Back to Journals » Journal of Inflammation Research » Volume 9
Familial Mediterranean fever: current perspectives
Authors Sonmez H, Batu E, Ozen S
Received 13 October 2015
Accepted for publication 21 November 2015
Published 17 March 2016 Volume 2016:9 Pages 13—20
DOI https://doi.org/10.2147/JIR.S91352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Hafize Emine Sönmez,* Ezgi Deniz Batu,* Seza Özen
Department of Pediatrics, Division of Rheumatology, Hacettepe University Faculty of Medicine, Ankara, Turkey
*These authors contributed equally to this work
Abstract: Familial Mediterranean fever (FMF) is the most frequent monogenic autoinflammatory disease, and it is characterized by recurrent attacks of fever and polyserositis. The disease is associated with mutations in the MEFV gene encoding pyrin, which causes exaggerated inflammatory response through uncontrolled production of interleukin 1. The major long-term complication of FMF is amyloidosis. Colchicine remains the principle therapy, and the aim of treatment is to prevent acute attacks and the consequences of chronic inflammation. With the evolution in the concepts about the etiopathogenesis and genetics of the disease, we have understood that FMF is more complicated than an ordinary autosomal recessive monogenic disorder. Recently, recommendation sets have been generated for interpretation of genetic testing and genetic diagnosis of FMF. Here, we have reviewed the current perspectives in FMF in light of recent recommendations.
Keywords: familial Meditarranean fever, recommendation, child
Introduction
Familial Mediterranean fever (FMF) is the most common autoinflammatory disease (AID) mainly affecting the populations of eastern Mediterranean descent.1,2 It is characterized by recurrent, self-limited flares of fever associated with polyserositis.3 The gene mutated in patients with FMF is the MEFV gene, which encodes pyrin, which forms an element of the NLRP3 inflammasome complex.2 Mutations in the MEFV gene are associated with increased interleukin-1β (IL-1β), which causes excess inflammation.4 The majority of FMF patients demonstrate a Mendelian autosomal recessive pattern of inheritance.5–7 However, since the MEFV gene was found to underlie FMF in 1997,5,6 the concepts about the etiopathogenesis and genetics of the disease have evolved. Progress in molecular biology suggested that this AID was more complicated than we had anticipated.
Colchicine still constitutes the mainstay of FMF treatment,8 and the aim of the treatment should be preventing acute attacks and amyloidosis, decreasing the chronic inflammation, and providing an acceptable quality of life. Recent insights into the pathogenesis of FMF have made anti-IL 1 treatments important in colchicine-resistant or -intolerant patients. The most severe complication of FMF is secondary amyloidosis;9 however, it is less common in the colchicine and anti-IL 1 era.
In 2012, Shinar et al10 proposed recommendations for interpretation of genetic testing in AIDs. Most recently, the SHARE (a pediatric initiative to develop better care and management for rheumatology patients; Single Hub and Access point for pediatric Rheumatology in Europe) initiative has developed evidence-based recommendations for genetic diagnosis of FMF.11
In this paper, we have reviewed the current perspectives in FMF in the light of recent recommendations.
Etiopathogenesis
The MEFV gene encodes the protein pyrin, which is a part of the inflammasome complex, NLRP3, an intracellular organelle required for the production of IL-1β.12 Thus, FMF may be classified as an inflammasomeopathy.2 Pyrin has been suggested to interact with the inflammasome adaptor protein ASC, and this results in increased caspase-1 activation and IL-1β processing.13
In 2007, Yu et al14 have shown that activated pyrin interacts with ASC and PSTPIP1 to form a trimolecular complex that directly activates caspase-1 and then leads to the secretion of active IL-1β. Booty et al15 demonstrated a significant increase in pyrin expression in FMF patients in comparison with healthy controls. Supporting the hypothesis of a gain-of-function model, an elegant study by Chae et al16 revealed that FMF-associated B30.2 mutations knock-in mice, but not pyrin deficient ones, showed severe spontaneous inflammatory phenotype.
On the other hand, Papin et al17 showed that pyrin knockdown resulted in increased caspase-1 activation and IL-1β secretion. In another study by Hesker et al,18 enhanced IL-1β release by macrophages was demonstrated in response to a spectrum of inflammatory stimuli in a mouse line lacking the MEFV gene. The findings of these two studies suggest a loss-of-function model.
