")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Facile and green fabrication of electrospun poly(vinyl alcohol) nanofibrous mats doped with narrowly dispersed silver nanoparticles
Authors Lin S, Wang R, Yi Y, Wang Z, Hao L, Wu J, Hu G, He H
Received 27 March 2014
Accepted for publication 24 May 2014
Published 18 August 2014 Volume 2014:9(1) Pages 3937—3947
DOI https://doi.org/10.2147/IJN.S64985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Song Lin,1,2,* Run-Ze Wang,1,2 Ying Yi,1 Zheng Wang,2,* Li-Mei Hao,2 Jin-Hui Wu,2 Guo-Han Hu,3 Hua He3,*
1National Bio-Protection Engineering Centre, Tianjin, People’s Republic of China; 2Institute of Medical Equipment, Academy of Military Medical Sciences, Tianjin, People’s Republic of China; 3Department of Neurosurgery, Changzheng Hospital, Second Affiliated Hospital of Second Military Medical University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Submicrometer-scale poly(vinyl alcohol) (PVA) nanofibrous mats loaded with aligned and narrowly dispersed silver nanoparticles (AgNPs) are obtained via the electrospinning process from pure water. This facile and green procedure did not need any other chemicals or organic solvents. The doped AgNPs are narrowly distributed, 4.3±0.7 nm and their contents on the nanofabric mats can be easily tuned via in situ ultraviolet light irradiation or under preheating conditions, but with different particle sizes and size distributions. The morphology, loading concentrations, and dispersities of AgNPs embedded within PVA nanofiber mats are characterized by transmission electron microscopy, scanning electron microscopy, energy dispersive X-ray spectroscopy, ultraviolet-visible spectra, X-ray photoelectron spectroscopy, and X-ray diffraction, respectively. Moreover, the biocidal activities and cytotoxicity of the electrospun nanofiber mats are determined by zone of inhibition, dynamic shaking method, and cell counting kit (CCK)-8 assay tests.
Keywords: electrospinning, silver nanoparticles, polyvinyl alcohol, nanocomposites, bioactivity
Introduction
Homogeneous distribution of controlled nanoparticle assemblies in a homopolymer matrix is still considered an important challenge in material science today, especially at a low volume fraction.1 Among them, silver nanoparticles (AgNPs) are well-known and applied in many hygienic fields.2–4 Their predominant antimicrobial activity can be attributed to a strong interaction with bacterial cells,5 but relatively low toxicity to human cells.6 Nevertheless, such dispersed metallic nanoparticles often tend to congregate and become unstable, and lose their desired qualities. Moreover the size and size distribution are two of the crucial factors accounting for AgNPs-induced toxicity and biologic responses,7–11 thus many efforts have been made to design stable AgNPs of a smaller size.
Several groups have utilized a variety of stabilizing agents and methods to create small and stable AgNPs, which include Daxad 19® (GEO Specialty Chemicals) (12 nm in diameter);12 Myramistin® (10.0±1.8 nm in diameter);13 and hyperbranched poly(amidoamine) with AgNP diameters ranging from approximately 7.1 to 1.0 nm.14 Recently, a series of monodispersed AgNPs with 25, 35, 45, 60 and 70 nm sizes was reported by using poly(vinyl pyrrolidone) (PVP) as the surfactant.15 Also AgNPs can be incorporated within biodegradable poly(lactic acid)16 or deposited onto modified titanium surfaces17 as an antibacterial scaffold for tissue engineering and medical applications. It was found that the AgNPs with a smaller size have a stronger affinity for cells because of the larger surface area. However, producing monodispersed AgNPs with ultrasmall sizes,18 ie, <5 nm, as well as long-term dispersion stability has always been a primary challenge.8,9 Recently, Destaye et al prepared nanofibrous poly(vinyl alcohol) (PVA) mats with AgNPs formed in situ under a glutaraldehyde vapor atmosphere.10
The electrospinning technique has been widely used in filtration membranes, catalysis reaction, protective clothing, molecular templates, tissue scaffolds, and wound dressings,19–21 this procedure is easy-to-handle and cost-effective to fabricate man-made fibers with AgNPs.22–26 Herein, we demonstrate a one-step method to prepare stable and narrowly dispersed AgNPs size of approximately 4.3±0.7 nm doped along within PVA nanofibers via electrospinning process. Our method can be achieved by the mixing of AgNO3 and PVA in an aqueous solution before applying to the electrospinning apparatus without any other chemical additions.
Interestingly, the AgNPs generated via such electrospinning processes are found to form aligned arrays along the orientation of the PVA nanofibers. In addition to its simplicity and green chemistry, the contents of AgNPs on nanofabric mats can be easily tuned by the exposure of post-ultraviolet (UV) irradiation or the preheating treatment (Figure 1). Moreover, since PVA has been reported as an effective stabilizer of AgNPs, by preventing them from aggregation,27 this approach opens an alternative possibility of creating dispersed and aligned nanostructures of AgNPs in PVA nanofiber matrix for wide applications such as biological sensors,28–30 conductive interconnects,31,32 optoelectronic devices,33–35 and effective bioactive materials, as we demonstrate to follow.
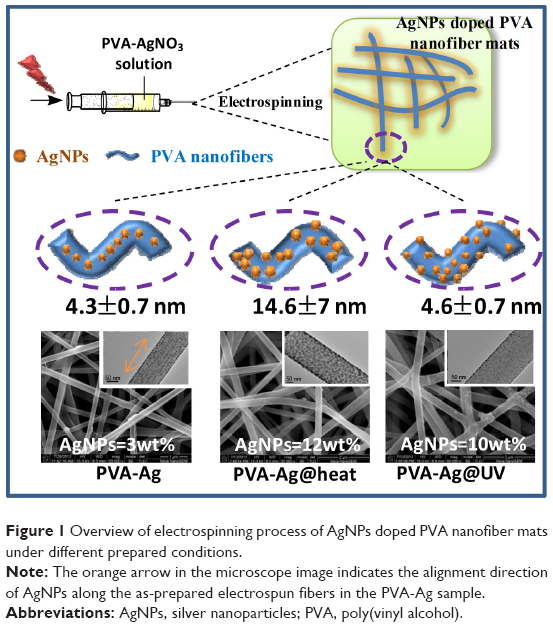 | Figure 1 Overview of electrospinning process of AgNPs doped PVA nanofiber mats under different prepared conditions. |
Materials and methods
PVA 1799 (99% alcoholysized, Mw =75,000) was purchased from Aladdin Industrial Co., Shanghai, People’s Republic of China. AgNO3 (99.8%) was purchased from Yingda Rare Chemical Reagents Factory, Tianjin, People’s Republic of China. Nonwoven textile (50 g·m−2), made of polypropylene and polyethylene terephthalate fabrics, was obtained from Liyang New Material Development Co., Ltd, Nantong, People’s Republic of China. Test bacteria such as Staphylococcus aureus (ATCC 6538) and Escherichia coli (8099) were purchased from the Institute of Microbiology and Epidemiology, Academy of Military Medical Science, People’s Republic of China. Nutrient broth (NB) and agar powder were provided by Aoboxing Bio-tech Co., Ltd., Beijing, People’s Republic of China. Deionized water was used as the solvent. The cell line PA317 was purchased from ATCC (Manassas, VA, USA).
