")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Familial Dyskeratotic Comedones: A Case Report and Literature Review
Authors Tejapira K , Suchonwanit P
Received 10 May 2023
Accepted for publication 27 June 2023
Published 3 July 2023 Volume 2023:16 Pages 1729—1735
DOI https://doi.org/10.2147/CCID.S420723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Anne-Claire Fougerousse
Kasama Tejapira, Poonkiat Suchonwanit
Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Correspondence: Poonkiat Suchonwanit, Division of Dermatology, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, 270 Rama VI Road, Ratchathewi, Bangkok, 10400, Thailand, Tel +66-2-2011141, Fax +66-2-201-1211 ext 4, Email [email protected]
Abstract: Familial dyskeratotic comedones (FDC) is an autosomal dominant inherited skin disorder characterized by generalized multiple discrete comedone-like hyperkeratotic papules. The disease demonstrates a distinct histopathologic feature of dyskeratosis of the crater-like invaginated epidermis or follicle-like structures with or without acantholysis. Despite its asymptomatic and benign course, the condition is refractory to treatment. Herein, we report a case of a 54-year-old female presenting with progressively developed generalized multiple hyperkeratotic papules with central keratin plugs on the trunk and extremities for 20 years. A definite diagnosis was made by clinical manifestations and histopathological examination. The lesions were slightly improved after 3 months of topical retinoids and urea cream treatments. Besides, we first describe dermoscopic findings of FDC and reviewed 21 previously reported FDC cases from 11 families in the literature.
Keywords: acne, autosomal dominant, comedone-like lesions, dermoscopy, dyskeratosis, hyperkeratosis
Introduction
Familial dyskeratotic comedones (FDC) is a rare autosomal dominant genodermatosis presenting generalized symmetrically scattered hyperkeratotic papules resembling comedones.1 Its prevalence remains unknown and probably underestimated due to its benign and asymptomatic course and patients’ ignorance.2 Skin lesions usually develop initially on the trunk or extremities and progressively spread to other body parts while sparing mucosal surfaces, palms, and soles.1–4 The disease onset is typically around puberty, and its course is chronic and persistent.5 Here, we present a case of a 54-year-old woman with extensive FDC on her trunk and extremities since the age of 12. Besides, we further review previously published articles on 21 FDC cases from 11 separate families.
Case Presentation
A 54-year-old Thai woman visited the outpatient department with asymptomatic multiple, widespread comedone-like papules over her trunk and extremities for almost 40 years. The lesions first developed as pinpoint dark spots on her legs while she was 12 and insidiously spread to her trunk when she grew older. The lesions became comedone-like papules when fully formed. The patient occasionally had redness and mild pruritus at the lesions without any specific precipitating causes. She sometimes forcefully removed the central plug, but the lesions were later filled back. She previously received topical treatments from other hospitals with no improvement.
A review of other systems was unremarkable. Her medical history included non-alcoholic fatty liver disease, bilateral carpal tunnel syndrome, and functional dyspepsia. She had undergone a hysterectomy with oophorectomy 10 years ago due to endometrioma. She denied a history of smoking and alcohol drinking. Her mother, one sister, and two brothers had similar skin lesions, first appearing around their age of 10 and mainly located on extremities with occasional pruritus. The pedigree chart of her family is shown in Figure 1.
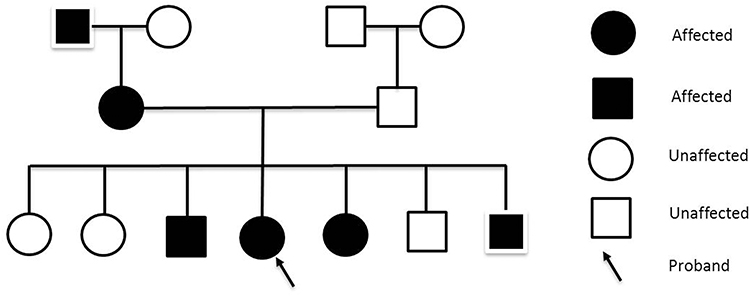 |
Figure 1 Pedigree chart of the patient (arrow indicates the proband). |
Dermatological examination revealed generalized multiple discrete comedone-like hyperkeratotic brownish papules distributed symmetrically on the trunk and extremities, sparing palms and soles (Figure 2). The dermoscopic evaluation showed brownish, slightly elevated border follicular papules with central keratin plugs (Figure 3). Other systems were unremarkable except for her body mass index of 28.6, which corresponds to being overweight.