However, it is still controversial whether MEFV mutations cause loss of function or gain of function.
A recent study by Xu et al19 has elegantly shown that pyrin is a specific immune sensor for bacterial modifications of Rho GTPases, and responds to Clostridium difficile, which is a frequent cause of nosocomial diarrhea. Pyrin does not directly recognize the microbial products but detects pathogen virulence activity. This recent finding has shed some light on FMF pathogenesis.
Diagnosis
Since FMF usually requires lifelong treatment, it is crucial to establish a timely, correct diagnosis. The diagnosis of FMF relies mainly on clinical findings, and molecular analysis of the MEFV gene provides genetic confirmation.20
There are different sets of classification and diagnostic criteria for FMF. The first set of criteria was created for adults by the experts in Tel Hashomer Hospital (Table 1).21 Livneh et al22 validated the new criteria in 1997, excluding some manifestations of the Tel Hashomer criteria such as amyloidosis (Table 1). However, there were certain differences between adult and pediatric FMF cases (such as shorter attacks in children, lack of unilateral characteristic of chest pain in some pediatric cases, more febrile attacks or even fever-only attacks in some children, and inability of some pediatric patients to express the severity and exact location of the pain), and some of the Tel Hashomer criteria were of less relevance to pediatric FMF patients. Thus, in 2009, our group attempted to define criteria for children as well (Table 1).23 Among Turkish children, the criteria (two out of five criteria for diagnosis) reached a sensitivity and specificity of 88.8% and 92.2%, respectively.23 In French children, the presence of three instead of two criteria yielded a better specificity of 95%.24 Validation of these criteria in a larger and genetically more heterogeneous group is crucial.
 | Table 1 The clinical criteria sets for FMF diagnosis |
It is important to keep in mind that FMF diagnosis is a clinical one, and if the phenotype is consistent with FMF, the physician should not exclude the diagnosis when there is no genetic confirmation.
Genetics
Variants to be checked in the MEFV gene
When the genetic association of FMF with the MEFV gene was first described in 1997,5,6 only a few mutations had been reported. Since the number of variants recognized associated with FMF has increased, concerns emerged about the adequacy of checking for four to six mutations only. Booty et al15 sequenced the MEFV gene in heterozygote FMF patients and demonstrated that additional sequencing of the whole gene did not yield a second mutation in any of the screened patients. Currently, all reported MEFV variants and the associated phenotypes are recorded in the INFEVERS database (http://fmf.igh.cnrs.fr/ISSAID/infevers/), and there are approximately 300 known sequence variants of MEFV.11 The most common disease-associated pathogenic variants are M694V, V726A, M680I, and M694I, whereas E148Q is the most frequent variant among carriers.25,26 An agreed set of best practice guidelines was proposed for genetic diagnostic testing of hereditary recurrent fevers including FMF in 2012.10 The consensus was to test for a total of 14 variants (the first nine are defined as clearly pathogenic, while the remaining five variants have unknown significance): M694V, M694I, M680I, V726A, R761H, A744S, E167D, T267I, I692del, K695R, E148Q, P369S, F479L, and I591T. We now suggest testing for these variants only.
Heterozygotes
FMF is an autosomal recessive disease. Thus, one would expect a heterozygote individual to be a carrier and lack the FMF phenotype. However, as FMF patients were genotyped, it has become evident that the mutation in the second allele could not be demonstrated in about one-quarter of patients with clinically confirmed FMF.10 As we previously mentioned, resequencing of the entire MEFV genomic region did not provide additional benefit.15
In 2009, Marek-Yagel et al27 studied the clinical and genetic characteristics of heterozygous FMF patients and performed haplotype studies. They concluded that although the heterozygotes tend to have a relatively mild disease, the disease could not be distinguished from that of homozygous patients and that FMF could be viewed as a dominant condition with low penetrance in some cases.27
In addition, convincing data indicate that heterozygous individuals have an increased propensity to display signs of inflammation. In the eastern Mediterranean population, the carrier rate is higher among the patients with rheumatic diseases than in the general population.28,29 Kalyoncu et al30 revealed that rheumatoid arthritis, acute rheumatic fever, arthralgia, and febrile episodes of ≥4 times/year were significantly more common in asymptomatic heterozygous parents of children with FMF than healthy controls. Along the same lines, Lachmann et al31 showed greater basal and peak acute-phase reactant concentrations in MEFV heterozygotes in comparison with wild-type controls which suggest a proinflammatory phenotype in carriers.
