Back to Journals » Cancer Management and Research » Volume 18

Fatal Disease Progression Driven by Acquired MET Amplification After EGFR-TKI Therapy in EGFR- and RBM10-Mutant Lung Adenocarcinoma
Received 8 August 2025
Accepted for publication 21 January 2026
Published 29 January 2026 Volume 2026:18 559385
DOI https://doi.org/10.2147/CMAR.S559385
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bilikere Dwarakanath
Jiwon Kim,1 Kiyong Na,2 Seung Hyeun Lee1,3
1Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Internal Medicine, Kyung Hee University College of Medicine, Kyung Hee University Hospital, Seoul, South Korea; 2Department of Pathology, Kyung Hee University College of Medicine, Kyung Hee University Hospital, Seoul, South Korea; 3Department of Precision Medicine, Graduate School Kyung Hee University, Seoul, South Korea
Correspondence: Seung Hyeun Lee, Division of Pulmonary, Allergy, and Critical Care Medicine, Department of Internal Medicine, Kyung Hee University College of Medicine, Kyungheedae-ro 23, Dongdaemun-gu, Seoul, 02447, South Korea, Tel +82 2 958 8511, Fax +82 2 968 1848, Email [email protected]
Abstract: Although mesenchymal–epithelial transition proto-oncogene (MET) amplification is a common resistance mechanism in targeted therapy for lung cancer, rapid disease progression associated with this resistance mechanism in patients with epidermal growth factor receptor (EGFR) mutation has scarcely been reported. Herein, we report a fatal case of lung adenocarcinoma that rapidly progressed after failure of EGFR-tyrosine kinase inhibitor treatment following the emergence of MET amplification. A 62-year-old man was diagnosed with metastatic lung adenocarcinoma containing mutations in EGFR exon L858R and RNA-binding motif 10. He received afatinib as frontline treatment and showed a partial response; however, the right lung lesion progressed after 14 months of treatment. Although the drug was maintained after salvage segmentectomy of the lesion, lung nodules and pleural effusion developed shortly thereafter. Because EGFR testing using resected tissue showed only the original mutation, we switched his regimen to pemetrexed and carboplatin. However, the disease rapidly progressed with a very large mass in the right lung and massive pleural effusion, which led to death within 7 weeks of treatment. Next-generation sequencing was performed at the time of first progression and second progression revealed acquired MET amplification (copy number gain 15.5 and 9.1, respectively) in addition to baseline mutations. Although an association between MET amplification and rapidly progressive lung cancer has been predicted previously, to the best of our knowledge, this is the first report on the potential contribution of other mutations, such as those in RNA-binding motif 10, during MET-driven rapid progression. Our report highlights the importance of more active utilization of molecular profiling for the emergence of resistance during tyrosine kinase inhibitor use and the early identification of MET amplification and timely initiation of MET-targeted therapy, such as MET inhibitors in combination with EGFR-TKIs, to potentially mitigate rapid disease progression and clinical deterioration.
Keywords: lung cancer, EGFR-TKI, MET amplification, RBM10, next-generation sequencing
Introduction
Recent advancements in targeted therapies have substantially improved the outcomes of patients with advanced non-small cell lung cancer (NSCLC) harboring genetic driver alterations. Epidermal growth factor receptor (EGFR) is the most common targetable driver gene, with a mutation incidence of up to 60% in Asian populations with NSCLC.1 Targeted therapies can selectively inhibit oncogenic driver mutations.2 However, acquired resistance inevitably develops, limiting long-term treatment efficacy, and primary (intrinsic) resistance hinders the clinical benefits of tyrosine kinase inhibitors (TKIs) in some cases. Among the several resistance mechanisms, mesenchymal–epithelial transition proto-oncogene (MET) amplification is a key mechanism in bypass pathway activation and is commonly observed in patients receiving TKIs targeting anaplastic lymphoma kinase (ALK), c-ros oncogene 1 (ROS1), rearranged during transfection (RET), and EGFR.3 This alteration enables sustained oncogenic signaling through activation of the downstream PI3K/AKT and MAPK pathways independent of EGFR, thereby conferring therapeutic resistance.4 Beyond its role as a common mechanism of acquired resistance, emerging evidence suggests that MET amplification may also be associated with aggressive clinical phenotypes, including hyperprogression and disease flare, characterized by rapid tumor growth and abrupt clinical deterioration.5,6 Although MET-targeted therapies have shown promise in overcoming resistance, their optimal use and timing remain under investigation.
Herein, we present a case of EGFR and RNA-binding motif 10 (RBM10) dual mutant NSCLC with acquired MET amplification that led to fulminant disease progression following resistance to first-line EGFR-TKI therapy. RBM10 mutations have been reported in approximately 5–10% of NSCLC and are more frequently observed in EGFR-mutant tumors, particularly those harboring the L858R mutation.7 Although RBM10 is not considered a primary oncogenic driver, its loss-of-function has been associated with altered apoptotic signaling and reduced sensitivity to EGFR-tyrosine kinase inhibitors. Although the molecular mechanisms underlying MET amplification and RBM10 loss-of-function have been individually described, the clinical significance of their temporal co-occurrence in the setting of abrupt and fatal disease progression has not been well characterized. We explored the underlying genomic features using serial next-generation sequencing (NGS), and reviewed of the potential role of MET-directed therapies that may have altered the clinical outcomes. This case highlights the importance of proactive use of molecular reassessment and timely application of targeted strategies for the better management of rapidly progressive NSCLC.