Characterization
The X-ray diffraction (XRD; MiniFlex 600, Rigaku Corporation, Tokyo, Japan) patterns of the electrospun nanofiber mats were recorded with the Cu-Ka radiation (40 kV and 15 mA) at a scan rate of 2°·min−1 ranging 5° to 60° (2θ).
The morphology of electrospun PVA nanofiber mats doped with AgNPs was observed with scanning electron microscopy (SEM) (FEI Nova Nano, SEM 230; FEI, Hillsboro, OR, USA) after gold coating under high vacuum (EMITECH K550x, Quorum Technologies Limited, East Grinstead, UK). The average diameter of the electrospun fibers was measured by Image-Pro Plus software (Media Cybernetics, Inc., Rockville, MD, USA) from the SEM images; and at least 50 counts of fibers from two separate images of each sample were measured. Energy dispersive X-ray spectroscopy (EDS) with accelerating voltage of 200 kV was measured from the SEM images.
Transmission electron microscopy (TEM) analysis of the electrospun samples was conducted on Tecnai G2 F20 (FEI) equipped with electron diffractometer by using Lacey formvar/carbon film-coated copper grids (230 mesh, Beijing Zhongjing-keyi Technology Co., Ltd, Beijing, People’s Republic of China) as the supporter. The size of the AgNPs was measured using Image Pro Plus software from the TEM images (n=200).
The size of AgNPs in solution was measured using dynamic light scattering (DLS) with a zetasizer (Nano ZS, Malvern Instruments Ltd., Malvern, UK). Diluted samples (0.2% w/v of Ag) were used to avoid multiple scattering. The analyses was performed with He–Ne laser (633 nm) at scattering angle of 175° at 25°C. The mean hydrodynamic diameter (Z-average) was calculated from the autocorrelation function of the intensity of light scattered from the particles. The analysis software used was Zetasizer software version 6.34, supplied by the manufacturer (Malvern Instruments Ltd.).
The X-ray photoelectron spectroscopy (XPS) measurements were conducted on a Quanta200 spectrometer (FEI) using monochromated Al-Ka radiation generated from an electron beam operating at pass energy of 93.9 eV. Survey spectra were run in the binding energy range 0–1,000 eV, followed by high-resolution spectra of the C1s, O1s, and Ag3d region. Binding energies (BEs) were calibrated using the containment carbon (C1s =285.0 eV).
Electrospinning of AgNPs-containing nanofiber mats (PVA-Ag)
The electrospinning set-up was purpose built by the authors (Figure 2). Firstly 15% (w/v) PVA in deionized water was stirred in water-bath at 85°C for at least 2 hours to obtain the viscous solution. Then after cooling to room temperature, 1.0 g of AgNO3 in water was added to the solution above to achieve the final concentration of 1% (w/v) of AgNO3 and 12% (w/v) PVA mixture. The mixture was stirred vigorously in the dark for 30 minutes at room temperature to obtain an isotropic solution, and then was pumped into a disposable syringe capped with an 18 gauge stainless steel cannula with a blunt end tip. During the electrospinning process, a high voltage (15 kV) was applied to the needle and solution was jetted at a flow rate of 1 mL·h−1 controlled by a syringe pump. The tip-to-collector distance (TD) was 15 cm. The collector plate was a piece of electronically grounded aluminum drum covered by a piece of nonwoven textile as supporting material (Figure 2). After electrospinning, the as-prepared material was removed from the collector plate and stored in the dark until needed.
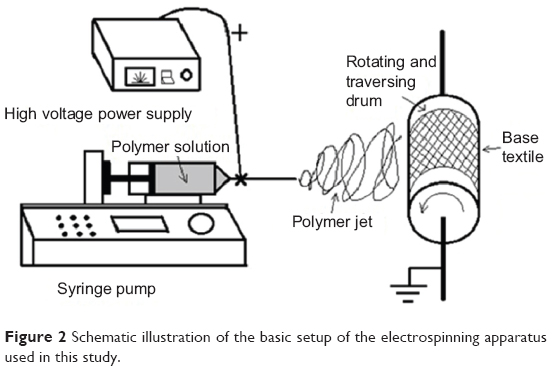 | Figure 2 Schematic illustration of the basic setup of the electrospinning apparatus used in this study. |
Electrospinning of PVA-Ag on preheating conditions (PVA-Ag@heat)
The prepared concentration of 1% (w/v) of AgNO3 and 12% (w/v) PVA mixture was refluxed for 1 hour at 105°C before applying it to the electrospinning process under the same condition described in the Electrospinning of AgNPs-containing nanofiber mats section.
Electrospinning of PVA-Ag with in situ UV irridiation (PVA-Ag@UV)
The as-prepared PVA-Ag sample mentioned in the Electrospinning of AgNPs-containing nanofiber mats section was put under a UV lamp at 365 nm for 3 hours at an intensity of 700 mw·cm−2.
Inhibition zone assay
The bacterial suspensions, S. aureus (ATCC 6538) and E. coli (8099), were diluted to 1×107 colony forming units (CFU)·mL−1 in 0.03 M phosphate buffered saline separately, and 100 μL of the bacterial suspension was then spread evenly with a sterilized glass spreader on agar plates. Samples of nanofiber mats were cut into circular shaped sheets of 25 mm diameter. Each circular sheet was pressed tightly on the center of the plate in contact with the mat side. After 24 hours at 37°C incubation, the inhibition zones were observed visually. The pure PVA nanofiber mats without AgNPs were used as negative control.