One explanation for heterozygote individuals displaying FMF phenotype may be the effect of other modifier genes associated with inflammation.15 One example is the serum amyloid A (SAA) gene, encoding an acute-phase protein, SAA, which activates NLRP3 inflammasome causing increased secretion of active IL-1β.32
We also know that epigenetics and environment may affect the disease phenotype, and this will now be discussed later in the paper.
Genotype–phenotype correlation
Since the first description of the MEFV mutations, most experts agree that M694V was related with a severe disease phenotype.5,6,33 Ozturk et al34 demonstrated that both patients homozygous and compound heterozygous for M694V were at increased risk for severe disease in comparison with patients with one mutant allele and patients not displaying M694V. Giaglis et al35 and Mattit et al36 confirmed these findings in two separate studies. Giaglis et al35 also showed that a more severe phenotype was evident in patients homozygous for M694V as well as mutations at position 680–694 on exon 10. In evidence-based recommendations, the experts concluded that FMF patients homozygous or compound heterozygous for M694V or with mutations at position 680–694 on exon 10 must be considered at risk of having a more severe and early-onset disease.11 In addition, the individuals homozygous for M694V without disease manifestations should be evaluated and followed closely in order to decide about therapy.11
Besides issues about mutations at position 680–694 on exon 10, in a recent study, Federici et al37 demonstrated that the frequency of FMF-like symptoms increased from patients carrying a single low penetrance mutation toward patients with two high penetrance mutations, suggesting a “dose effect” associated with the mutations.
The pathogenic role of E148Q on exon 2 (one of the most common alterations in the MEFV gene) still remains controversial.11 It might be a polymorphism since it is present in >1% of the healthy population. It has been reported to be present as high as 21% of healthy in some eastern Asian populations.38 A consensus conference led by Shinar et al10 has defined E148Q as a variant of unknown significance. According to the recent recommendations by SHARE, as the only MEFV variant, E148Q does not support the diagnosis of FMF.11 However, there are rare reports of patients with homozygous E148Q variation having FMF phenotype.26 Furthermore, patients carrying E148Q with a clearly pathogenic mutation on the other allele often display disease phenotype.2
The recent evidence-based recommendations for genetic diagnosis of FMF are as follows:11
- FMF is a clinical diagnosis, which can be supported but not excluded by genetic testing.
- Consider patients homozygous for M694V at risk of developing, with very high probability, a severe phenotype.
- FMF patients carrying two of the common mutated alleles (homozygotes or compound heterozygotes), especially for M694V mutation or mutations at position 680–694 on exon 10, must be considered at risk of having a more severe disease.
- The E148Q variant is common, of unknown pathogenic significance, and, as the only MEFV variant, does not support the diagnosis of FMF.
- Patients homozygous for M694V mutation are at risk of early-onset disease.
- Individuals homozygous for M694V who are not reporting symptoms should be evaluated and followed closely to consider therapy.
- For individuals with two pathogenic mutations for FMF who do not report symptoms, if there are risk factors for AA amyloidosis (such as the country, family history, and persistently elevated inflammatory markers, particularly serum amyloid A protein), close follow-up should be started and treatment considered.
- Consultation with an AID specialist may be helpful to aid in the indication and interpretation of the genetic testing and diagnosis.
Epigenetics and environment
FMF patients with similar genotype may express different disease phenotypes. This difference may be due to other modifier genes, epigenetics, or the effect of the environment. The first clue suggesting the effect of environment on FMF was the observation of lack of secondary amyloidosis among Armenian FMF patients in the USA.39 We also know that the eastern Mediterranean patients had a less severe disease if they migrated to Europe.9 Touitou et al40 demonstrated that country of recruitment, rather than MEFV genotype, is the likely key risk factor for development of amyloidosis. We had previously shown that Turkish children with FMF living in Germany expressed a less severe disease phenotype in comparison with the ones living in Turkey.41 These findings emphasize the effect of environment on FMF disease severity.