Case Presentation
A 62-year-old man presented to our institution in March 2022 with a one-month history of progressive dyspnea. He had no significant past medical history or regular medication use but had a smoking history of 40 pack-years. His vital signs were stable; however, right lung sounds were markedly decreased. Chest radiography revealed massive pleural effusion in the right hemithorax (Figure 1A). We drained the effusion using percutaneous catheter insertion, and effusion analysis revealed a lymphocyte-dominant exudate without adenosine deaminase elevation suggesting malignant pleural effusion. Chest computed tomographic (CT) scan revealed a large mass (8 × 10 cm) in the right middle lobe; enlargement and conglomeration of the right hilar, interlobar, and paratracheal lymph nodes; and pleural thickening (Figure 1B). Bronchoscopy revealed near-total obstruction due to a fungating mass at the orifice of the right bronchus intermedius. Bronchoscopic biopsy confirmed the diagnosis of lung adenocarcinoma with a micropapillary pattern (Figure 1C).
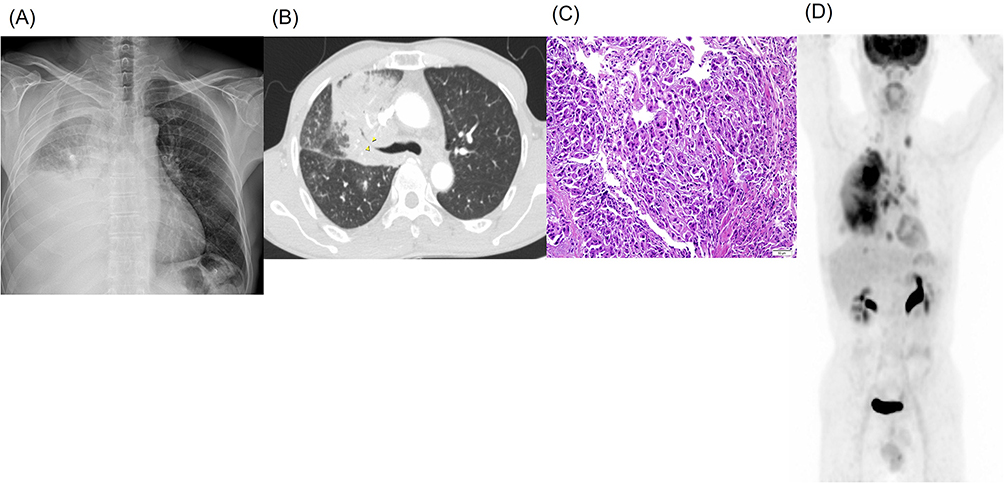 |
Figure 1 Imaging at diagnosis. The initial chest x-ray showed a massive pleural effusion in the right hemithorax (A), and chest CT revealed a large mass in the right middle lobe with luminal narrowing due to external compression (yellow arrowheads) (B). Bronchoscopic biopsy confirmed primary lung adenocarcinoma with a micropapillary pattern (H&E staining, x200) (C). Positron emission tomography demonstrated a lung mass in the right lung, mediastinal lymph node metastases, and pleural seeding, consistent with clinical stage IVA disease (D). |
Although brain magnetic resonance and bone scan were negative for metastasis, positron emission tomography (PET) confirmed metastatic involvement of the mediastinal lymph nodes and pleura. Thus, the final clinical stage was stage IVA (cT4N3M1a, Figure 1D) according to the criteria of the American Joint Committee on Cancer (AJCC) 8th edition. The tumor proportion score of programmed death-ligand 1 (PD-L1) expression using SP263 clone was 5% and molecular analysis using a peptide nucleic acid clamping-based real-time PCR (PANAMutyper™, PANAGENE, Seoul, Korea) revealed an EGFR L858R mutation. NGS was conducted employing the IonTorrent S5 XL, alongside a control cell line mixture (Horizon Discovery, Cambridge, UK), and a targeted gene panel, the Oncomine Comprehensive Assay Plus (Thermo Fisher Scientific). This panel facilitates the identification of single nucleotide variants and copy number variation from a pool of 500 unique cancer genes. For genomic data analysis and variant calling the Ion Reporter software v5.2 was employed (Thermo Fisher Scientific). The criteria for identifying mutant alleles were as follows: minimum depth of coverage, 500×; mutant allele frequency, >5%. Following manufacture’s guideline, gene copy number above 6 and below 0.5 was categorized to amplification and deletion, respectively. With EGFR L858R mutation, NGS also identified RBM10 mutations, and amplification of PDGFRA, KIT and EGFR (Table 1, primary tumor, formalin-fixed paraffin-embedded tumor tissues). Based on these findings, afatinib 40 mg daily was initiated as first-line treatment.
