Back to Journals » Journal of Pain Research » Volume 15
Familial Episodic Pain Syndromes
Authors Shen Y ![]() , Zheng Y, Hong D
, Zheng Y, Hong D
Received 18 May 2022
Accepted for publication 26 July 2022
Published 26 August 2022 Volume 2022:15 Pages 2505—2515
DOI https://doi.org/10.2147/JPR.S375299
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Yu Shen,1 Yilei Zheng,1 Daojun Hong1,2
1Department of Neurology, The First Affiliated Hospital of Nanchang University, Nanchang, 330006, People’s Republic of China; 2Department of Medical Genetics, The First Affiliated Hospital of Nanchang University, Nanchang, 330006, People’s Republic of China
Correspondence: Daojun Hong, Department of Neurology, The First Affiliated Hospital of Nanchang University, 17# Yongwaizheng St, Nanchang, 330006, People’s Republic of China, Tel/Fax +86-791-8869-2511, Email [email protected]
Abstract: Over the past decades, advances in genetic sequencing have opened a new world of discovery of causative genes associated with numerous pain-related syndromes. Familial episodic pain syndromes (FEPS) are one of the distinctive syndromes characterized by early-childhood onset of severe episodic pain mainly affecting the distal extremities and tend to attenuate or diminish with age. According to the phenotypic and genetic properties, FEPS at least includes four subtypes of FEPS1, FEPS2, FEPS3, and FEPS4, which are caused by mutations in the TRPA1, SCN10A, SCN11A, and SCN9A genes, respectively. Functional studies have revealed that all missense mutations in these genes are closely associated with the gain-of-function of cation channels. Because some FEPS patients may show a relative treatability and favorable prognosis, it is worth paying attention to the diagnosis and management of FEPS as early as possible. In this review, we state the common clinical manifestations, pathogenic mechanisms, and potential therapies of the disease, and provide preliminary opinions about future research for FEPS.
Keywords: familial episodic pain syndromes, voltage-gated sodium channel, transient receptor potential A1, dorsal root ganglia, nociceptive pain
Introduction
Pain is an intrinsic alert system that defends organisms against further injuries.1 Pain originates from particular stimuli acting on the nociceptive receptors and triggering the depolarization of membranes that transmits sensory information to corresponding regions of the central nervous system (CNS).2–6 According to its anatomical and pathophysiological properties, pain can be divided into nociceptive and neuropathic pain, both of which sometimes overlap in clinical practice.7
Sensory neurons of the dorsal root ganglia (DRG) are a group of heterogeneous neurons with distinct functional and histochemical properties and play a vital role in transmitting and integrating pain sensation.8 DRG neurons can be sensitized by various inflammations and injuries, leading to hyperactivity of the voltage-gated sodium channel (VGSC).9–13 DRG neurons express multiple VGSCs, including Nav1.1, Nav1.3, Nav1.6, Nav1.7, Nav1.8, and Nav1.9, which are essential for their ability to fire action potentials.14–17 VGSCs contain large pore-forming α-subunits and smaller β-subunits that structure sophisticated heteromultimers.18–21 As for the adaptive evolution and tissue specificity in vertebrates, there are ten subtypes of α-subunits (Nav1.1–1.9 and Nav2.1) with distinct electrophysiological and pharmacological properties.22 Each α-subunit is composed of four homologous membrane-spanning domains (DI-DIV). Each domain consists of six transmembrane segments (S1-S6).23,24
Genetic variants in Nav1.7, Nav1.8, and Nav1.9 have been associated with human Mendelian pain-related disorders, including familial episodic pain syndromes (FEPS), small fiber neuropathy (SFN), paroxysmal extreme pain disorder (PEPD), primary erythromelalgia (PEM), and congenital insensitivity to pain (CIP).25–30 FEPS is an autosomal dominant inherited disorder characterized by recurrent, stereotyped, intense episodic pain, occurring predominantly in the distal lower limbs and sometimes affecting the upper body in several members of a family.29,30 The pain can be triggered or exacerbated by fatigue, cold exposure, fasting, weather changes and/or physical stress or exertion. Sweating and other manifestations, such as tachycardia, breathing difficulties and generalized pallor, may be associated. The attacks may or may not diminish with age and be relieved by anti-inflammatory medicine.31,32 Although FEPS shows a unique clinical phenotype, there is no clear definition of disease so far, and it is often confused or overlapped with small fiber neuropathy due to the common VGSC-related pathogenic genes. In this review, we will discuss the subtypes of FEPS, summarize recent findings on FEPS, and highlight the similarities and differences between these diseases. We also discuss the pathogenic mechanism across different FEPS, potential therapeutic strategies, and challenges.
Subtypes of FEPS
According to the Online Mendelian Inheritance in Man (OMIM) classification system, FEPS includes three subtypes: FEPS1, FEPS2, and FEPS3. FEPS1 (OMIM#615040) is caused by a heterozygous variant in the transient receptor potential A1 (TRPA1) gene on chromosome 8q21;33 FEPS2 (OMIM#615551) is caused by variants in the sodium voltage-gated channel alpha subunit 10 (SCN10A) gene on chromosome 3p22;26 and FEPS3 (OMIM#615552) is caused by variants in the sodium voltage-gated channel alpha subunit 11 (SCN11A) gene on chromosome 3p22.29,30 However, there are still many patients who present with typical episodic pain syndromes, of which the genetic causes are unknown. Herein, we will propose a new subtype, FEPS4, in this review, and will discuss the reasons for this classification and its related mechanisms.