Biocidal kinetic study
S. aureus suspension, 2 mL of 1.4×106 CFU·mL−1 were added to the sterilized tube containing 4.0 mg of each sample. After vortexing for 10 seconds, all the mixtures were incubated with gentle shaking at room temperature. At the determined time point (5 min/10 min/20 min/30 min/60 min), 100 μL of the bacterial suspension was withdrawn and spread evenly with a sterilized glass spreader on the prepared agar plates, colony counting occurred after being cultured at 37°C for 24 hours.
Cytotoxicity of membranes by cell counting kit-8 assay
Each membrane, 10×20 mm, was incubated in 50 mL Dulbecco’s Modified Eagle’s Medium (DMEM) to obtain extract solution after 1 day of incubation with shaking in a 37°C humidified environment. Extracted solutions were filtered with 0.20 μm filter for sterilization and then diluted with fresh media to obtain different fractional concentrations- ie, nondiluted (100%) and diluted (1:7 [12.5%], 1:3 [25%], 1:1 [50%]) extract solutions with DMEM, respectively. Fresh media without the extract solutions was designated as negative control (0%). Diluted and nondiluted extract solutions were then used to treat the PA317 cell line, 104 cells/mL, grown for 24 hours in the growth medium (DMEM with 10% fetal bovine serum [FBS]). Cell viability was quantified by cell counting kit (CCK)-8 kit following the protocol of the manufacturer (Dojindo Inc., Rockville, MD, USA). CCK-8 is a very convenient assay utilizing a highly water-soluble tetrazolium salt–WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt]. It allows sensitive colorimetric assays for the determination of the number of viable cells in cell proliferation and cytotoxicity assays superior to the MTT method.
Results and discussion
Since the PVA molecule is consist of many active OH groups on its side chains, it can serve as a chemical reducer and a template agent for electrospinning simultaneously. The reaction between the R-OH and Ag+ can be expressed as follows in Equation 1 (where , R-OH, R=O represents the monomer before and after partially oxidized in PVA molecules with the addition of AgNO3 in solution [Ag+] to produce AgNPs [Ag0]):
R-OH + Ag+ → R=O + Ag0 + H+ | (1) |
Such a process is known to be achieved by heating,36 chemical reduction,37 or photoreduction.38 Herein, it is found that AgNPs in the PVA nanofibers can also be easily obtained either by the single electrospinning process or when left under mild conditions, but bearing with distinct sizes and size distributions. The as-prepared PVA-Ag electrospun nanofibrous mats were observed under TEM (shown in top row of Figure 3A), and interestingly the approximately spherical AgNPs are formed in aligned arrays along the orientation of the fibers. The high voltage (15 kV) applied to the PVA-Ag solution during the electrospinning process is believed to contribute to this unique linear arrangement as well as electron transfer of oxido-reduction reaction between the R-OH and Ag+ to produce final AgNPs. When a high voltage was applied to the solution of PVA-Ag in the syringe, the pendent droplet at the nozzle of the spinneret became highly electrified. Under the strong action of Coulombic force exerted by the external electric field, the liquid droplet was distorted into the formation of the Taylor cone,39 leading to a stretching and alignment distribution of silver ions along the electric field in solution simultaneously. During elongation of the jet and evaporation of water, the redox reaction between R-OH and Ag+ occurs. Thus aligned AgNPs along the PVA nanofibers were directly formed and collected as non-woven mats. The size distribution of such AgNPs in PVA-Ag sample is uniformly distributed, ie, 4.3±0.7 nm (bottom row in Figure 3A). The AgNPs are believed to be distributed mostly within the fiber, since the corresponding SEM image of PVA-Ag (top row in Figure 4B) shows that few Ag clusters can be observed on the surface of the fibers. EDS analysis indicates that the content of Ag is approximately 2.6 wt% (bottom row in Figure 4B), while there is no existence of Ag detected in the pure PVA nanofibers (Figure 4A). The AgNPs within the electrospun PVA nanofibers are stable for at least 3 months, as the obtained nanofiber mats are usually in a dry state, which allows the AgNPs to be “trapped” in a stable state. Thus, the AgNPs are sufficiently dispersed that they do not agglomerate with each other. In order to verify the growth mechanism of AgNPs in PVA, we performed the reaction between AgNO3 and PVA in a solution by stirring in the dark at room temperature for a longer time (96 hours) with in situ UV and DLS monitoring.40 It was found that the reaction still occurs under such mild conditions, and even after reaction of 0.5 hour, oligomeric clusters of silver ions start to form as the UV spectrum shows an arising absorption (Abs) peak at 295 nm (Figure 5).41 The intensity of the Abs peak at 295 nm increased due to larger amounts of the oligomeric clusters of Ag+ produced within 12 hours. After stirring for 96 hours at room temperature, the original PVA-Ag solution turned from a colorless solution into a brown colored solution (inset Figure 5), and the average size of the AgNPs are measured to change from 29.7 nm (12 hours) to approximately 430 nm (96 hours). Additionally, a new absorbance peaks occurred in the UV spectra at 430 nm, indicating the formation of larger AgNPs.42 This is because these clusters formed final NPs due to coalescence effect after certain time period during the growth process.43 Although, extending the reaction time scale in solution can increase the amount of AgNPs formed under mild conditions, it is inappropriate to obtain controllable AgNPs this way, especially for the ultrasmall and narrowly dispersed ones. The ultrasmall particle size bears an ultralarge surface area per mass which leads to the AgNPs immediate contact with ambient AgNPs in aqueous solution, and eventual aggregation of AgNPs in the aqueous solution. On the other hand, this reaction process could also be accelerated thermally. By heating one portion of AgNO3 and PVA solution for 1 hour at 105°C, then applying the electrospinning process, PVA-Ag@heat sample can be obtained. TEM images (Figure 3C) and EDS results (Figure 4C) show that PVA nanofibers loaded with a higher content of AgNPs (12.4 wt%) could be obtained, but with a larger size and wider distribution, ie 14.6±7 nm, compared with that of the PVA-Ag sample (4.3±0.7 nm).