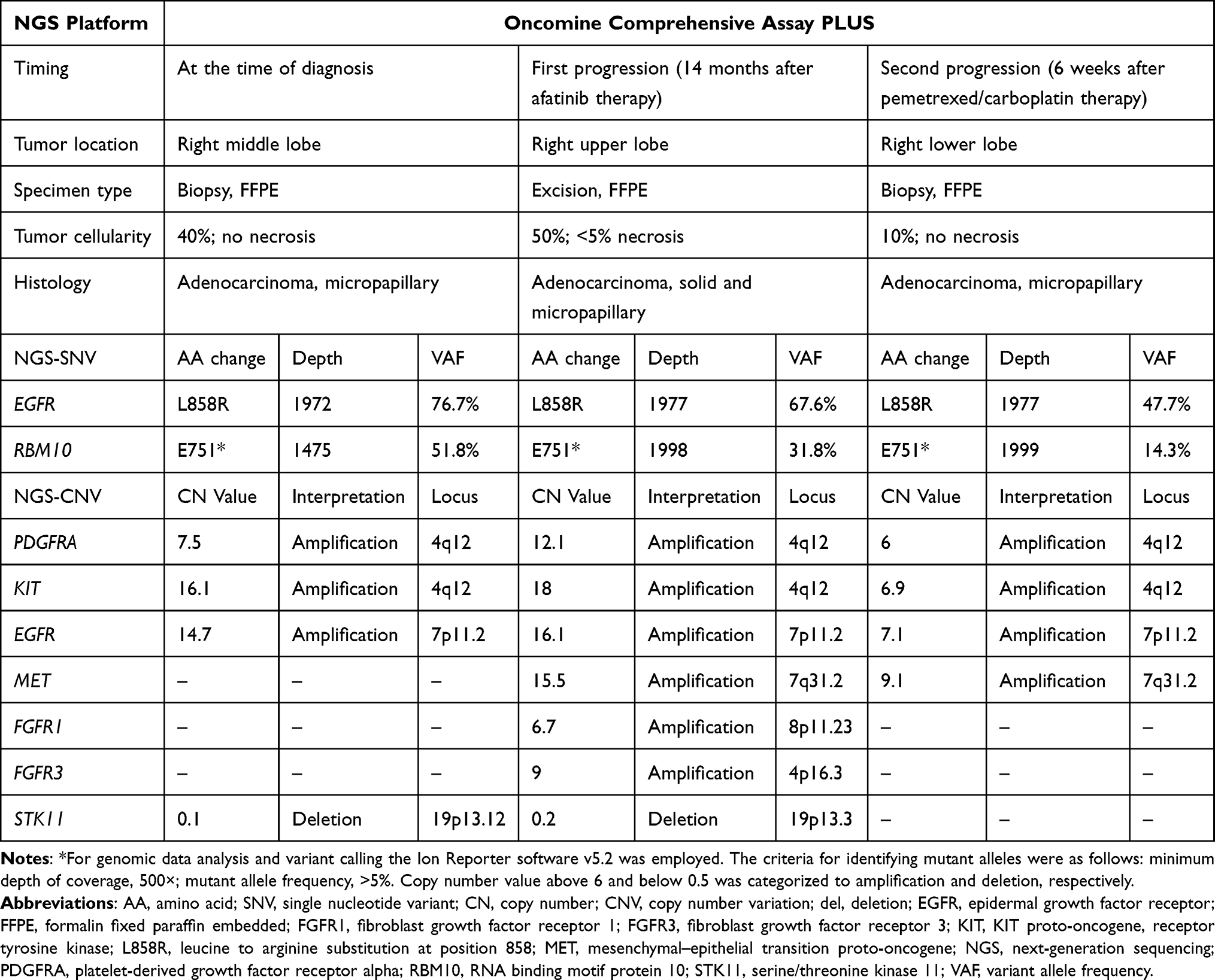 |
Table 1 Sequential Next-Generation Sequencing Results in Tumor Samples During Treatment |
The patient experienced mild diarrhea, acneiform dermatitis, and stomatitis, which did not require dose reduction; thus, full-dose treatment was maintained. Table 2 summarizes key clinical events, treatments, molecular assessments, and outcomes of this case. In addition, Figure 2 summarizes serial chest radiographs, CT images, and NGS results in chronological order according to the treatment regimen. The first response evaluation CT scan performed 2 months after treatment initiation demonstrated a partial response, and CT performed 4 months later showed a nearly complete response. After 14 months of afatinib treatment, follow-up CT showed sustained partial response in most lesions; however, the right upper lobe mass was increased in size (red asterisk, Figure 2). For this oligoprogressive disease confined to a single pulmonary lesion with otherwise sustained systemic disease control under EGFR-TKI therapy, salvage segmentectomy was performed, and pathology confirmed lung adenocarcinoma. Persistent EGFR L858R positivity and no acquired mutations, such as EGFR T790M, were evident in real-time PCR. According to treatment guidelines, afatinib was continued as a beyond-progression strategy.