FEPS1
FEPS1 has only been reported in one family, Colombia in South America.33 The family has a total of 21 affected members over four generations, which accords with autosomal dominant inherited pattern. Their clinical attacks are characterized by the onset in infancy of episodic debilitating upper body pain usually triggered by fasting, fatigue, cold, illness, and physical exertion. The episodes typically last for about 1.5 hours, have a prodromal phase during which the episode could sometimes be aborted, and are followed by a period of exhaustion and deep sleep. Episodes of intense pain are sometimes accompanied by some autonomic symptoms such as dyspnea, tachycardia, sweating, generalized pallor, peribuccal cyanosis, and abdominal wall stiffness. However, no altered pain sensitivity outside the episodes has been observed in the affected individuals.
Quantitative sensory testing (QST) has no differences between affected and unaffected individuals. Skin biopsies of affected individuals show no abnormalities of intraepidermal nerve fibers. Tests with mustard oil known to activate TRPA1 receptors show that some affected individuals have a higher flare response and secondary hyperalgesia compared to unaffected individuals. Genetic screening revealed a heterozygous variant (c.2564A>G, p.N855S) at exon 22 of TRPA1 (Figure 1). The variant was observed in all affected individuals but not in unaffected members, suggesting a family co-segregation. TRPA1 is a nonselective cation channel permeable to Ca(2+), Na(+), and K(+) and involves a subpopulation of Aδ- and C-fiber nociceptive sensory neurons and is also in other sensory cells. The mutation leads to a gain-of-function of the TRPA1 gate and is associated with enhanced secondary nociceptive hyperalgesia.34,35
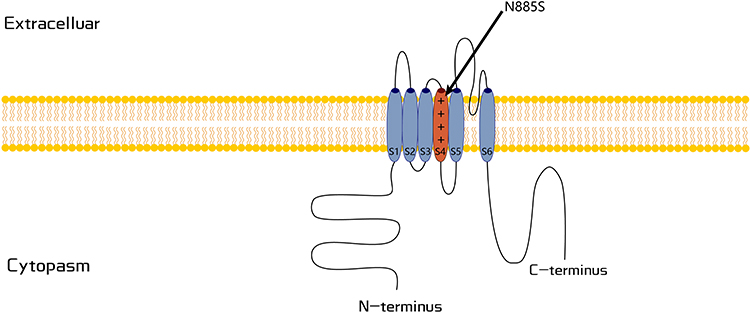 |
Figure 1 Schematic representation of the TRPA1 channel. The variant associated with represents as p.N855 in putative transmembrane S4. |
FEPS2
FEPS2 has been reported in only three patients from two independent families with atypical episodes of pain.26 The first case is a man who developed a burning and intense paroxysmal itch in his feet at age 57. His son developed a similar phenotype with allodynia and hyperalgesia at age 39. The third case is an unrelated woman who developed stabbing pain in her feet, lower legs, and hands at age 65. Her soles occasionally show red discoloration. The pain attack of FEPS2 is not associated with cold or other triggering factors, while warmth sometimes can relieve the attack.
Nerve conduction studies showed normal results in the father and unrelated woman, while the son showed decreased action potential amplitudes, suggesting the involvement of larger fibers. Skin biopsies were conducted on all three patients, and two patients (the father and son) had a partial or complete loss of intraepidermal nerve fiber density. In the father and son, a heterozygous missense mutation (c.1661T>C, p.L554P) in the SCN10A gene was identified (Figure 2). The unrelated woman carried a heterozygous mutation (c.3910G>A, p.A1304T). The SCN10A gene encodes the tetrodotoxin-resensitive channel Nav1.8 that regulates sensory neuron excitability and contributes to several sensory modalities. Functional studies in vitro indicate that the gain-of-function of Nav1.8 is associated with episodic pain attacks.26
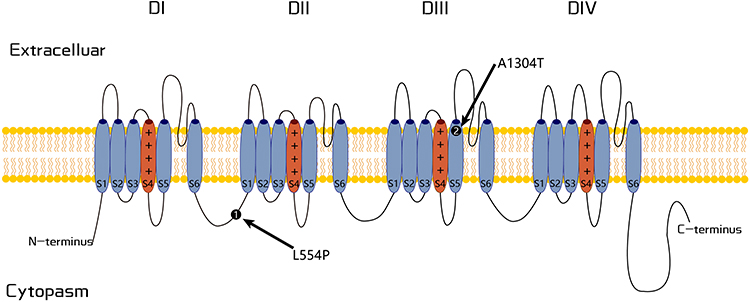 |
Figure 2 Schematic representation of the Nav1.8 channel. The positions of variants p.L544P and p.A1304T associated with FEPS2 are illustrated. |
FEPS3
Unlike FEPS1 and FEPS2, FEPS3 has been reported in over 20 families.29–32,36–41 We summarized the literature and found that almost all FEPS3 cases came from large autosomal dominant inherited families, except for one sporadic case originating from her asymptomatic father.32 The episodic pain syndrome with onset in infancy or early childhood is length-dependent and primarily involves in the distal joints, including the knees and ankles, and sometimes moves up to wrists, elbows, and palms, and occasionally affects the proximal limbs and neck. The affected lesions do not have redness, swelling, fever, rashes, or destruction, while a feeling of extreme cold is usually experienced in the pain region. Episodic pain appears late in the day and occurs in cycles. Pain attacks typically last for tens of minutes, and an episodic cycle consists of several attacks. An episodic attack is closely exacerbated by weather changes or stressful events such as rainy days, cold temperatures, fatigue, and fever. Disturbed symptoms of autonomic nerves such as hyperhidrosis, constipation, and intestinal spasm can be concomitantly observed in some patients. The pain can sometimes be relieved by nonsteroidal anti-inflammatory drugs (NSAIDs). Most affected individuals report that the severe pain episodes diminish with age.
