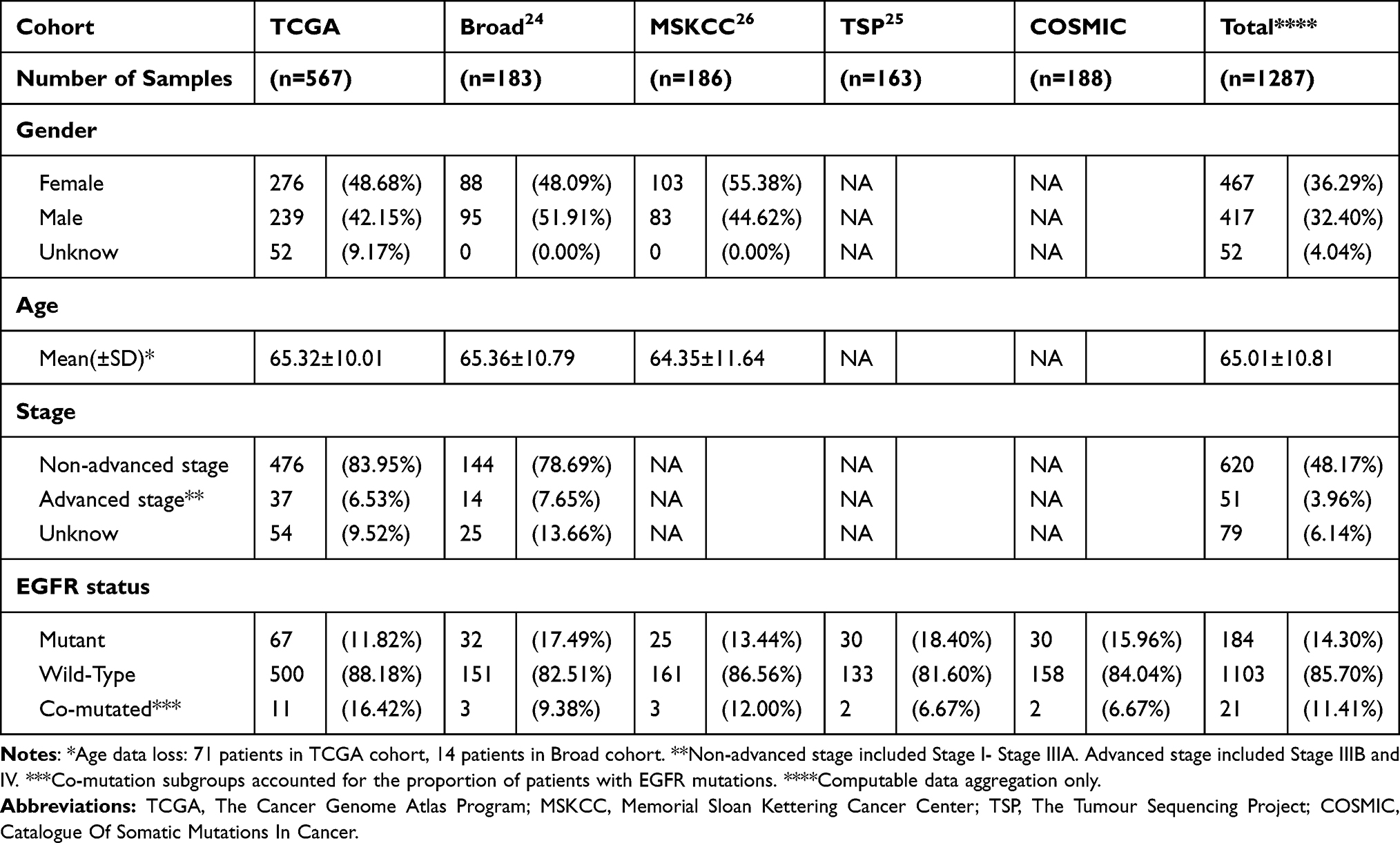 |
Table 1 Patient Baseline Disease Characteristics |
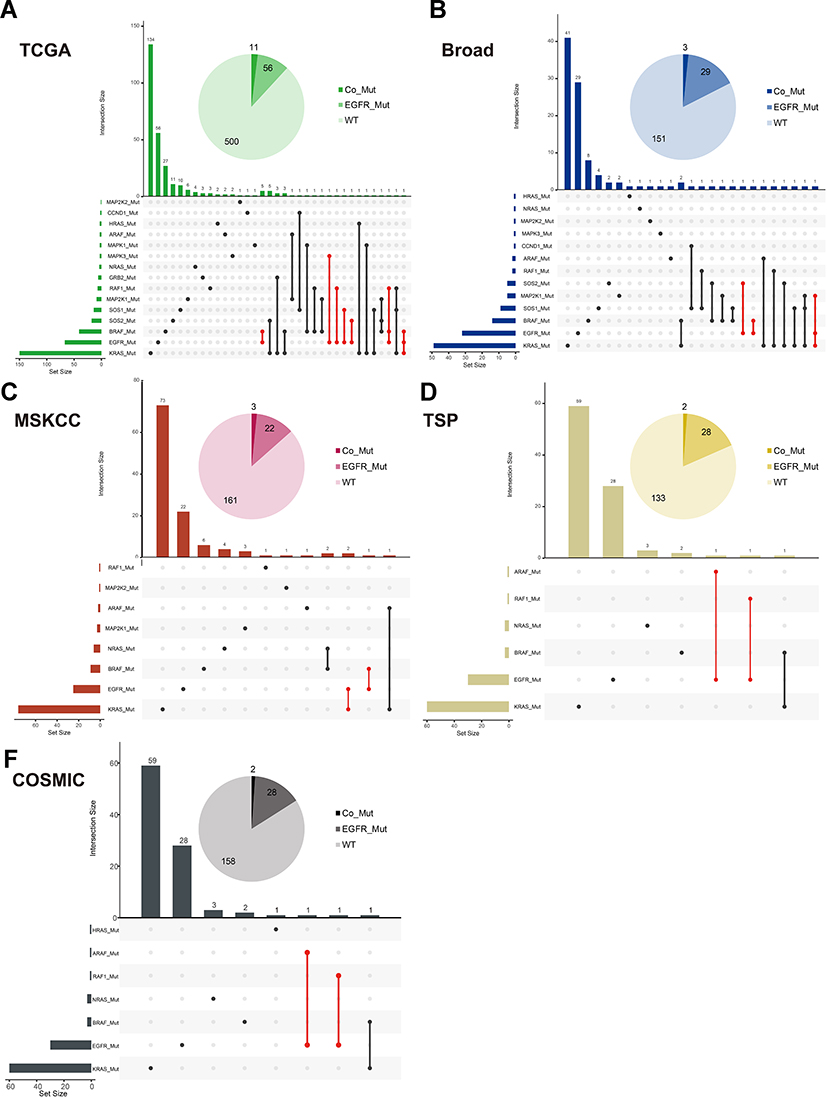 |
Figure 4 Mutation Distribution of EGFR-MAPK Co-mutation in Each Cohort. (A–E) showed the mutation distribution of TCGA-LUAD, Broad, MSKCC, TSP, COSMIC, respectively. The upset plot showed that the distribution of combinations of EGFR-MAPK co-mutation. The bar chart above represented the number of mutation frequencies contained in each type. The pie chart showed the number of co-mutated, EGFR-mutated, and wild-type patients. Abbreviations: TCGA, The Cancer Genome Atlas Program; MSKCC, Memorial Sloan Kettering Cancer Center; TSP, The Tumour Sequencing Project; COSMIC, Catalogue Of Somatic Mutations In Cancer. |
More Favorable Immune Microenvironment in Patients with EGFR-MAPK Co-Mutation
To further understand the changes in the immune microenvironment caused by EGFR-MAPK co-mutation, we compared the abundance of immune infiltrates among different mutation types. The different mutation types had distinct distributions of DC (p-value = 0.043), iDC (p-value = 0.012), NK CD56dim cells (p-value = 0.011), NK cells (p-value = 0.019), Th2 cells (p-value = 0.002), Tgd cells (p-value = 0.002), and cytotoxic cells (p-value = 0.017) (Figure 5A, Figure S10). Patients with EGFR mutations had higher iDC and DC infiltration and lower Th2, Tgd, Cytotoxic, and NK CD56dim cell infiltration (Figure 5B).