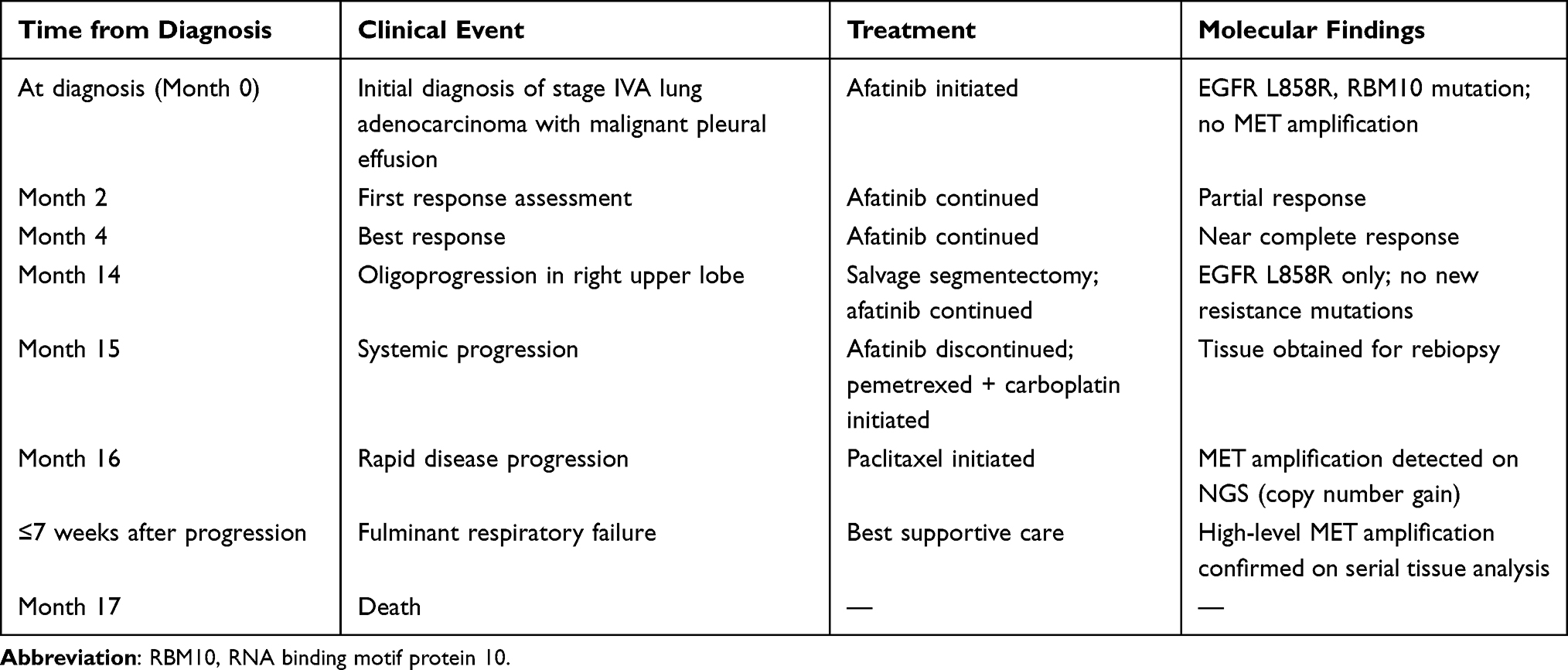 |
Table 2 Timeline Summarizing Key Clinical Events, Treatments, Molecular Assessments, and Outcomes, Illustrating the Temporal Association Between EGFR-TKI Exposure, Emergence of MET Amplification |
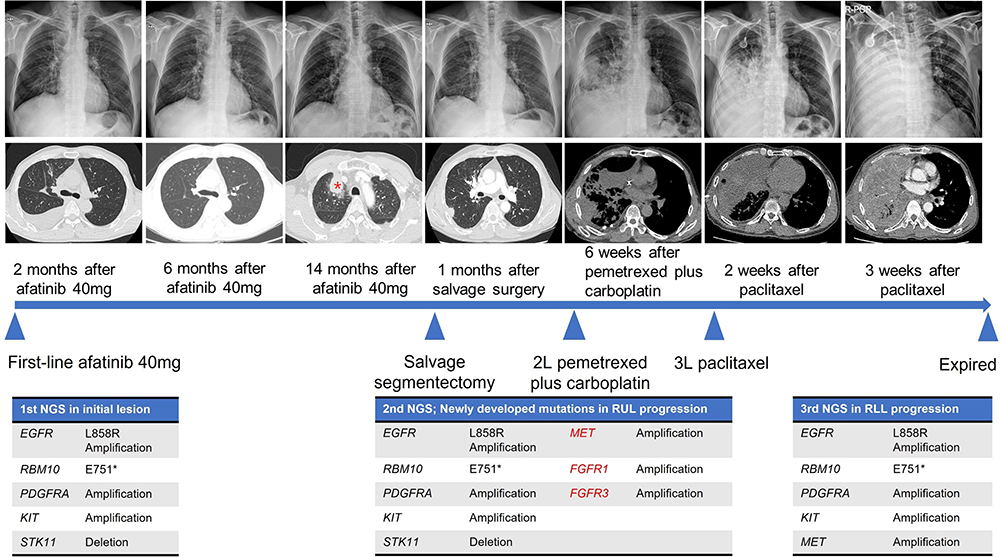 |
Figure 2 Clinical course with subsequent treatment and NGS results after first-line Afatinib treatment. The patient showed an initial partial response but developed oligo-progression in the right upper lobe (RUL) after 14 months of afatinib. Salvage surgery was performed for the regrowing RUL mass (red asterisk), and afatinib was maintained as a beyond-progression strategy based on the mutation test using resected tissue. However, a new lesion in the right lower lobe developed within 1 month after surgery. Rapid disease progression ensued despite second- and third-line chemotherapy. At 2 months after the switching treatment, the tumor occupied the whole right hemithorax, and the patient ultimately died from fulminant respiratory failure. E751* indicates a nonsense mutation in which the glutamic acid at amino acid position 751 is substituted by a stop codon. Genes shown in red (MET, FGFR1, and FGFR2) were newly amplified at first progression. |
One month later, a new lesion developed in the right lower lobe, indicating systemic progression; afatinib was discontinued, and second-line therapy with pemetrexed plus carboplatin was initiated. However, a follow-up CT scan after 2 cycles of chemotherapy revealed rapid progression of the right lower lobe mass with the development of pleural effusion. The right lower lobe lesion was rebiopsied for molecular reassessment, showing the same histology with EGFR L858R mutation. Third-line therapy with paclitaxel was initiated with NGS profiling of the rebiopsied tissue obtained from right lower lobe. Two weeks after initiating third-line treatment, the patient presented to the emergency department with worsening dyspnea. Imaging revealed further enlargement of the right lung mass, and the patient complained of respiratory distress, with a respiratory rate of 35 breaths per minute and persistent hypoxemia. Follow-up CT and PET scans taken 1 week later demonstrated a hypermetabolic mass occupying the entire right lung, and hypoxemia worsened despite oxygen supplementation via high-flow nasal cannula with 100% FiO2. Despite transfer to the intensive care unit and mechanical ventilation, the patient developed hypotension and died on day 3 of admission. The NGS profile, which became available after his death, revealed the emergence of MET amplification (copy number gain 9.1) in addition to previously detected mutations (Table 1, second progression). To determine possible changes in molecular profiles during the clinical course, we performed postmortem NGS using right upper lobe lobectomy specimens. The results confirmed the presence of MET amplification (copy number gain 15.5) along with preexisting genetic alterations (Table 1, first progression), suggesting that the exceptionally aggressive disease progression observed in this patient was driven by MET amplification.