Nerve conduction studies were normal in most patients, while QST showed abnormal cold and warm temperature thresholds in some patients. Skin biopsies in some FEPS3 patients have revealed different degrees of reduction of intraepidermal nerve fiber density and vacuolar degeneration of unmyelinated axons, indicating small-fiber neuropathy.36 Mild loss of myelinated nerve fibers has been observed in a patient with sensory axonal neuropathy with sharp episodes of pain caused by the p.N820Y mutation. Intriguingly, long-term dysfunction of the Nav1.9 channel may cause degeneration of the unmyelinated fibers in FEPS3 patient with pain remission. Genetic screening identified nine different variants in the SCN11A gene (Figure 3), which encodes channel Nav1.9, which is best known for its high expression levels in peripheral sensory neurons and is thought to be associated with pain-related signaling.32 The positively charged residues in the voltage sensor of transmembrane segment S4 could be variant hotspots (p.R222H/S and p.R225C) for FEPS3. Functional studies suggest that gain-of-function of Nav1.9 is associated with episodic pain.29,30,36,38
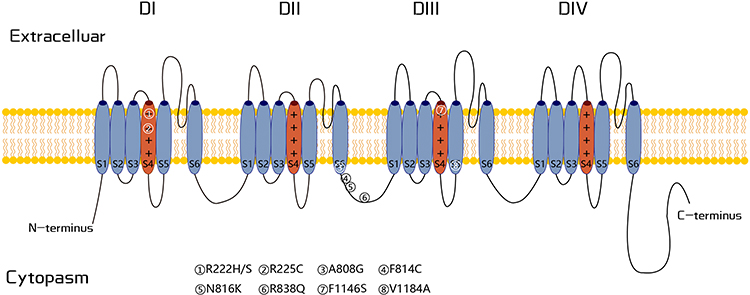 |
Figure 3 Schematic representation of the Nav1.9 channel. All variants associated with FEPS3 are marked in the framework. |
FEPS4
The current OMIM classification of FEPS only includes three subtypes. A recent study indicated that a patient with a mutation (c.130G>C, p.E44Q) in the SCN9A gene experienced paroxysmal knee pain resembling SCN11A-related FEPS rather than other classic SCN9A-related pain disorders (Figure 4).42 Therefore, we propose that the phenotype associated with SCN9A mutation should be called FEPS type 4 (FEPS4). The index patient is a 53-year-old male who experienced episodes of paroxysmal knee pain at age 4. The pain is dull and lasts for several hours to a few days, at a frequency of four times per year. Pain episodes were induced by fatigue, weather changes, and seasonal changes, especially when the temperature and atmospheric pressure drop, and never occurred in the summer or on a hot day. Pain can be relieved by warming. Paroxysmal knee pain episodes started to decrease at age 12, where the pain lasted for a few hours to several days and improved with NSAIDs. His father experienced similar symptoms (knee pain) in his childhood. Additionally, the patient and his father have trigeminal neuralgias. Functional studies indicate that gain-of-function mutation is responsible for this pain phenotype.
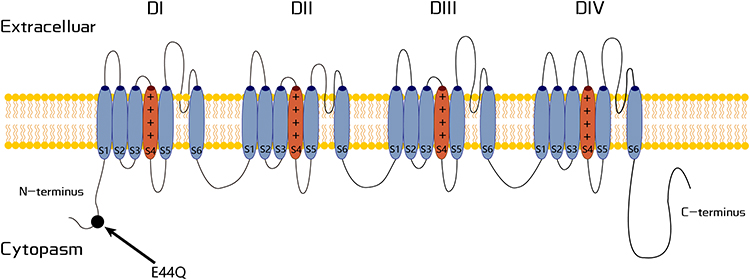 |
Figure 4 Schematic representation of the Nav1.7 channel. The variant related to FEPS4 is labeled in the N-terminus. |
Differential Diagnosis of FEPS
Similarities and Differences Between FEPS
Episodic pain attack is a common symptom among the different FEPS subtypes. Affected individuals have pain attacks only in the acute period, and no clinical symptoms are observed in the remission period. Pain episodes occur abruptly and last for several minutes to several hours, can be self-relieved after several episodes, and tend to attenuate or diminish with age. The pain typically affects the joints of limbs, while no fever, redness, swelling, rashes, or destruction is experienced in the pain regions. Episodic attacks can sometimes be triggered by weather changes or stressful events and can be relieved by warmth. Some concomitant symptoms, especially autonomic nerve disorders, have been observed in a few affected individuals. No abnormal signs have been identified upon physical examination. Nerve conduction studies are normal, while varying reductions in intraepidermal nerve fiber density and the degeneration of unmyelinated axons have been observed in the skin. Collectively, the clinical features of FEPS show unique characteristics that can make it an independent disease entity (Table 1).