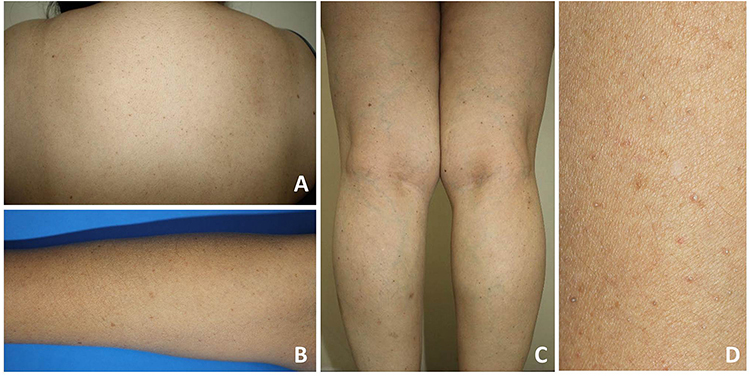 |
Figure 2 Clinical presentations: multiple discrete comedone-like hyperkeratotic brownish papules distributed symmetrically on the trunk (A) and extremities (B and C); close-up image of the lesions (D). |
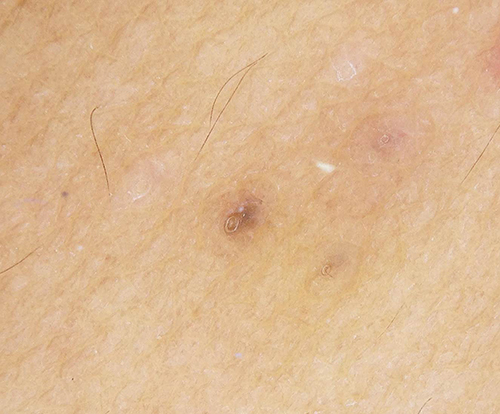 |
Figure 3 Dermoscopic features: brownish, slightly elevated border follicular papules with central keratin plugs (original magnification x20). |
A skin biopsy was performed on a lesion on her left thigh. Histologic examination revealed cystic dilatation with keratotic follicular plugging and multiple zones of focal acantholytic dyskeratosis in the cystic epithelium (Figure 4). Based on the patient’s history, clinical characteristics, and histologic examination, a definite diagnosis of FDC was made. The patient was reassured that the disease course was benign, and inflammation could occasionally occur. She was then given 10% topical urea cream, 0.025% topical retinoic acid, and an oral antihistamine for the treatment. After 3 months, the patient reported slight improvement.