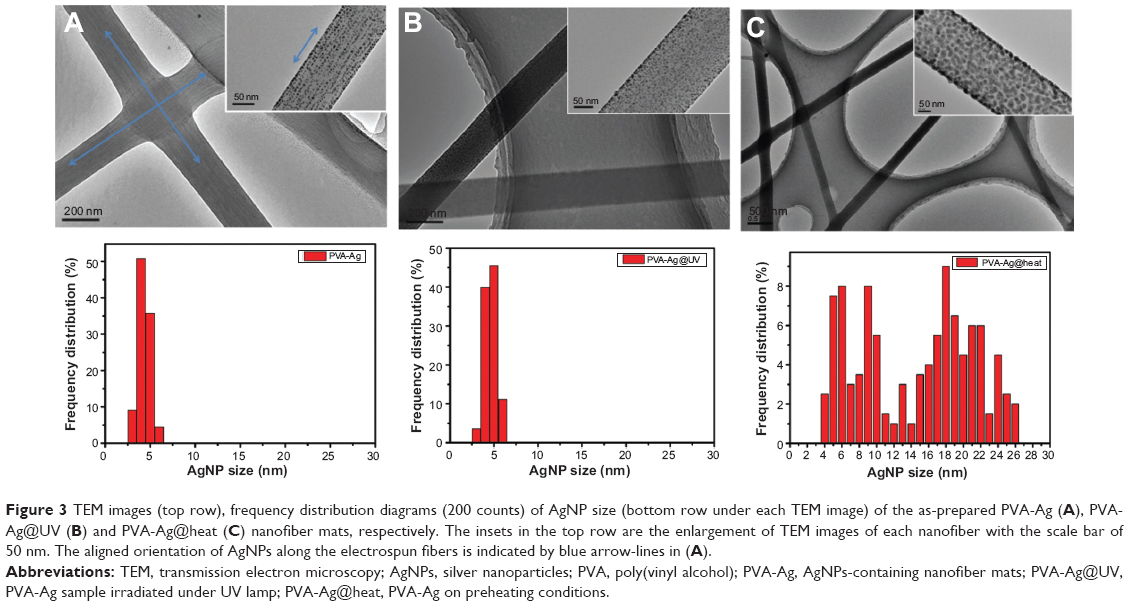 | Figure 3 TEM images (top row), frequency distribution diagrams (200 counts) of AgNP size (bottom row under each TEM image) of the as-prepared PVA-Ag (A), PVA-Ag@UV (B) and PVA-Ag@heat (C) nanofiber mats, respectively. The insets in the top row are the enlargement of TEM images of each nanofiber with the scale bar of 50 nm. The aligned orientation of AgNPs along the electrospun fibers is indicated by blue arrow-lines in (A). |
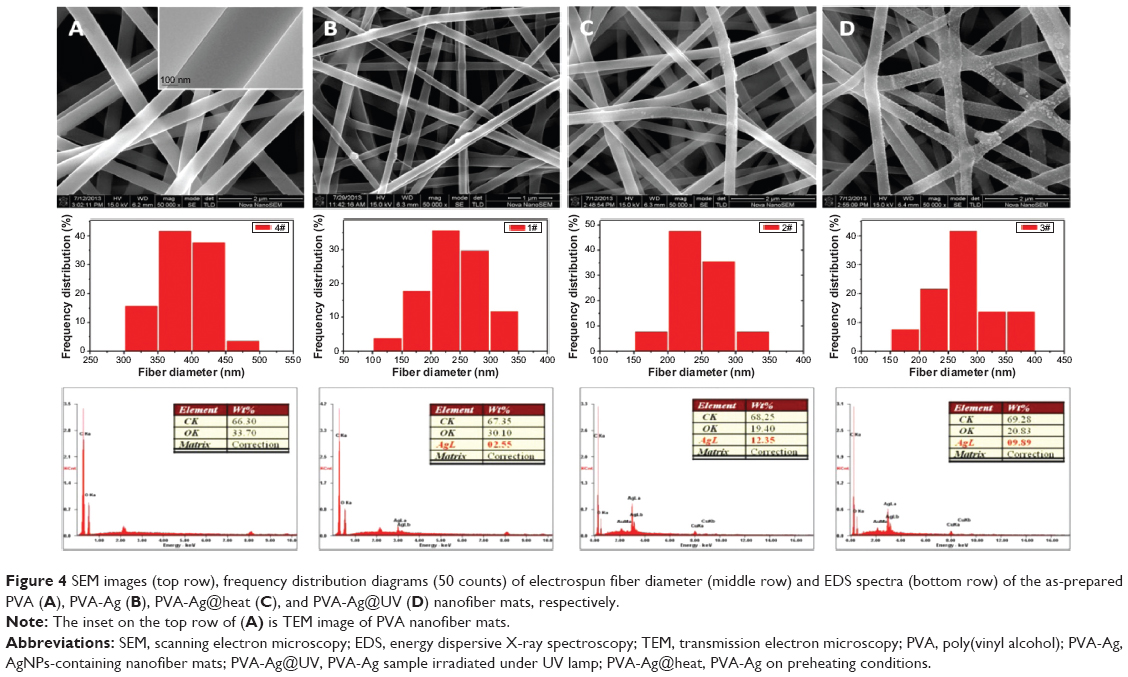 | Figure 4 SEM images (top row), frequency distribution diagrams (50 counts) of electrospun fiber diameter (middle row) and EDS spectra (bottom row) of the as-prepared PVA (A), PVA-Ag (B), PVA-Ag@heat (C), and PVA-Ag@UV (D) nanofiber mats, respectively. |
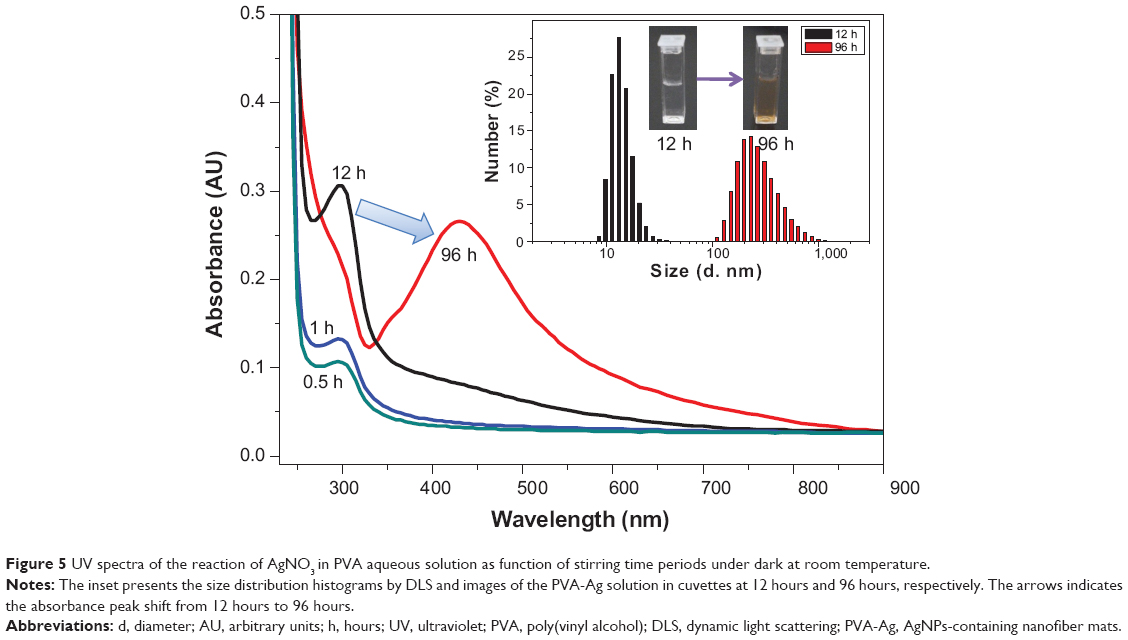 | Figure 5 UV spectra of the reaction of AgNO3 in PVA aqueous solution as function of stirring time periods under dark at room temperature. |
In order to solve such a dilemma, the as-prepared electrospun PVA-Ag sample was irradiated under UV lamp (PVA-Ag@UV). The loading levels of AgNPs could be greatly enhanced by such in situ UV irradiation, with ultrasmall size and narrowly-dispersed AgNPs compared to those prepared in the PVA-Ag sample. After the treatment, the statistic size of AgNPs in PVA-Ag@UV sample is measured to be 4.6±0.7 nm (bottom row in Figure 3B), similar to that in the PVA-Ag sample but with a higher loading content of approximately 9.9 wt% (bottom row in Figure 4D), almost four-fold that compared to the PVA-Ag sample. From the SEM image of PVA-Ag (top row in Figure 4D), it can be seen that most of the AgNPs are laid on the surface of the PVA nanofibers randomly, which is attributed to the UV irradiation. Moreover, the diameters of the PVA nanofibers are similar for all the AgNPs containing samples, ie, 235±50 nm for PVA-Ag, 245±40 nm for PVA-Ag@heat, and 270±55 nm for PVA-Ag@UV, indicating that the posttreatments (UV irradiation and heating) have little effect on the diameters of the PVA nanofibers. However, the diameters of pure PVA electrospun nanofibers are a bit higher, ie, 385±40 nm (middle row in Figure 4). The reason for this could be due to the addition of silver ions in the matrix, which changed the electric conductivity of the starting solution, thus achieving smaller diameters in the AgNPs containing products.