 |
Table 1 The Clinical and Laboratory Summarization of Different FEPS |
Apart from different causative genes for the four FEPS types, there are some distinctive manifestations among these FEPS. Most patients with FEPS experience a similar onset of the disease, except for the FEPS2, which occurs in late adulthood. The pain regions of FEPS2/3/4 are length-dependent and mainly involve the distal joints, including the knees and ankles, and sometimes move up to the wrists, elbows, and palms. In contrast, FEPS1 pain attacks are primarily concentrated in the arms, shoulder, and thorax. The pain symptoms of FEPS1/3/4 differ from FEPS2, in which the affected individuals have intense burning, itching, and stabbing pain. Additionally, the triggers, frequency, duration, and circadian rhythm of pain attacks are also different between the four types of FEPS. FEPS1 or FEPS3 patients can experience symptoms of autonomic dysfunction such as breathing difficulties, tachycardia, hyperhidrosis, generalized pallor, constipation, and intestinal cramps. FEPS1 is more likely to involve the cardiopulmonary system, while gastrointestinal symptoms are emphasized in FEPS3 patients.32,36,38
Since FEPS3 has been reported more frequently, some rare clinical manifestations have also been found in FEPS3 patients. For example, in a large FEPS3 Chinese family, affected individuals exhibit a typical phenotype of an episodic pain attack, while all patients simultaneously have essential tremors, which is the first association of Nav1.9 with essential tremor.31 Another report demonstrates that patients with the p.R838Q mutation can exhibit various clinical manifestations such as recurrent exertional rhabdomyolysis with hematuria and proteinuria and paroxysmal pain.40 The clinical manifestations related to the Nav1.9 mutations are increasingly associated with rare symptoms and common pain.
Other Paroxysmal Pain Syndromes
Aside from the discrimination between the different subtypes of FEPS, several other syndromes with episodic pain should be distinguished from FEPS.43 Changes in skin color and pain locations are important biomarkers for differentiating between PEM and PEPD. PEM results in a paroxysmal burning sensation and erythema in the arms and legs. Concomitant pain begins in childhood, does not decrease with age, and will persist for a lifetime. The triggering factor of PEM is usually high temperature, and cooling can reduce or relieve the pain attack. Clinical symptoms of PEPD are characterized by attacks with sudden burning and lancinating pain at the proximal regions of the body, especially in the rectal, pubic, and facial areas, rather than the distal of the limbs.44,45 PEPD attacks last from a few seconds to several hours and are accompanied by sharp color changes in the skin. The pain can be so severe that it can accompany cardiopulmonary dysfunctions such as apnea, high blood pressure, asystole, or epileptic seizures. Most often, PEPD attacks can be triggered by defecation, eating, micturition, gynecological or rectal examination, and even touch, and partially respond to carbamazepine.46,47
In some cases, it is difficult to distinguish between paroxysmal pain. For example, two 63-year-old female patients show severe paroxysmal pain accompanied by skin changes resembling PEM-like pain, while the causative variants can be attributed to p.N1169S and p.I1293V in the SCN11A gene.48 A 50-year-old male patient with p.I381T mutation in SCN11A shows an SFN phenotype, while he simultaneously presents with typical episodic pain and red discoloration of the skin.49 Therefore, the clinical spectrum of episodic pain significantly overlaps with the phenotypes of NaV1.7, NaV1.8, and NaV1.9. Additionally, primary or secondary SFN, vasculitis, thrombotic diseases, and rheumatic diseases should be considered when making a differential diagnosis of paroxysmal pain.
Pathogenic Mechanisms of FEPS
Various stimuli can fire DRG neurons through the hyperactivity of ion channels on membranes. All variants associated with FEPS are heterozygous missense mutations associated with the gain-of-function of cation channels rather than dominant-negative mutants or haploinsufficiency. However, functional studies have demonstrated that different mutations cause distinctive changes in the transmembrane potential.
TRPA1-Related FEPS1
TRP is one of the most important components of the nociceptive pathway and is exclusive to pain sensation.50 TRPA1 is the only member of the TRPA family in mammals and is primarily expressed in small-diameter C- or Aδ-fibers of sensory ganglia, including DRG and trigeminal ganglia.51–54 This provides a basis for functional experiments to verify the association of TRPA1 with FESP1.
In vitro studies on HEK293 cells have demonstrated that the current-voltage of wild-type (WT) TRPA1 is the same as the mutant (p.N855S) in the absence of an agonist, while the mutant channels have a 5.4-fold greater current with agonist. More than a 4-fold increase in current is observed at normal resting potentials, but no differences in current density have been recorded between WT and mutant channels.33 Interestingly, cooling is more likely to activate the mutant channel and induce a greater inward current shift under physiological temperature. Several studies have demonstrated that voltage-dependent thermal TRPs are based on Ca2+ dependence.34,35 Therefore, the mutant p.N855S is thought to be associated with increased inward current and has cold activation properties, which explains why warmth can relieve pain symptoms.
Nav1.8-Related FEPS2
Nav1.8 is indispensable in the sensory pathway since it is responsible for most of the sodium inward flow (58–90%) and is essential for the depolarization phase of the action potential.55,56 Additionally, deletion or knockout of Nav1.8 in neurons results in cold nociceptive deficits and inflammatory damage.57–59
To investigate the pathogenic basis of the two mutants (p.L554P and p.A1304T) on the Nav1.8 channel, in vitro experiments were performed on DRG neurons. Mutant p.L554P shows no significant differences in resting potential, depolarization voltage, and inactivation compared with WT. However, it has great enhancement and increases the excitability of DRG neurons during the recovery process after inactivation (ramp effect), and the response to slow ramp stimuli is one-third larger at its peak for p.L554P compared with the WT channels.60,61 Additionally, the p.L554P mutant also shows more action potential under a series of graded stimuli (500ms steps at 1×, 2×, and 3× current threshold) and displays more spontaneous firing. The p.A1304T mutant is similar to the L554P and has a little difference in its response magnitude to slow ramp stimulation and reflection in the peak ramp current. Functional experiments confirm that both mutants cause episodic pain and are primarily dependent on faster potential recovery and more action potential.