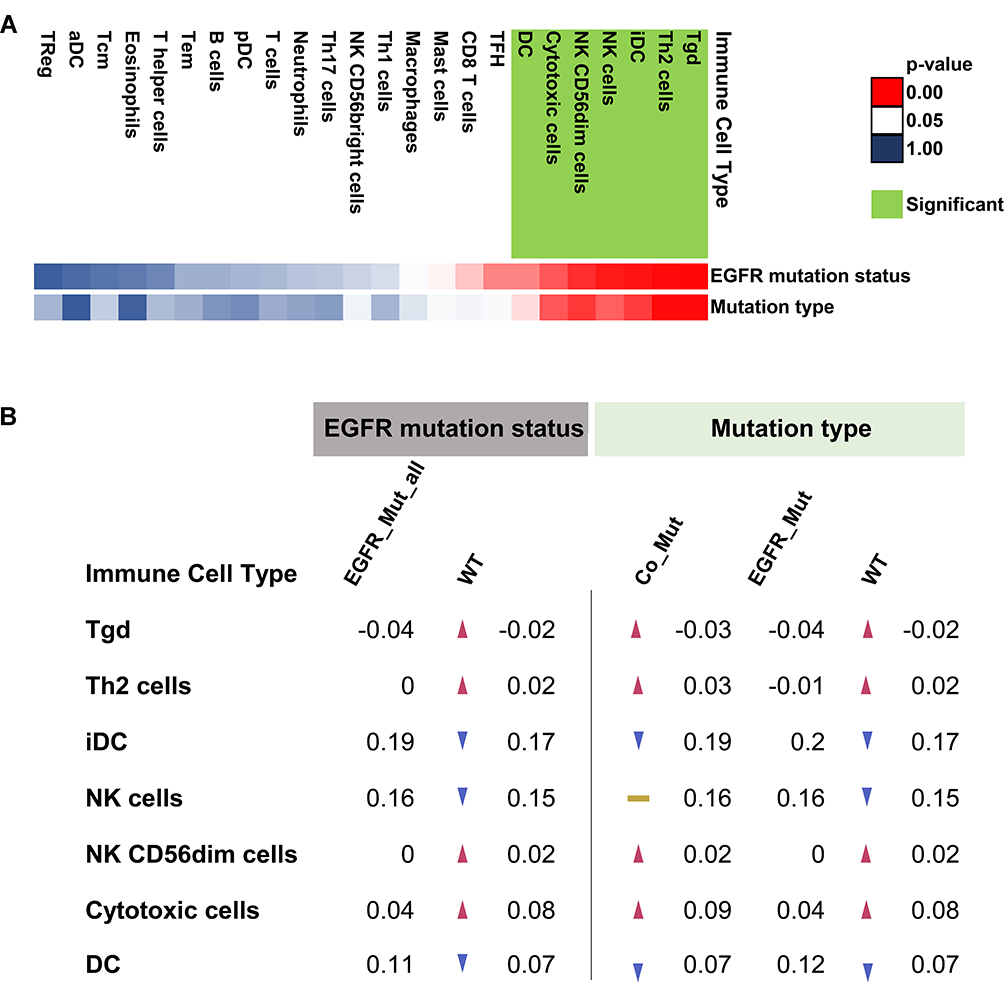 |
Figure 5 Summary of Immune Infiltrates in Different Types of Lung Adenocarcinoma. (A) The green tag represented significant changes in the immune microenvironment compared to patients with EGFR mutations. Red represented 0<p-value<0.05, white represented p-value=0.05, blue represented 0.05<p-value<1.00. (B) Compared to the EGFR-mutated patients’ level of immune infiltration, red arrows represent increased infiltration of that cell within the group, and blue arrows represent decreased infiltration of that cell within the group. The yellow band indicates that the level of cell infiltration is similar to that of the EGFR-mutated patients. The trend of infiltration was compared according to the average values of infiltration in each group. |
Functional Enrichment of LUAD Patients with EGFR-MAPK Co-Mutation
Compared with EGFR single-mutation group, two functions were generally upregulated in the EGFR-MAPK co-mutation group; namely, cancer phenotype (“KEGG_CHRONIC_MYELOID_LEUKEMIA”, “KEGG_GLIOMA”, “KEGG_NON_SMALL_CELL_LUNG_CANCER”, “KEGG_PANCREATIC_CANCER”, “KEGG_RENAL_CELL_CARCINOMA”) and immune-related pathway (“KEGG_B_CELL_RECEPTOR_SIGNALING_PATHWAY”, “KEGG_FC_GAMMA_R_MEDIATED_PHAGOCYTOSIS”, “KEGG_T_CELL_RECEPTOR_SIGNALING_PATHWAY”) (Figure 2B).
Progression-Free Survival Analysis in Checkpoint Inhibitor Treatment
Compared with the EGFR-mutated group, the wild-type group and the co-mutated group had longer progression-free survival (PFS) (EGFR-Mut: WT: Co-Mut = 1.87: 3.77: 7.82 [months], p-value = 0.03). (Figure S11).
Discussion
The present study investigated whether patients with EGFR mutations benefited from ICI therapy. The KEYNOTE-001 study results suggested that patients with EGFR mutations were not suitable for ICI treatment; however, nine EGFR-mutated patients (9/74) benefitted from this treatment.8 Via mutation relationships between EGFR and EGFR downstream pathway genesets, we isolated a subset of EGFR-MAPK co-mutated patients from patients with EGFR mutations. The co-mutated patients had higher TMB and PD-L1 protein levels and a more favorable immune microenvironment. The probability of co-mutation was about 11.41% of the EGFR mutation population. In the present study, L858R patients were more prone to co-mutation than those with exon 19 deletion. TMB and PD-L1 protein levels were used to predict the therapeutic efficacy of ICI.35 Therefore, the EGFR-mutated label concealed that some EGFR-mutated patients benefit from ICI treatment. What’s more, in the first- or second-line ICI monotherapy group, the EGFR-MAPK co-mutated patients showed no less efficacy than the wild-type patients.