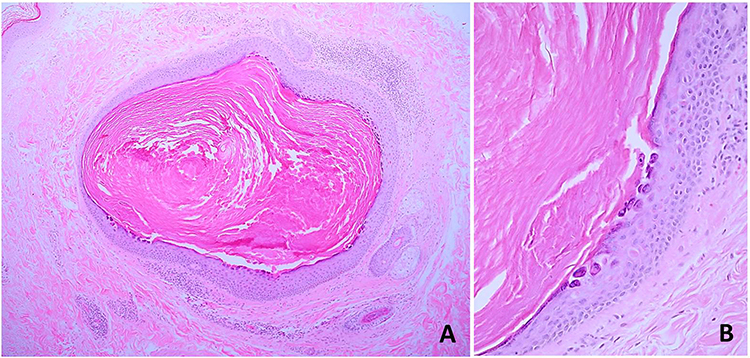 |
Figure 4 Histopathological findings: (A) cystic dilatation with keratotic follicular plugging (hematoxylin-eosin, original magnification x10) and (B) focal acantholytic dyskeratosis in the cystic epithelium (hematoxylin-eosin, original magnification x100). |
Discussion
FDC is a rare autosomal dominant penetrance disease with unknown etiology. Its prevalence is probably underestimated due to its benign and asymptomatic nature and patients’ unawareness. It was first described by Carneiro et al in 1972 in a patient with widespread papules over the trunk and extremities, resembling open comedones.1 Since then, to our knowledge, a few additional FDC cases have been described throughout the literature, with 21 affected individuals from 11 different families. Table 1 summarizes all previously reported FDC cases.1–11
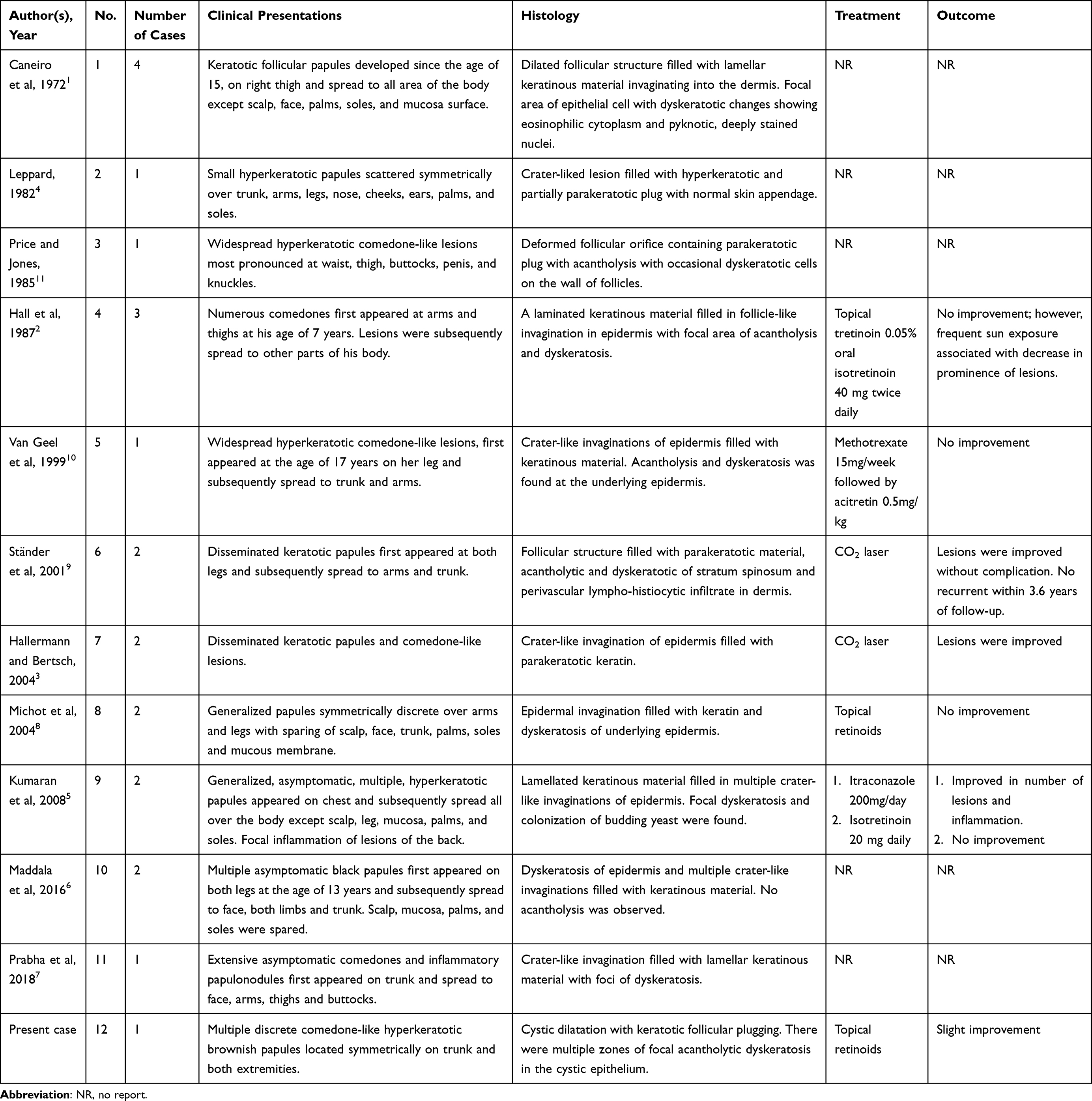 |
Table 1 Previously Reported Cases of Familial Dyskeratotic Comedones in the Literature |
FDC can be suspected in a patient with the following distinctive features: (1) lesions clinically resembling comedones; (2) occurrence in family members; and (3) presence of dyskeratotic features on histology.6 The disease reveals an asymptomatic, noninflammatory, and refractory disease course.1 Its onset is typically during childhood to early puberty, with a gradual increase in the number of lesions by age. Clinically, lesions are scattered symmetrically distributed over the trunk, face, scalp, and extremities with palms, soles, and mucosal surfaces sparing; however, the genital area has also been reported.4 Dermoscopic findings of FDC have never been reported. Brownish, slightly elevated border follicular papules with central keratin plugs were first demonstrated for FDC in this article. These described features may help differentiate FDC from its mimicking conditions.