The epigenetic mechanisms such as histone modification, methylation, and microRNAs may play role in the pathogenesis of FMF. Kirectepe et al42 demonstrated a slightly higher methylation level of exon 2 of MEFV in FMF patients when compared to healthy controls.
MicroRNAs (miRNAs) are small, noncoding RNAs that regulate gene expression at a posttranscriptional level by degrading mRNA molecules or blocking their translation.43 Their circulating levels have already been described in inflammatory disorders such as rheumatoid arthritis, systemic lupus erythematosus, and tumor necrosis factor (TNF) receptor–associated periodic syndrome.44–47 Circulating miRNAs in FMF have not been investigated, but this might have a role in the disease pathogenesis of FMF as well.
Microorganisms may affect FMF since pyrin is a component of NLRP3, which is a pathogen recognition receptor.9 Pyrin has also been shown to detect virulent pathogenic activity.19 The cross-talk between the innate immune system and commensal gut bacteria (microbiota) may affect (or may be affected by) the inflammatory status of the patient, as well. In a study by Khachatryan et al,48 they showed that the composition and divergence of microbiota were different during attack and attack-free periods as well as between FMF patients and healthy controls.
Further studies on the effect of epigenetics, microbiota, and environment on FMF may enable us to answer more unsolved questions about the pathogenesis.
Treatment
Colchicine is still the main form of treatment for patients with FMF. Since 1972, colchicine has been used for treatment in FMF patients.8 It decreases attack frequency and increases the quality of life.49 Furthermore, Livneh et al50 have shown that colchicine can effectively prevent the complication of secondary amyloidosis in FMF patients.
In pediatric FMF patients, colchicine was shown to be a well-tolerated drug, even when given in infancy.51 The commonest side effects of colchicine are gastrointestinal, with vomiting, diarrhea, and transient elevation of transaminases. These mild side effects can occur in 5%–10% of patients even at recommended doses. It has a narrow therapeutic index; colchicine doses of 0.5–0.8 mg/kg are highly toxic, and doses of more than 0.8 mg/kg are typically lethal. In fact, the US Food and Drug Administration withdrew approval for intravenous colchicine to reduce the risk of irreversible bolus overdose.52,53
Colchicine is undoubtedly the main treatment for FMF, but approximately one-third of the patients treated with colchicine have a partial remission, and approximately 5% are nonresponders; another 2%–5% do not tolerate the drug mainly due to gastrointestinal symptoms.54 Lidar et al55 have suggested that colchicine efficacy differs between patient groups depending on the MEFV genotype. M694V homozygotes showed a more severe disease and were treated with higher doses of colchicine compared to patients with the V726A genotype.
In recent years, biologic agents have been used in treatment of FMF patients unresponsive to colchicine therapy.56 Pyrin, the mutated protein in FMF, plays an important role in the regulation of interleukin-1 (IL-1) activation; anti-IL-1 treatment has proven beneficial in suppressing inflammation in colchicine-resistant FMF patients. Anakinra, a recombinant, human IL-1 receptor antagonist that competitively inhibits binding of IL-1α and IL-1β to the IL-1 receptor; canakinumab, a human IgG1 monoclonal antibody directed against IL-1β; and rilonacept a fully human dimeric fusion protein that binds the extracellular domains of IL-1α and IL-1β were used in FMF patients.57 There are small series or trials about anti-IL-1 treatment in colchicine-resistant FMF patients.56,58 The only randomized controlled study was with rilonacept, in 14 colchicine-resistant FMF patients.59 Recently, a 6-month Phase II, open-label, single-arm pilot study showed the efficacy of canakinumab in pediatric patients with colchicine-resistant FMF.60 The major limitation of this study was the small sample size. A larger controlled study is needed to explain the benefit, optimal dose, and side effect of canakinumab better in FMF patients.