Firstly, we confirmed that the TMB or PD-L1 protein levels were lower in EGFR-mutated patients in the TCGA and Broad datasets. Previous studies focused on the impact of clinically-driven mutations on ICI treatment; thus, the link between these driving changes has not been clear. As a representation of cancer development, the driving mutation was difficult to characterize systematically. However, signal transduction is required for EGFR to perform various functions. The NSCLC signaling pathway in the KEGG database showed that the executive role of EGFR was mainly through its three downstream pathways. Thus, it was feasible and meaningful to explore the EGFR mutation mode utilizing prior knowledge. Finally, we found that EGFR-MAPK co-mutated patients have both high TMB and high PD-L1 protein levels.
Furthermore, Zhang et al showed that the EGFR/MAPK pathway is involved in immune evasion of pancreatic cancer.36 Previous studies had shown that patients with L858R mutations benefited more from ICI treatment than those with exon 19 deletions.15 A survey by Sutiman et al showed a longer PFS in patients with EGFR exon 19 deletions treated with EGFR-TKI than that in patients with the L858R mutation.37 Previous studies have shown an association between L858R and high TMB; however, whether this phenomenon is associated with the co-mutation of downstream signaling pathways requires further analysis. Our results suggested that L858R is more likely to co-mutate than exon 19 deletions to achieve better ICI efficacy. A study by Hong et al showed that patients with exon 21 mutations have more concomitant mutations than others, and concomitant mutations negatively affect response and survival in patients treated with EGFR TKIs.38 Offin et al showed that high TMB levels were not conducive to EGFR-TKI treatment but were beneficial to ICI therapy.16 Therefore, most patients with EGFR mutations were suitable for EGFR-TKI therapy, and patients with EGFR- MAPK co-mutations were potential beneficiaries of ICI treatment.