Nav1.9-Related FEPS3
The Nav1.9 channel is preferentially expressed in the nociceptive neurons of DRG, trigeminal ganglia, and myenteric plexus in the gut.62,63 In contrast to other VGSCs, the Nav1.9 channel is thought to be associated with a tetrodotoxin-resistant Na+ current, which shows unique biophysical properties. Additionally, this channel plays an important role in neuronal inflammatory hyperalgesia and is thought to be closely related to the threshold potential during depolarization.22,64,65
The FEPS3 phenotype has been associated with nine heterogeneous mutations in SCN11A, most of which are positively charged residues in the voltage sensor of the transmembrane segment S466. It is believed that these mutations alter the gating properties of the mutant channel and lower the threshold for action potentials. The p.R222H mutation has been reported in several independent families, indicating that the position may be a variant hotspot. Therefore, we took the mutation as an example. DRG neurons with p.R222H expression have revealed that the mutation alters the current density and gating properties of the Nav1.9 channel. It shows no significant difference in the current density, but it has a significant shift of 6.4mV in the half-maximal activation voltage and a larger slope factor for the p.R222H mutant channel, which is consistent with more channels opening at hyperpolarized voltages compared to WT channels, and an increase in the window current. The other eight mutations have different pathogenic effects on the Nav1.9 channel, while they all show a gain-of-function for action potentials by increasing the Na+ influx under resting potential, accelerating the channel opening, or decelerating the channel closing. Nav1.9 is also expressed in the muscle and submucosa of the normal human colon. One study found reduced expression of Nav1.9 in the ganglion tissue of patients with congenital megacolon,67,68 which could explain the unique gastrointestinal symptoms of FEPS3 patients.
Nav1.7-Related FEPS4
Nav1.7 is highly expressed in peripheral neurons, especially in various DRG neurons.11 Nav1.7 defects have been associated with various painful disorders, which originate from the dysfunction of its role in nociceptive processing. Currently, only one mutant (p.E44Q) is associated with FEPS4, of which the symptoms are more like Nav1.9-related FEPS3. Compared with WT, the Nav1.7 channel with p.E44Q shows no differences in sodium current of activation, inactivation, and depolarization threshold. However, the current amplitude caused by slow-slope depolarization is significantly larger than the WT. The percentage of peak currents associated with slow ramp depolarizations is also significantly larger than that of the WT, more than 2 times.42 Therefore, p.E44Q is thought to affect the slope and peak during channel depolarization, and enhanced excitability is related to episodic pain.
Therapeutic Approaches for FEPS
FEPS is a severe and disabling pain syndrome, and currently lacks effective treatments. Fortunately, some patients with VGSC-related FEPS experience pain attacks that can gradually weaken and disappear with age.30,36,37 A similar age-dependent reduction in painful events is reported in patients suffering from TRPA1-related FEPS1. The underlying pathogenesis could be related to the physiological expression level of ion channels with age. In contrast, episodic pain does not gradually weaken in patients with Nav1.8-related FEPS2. Pain episodes can continue in some patients with Nav1.9-related FEPS3, which indicates that environmental factors or modifier genes can play a role in the phenotype as the ages of patient.
Multiple studies have demonstrated that VGSCs (Nav1.7, Nav1.8, and Nav1.9) are involved in the development of inflammatory pain, which is consistent with the effect of inflammatory mediators on modulating current density and gating properties altering the firing properties of DRG neurons. The local release of inflammatory mediators by immune and injured cells sensitizes DRG neurons, which contributes to the pain phenotype. Therefore, it is reasonable that some patients with Nav1.7- and Nav1.9-related FEPS respond favorably to NSAID treatments, while it is puzzled that patients with Nav1.8-related FEPS are irresponsive to NSAIDs. Additionally, FEPS3 patients with the p.R222H mutation from South America do not respond to NSAIDs. It is not surprising that these individuals respond differently to NSAID treatments since these medications do not act directly on the channel. Their molecular cascades and neural circuitry likely differ in different patients.
Medications such as mexiletine, lidocaine, and carbamazepine are nonselective sodium channel blockers and are currently used as analgesics. Some efficacy has been shown in certain pain channelopathies associated with VGSC mutations (such as Nav1.7-related PEPD). Other anti-epilepsy medications with varying effectiveness include lamotrigine, topiramate, tiagabine, and sodium valproate. However, no positive outcomes for these anti-epilepsy nonselective blockers have been observed in FEPS patients. Additionally, analgesic efficacy is accompanied by dose-dependent side effects on the CNS, including seizures, ataxia, confusion, and sedation.69,70
Current small-molecule sodium channel blockers in clinical use all lack isoform selectivity.69 Therefore, it is important for VGSC blockers to develop subunit-specific blockers during chronic pain. For example, A-803467, an inhibitor for Nav1.8 with 100-fold selectivity over other Nav isoforms, has demonstrated that the acute and selective pharmacological blockade of Nav1.8 can produce significant analgesia in rodent models with neuropathic and inflammatory pain.71 Additionally, considering that nociceptors express at least two of the peripheral VGSC subunits (Nav1.7, Nav1.8 and Nav1.9), an effective analgesic strategy can result from a combination of blockers against these subunits, which may have additive and synergistic effects on nociceptors.
Prospects and Challenges
VGSCs are required for firing action potentials in DRG neurons and contribute to increased sensitization of primary transduction channels such as TRP channels. Therefore, it is necessary to explore the clinical and pathogenic mechanisms of FEPS, which is one of the important forms of pathological pain. The clinical phenotype of FEPS has its unique characteristics, but there are significant differences between different FEPS subtypes. Our study recently revealed that the long-term dysfunction of the Nav1.9 channel could cause the degeneration of unmyelinated fibers in FEPS3 patients with pain remission, indicating that the persistent dysfunction of VGSC channels could be the pathological basis of SFN.41 Therefore, the dysfunction of ion channels will induce time-dependent neuropathological changes, which form a common basis of the clinical spectrum for various pain syndromes.