Recently, French and Israeli experts have suggested evidence-based recommendations for the management of FMF.49 The treatment recommendations are about colchicine dosage, maximum dosage of colchicine in children and adults, definition of colchicine resistance, and treatment alternatives in colchicine-resistant patients. Colchicine dose should be increased if patients have more than one FMF attack every 3 months and/or persistent elevation of inflammatory markers during the attack-free period. If a patient suffers from more than six typical FMF attacks per year, the patient should be accepted to be resistant to colchicine. The experts have suggested alternative treatment, such as IL-1 inhibitors, if there are persistent attacks despite the maximum doses of colchicine (2 mg in children; 3 mg in adults).49
TNF blockade with etanercept, infliximab, or adalimumab has been studied especially in FMF patients. Anti-TNF treatment can have beneficial effects for controlling FMF attacks in FMF patients with chronic arthritis and/or sacroiliitis.61
Monitoring disease activity and response to treatment
The AutoInflammatory Disease Activity Index (AIDAI) is used to assess disease activity in FMF and the other common monogenic AIDs. AIDAI is a patient-based symptom diary that consists of 13 items as follows: fever, overall symptoms, abdominal pain, nausea/vomiting, diarrhea, headaches, chest pain, painful nodes, arthralgia or myalgia, swelling of the joints, eye manifestations, skin rash, and pain relief. It is scored daily as yes (1 point) or no (0 point) by patients or parents. All items but pain relief are used in the calculation of the activity score (Figure 1). Validation of AIDAI revealed a cutoff score of ≥9, discriminating active from inactive patients with a sensitivity of 89% and specificity of 92%.9,62
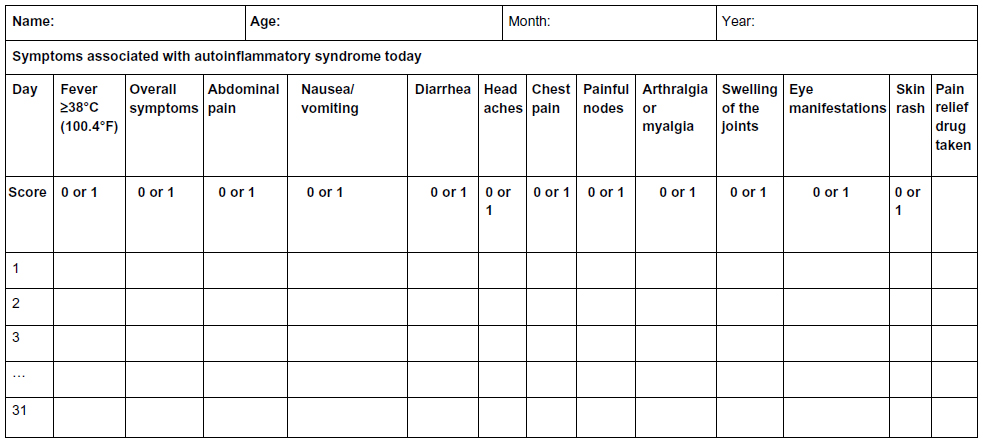 | Figure 1 AID activity index diary. |
Turkish physicians have suggested the FMF50 score to assess the response to treatment, given the patient is compliant and is receiving the maximum tolerated dose of the drug. The items of this score are: percentage change in the frequency and duration of attacks with the treatment, patients/parents’ and physicians’ global assessment of disease severity (10 cm visual analog scale), percentage change in arthritis attacks with the treatment, percentage change in C-reactive protein (CRP), erythrocyte sedimentation rate, or serum amyloid A level with the treatment. At least 50% improvement in five of six criteria, without worsening in any one, was defined as response to treatment with very high sensitivity and specificity.63 However, this score needs further validation before use in clinical trials.
Prognosis
The prognosis of a compliant FMF patient is now excellent thanks to the advances in management of FMF and the use of biologics, especially anti-IL-1 drugs. Secondary amyloidosis was the most serious complication. However, because of the advances in the management of AIDs, this complication is now very rare. Many studies have focused on the role of factors affecting the development of amyloidosis in patients with FMF, such as male sex, M694V homozygosity, and SAA1.1α/α genotype.64–66 Colchicine treatment prevents the development of amyloidosis. However, some patients are refractory to colchicine treatment. Biologic treatments, including anti-TNF agents and IL-1 and IL-6 antagonists, have been suggested to be effective in the treatment of amyloidosis.67,68
There are limited data on long-term comorbidities and mortality among patients with FMF. Twig et al69 studied long-term comorbidities and mortality among patients with FMF and found that FMF causes increased risk of mortality associated with amyloidosis, but does not have an increased incidence of cancer.