Our results showed that a significant increase in iDC and a significant decrease in NK-CD56dim, T gamma delta (Tgd), cytotoxic, and Th2 cells were significantly decreased in EGFR-mutated patients. However, the immune microenvironments of EGFR-MAPK co-mutated patients were similar to those seen in wild-type patients. iDCs could promote immune tolerance and reduce CD4+ T cells infiltration.39 Moreover, NK CD56dim cells, in addition to their cytolytic activity and target cell killing, were the primary sources of proinflammatory and chemokines.40 Th2 cells enhance B cell activity and have complex regulation of the immune environment.41,42 Tgd cells have an antitumor effect, and the number of Tgd could be used as a prognostic indicator of NSCLC.43
The functional changes in EGFR mutations were mainly in glycosaminoglycan (GAG) metabolism compared to wild-type LUAD. GAG, as the main component of the extracellular matrix or the surface of cells, combines many kinds of cytokines and chemokines to regulate immune function. Heparin sulfate (HS) is the primary type of GAG, and the active degradation of HS can activate the immune system; HS on the surface of white blood cells could also regulate immune cell activation. Nishio et al showed that high serum HS concentrations were associated with shorter PFS and overall survival (OS) in EGFR-mutated NSCLC.44 iDCs, NK cells, and Tgd belong to the innate immune system activated by chemokines. GAGs can protect chemokines from degradation, but it also interfered with the action of chemokines.45 These results suggest that an inadequate immune response in EGFR-mutated patients might be due to the accumulation of GAG, leading to an inhibitory immune microenvironment. Compared to patients with only EGFR mutation, patients with co-mutations had lower Syndecan-2 (SDC2) expression levels. SDC2 could promote the clearance of TCR/CD3 complex on the surface of T cell46 and downregulate immune activity,47 suggesting that SDC2 is involved in the inhibitory microenvironment of patients with EGFR mutations.
Meanwhile, functional changes between co-mutated and EGFR-mutated patients were mainly concentrated on cancer phenotype and immune function. The cancer phenotypes of co-mutated patients were closer to NSCLC, pancreatic cancer, chronic myeloid leukemia, renal cell carcinoma, and gliomas, which shown promise and even improved survival in these cancers by treated with ICI. Immune-related pathways, including T cells, B cells, and FCγR-mediated phagocytosis, were upregulated in EGFR-MAPK co-mutated patients.
Our study had several limitations. Our results suggested that enhanced FCγR-mediated phagocytosis in co-mutated patients. However, FCγR activation decreased the curative effect of the IgG4-based PD-1 antibody. While FCγR activation reduces ICI antibody utilization, techniques have been developed to improve the impact of this effect.48 Meanwhile, FCγR activation enhanced the efficacy of anti-PD-L1.49 Patients with EGFR mutations are excluded from most ICI studies because they generally do not benefit from ICI treatment.8 However, we note that there are still patients with EGFR mutations who benefit from ICI therapy. In limited data, patients with EGFR-MAPK co-mutations can benefit from ICI therapy. Although this study is an interesting result based on limited data, our results reveal why EGFR-mutated patients generally do not benefit from ICI treatment. What’s more, we have screened out EGFR-MAPK co-mutated patients who may benefit from ICI treatment, which will be of concern in further clinical studies.
In conclusion, the results of our study showed that most of EGFR-mutated patients had higher GAGs and an inhibitory immune microenvironment than EGFR wild-type patients. The EGFR-MAPK co-mutated patients, with higher TMB, PD-L1 protein levels, and favorable immune microenvironment, were different from EGFR-mutated-only patients. Thus, the EGFR mutational labels might mask some patients, such as those with co-mutations, who missed out on ICI treatment. The results of this study are worth undertaking a larger cohort to validate and further reveal distinctive and heterogeneous EGFR mutational labels. Both targeted agents and ICI therapy have led to significant advances in cancer treatment, and our findings can contribute to clinical treatment decisions, which provide more precise treatment options for patients with EGFR mutations.