Histopathological features of FDC include crater-like invagination or follicle-like structures filled with keratinous material that contains a mixture of parakeratotic cells and melanin. Dyskeratotic changes (increased eosinophilic keratinous cells containing vesicles with an occasionally seen eccentric nucleus and small nucleolus) of underlying 3–12 layers of epithelial cells are also observed.1,10 Early-developed lesions may show only acanthosis and dyskeratosis in the epidermis.4 Acantholysis is also often seen and mostly starts at the base of stratum spinulosum.2,3,10 Hypoplastic sebaceous glands, rudimentary follicle structures, and normal sweat glands can be observed near the lesion.1,4 Attenuated surrounding connective tissues are found with an absence of arrector pili muscle. Perivascular lymphocytic infiltration is occasionally seen.1,4 Electron microscopic examination of the lesions showed decreased desmosomal attachments in stratum malpighii and separation of tonofilaments from normal insertion sites at the desmosome.2
Differential diagnoses of FDC include Darier's disease, keratosis pilaris, Grover disease, and nevus comedonicus, which can all be distinguished by clinical presentations. Despite the fact that Darier disease and FDC share the histopathologic hallmark of acantholytic dyskeratosis, lesions of Darier's disease have a predilection on the seborrheic area, and nail abnormality is usually observed.11–13 Keratosis pilaris presents follicular hyperkeratotic papules without comedone-like lesions distributed on the extensor surface of the proximal extremities. Grover disease reveals papulovesicular rash commonly presenting on the trunk with a preference on the anterior chest and upper back.14,15 Lesions are typically spontaneously resolved within a year, and its prevalence is among Caucasians.14 Nevus comedonicus usually appears at birth, typically presenting as comedone-like lesions with lamellate keratinocytic material plugs arranged along Blaschko’s lines.16–18 All clinical features of the above-mentioned help distinguish them from FDC.
Therapeutic options for FDC remain limited. Several reports found the disease refractory to treatment.5 Throughout the literature, FDC appeared ineffective with methotrexate, topical, and systemic retinoids. Some reports showed successful treatment with CO2 laser,3 itraconazole,5 and repeated sun exposure.2 CO2 laser minimizes the appearance of lesions by its destructive ability, while itraconazole may help the condition by decreasing local yeast colonization.2 However, the mechanism of frequent sun exposure in improving FDC is still unexplored.
Conclusion
We report a case of FDC in a Thai woman who developed since childhood with a delayed diagnosis. We also describe, for the first time, the dermoscopic features of this condition. Our report underlines the importance of disease recognition for prompt diagnosis. Despite the rarity of the disease, it could be underestimated due to its benign and asymptomatic course. Appropriate history taking and physical examination are essential to avoid unnecessary invasive diagnostic procedures that benefit patients’ management. Additionally, patients should be informed regarding disease course and treatment modalities since lesions are usually persistent and resistant to treatment.
Ethics Approval and Consent to Participate
This article was performed in accordance with the principles of Declaration of Helsinki. Ethical review and approval was not required to publish the case details in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the patient for publication of this case report and any accompanying images as per our standard institutional rules.