The XRD results of all the samples show broad reflection peaks at approximately 19.6° of 2θ originated from the PVA matrix (Figure 6), due to the semi-crystalline property of the polymer.44 Moreover, the XRD peaks for the AgNPs are found at 38.2° and 44.6° of 2θ, which are indexed to the (111) and (200) planes with cubic symmetry, respectively.45 This indicates that the as-prepared AgNPs crystals were stabilized in these samples due to the existence of PVA molecules, even for the ultrasmall ones. However, the peaks of the silver nanocrystals from the PVA-Ag sample, which contains the lowest content of AgNPs, can barely been detected due to the resolution limit of XRD analysis. Figure 7 shows the XPS spectra of each sample, from C1s peaks (Figure 7A) it can be seen that all the peaks were shifted to a higher binding energy after the loading of AgNPs. With an increase in the AgNPs content, ie, in the samples of PVA-Ag@heat and PVA-Ag@UV, the C1s peak at 286.5 eV rises accordingly, which is attributed to the stronger affinity of AgNPs on the C-O-H bonds in the PVA nanofiber mats. Similar trends can be found in Figure 7B, as the O1s photoemission spectra moved from 532.2 eV to 532.4 eV for all the three AgNPs containing samples compared with the pure PVA sample. This indicates that the electron density of the hydroxyl oxygen atoms in the PVA is decreased due to the stronger interaction with AgNPs.24 Meanwhile, there are almost no differences in the O1s peaks among these three AgNP containing samples, which means the as-prepared AgNPs interacted with the hydroxyl oxygen atoms in the same manner despite the different method that they were produced in. The Ag3d peaks in Figure 7C at 368.4 eV (3d5/2) and 374.5 eV (3d3/2) are assigned to be the pure metal Ag in the valence state of zero (Ag0).46 Compared with the reported binding energy peak of AgNO3, ie, 370.3 eV (Ag3d5/2),47 it can be concluded that no residual silver ions can be found on the surface of the PVA nanofibers. The distinct biomodal distribution of the Ag3d3/2 in the sample of PVA-Ag@heat could probably be due to the size effects of the AgNPs. Increasing the sizes of the AgNPs leads to a decrease in the specific surface area and hence, to a decline in the interaction with the PVA.48
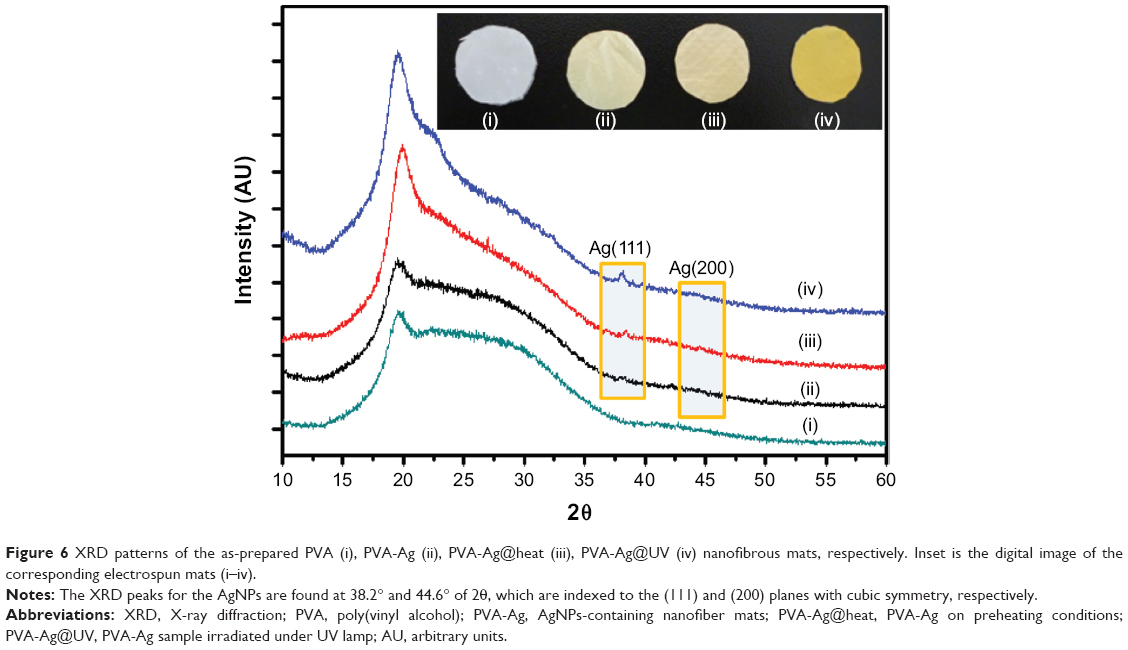 | Figure 6 XRD patterns of the as-prepared PVA (i), PVA-Ag (ii), PVA-Ag@heat (iii), PVA-Ag@UV (iv) nanofibrous mats, respectively. Inset is the digital image of the corresponding electrospun mats (i–iv). |
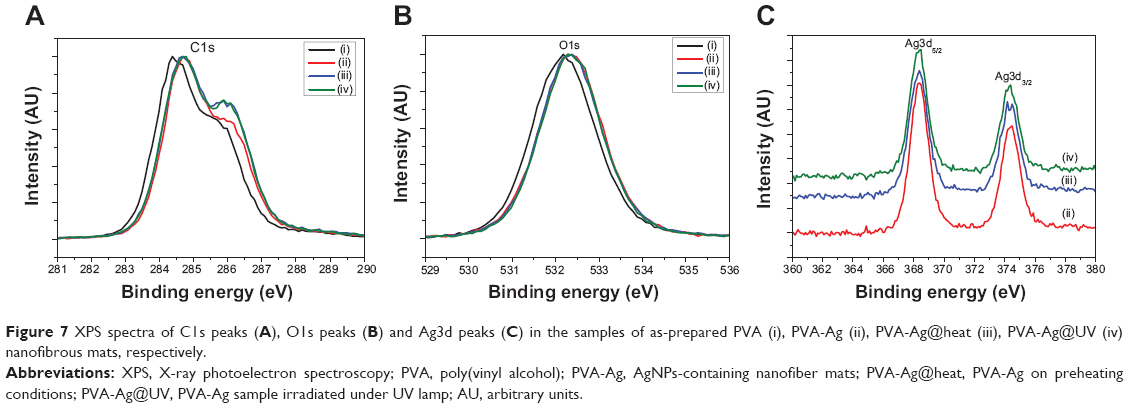 | Figure 7 XPS spectra of C1s peaks (A), O1s peaks (B) and Ag3d peaks (C) in the samples of as-prepared PVA (i), PVA-Ag (ii), PVA-Ag@heat (iii), PVA-Ag@UV (iv) nanofibrous mats, respectively. |