Advances in next-generation sequencing technology and bioinformatic analysis have allowed for significant progress in genetic diagnostic workflows over the last 20 years. This has led to identifying disease-related mutations in all of the VGSC subunits selectively expressed in DRG-related nociceptors (Nav1.7, 1.8, and 1.9). This progress is likely to continue since many patients with clinical symptoms of episodic pain have no clear genetic cause. Several genes related to action potential should be investigated, including SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN7A, SCN8A, SCN1B, SCN2B, SCN3B, SCN4B, TRPV1, TRPM8, NGF, NGFR, BDNF, GDNF, NTRK1, CACNG2, KCNS1, COMT, P2RX3, TAC1, TACR1, and WNK1.
Studies on these pain-related diseases have provided important insights into the normal function and pathophysiological role of these ion channels. However, it is difficult to find small-molecule blockers that show subunit specificity to avoid CNS and cardiac side effects. Improved structural modeling through artificial intelligence may hopefully be overcome the tremendous search work. Personalized medicine may also enable us to integrate a combination of blockers to produce synergistic effects. The development of novel small-molecule inhibitors of sodium channels or other approaches, including peptide toxins, monoclonal antibodies against specific channels, and gene therapy that target these channels on peripheral neurons, could provide effective pain treatments with fewer adverse effects. Clinical trials of some of these medicines are ongoing and can hopefully, within the next decade, provide specific medicines that can effectively treat chronic pain.
Conclusion
As discussed in this review, the clinical spectrum of FEPS is variable, but mainly characterized by severe episodic pain involving the distal limbs in young patients. The disease spectrum may be further enlarged in the future due to the increasing attention on pain-related pathway and the advances of DNA sequencing methods. However, a well-explained gain-of-function pathogenesis underlying these different disease phenotypes will still be a huge challenge. Future investigations of small-molecule blockers to specific subunits of sodium channels may reveal more effective downstream targets of VGSCs, and provide us with a more complete picture to better understand, diagnose, and treat the diseases.
Abbreviations
CNS, central nervous system; CIP, congenital insensitivity to pain; DRG, dorsal root ganglia; FEPS, familial episodic pain syndrome; NSAIDs, anti-inflammatory drugs; OMIM, Online Mendelian Inheritance in Man; PEPD, paroxysmal extreme pain disorder; PEM, primary erythromelalgia; QST, Quantitative sensory testing; SFN, small fiber neuropathy; TRPA1, transient receptor potential A1; VGSC, voltage-gated sodium channel; WT, wild-type.
Author Contributions
All authors made a significant contribution to the work including the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (Grants 81460199 and 82160252), Science and technology project of Jiangxi Health Commission (202110028), Double thousand talents program of Jiangxi province (jxsq2019101021).
Disclosure
The authors declare that they have no competing interests.
References
1. Raouf R, Quick K, Wood JN. Pain as a channelopathy. J Clin Invest. 2010;120(11):3745–3752. doi:10.1172/JCI43158
2. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68(1):619–647. doi:10.1146/annurev.physiol.68.040204.100431
3. Gatchel RJ, Reuben DB, Dagenais S, et al. Research agenda for the prevention of pain and its impact: report of the work group on the prevention of acute and chronic pain of the federal pain research strategy. J Pain. 2018;19(8):837–851. doi:10.1016/j.jpain.2018.02.015
4. Gatchel RJ, Bevers K, Licciardone JC, Su J, Du Y, Brotto M. Transitioning from acute to chronic pain: an examination of different trajectories of low-back pain. Healthcare. 2018;6(2). doi:10.3390/healthcare6020048
5. Cairns BE, Arendt-Nielsen L, Sacerdote P. Perspectives in pain research 2014: neuroinflammation and glial cell activation: the cause of transition from acute to chronic pain? Scand J Pain. 2015;6(1):3–6. doi:10.1016/j.sjpain.2014.10.002
6. Schnabel A. Acute neuropathic pain and the transition to chronic postsurgical pain. Pain Manag. 2018;8(5):317–319. doi:10.2217/pmt-2018-0026
7. Breivik H, Collett B, Ventafridda V, Cohen R, Gallacher D. Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10(4):287–333. doi:10.1016/j.ejpain.2005.06.009
8. Berta T, Qadri Y, Tan PH, Ji RR. Targeting dorsal root ganglia and primary sensory neurons for the treatment of chronic pain. Expert Opin Ther Targets. 2017;21(7):695–703. doi:10.1080/14728222.2017.1328057
9. Beyak MJ, Vanner S. Inflammation-induced hyperexcitability of nociceptive gastrointestinal DRG neurons: the role of voltage-gated ion channels. Neurogastroenterol Motil. 2005;17(2):175–186. doi:10.1111/j.1365-2982.2004.00596.x
10. Rush AM, Cummins TR, Waxman SG. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J Physiol. 2007;579(Pt 1):1–14. doi:10.1113/jphysiol.2006.121483
11. Black JA, Dib-Hajj S, McNabola K, et al. Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res Mol Brain Res. 1996;43(1–2):117–131. doi:10.1016/S0169-328X(96)00163-5
12. Fukuoka T, Noguchi K. Comparative study of voltage-gated sodium channel α-subunits in non-overlapping four neuronal populations in the rat dorsal root ganglion. Neurosci Res. 2011;70(2):164–171. doi:10.1016/j.neures.2011.01.020
13. Ho C, O’Leary ME. Single-cell analysis of sodium channel expression in dorsal root ganglion neurons. Mol Cell Neurosci. 2011;46(1):159–166. doi:10.1016/j.mcn.2010.08.017
14. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi:10.1146/annurev-neuro-060909-153234
15. Baker MD, Chandra SY, Ding Y, Waxman SG, Wood JN. GTP-induced tetrodotoxin-resistant Na+ current regulates excitability in mouse and rat small diameter sensory neurones. J Physiol. 2003;548(Pt 2):373–382. doi:10.1113/jphysiol.2003.039131
16. Herzog RI, Cummins TR, Waxman SG. Persistent TTX-resistant Na+ current affects resting potential and response to depolarization in simulated spinal sensory neurons. J Neurophysiol. 2001;86(3):1351–1364. doi:10.1152/jn.2001.86.3.1351
17. Waxman SG, Zamponi GW. Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci. 2014;17(2):153–163. doi:10.1038/nn.3602
18. Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. J Physiol. 1952;116(4):473–496. doi:10.1113/jphysiol.1952.sp004718
19. Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol. 1952;116(4):449–472. doi:10.1113/jphysiol.1952.sp004717
20. Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol. 1952;116(4):497–506. doi:10.1113/jphysiol.1952.sp004719
21. Hodgkin AL, Huxley AF. Propagation of electrical signals along giant nerve fibers. Proc R Soc Lond B Biol Sci. 1952;140(899):177–183.
22. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in Pain signaling. Physiol Rev. 2019;99(2):1079–1151. doi:10.1152/physrev.00052.2017
23. Noda M, Ikeda T, Suzuki H, et al. Expression of functional sodium channels from cloned cDNA. Nature. 1986;322(6082):826–828. doi:10.1038/322826a0
24. Noda M, Shimizu S, Tanabe T, et al. Primary structure of electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312(5990):121–127. doi:10.1038/312121a0
25. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi:10.1038/nature05413
26. Faber CG, Lauria G, Merkies IS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci USA. 2012;109(47):19444–19449. doi:10.1073/pnas.1216080109
27. Lauria G. Small fibre neuropathies. Curr Opin Neurol. 2005;18(5):591–597. doi:10.1097/01.wco.0000177330.35147.70
28. Yang Y, Wang Y, Li S, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–174. doi:10.1136/jmg.2003.012153
29. Zhang XY, Wen J, Yang W, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93(5):957–966. doi:10.1016/j.ajhg.2013.09.016
30. Okuda H, Noguchi A, Kobayashi H, et al. Infantile pain episodes associated with novel Nav1.9 mutations in familial episodic pain syndrome in Japanese families. PLoS One. 2016;11(5):e0154827. doi:10.1371/journal.pone.0154827
31. Leng XR, Qi XH, Zhou YT, Wang YP. Gain-of-function mutation p.Arg225Cys in SCN11A causes familial episodic pain and contributes to essential tremor. J Hum Genet. 2017;62(6):641–646. doi:10.1038/jhg.2017.21
32. Huang J, Estacion M, Zhao P, et al. A novel gain-of-function Nav1.9 mutation in a child with episodic pain. Front Neurosci. 2019;13:918. doi:10.3389/fnins.2019.00918
33. Kremeyer B, Lopera F, Cox JJ, et al. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron. 2010;66(5):671–680. doi:10.1016/j.neuron.2010.04.030
34. Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V, Nilius B. The principle of temperature-dependent gating in cold- and heat-sensitive TRP channels. Nature. 2004;430(7001):748–754. doi:10.1038/nature02732
35. Voets T, Owsianik G, Janssens A, Talavera K, Nilius B. TRPM8 voltage sensor mutants reveal a mechanism for integrating thermal and chemical stimuli. Nat Chem Biol. 2007;3(3):174–182. doi:10.1038/nchembio862
36. Leipold E, Hanson-Kahn A, Frick M, et al. Cold-aggravated pain in humans caused by a hyperactive NaV1.9 channel mutant. Nat Commun. 2015;6(1):10049. doi:10.1038/ncomms10049
37. Kabata R, Okuda H, Noguchi A, et al. Familial episodic limb pain in kindreds with novel Nav1.9 mutations. PLoS One. 2018;13(12):e0208516. doi:10.1371/journal.pone.0208516
38. Han C, Yang Y, Te Morsche RH, et al. Familial gain-of-function Na(v)1.9 mutation in a painful channelopathy. J Neurol Neurosurg Psychiatry. 2017;88(3):233–240. doi:10.1136/jnnp-2016-313804
39. Castoro R, Simmons M, Ravi V, et al. SCN11A Arg225Cys mutation causes nociceptive pain without detectable peripheral nerve pathology. Neurol Genet. 2018;4(4):e255. doi:10.1212/NXG.0000000000000255
40. Sambuughin N, Ren M, Capacchione JF, et al. Multifactorial origin of exertional rhabdomyolysis, recurrent hematuria, and episodic pain in a service member with sickle cell trait. Case Rep Genet. 2018;2018:6898546. doi:10.1155/2018/6898546
41. Zheng Y, Huang P, Li S, et al. Pathological changes of the sural nerve in patients with familial episodic pain syndrome. Neurol Sci. 2022;43(9):5605–5614. doi:10.1007/s10072-022-06107-7
42. Takahashi K, Ohba T, Okamoto Y, et al. E44Q mutation in Na(V)1.7 in a patient with infantile paroxysmal knee pain: electrophysiological analysis of voltage-dependent sodium current. Heliyon. 2021;7(6):e07396. doi:10.1016/j.heliyon.2021.e07396
43. Wadhawan S, Pant S, Golhar R, et al. Na(V) channel variants in patients with painful and nonpainful peripheral neuropathy. Neurol Genet. 2017;3(6):e207. doi:10.1212/NXG.0000000000000207
44. Hayden R, Grossman M. Rectal, ocular, and submaxillary pain; a familial autonomic disorder related to proctalgia fugaz: report of a family. AMA J Dis Child. 1959;97(4):479–482. doi:10.1001/archpedi.1959.02070010481013
45. Fertleman CR, Ferrie CD. What’s in a name – familial rectal pain syndrome becomes paroxysmal extreme pain disorder. J Neurol Neurosurg Psychiatry. 2006;77(11):1294–1295. doi:10.1136/jnnp.2006.089664