Discussion
To the best of our knowledge, this is among the first reports to highlight fatal hyperprogressive disease associated with acquired MET amplification in an EGFR-mutant lung adenocarcinoma harboring an RBM10 co-mutation. The MET proto-oncogene encodes a receptor tyrosine kinase known as the hepatocyte growth factor (HGF) receptor. Together with its ligand, HGF, the HGF/MET axis plays a crucial role in regulating cell survival, proliferation, motility, and migration. Aberrant MET signaling is implicated in multiple cancers through various mechanisms, such as MET-activating mutations, HGF overexpression, MET amplification or copy number gain, and MET fusion.8 MET amplification occurs in 1–6% of NSCLC cases and is considered a negative prognostic factor.9 Among the acquired resistance mechanisms to EGFR-TKIs, MET amplification ranks second in frequency following the T790M resistance mutation, and the frequency of secondary MET amplification is 5–15% among those previously treated with EGFR-TKIs.3,4 MET amplification has also been identified as a common resistance mechanism in patients with other actionable genomic alterations, including ALK, RET, and ROS1 fusions and kirsten rat sarcoma viral oncogene homolog (KRAS) G12C mutation.4 However, its role in rapid disease flares and fulminant progression was only suggested recently. In our case, the patient exhibited catastrophic clinical deterioration following the emergence of MET amplification after afatinib failure, with death occurring within two months of treatment change, despite standard second- and third-line chemotherapy. Although the confirmation of MET amplification using fluorescence in situ hybridization or immunohistochemistry could not be performed due to limited tissue availability and the rapid clinical deterioration of the patient, this unusually aggressive clinical course indicates that MET amplification not only confers therapeutic resistance but also drives tumor hyperproliferation.
A similar phenomenon was described in previous case reports and cohort studies. Reported cases on MET amplification-related rapid progression or disease flares is summarized in Table 3. Peng et al reported a case of lung adenocarcinoma with high PD-L1 expression and no driver genetic alterations which showed hyperprogressive disease.6 They performed whole-exome sequencing using primary tumors and found that the tumor was MET-amplified. The patient showed a partial response to subsequent treatment with crizotinib, a MET inhibitor, suggesting that MET amplification contributes to hyperprogressive disease and primary resistance. Additionally, Long et al reported a case of ALK-positive lung adenocarcinoma with rapid progression to liver metastasis, whereas other lesions showed a partial response to alectinib.5 Targeted NGS using both plasma circulating tumor DNA (ctDNA) and liver metastasis revealed newly emerged MET amplification, in addition to baseline EML4-ALK fusion. Despite subsequent co-treatment with ALK and MET inhibitors, the disease rapidly progressed, and the patient died from respiratory failure.5 Very recently, Wang et al reported a case of lung adenocarcinoma which was progressed after 8-months treatment with first-line gefitinib and refractory to the osimertinib treatment despite the detection of acquired T790M mutation. NSG revealed that the tumor acquired MET amplification, and partial response was achieved after savolitinib and osimertinib combination.10 In a cohort study, Han et al investigated the underlying genomic characteristics of hyperprogressive disease in 117 patients with lung cancer who were treated with immune checkpoint blockade and identified MET amplification as the causative genomic alteration.11
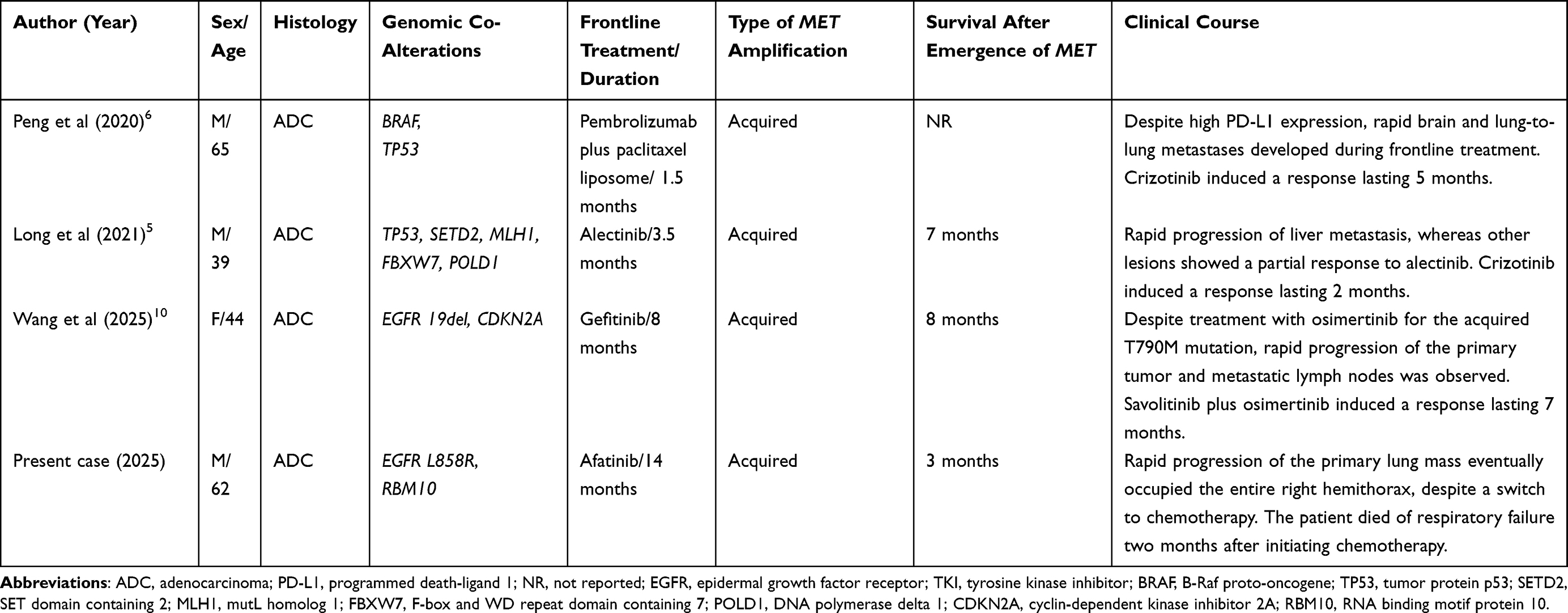 |
Table 3 Summary of Reported Cases of Rapid Progression or Disease Flare Associated with MET Amplification in Non-Small Cell Lung Cancer |