Acknowledgments
The results here were in part based upon data generated by the TCGA Research Network, TCPA, Broad, MSKCC, TSP, and COSMIC. We thank “FigureYa” for intellectual contribution.
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Midha A, Dearden S, McCormack R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII). Am J Cancer Res. 2015;5(9):2892–2911.
3. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi:10.1056/NEJMoa050753
4. Wu Y-L, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18(11):1454–1466. doi:10.1016/S1470-2045(17)30608-3
5. Lee CK, Man J, Lord S, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non-small cell lung carcinoma: a systematic review and meta-analysis. JAMA Oncol. 2018;4(2):210–216. doi:10.1001/jamaoncol.2017.4427
6. Tohmé R, Izadmehr S, Gandhe S, et al. Direct activation of PP2A for the treatment of tyrosine kinase inhibitor-resistant lung adenocarcinoma. JCI Insight. 2019;4(4):e125693. doi:10.1172/jci.insight.125693
7. Deng -L-L, Gao G, Deng H-B, Wang F, Wang Z-H, Yang Y. Co-occurring genetic alterations predict distant metastasis and poor efficacy of first-line EGFR-TKIs in EGFR-mutant NSCLC. J Cancer Res Clin Oncol. 2019;145(10):2613–2624. doi:10.1007/s00432-019-03001-2
8. Leighl NB, Hellmann MD, Hui R, et al. Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label, Phase 1 study. Lancet Respir Med. 2019;7(4):347–357. doi:10.1016/S2213-2600(18)30500-9
9. Castellanos E, Feld E, Horn L. Driven by mutations: the predictive value of mutation subtype in EGFR-mutated non-small cell lung cancer. J Thorac Oncol. 2017;12(4):612–623. doi:10.1016/j.jtho.2016.12.014
10. Negrao MV, Lam VK, Reuben A. PD-L1 expression, tumor mutational burden, and cancer gene mutations are stronger predictors of benefit from immune checkpoint blockade than HLA class I genotype in non-small cell lung cancer. J Thorac Oncol. 2019;14(6):1021–1031. doi:10.1016/j.jtho.2019.02.008
11. Singal G, Miller PG, Agarwala V, et al. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non-small cell lung cancer using a clinicogenomic database. JAMA. 2019;321(14):1391–1399. doi:10.1001/jama.2019.3241
12. Rangachari D, VanderLaan PA, Shea M. Correlation between classic driver oncogene mutations in EGFR, ALK, or ROS1 and 22C3-PD-L1 ≥50% expression in lung adenocarcinoma. J Thorac Oncol. 2017;12(5):878–883. doi:10.1016/j.jtho.2016.12.026
13. Edlund K, Madjar K, Mattsson JSM, et al. Prognostic impact of tumor cell programmed death ligand 1 expression and immune cell infiltration in NSCLC. J Thorac Oncol. 2019;14(4):628–640. doi:10.1016/j.jtho.2018.12.022
14. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585–4593. doi:10.1158/1078-0432.CCR-15-3101
15. Hastings K, Yu HA, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small cell lung cancer. Ann Oncol. 2019;undefined(undefined):undefined.