As previously reported,49 the size of the silver particles plays an important role in determining their biocidal efficacy. Smaller AgNPs bear stronger antibacterial properties, due to their larger surface area and faster silver ion release.50,51 Recently, Agnihotri et al52 reported immobilized or entrapped AgNPs on an amine-functionalized silica surface and studied their superior biocidal efficacy. Similarly, the AgNPs herein also show excellent antibacterial activities. The bacteria zone of inhibition tests53 in Figure 8A show that all the AgNPs-containing samples have excellent inhibition activities against both S. aureus and E. coli. However, the pure electrospun PVA sample (negative controls) did not show inhibition zones (Figure 8A [i]). As for the S. aureus tests, a more clear and larger circular zone of inhibition was observed, an approximate 2-fold increase in diameter of final inhibition zones compared to the initial mats for PVA-Ag, PVA-Ag@heat and PVA-Ag@UV samples. This is explained by the diffusion of AgNPs out of the fibrous mat, which killed bacteria within a certain diameter (top row Figure 8A [ii, iii and iv]). However, for the E. coli tests, the diameter of the inhibition zone for each PVA-Ag, PVA-Ag@heat and PVA-Ag@UV sample is approximately 1.4-fold longer than the original samples (see bottom row in Figure 8A [ii, iii, and iv]). These results show that AgNPs have a superior inhibition activity with S. aureus, compared to E. coli. The reason is believed to be the sophistication of the cell wall structures between gram-positive (ie, S. aureus) and gram-negative (ie, E. coli) bacteria.54,55 In order to quantitatively determine the kinetics of the as-prepared samples, a time-kill study was performed against S. aureus suspension. As seen in Figure 8B, the surviving cell numbers of S. aureus are time-dependent. The fractional survival descends sharply with the increase in contacting time. Moreover, due to the size effect, the PVA-Ag@UV sample shows the strongest antibacterial effects with the highest biocidal rates against S. aureus, as no bacterial colonies was detected only after 30 minutes of incubation, ie, 6-fold log reduction, while approximately 0.04‰ and 0.6‰ of remaining colonies were still found in the PVA-Ag@heat and PVA-Ag samples at the same time, respectively (see inset in Figure 8B). The PVA-Ag@heat sample shows medium antibacterial activity, due to its larger size and higher content of AgNPs. Although PVA-Ag shows the weakest biocidal activity among the three AgNPs-containing electrospun samples, it is still believed to bear relatively good antibacterial activity because colonies could not be detected after 1 hour; while for the pure PVA samples no antibacterial effects were found, as the bacterial colonies showed a 1.7-fold log increase compared to the starting one after 1 hour of incubation.