The underlying mechanism by which MET amplification leads to EGFR-TKI resistance may be associated with phosphorylation of ErbB3 (HER3), which functions as a key activator of the PI3K/AKT and MEK/MAPK pathways and provides bypass signaling in the presence of EGFR-TKIs.4 Yarden et al reported that the combination of an anti-HER3 antibody with cetuximab and osimertinib markedly reduced HER3 and MET expression.12 In addition to its role as a resistance mechanism, MET-mediated bypass signaling not only maintains survival but also may create a synergistic proliferative signal when EGFR activity is blocked, potentially accelerating tumor growth.13 Indeed, a study of a preclinical model showed that MET overexpression increases tumor invasiveness, angiogenesis, and metastatic potential.8
It should be noted that additional copy number gains involving genes such as KIT and PDGFRA were also identified in the NGS using primary mass (Table 1), reflecting a broader chromosomal amplification event. However, unlike MET, these alterations have not been established as oncogenic drivers or mediators of aggressive clinical behavior in NSCLC, and are generally considered passenger events.14 We therefore interpret MET amplification as the dominant driver of the observed hyperaggressive disease course, while recognizing that the contribution of co-amplified genes cannot be entirely excluded and warrants further investigation.
Notably, our patient harbored an RBM10 mutation identified in initial NGS analysis, whereas MET amplification was observed during afatinib treatment. RBM10, a member of the RBM gene family, is involved in pre-mRNA splicing and posttranscriptional regulation.15 Previous studies showed that RBM10 overexpression inhibits the malignant behavior of lung adenocarcinoma, including cell viability and cell cycle progression, suggesting that this protein acts as a tumor suppressor.16 In an in vitro study, Jung et al demonstrated that RBM10 induces apoptosis and inhibits the growth of cancer cells, in part by blocking the MDM2-p53 feedback loop and consequently activating the p53 pathway.17
The clinical relevance of RBM10 in this context lies not in its role as a primary oncogenic driver, but in its reported function as a context-dependent modifier in EGFR-mutant NSCLC, where RBM10 loss-of-function has been associated with attenuated apoptotic responses to EGFR-TKIs and reduced therapeutic sensitivity. Increasing evidence has identified a critical role for RBM10 in EGFR-mutant NSCLC. Foggetti et al investigated the effects of inactivating 10 putative tumor suppressor genes in EGFR-mutant lung cancer. They found that RBM10 inactivation strongly promoted tumor growth in a mouse model of EGFR-driven Trp53-deficient lung adenocarcinoma.18 The incidence of RBM10 truncating mutations was 8% among EGFR-mutant lung adenocarcinomas,7,19 which is more frequently observed in the EGFR L858R subtype than in tumors harboring EGFR exon 19 deletions (15% vs 3%, p < 0.01).7 Frequent RBM10 mutations can partly explain the limited therapeutic response in patients with L858R mutation. Nanjo et al reported that RBM10 mutation attenuated the apoptotic response to EGFR-TKIs, resulting in decreased sensitivity to these agents without modulating the oncoprotein target itself-namely, mutant EGFR.7 They showed that RBM10 deficiency altered Bcl-x splicing to increase the relative abundance of its anti-apoptotic isoform, Bcl-xL, to limit apoptosis upon EGFR-TKI treatment.
The RBM10 mutation in the present case (p.E751*) was a nonsense mutation resulting in loss-of-function of RBM10. Interestingly, the tumor initially responded to frontline TKI treatment and the response was maintained for 14 months, although the duration of response was slightly shorter than that in previous studies of frontline afatinib (median 16–18 months of treatment).20,21 This treatment response was in accordance with an aforementioned study that reported that the RBM10 mutation induced an attenuated apoptotic response to EGFR-TKIs rather than being associated with primary resistance.7 Although RBM10 loss-of-function may have attenuated apoptotic responses and potentially facilitated aggressive tumor behavior, its direct causal contribution to the fatal outcome cannot be established from a single case, and should be interpreted as a modifying co-alteration rather than a primary oncogenic driver.