16. Offin M, Rizvi H, Tenet M. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. 2019;25(3):1063–1069. doi:10.1158/1078-0432.CCR-18-1102
17. Wu C-K, Kao S-J, Lai H-C. Targeted therapy and immunotherapy lead to rapid regression of advanced non-small cell lung cancer with multiple driver mutations. J Thorac Oncol. 2018;13(6):e103–e105. doi:10.1016/j.jtho.2018.01.025
18. Chen M-JM, Li J, Wang Y, et al. TCPA v3.0: an integrative platform to explore the pan-cancer analysis of functional proteomic data. Mol Cell Proteomics. 2019;18(8 suppl 1):S15–S25. doi:10.1074/mcp.RA118.001260
19. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
20. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
21. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47(D1):D941–D947. doi:10.1093/nar/gky1015
22. Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2019. doi:10.1093/nar/gkz1031
23. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi:10.1093/nar/gkw1092
24. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi:10.1016/j.cell.2012.08.029
25. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi:10.1038/nature07423
26. Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–641. doi:10.1200/JCO.2017.75.3384
27. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016;44(8):e71. doi:10.1093/nar/gkv1507
28. Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–1756. doi:10.1101/gr.239244.118
29. Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi:10.1158/0008-5472.CAN-17-0307
30. Li B, Severson E, Pignon JC, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. doi:10.1186/s13059-016-1028-7
31. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi:10.1186/1471-2105-14-7
32. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi:10.1016/j.immuni.2013.10.003
33. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
34. Mootha VK, Lindgren CM, Eriksson K-F, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi:10.1038/ng1180
35. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16(11):2598–2608. doi:10.1158/1535-7163.MCT-17-0386
36. Zhang Y, Velez-Delgado A, Mathew E, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2017;66(1):124–136. doi:10.1136/gutjnl-2016-312078
37. Sutiman N, Tan SW, Tan EH, et al. EGFR mutation subtypes influence survival outcomes following first-line gefitinib therapy in advanced Asian NSCLC patients. J Thorac Oncol. 2017;12(3):529–538. doi:10.1016/j.jtho.2016.11.2225
38. Hong S, Gao F, Fu S, et al. Concomitant genetic alterations with response to treatment and epidermal growth factor receptor tyrosine kinase inhibitors in patients with EGFR-mutant advanced non-small cell lung cancer. JAMA Oncol. 2018;4(5):739–742. doi:10.1001/jamaoncol.2018.0049
39. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. 2002;23(9):445–449. doi:10.1016/S1471-4906(02)02281-0
40. Fauriat C, Long EO, Ljunggren H-G, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood. 2010;115(11):2167–2176. doi:10.1182/blood-2009-08-238469
41. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830.e814.
42. Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16(1):45–56. doi:10.1038/ni.3049
43. Izumi H, Yamasaki A, Takeda K, et al. Acute-phase reaction induced by zoledronate and its effect on prognosis of patients with advanced non-small cell lung cancer. Lung Cancer. 2018;122:200. doi:10.1016/j.lungcan.2018.06.022
44. Nishio M, Yamanaka T, Matsumoto K, et al. Serum heparan sulfate concentration is correlated with the failure of epidermal growth factor receptor tyrosine kinase inhibitor treatment in patients with lung adenocarcinoma. J Thorac Oncol. 2011;6(11):1889–1894. doi:10.1097/JTO.0b013e3182286d41
45. Metzemaekers M, Mortier A, Janssens R, et al. Glycosaminoglycans regulate CXCR3 ligands at distinct levels: protection against processing by dipeptidyl peptidase IV/CD26 and interference with receptor signaling. Int J Mol Sci. 2017;18(7):1513. doi:10.3390/ijms18071513
46. Rovira-Clavé X, Angulo-Ibáñez M, Noguer O, Espel E, Reina M. Syndecan-2 can promote clearance of T-cell receptor/CD3 from the cell surface. Immunology. 2012;137(3):214–225. doi:10.1111/j.1365-2567.2012.03626.x
47. Teixé T, Nieto-Blanco P, Vilella R, Engel P, Reina M, Espel E. Syndecan-2 and −4 expressed on activated primary human CD4+ lymphocytes can regulate T cell activation. Mol Immunol. 2008;45(10):2905–2919. doi:10.1016/j.molimm.2008.01.033
48. Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood. 2016;127(9):1097–1101. doi:10.1182/blood-2015-09-625343
49. Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell. 2015;28(3):285–295. doi:10.1016/j.ccell.2015.08.004
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.