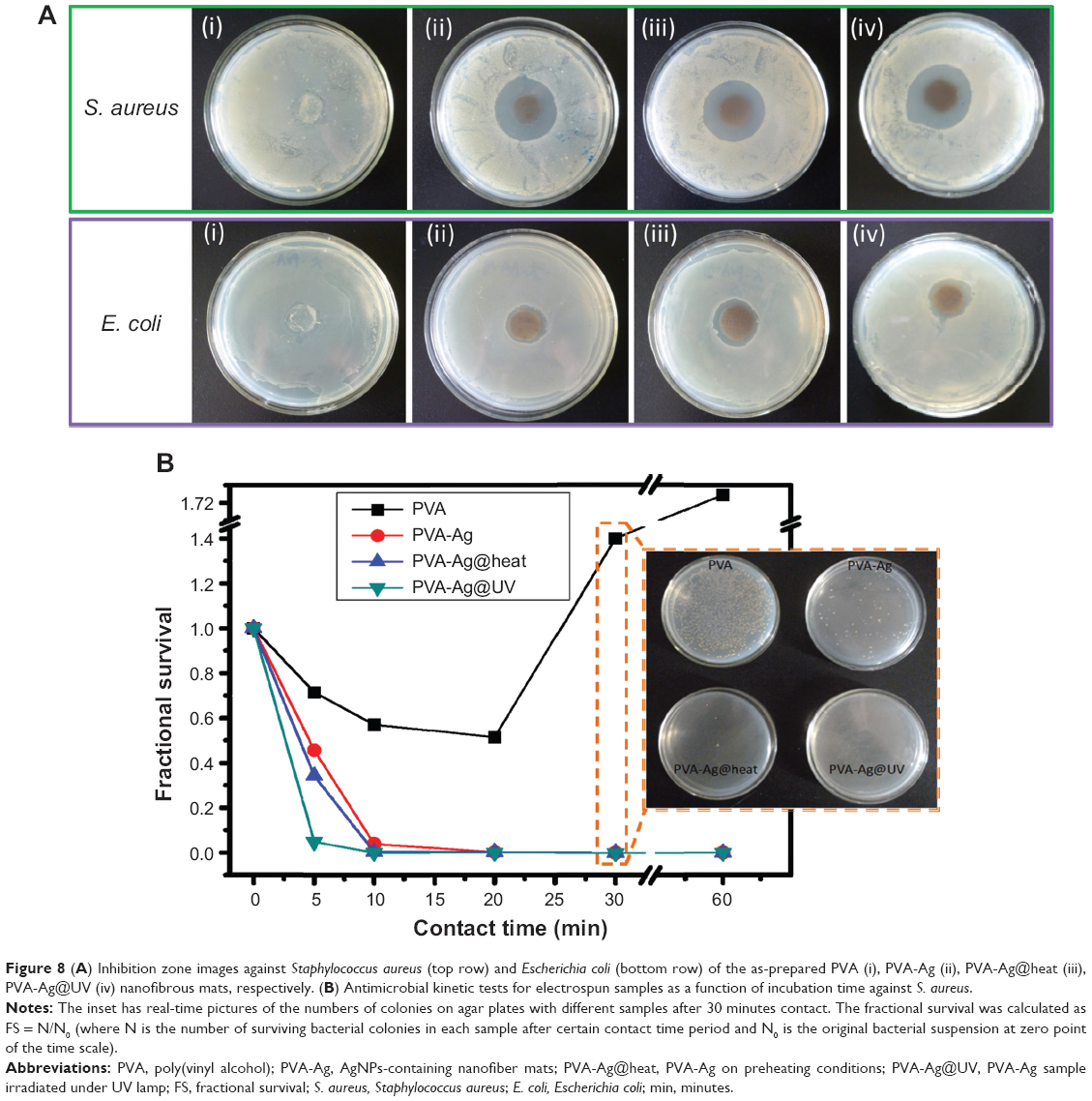 | Figure 8 (A) Inhibition zone images against Staphylococcus aureus (top row) and Escherichia coli (bottom row) of the as-prepared PVA (i), PVA-Ag (ii), PVA-Ag@heat (iii), PVA-Ag@UV (iv) nanofibrous mats, respectively. (B) Antimicrobial kinetic tests for electrospun samples as a function of incubation time against S. aureus. |
Furthermore, the cell viability of the PVA, and PVA doped with AgNPs mats were evaluated using the CCK-8 assay. CCK-8 is a cell viability assay reagent with a higher sensitivity and a better reproducibility than traditional MTT assay.56 As shown in Figure 9, the PVA nanofibrous mat shows almost no cytotoxicity at various concentrations, assuring its good biocompatibility. Similar trends can also been found in PVA-Ag@UV and PVA-Ag@heat samples, since approximately 85% and 80% of cells remain alive when incubated with the non-diluted (100%) extract solutions, respectively. However, the PVA-Ag samples indicate a concentration-dependent toxicity, and the cell viability decreases from 85% to 70% with the increase of the extracted concentration. Such toxic effect is probably due to the residual and unreactive silver ions in the PVA-Ag nanofibrous mat. Although the amount of AgNPs in the PVA-Ag@UV and PVA-Ag@heat is much higher than that in the PVA-Ag, the silver ions are more cytotoxic than AgNPs. This is consistent with earlier results reported by Panácek et al57 who have shown that the LC100 (the concentrations of silver lethal to 100% of the cells) on human fibroblasts of AgNPs is 30-fold higher than silver ions.