The novelty of this case lies not in the identification of new molecular mechanisms, but in the clinical observation of rapidly fatal progression coinciding with the temporal emergence of MET amplification in a tumor harboring an RBM10 co-mutation, underscoring the need for heightened clinical vigilance and further investigation. In addition, serial NGS showing declining RBM10 variant allele frequency (51.8%→14.3%) alongside emerging MET amplification may provide novel genomic insights. To date, there are no data on the possible interactions between RBM10 and MET amplification. Whether the aggressive and fatal clinical behavior observed in our case was attributable to MET amplification or the interplay of the two genetic alterations is uncertain. However, there are two possible scenarios. First, although there is no direct evidence that RBM10 mutations induce MET amplification, RBM10 loss-of-function may contribute to a permissive cellular environment where such amplification is more likely to occur. Second, the unchecked tumor growth caused by MET amplification may be accelerated by attenuated apoptosis induced by RBM10. Further studies are needed to determine whether RBM10 mutations act synergistically with MET amplification or independently contribute to fulminant progression.
MET-targeted therapy is a feasible option, with a scientific rationale for MET-associated acquired resistance during TKI treatment. Particularly, recent data support the idea that dual EGFR/MET inhibition is effective against MET-driven resistance in EGFR-mutant NSCLC.22–24 A phase Ib/II study of combinational treatment with capmatinib and gefitinib demonstrated a high objective response rate (47%) in the subgroup population with a high degree of MET amplification (copy number gain ≥6).23 Similarly, in the TATTON phase Ib trial, the osimertinib and savolitinib combination achieved an objective response rate of 33–67% in the dose-escalation cohorts (Part B) and 62% in the expansion cohort (Part D), with median progression-free survival times of 5.5–11.1 and 9.0 months, respectively. Notably, patients with high MET amplification (copy number ≥10) derived greater benefit, supporting the clinical application of MET-targeted therapy to overcome TKI- resistance.22 Very recently, the Phase II trial (INSIGHT 2) of tepotinib plus osimertinib demonstrated a high objective response rate (50%) and durable response (8.5 months) in EGFR-mutant patients who progressed on first-line osimertinib and had MET amplification as a resistance mechanism.24 Current international guidelines recommend MET-targeted therapies, including capmatinib, tepotinib, and crizotinib, for high-level MET amplification.25 Although we performed NGS using rebiopsy tissue at the time of the second progression, the results were not obtained until clinical deterioration. Thus, unfortunately, the patient could not be treated with MET-targeted therapy that may have improved clinical outcomes. We should mention that delayed turnaround of tissue-based NGS may have adversely affected the clinical outcome in this patient. In routine practice, comprehensive tissue NGS often requires approximately 2–3 weeks for result reporting. In the present case, the patient experienced fulminant clinical deterioration and died while awaiting NGS results, and actionable molecular information, including MET amplification, became available only after death. These findings underscore the importance of minimizing delays in molecular profiling in cases of abrupt progression. ctDNA-based liquid NGS offers a practical approach to shorten diagnostic latency and provide real-time molecular information. Early and proactive use of blood-based liquid biopsy should therefore be considered in rapidly progressive disease, where timely identification of resistance mechanisms and early targeted therapy based on the NGS results may directly influence clinical outcomes.
In summary, this case highlights the potential for MET amplification to drive not only resistance to EGFR-TKIs, but also rapid clinical deterioration in patients with NSCLC. The temporal emergence of MET amplification and an aggressive disease course suggests that this alteration acts as a potent oncogenic driver in select cases. Furthermore, co-mutations such as RBM10 may represent potential biological modifiers; however, this case does not provide direct evidence that they contributed to tumor progression, and further mechanistic studies are required. Early identification of MET amplification may allow timely initiation of MET-targeted strategies, including combination therapies such as tepotinib plus osimertinib, which have shown clinical efficacy in this setting. In cases of abrupt or hyperaggressive disease progression, the timeline of molecular testing is particularly critical for clinical decision-making. In this case, reliance on tissue-based NGS resulted in a delay between disease progression and availability of molecular results. Had plasma-based ctDNA analysis been performed, molecular alterations such as MET amplification might have been identified more rapidly, potentially allowing earlier consideration of targeted therapeutic options. Future studies using preclinical models, such as EGFR-mutant NSCLC cell lines or patient-derived xenografts with RBM10 loss-of-function and MET amplification, may help clarify whether RBM10 modulates MET-driven signaling, apoptotic resistance, or the aggressive clinical phenotypes observed in select cases.
Data Sharing Statement
All data generated or analyzed during this study are available in this manuscript.
Ethics Statement
The publication of this case report was approved by the Institutional Review Board at Kyung Hee University Hospital (KHUH 2022-02-020).
Consent for Publication
Written informed consent was obtained from the patient’s wife for the publication of both the case report and its accompanying images.