Rheumatic diseases may be risk factors for atherosclerosis. Studies have shown that inflammatory processes had played a significant role in atherosclerosis and have demonstrated an important association between high CRP levels and risk of atherosclerosis.70 Several studies have shown that FMF might be a risk factor for early atherosclerosis because of its inflammatory nature.71–73 However, a recent study from Israel showed that there were lower rates of metabolic syndrome compared to normal subjects, unlike other inflammatory diseases.74
Thus, FMF is a disease in which we can promise our compliant patients a normal life expectancy and remission with medication with a good quality of life.
Disclosure
The authors report no conflicts of interest in this work.
References
Aksentijevich I, Kastner DL. Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol. 2011;7:469–478. | |
Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol. 2014;10:135–147. | |
Tunca M, Akar S, Onen F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore). 2005;84:1–11. | |
Chae JJ, Wood G, Masters SL, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1β production. Proc Natl Acad Sci U S A. 2006;103:9982–9987. | |
Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell. 1997;90:797–807. | |
French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17:25–31. | |
Stoffels M, Szperl A, Simon A, et al. MEFV mutations affecting pyrin amino acid 577 cause autosomal dominant autoinflammatory disease. Ann Rheum Dis. 2014;73:455–461. | |
Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287:1302. | |
Ozen S, Batu ED. The myths we believed in familial Mediterranean fever: what have we learned in the past years? Semin Immunopathol. 2015;37:363–369. | |
Shinar Y, Obici L, Aksentijevich I, et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis. 2012;71: 1599–1605. | |
Giancane G, Nienke ter H, Wulffraat N, Vastert B, et al. Evidence based recommendations for genetic diagnosis of Familial Mediterranean Fever. Pediatr Rheumatol. 2014;12(Suppl 1):241. | |
Chae JJ, Komarow HD, Cheng J, et al. Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol Cell. 2003;11:591–604. | |
Park H, Bourla AB, Kastner DL, Colbert RA, Siegel RM. Lighting the fires within: the cell biology of autoinflammatory diseases. Nat Rev Immunol. 2012;12:570–580. | |
Yu JW, Fernandes-Alnemri T, Datta P, et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28:214–227. | |
Booty MG, Chae JJ, Masters SL, et al. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 2009;60:1851–1861. | |
Chae JJ, Cho YH, Lee GS, et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity. 2011;34:755–768. | |
Papin S, Cuenin S, Agostini L, et al. The SPRY domain of Pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1β processing. Cell Death Differ. 2007;14:1457–1466. | |
Hesker PR, Nguyen M, Kovarova M, Ting JP, Koller BH. Genetic loss of murine pyrin, the Familial Mediterranean Fever protein, increases interleukin-1β levels. PloS One. 2012;7:e51105. | |
Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513:237–241. | |
Soriano A, Manna R. Familial Mediterranean fever: new phenotypes. Autoimmun Rev. 2012;12:31–37. | |
Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. 1967;43: 227–253. | |
Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40:1879–1885. | |
Yalcinkaya F, Ozen S, Ozcakar ZB, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford). 2009;48:395–398. | |
Kondi A, Hentgen V, Piram M, Letierce A, Guillaume-Czitrom S, Kone-Paut I. Validation of the new pediatric criteria for the diagnosis of familial Mediterranean fever: data from a mixed population of 100 children from the French reference centre for auto-inflammatory disorders. Rheumatology (Oxford). 2010;49:2200–2203. | |
Touitou I. The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet. 2001;9:473–483. | |
Yilmaz E, Ozen S, Balci B, et al. Mutation frequency of familial Mediterranean fever and evidence for a high carrier rate in the Turkish population. Eur J Hum Genet. 2001;9:553–555. | |
Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 2009;60:1862–1866. | |
Kogan A, Shinar Y, Lidar M, et al. Common MEFV mutations among Jewish ethnic groups in Israel: high frequency of carrier and phenotype III states and absence of a perceptible biological advantage for the carrier state. Am J Med Genet. 2001;102:272–276. | |
Ozen S, Bakkaloglu A, Yilmaz E, et al. Mutations in the gene for familial Mediterranean fever: do they predispose to inflammation? J Rheumatol. 2003;30:2014–2018. | |
Kalyoncu M, Acar BC, Cakar N, et al. Are carriers for MEFV mutations “healthy”? Clin Exp Rheumatol. 2006;24:S120–S122. | |
Lachmann HJ, Sengul B, Yavuzsen TU, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford). 2006;45:746–750. | |
Niemi K, Teirila L, Lappalainen J, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–6128. | |
Balow JE Jr, Shelton DA, Orsborn A, et al. A high-resolution genetic map of the familial Mediterranean fever candidate region allows identification of haplotype-sharing among ethnic groups. Genomics. 1997;44:280–291. | |
Ozturk C, Halicioglu O, Coker I, et al. Association of clinical and genetical features in FMF with focus on MEFV strip assay sensitivity in 452 children from western Anatolia, Turkey. Clin Rheumatol. 2012;31: 493–501. | |
Giaglis S, Papadopoulos V, Kambas K, et al. MEFV alterations and population genetics analysis in a large cohort of Greek patients with familial Mediterranean fever. Clin Genet. 2007;71:458–467. | |
Mattit H, Joma M, Al-Cheikh S, et al. Familial Mediterranean fever in the Syrian population: gene mutation frequencies, carrier rates and phenotype-genotype correlation. Eur J Med Genet. 2006;49:481–486. | |
Federici S, Calcagno G, Finetti M, et al. Clinical impact of MEFV mutations in children with periodic fever in a prevalent western European Caucasian population. Ann Rheum Dis. 2012;71:1961–1965. | |
Booth DR, Lachmann HJ, Gillmore JD, Booth SE, Hawkins PN. Prevalence and significance of the familial Mediterranean fever gene mutation encoding pyrin Q148. QJM. 2001;94:527–531. | |
Schwabe AD, Peters RS. Familial Mediterranean fever in Armenians. Analysis of 100 cases. Medicine (Baltimore). 1974;53:453–462. | |
Touitou I, Sarkisian T, Medlej-Hashim M, et al. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum. 2007;56:1706–1712. | |
Ozen S, Aktay N, Lainka E, Duzova A, Bakkaloglu A, Kallinich T. Disease severity in children and adolescents with familial Mediterranean fever: a comparative study to explore environmental effects on a monogenic disease. Ann Rheum Dis. 2009;68:246–248. | |
Kirectepe AK, Kasapcopur O, Arisoy N, et al. Analysis of MEFV exon methylation and expression patterns in familial Mediterranean fever. BMC Med Genet. 2011;12:105. | |
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. | |
Ceribelli A, Yao B, Dominguez-Gutierrez PR, Nahid MA, Satoh M, Chan EK. MicroRNAs in systemic rheumatic diseases. Arthritis Res Ther. 2011;13:229. | |
Dai Y, Sui W, Lan H, Yan Q, Huang H, Huang Y. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. 2009;29:749–754. | |
Lucherini OM, Obici L, Ferracin M, et al. First report of circulating microRNAs in tumour necrosis factor receptor-associated periodic syndrome (TRAPS). PloS One. 2013;8:e73443. | |
Wang GK, Zhu JQ, Zhang JT, et al. Circulating microRNA: a novel potential biomarker for early diagnosis of acute myocardial infarction in humans. Eur Heart J. 2010;31:659–666. | |
Khachatryan ZA, Ktsoyan ZA, Manukyan GP, Kelly D, Ghazaryan KA, Aminov RI. Predominant role of host genetics in controlling the composition of gut microbiota. PloS One. 2008;3:e3064. | |
Hentgen V, Grateau G, Kone-Paut I, et al. Evidence-based recommendations for the practical management of Familial Mediterranean fever. Semin Arthritis Rheum. 2013;43:387–391. | |
Livneh A, Zemer D, Langevitz P, Shemer J, Sohar E, Pras M. Colchicine in the treatment of AA and AL amyloidosis. Semin Arthritis Rheum. 1993;23:206–214. | |
Padeh S, Gerstein M, Berkun Y. Colchicine is a safe drug in children with familial Mediterranean fever. J Pediatr. 2012;161:1142–1146. | |