46. Themistocleous AC, Ramirez JD, Serra J, Bennett DL. The clinical approach to small fibre neuropathy and painful channelopathy. Pract Neurol. 2014;14(6):368–379. doi:10.1136/practneurol-2013-000758
47. Choi JS, Boralevi F, Brissaud O, et al. Paroxysmal extreme pain disorder: a molecular lesion of peripheral neurons. Nat Rev Neurol. 2011;7(1):51–55. doi:10.1038/nrneurol.2010.162
48. Zhang Z, Schmelz M, Segerdahl M, et al. Exonic mutations in SCN9A (NaV1.7) are found in a minority of patients with erythromelalgia. Scand J Pain. 2014;5(4):217–225. doi:10.1016/j.sjpain.2014.09.002
49. Huang J, Han C, Estacion M, et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014;137(Pt 6):1627–1642. doi:10.1093/brain/awu079
50. Wang H, Woolf CJ. Pain TRPs. Neuron. 2005;46(1):9–12. doi:10.1016/j.neuron.2005.03.011
51. Barabas ME, Kossyreva EA, Stucky CL. TRPA1 is functionally expressed primarily by IB4-binding, non-peptidergic mouse and rat sensory neurons. PLoS One. 2012;7(10):e47988. doi:10.1371/journal.pone.0047988
52. Kobayashi K, Fukuoka T, Obata K, et al. Distinct expression of TRPM8, TRPA1, and TRPV1 mRNAs in rat primary afferent neurons with adelta/c-fibers and colocalization with trk receptors. J Comp Neurol. 2005;493(4):596–606. doi:10.1002/cne.20794
53. Nagata K, Duggan A, Kumar G, García-Añoveros J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci. 2005;25(16):4052–4061. doi:10.1523/JNEUROSCI.0013-05.2005
54. Story GM, Peier AM, Reeve AJ, et al. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell. 2003;112(6):819–829. doi:10.1016/S0092-8674(03)00158-2
55. Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86(2):629–640. doi:10.1152/jn.2001.86.2.629
56. Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22(23):10277–10290. doi:10.1523/JNEUROSCI.22-23-10277.2002
57. Akopian AN, Souslova V, England S, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–548. doi:10.1038/9195
58. Abrahamsen B, Zhao J, Asante CO, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321(5889):702–705. doi:10.1126/science.1156916
59. Zimmermann K, Leffler A, Babes A, et al. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447(7146):855–858. doi:10.1038/nature05880
60. Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18(23):9607–9619. doi:10.1523/JNEUROSCI.18-23-09607.1998
61. Waxman SG. Neurobiology: a channel sets the gain on pain. Nature. 2006;444(7121):831–832. doi:10.1038/444831a
62. Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG. NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci U S A. 1998;95(15):8963–8968. doi:10.1073/pnas.95.15.8963
63. Rugiero F, Mistry M, Sage D, et al. Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J Neurosci. 2003;23(7):2715–2725. doi:10.1523/JNEUROSCI.23-07-02715.2003
64. Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG. A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci. 1999;19(24):Rc43. doi:10.1523/JNEUROSCI.19-24-j0001.1999
65. Dib-Hajj SD, Tyrrell L, Cummins TR, Black JA, Wood PM, Waxman SG. Two tetrodotoxin-resistant sodium channels in human dorsal root ganglion neurons. FEBS Lett. 1999;462(1–2):117–120. doi:10.1016/S0014-5793(99)01519-7
66. Baker MD, Nassar MA. Painful and painless mutations of SCN9A and SCN11A voltage-gated sodium channels. Pflugers Arch. 2020;472(7):865–880. doi:10.1007/s00424-020-02419-9
67. O’Donnell AM, Coyle D, Puri P. Decreased Nav1.9 channel expression in Hirschsprung’s disease. J Pediatr Surg. 2016;51(9):1458–1461. doi:10.1016/j.jpedsurg.2016.05.007
68. Hockley JR, Boundouki G, Cibert-Goton V, et al. Multiple roles for NaV1.9 in the activation of visceral afferents by noxious inflammatory, mechanical, and human disease-derived stimuli. Pain. 2014;155(10):1962–1975. doi:10.1016/j.pain.2014.06.015
69. Nardi A, Damann N, Hertrampf T, Kless A. Advances in targeting voltage-gated sodium channels with small molecules. ChemMedChem. 2012;7(10):1712–1740. doi:10.1002/cmdc.201200298
70. Kemp MI. Structural trends among second-generation voltage-gated sodium channel blockers. Prog Med Chem. 2010;49:81–111.
71. Jarvis MF, Honore P, Shieh CC, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci USA. 2007;104(20):8520–8525. doi:10.1073/pnas.0611364104
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.