Acknowledgments
The authors thank the patient and his family for their permission to publish this case report along with the accompanying images.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–10. doi:10.1016/S0140-6736(21)00312-3
2. Tsuboi M, Herbst RS, John T, et al. Overall survival with osimertinib in resected EGFR-mutated NSCLC. N Engl J Med. 2023;389(2):137–147. doi:10.1056/NEJMoa2304594
3. Reita D, Pabst L, Pencreach E, et al. Molecular mechanism of EGFR-TKI resistance in EGFR-mutated non-small cell lung cancer: application to biological diagnostic and monitoring. Cancers. 2021;13(19):4926. doi:10.3390/cancers13194926
4. Yang M, Mandal E, Liu FX, O’Hara RM Jr, Lesher B, Sanborn RE. Non-small cell lung cancer with MET amplification: review of epidemiology, associated disease characteristics, testing procedures, burden, and treatments. Front Oncol. 2023;13:1241402. doi:10.3389/fonc.2023.1241402
5. Long X, Wu H, Yang C, Li F, Zhang M, Wu X. Complex genetic alterations contribute to rapid disease progression in an ALK rearrangement lung adenocarcinoma patient: a case report. Transl Cancer Res. 2021;10(6):3081–3086. doi:10.21037/tcr-20-3473
6. Peng Y, Zhang L, Zeng T, et al. Characterization of hyperprogression after immunotherapy in a lung adenocarcinoma patient with strong expression of programmed death ligand 1. J Thorac Oncol. 2020;15(1):e4–e8. doi:10.1016/j.jtho.2019.08.007
7. Nanjo S, Wu W, Karachaliou N, et al. Deficiency of the splicing factor RBM10 limits EGFR inhibitor response in EGFR-mutant lung cancer. J Clin Invest. 2022;132(13). doi:10.1172/JCI145099
8. Qin K, Hong L, Zhang J, Le X. MET amplification as a resistance driver to TKI therapies in lung cancer: clinical challenges and opportunities. Cancers. 2023;15(3):612. doi:10.3390/cancers15030612
9. Planchard D. Have we really MET a new target? J Clin Oncol. 2018;JCO2018793455. doi:10.1200/JCO.2018.79.3455
10. Wang J, Dong X, Liu Y, Lin K, Chen J. Copy number gain of MET gene with low level in a metastatic lung adenocarcinoma patient represents response to salvage treatment with savolitinib and osimertinib: a case report. Front Oncol. 2025;15:1507677. doi:10.3389/fonc.2025.1507677
11. Li J, Xiang C, Wang Y, et al. The genomic characteristics of different progression patterns in advanced non-small cell lung cancer patients treated with immune checkpoint inhibitors. Ann Transl Med. 2021;9(9):779. doi:10.21037/atm-20-6910
12. Romaniello D, Marrocco I, Belugali Nataraj N, et al. Targeting HER3, a catalytically defective receptor tyrosine kinase, prevents resistance of lung cancer to a third-generation EGFR kinase inhibitor. Cancers. 2020;12(9):2394. doi:10.3390/cancers12092394
13. Coleman N, Hong L, Zhang J, Heymach J, Hong D, Le X. Beyond epidermal growth factor receptor: MET amplification as a general resistance driver to targeted therapy in oncogene-driven non-small-cell lung cancer. ESMO Open. 2021;6(6):100319. doi:10.1016/j.esmoop.2021.100319
14. Disel U, Madison R, Abhishek K, et al. The pan-cancer landscape of coamplification of the tyrosine kinases KIT, KDR, and PDGFRA. Oncologist. 2020;25(1):e39–e47. doi:10.1634/theoncologist.2018-0528
15. Yang M, Sun H, Wang H, Zhang S, Yu X, Zhang L. Down-regulation of ribosomal protein L22 in non-small cell lung cancer. Med Oncol. 2013;30(3):646. doi:10.1007/s12032-013-0646-0
16. Cao B, Fang Z, Liao P, et al. Cancer-mutated ribosome protein L22 (RPL22/eL22) suppresses cancer cell survival by blocking p53-MDM2 circuit. Oncotarget. 2017;8(53):90651–90661. doi:10.18632/oncotarget.21544
17. Jung JH, Lee H, Cao B, Liao P, Zeng SX, Lu H. RNA-binding motif protein 10 induces apoptosis and suppresses proliferation by activating p53. Oncogene. 2020;39(5):1031–1040. doi:10.1038/s41388-019-1034-9
18. Foggetti G, Li C, Cai H, et al. Genetic determinants of EGFR-driven lung cancer growth and therapeutic response in vivo. Cancer Discov. 2021;11(7):1736–1753. doi:10.1158/2159-8290.CD-20-1385
19. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
20. Su VY, Yang KY, Huang TY, et al. The efficacy of first-line tyrosine kinase inhibitors combined with co-medications in Asian patients with EGFR mutation non-small cell lung cancer. Sci Rep. 2020;10(1):14965. doi:10.1038/s41598-020-71583-w
21. Huang YH, Tseng JS, Hsu KH, et al. The impact of different first-line EGFR-TKIs on the clinical outcome of sequential osimertinib treatment in advanced NSCLC with secondary T790M. Sci Rep. 2021;11(1):12084. doi:10.1038/s41598-021-91657-7
22. Hartmaier RJ, Markovets AA, Ahn MJ, et al. Osimertinib + savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discov. 2023;13(1):98–113. doi:10.1158/2159-8290.CD-22-0586
23. Wu YL, Zhang L, Kim DW, et al. Phase Ib/II study of Capmatinib (INC280) plus gefitinib after failure of Epidermal Growth Factor Receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J Clin Oncol. 2018;36(31):3101–3109. doi:10.1200/JCO.2018.77.7326
24. Wu YL, Guarneri V, Voon PJ, et al. Tepotinib plus osimertinib in patients with EGFR-mutated non-small-cell lung cancer with MET amplification following progression on first-line osimertinib (INSIGHT 2): a multicentre, open-label, Phase 2 trial. Lancet Oncol. 2024;25(8):989–1002. doi:10.1016/S1470-2045(24)00270-5
25. Park J, Chelakkot C, Nam JH, et al. MET alterations in cancer and MET-targeted therapy: detection strategies, treatment efficacy, and emerging technologies. Target Oncol. 2025;20(5):767–789. doi:10.1007/s11523-025-01166-0
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.