Terkeltaub RA. Colchicine update: 2008. Semin Arthritis Rheum. 2009;38:411–419. | |
Finkelstein Y, Aks SE, Hutson JR, et al. Colchicine poisoning: the dark side of an ancient drug. Clin Toxicol. 2010;48:407–414. | |
Kallinich T, Haffner D, Niehues T, et al. Colchicine use in children and adolescents with familial Mediterranean fever: literature review and consensus statement. Pediatrics. 2007;119:e474–e483. | |
Lidar M, Yonath H, Shechter N, et al. Incomplete response to colchicine in M694V homozygote FMF patients. Autoimmun Rev. 2012;12:72–76. | |
Meinzer U, Quartier P, Alexandra JF, Hentgen V, Retornaz F, Kone-Paut I. Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum. 2011;41: 265–271. | |
Grattagliano I, Bonfrate L, Ruggiero V, Scaccianoce G, Palasciano G, Portincasa P. Novel therapeutics for the treatment of familial Mediterranean fever: from colchicine to biologics. Clin Pharmacol Ther. 2014;95:89–97. | |
Eroglu FK, Besbas N, Topaloglu R, Ozen S. Treatment of colchicine-resistant Familial Mediterranean fever in children and adolescents. Rheumatol Int. 2015;35:1733–1737. | |
Hashkes PJ, Spalding SJ, Giannini EH, et al. Rilonacept for colchicine-resistant or -intolerant familial Mediterranean fever: a randomized trial. Ann Intern Med. 2012;157:533–541. | |
Brik R, Butbul-Aviel Y, Lubin S, et al. Canakinumab for the treatment of children with colchicine-resistant familial Mediterranean fever: a 6-month open-label, single-arm pilot study. Arthritis Rheumatol. 2014;66:3241–3243. | |
Bilgen SA, Kilic L, Akdogan A, et al. Effects of anti-tumor necrosis factor agents for familial mediterranean fever patients with chronic arthritis and/or sacroiliitis who were resistant to colchicine treatment. J Clin Rheumatol. 2011;17:358–362. | |
Piram M, Kone-Paut I, Lachmann HJ, et al. Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis. 2014;73:2168–2173. | |
Ozen S, Demirkaya E, Duzova A, et al. FMF50: a score for assessing outcome in familial Mediterranean fever. Ann Rheum Dis. 2014;73: 897–901. | |
Saatci U, Ozen S, Ozdemir S, et al. Familial Mediterranean fever in children: report of a large series and discussion of the risk and prognostic factors of amyloidosis. Eur J Pediatr. 1997;156:619–623. | |
Shohat M, Magal N, Shohat T, et al. Phenotype-genotype correlation in familial Mediterranean fever: evidence for an association between Met694Val and amyloidosis. Eur J Hum Genet. 1999;7:287–292. | |
Gershoni-Baruch R, Brik R, Zacks N, Shinawi M, Lidar M, Livneh A. The contribution of genotypes at the MEFV and SAA1 loci to amyloidosis and disease severity in patients with familial Mediterranean fever. Arthritis Rheum. 2003;48:1149–1155. | |
Bilginer Y, Akpolat T, Ozen S. Renal amyloidosis in children. Pediatr Nephrol. 2011;26:1215–1227. | |
Yilmaz S, Cinar M, Simsek I, Erdem H, Pay S. Tocilizumab in the treatment of patients with AA amyloidosis secondary to familial Mediterranean fever. Rheumatology (Oxford). 2015;54:564–565. | |
Twig G, Livneh A, Vivante A, et al. Mortality risk factors associated with familial Mediterranean fever among a cohort of 1.25 million adolescents. Ann Rheum Dis. 2014;73:704–709. | |
Pearson TA, Mensah GA, Alexander RW, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. | |
Kucuk A, Uslu AU, Arslan S, et al. Ischemia-modified albumin and atherosclerosis in patients with familial Mediterranean fever. Angiology. Epub 2015 Aug 3. | |
Ugurlu S, Seyahi E, Cetinkaya F, Ozbakir F, Balci H, Ozdogan H. Intima-media thickening in patients with familial Mediterranean fever. Rheumatology (Oxford). 2009;48:911–915. | |
Uluca U, Demir F, Ece A, et al. Assessment of epicardial adipose tissue thickness and the mean platelet volume in children with familial Mediterranean fever. Ital J Pediatr. 2015;41:15. | |
Twig G, Livneh A, Vivante A, et al. Cardiovascular and metabolic risk factors in inherited autoinflammation. J Clin Endocrinol Metab. 2014;99:E2123–E2128. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.