Funding
No sources of funding were used to prepare this manuscript.
Disclosure
The authors declare that this manuscript was prepared in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Carneiro SJ, Dickson JE, Knox JM. Familial dyskeratotic comedones. Arch Dermatol. 1972;105(2):249–251. doi:10.1001/archderm.1972.01620050059013
2. Hall JR, Holder W, Knox JM, Knox JM, Verani R. Familial dyskeratotic comedones. A report of three cases and review of the literature. J Am Acad Dermatol. 1987;17(5 Pt 1):808–814. doi:10.1016/S0190-9622(87)70267-9
3. Hallermann C, Bertsch HP. Two sisters with familial dyskeratotic comedones. Eur J Dermatol. 2004;14(4):214–215.
4. Leppard BJ. Familial dyskeratotic comedones. Clin Exp Dermatol. 1982;7(3):329–332. doi:10.1111/j.1365-2230.1982.tb02435.x
5. Kumaran MS, Appachu D, Jayaseelan E. Familial dyskeratotic comedones. Indian J Dermatol Venereol Leprol. 2008;74(2):142–144. doi:10.4103/0378-6323.39700
6. Maddala RR, Ghorpade A, Polavarpu M, Adulkar SA, Das M. Familial dyskeratotic comedones: a rare entity. Indian Dermatol Online J. 2016;7(1):46–48. doi:10.4103/2229-5178.174308
7. Prabha N, Chhabra N, Kulkarni S, Hussain N. Familial dyskeratotic comedones. Skinmed. 2018;16(4):273–274.
8. Michot C, Guilhou JJ, Bessis D. Familial dyskeratotic comedone. Ann Dermatol Venereol. 2004;131(8–9):811–813. doi:10.1016/s0151-9638(04)93766-0
9. Ständer S, Rütten A, Metze D. Familiäre dyskeratotische Komedonen Eine seltene Entität [Familial dyskeratotic comedones. A rare entity]. Hautarzt. 2001;52(6):533–536. German. doi:10.1007/s001050000114
10. Van Geel NA, Kockaert M, Neumann HA. Familial dyskeratotic comedones. Br J Dermatol. 1999;140(5):956–959. doi:10.1046/j.1365-2133.1999.02835.x
11. Price M, Jones RR. Familial dyskeratotic comedones. Clin Exp Dermatol. 1985;10(2):147–153. doi:10.1111/j.1365-2230.1985.tb00543.x
12. Suchonwanit P, McMichael AJ. Alopecia in association with malignancy: a review. Am J Clin Dermatol. 2018;19(6):853–865. doi:10.1007/s40257-018-0378-1
13. Kositkuljorn C, Suchonwanit P. Darier’s disease: report of a case with facial involvement. Case Rep Dermatol. 2019;11(3):327–333. doi:10.1159/000504925
14. Fantini F, Kovacs E, Scarabello A. Unilateral transient acantholytic dermatosis (Grover’s disease) along Blaschko lines. J Am Acad Dermatol. 2002;47(2):319–320. doi:10.1067/mjd.2002.120596
15. Suchonwanit P, Kositkuljorn C, Pomsoong C. Alopecia areata: an autoimmune disease of multiple players. Immunotargets Ther. 2021;10:299–312. doi:10.2147/itt.S266409
16. Suchonwanit P, Iamsumang W, Leerunyakul K. Topical finasteride for the treatment of male androgenetic alopecia and female pattern hair loss: a review of the current literature. J Dermatolog Treat. 2022;33(2):643–648. doi:10.1080/09546634.2020.1782324
17. Vano-Galvan S, Hernández-Martín A, Colmenero I, Torrelo A. Disseminated congenital comedones. Pediatr Dermatol. 2011;28(1):58–59. doi:10.1111/j.1525-1470.2010.01358.x
18. Ferrari B, Taliercio V, Restrepo P, Luna P, Abad ME, Larralde M. Nevus comedonicus: a case series. Pediatr Dermatol. 2015;32(2):216–219. doi:10.1111/pde.12466
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.