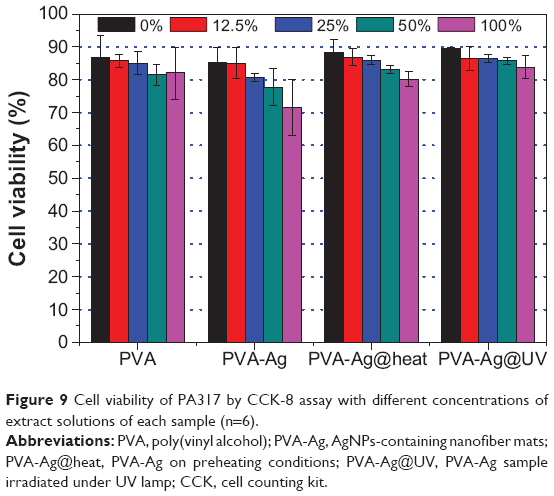 | Figure 9 Cell viability of PA317 by CCK-8 assay with different concentrations of extract solutions of each sample (n=6). |
Conclusion
In summary, the electrospun fabrication procedure described here produces stable PVA nanofabric mats doped with aligned AgNPs using a one-step methodology, and green manner. Moreover, the sizes and yields of doped AgNPs can be regulated either by exposure of UV irradiation or the preheating treatment. We have validated the potential of this functionalized fabric as a hygienic material by evaluating their excellent bactericidal effect on both gram-positive and gram-negative bacteria. In the meantime, reducing the local concentration of silver ions in the sample via in situ UV irradiation and preheating can greatly improve their biocompatibility as well as biocidal ability. Moreover, the proposed method herein is cost-effective, eco-friendly and suitable for broad applications as such as utilizing it as antimicrobial agents, wound dressings, and in water or air purification techniques, due to the up-to-date industrial progress of electrospinning technique.58
Acknowledgments
We gratefully acknowledge financial support by the Project of National Natural Science Foundation of China (Grant No. 50903090, 81302187 and 81172398) and Tianjin Research Program of Applied Foundation and Advanced Technology (Grant No. 13JCQNJC02500). S Lin acknowledges helpful discussion and communication on the manuscript with Dr Vinodh Rajendra (University of Toronto).
Disclosure
The authors report no conflicts of interest in this work.
References
Moffitt MG. Self-assembly of polymer brush-functionalized inorganic nanoparticles: from hairy balls to smart molecular mimics. J Phys Chem Let. 2013;4:3654–3666. | ||
Sun Y, Gates B, Mayers B, Xia Y. Crystalline silver nanowires by soft solution processing. Nano Lett. 2002;2:165–168. | ||
Lu H, Fan L, Liu Q, Wei J, Ren T, Du J. Preparation of water-dispersible silver-decorated polymer vesicles and micelles with excellent antibacterial efficacy. Polym Chem. 2012;3:2217–2227. | ||
Chen SC, Zhang ZH, Chen Q, et al. An efficient strategy to achieve hydrophilic polymeric silver(I) materials with exceptional antibacterial activity. Chem Commun. 2013;49:1270–1272. | ||
Ray S, Mohan R, Singh JK, et al. Anticancer and antimicrobial metallopharmaceutical agents based on palladium, gold, and silver N-hererocyclic carbine complexes. J A Chem Soc. 2007;129:15042–15043. | ||
Clement JL, Jarrett PS. Antibacterial silver. Met Based Drugs. 1994;1: 467–482. | ||
Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. | ||
Marambio-Jones C, Hoek EMV. A review of the antibacterial effects of silver nanomaterials and potential implications for human health and the environment. J Nanopar Res. 2010;12:1531–1551. | ||
Luo Z, Zheng K, Xie J. Learning from nature: introducing an epiphyte-host relationship in the synthesis of alloy nanoparticles by co-reduction methods. Chem Commun. 2014;50:5143–5155. | ||
Destaye AG, Lin CK, Lee CK. Glutaraldehyde vapor cross-linked nanofibrous PVA mat with in situ formed silver nanoparticles. ACS Appl Mater Interfaces. 2013;5:4745–4752. | ||
Gorth DJ, Rand DM, Webster TJ. Silver nanoparticle toxicity in Drosophila: size does matter. Int J Nanomedicine. 2011;6:343–350. | ||
Sondi I, Salopek-Sondi B. Silver nanoparticles as antimicrobial agent: a case study on E.coli as a model for gram-negative bacteria. J Colloid Interface Sci. 2004;275:177–182. | ||
Vertelov GK, Krutyako YA, Efremenkova OV, Olenin AY, Lisichkin GV. A versatile synthesis of highly bactericidal Myramistin stabilized silver nanoparticles. Nanotechnology. 2008;19:355707. | ||
Zhang Y, Peng H, Huang W, Zhou Y, Yan D. Facile preparation and characterization of highly antimicrobial colloid Ag or Au nanoparticles. J Coll Iinterface Sci. 2008;325:371–376. | ||
Li L, Sun J, Li X, et al. Controllable synthesis of monodispersed silver nanoparticles as standards for quantitative assessment of their cytotoxicity. Biomaterials. 2012;33:1714–1721. | ||
Shameli K, Bin Ahmad M, Yunus WMZW, et al. Silver/poly(lactic acid) nanocomposites: preparation, characterization, and antibacterial activity. Int J Nanomedicine. 2010;5:573–579. | ||
Juan L, Zhimin Z, Anchun M, et al. Deposition of silver nanoparticles on titanium surface for antibacterial effect. Int J Nanomedicine. 2010;5:261–267. | ||
Kim K, Sung W, Moon S, Choi J, Kim J, Lee D. Antifungal effect of silver nanoparticles on dermatophytes. J Microbiol Biotechnol. 2008;18:1482–1484. | ||
Reneker DH, Chun I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology. 1996;7:216–223. | ||
Frenot A, Chronakis IS. Polymer nanofibers assembled by electro-spinning. Curr Opin Colloid Interface Sci. 2003;8:64–75. | ||
Li D, Xia Y. Electrospinning of nanofibers: reinventing the wheel. Adv Mater. 2004;16:1151–1170. | ||
Sen R, Zhao B, Perea D, et al. Preparation of single-walled carbon nanotube reinforced polystyrene and polyurethane nanofibers and membranes by electrospinning. Nano Letters. 2004;4:459–464. | ||
Hong KH. Preparation and properties of electrospun poly(vinyl alcohol)-silver fiber web as wound dressings. Polymer Engineering and Science. 2007;43–49. | ||
Hong KH, Park JL, Sul IH, Youk JH, Kang TJ. Preparation of antimicrobial poly(vinyl alcohol) nanofibers containing silver nanoparticles. Journal of Polymer Science, Part B: Polymer Physics. 2006;44: 2468–2474. | ||
Dong G, Xiao X, Liu X, et al. Preparation and characterization of Ag nanoparticle-embedded polymer electrospun nanofibers. J Nanopart Res. 2010;12:1319–1329. | ||
Jin W, Jeong H, Kim J, Youk J. A study on the preparation of poly (vinyl alcohol) nanofibers containing silver nanoparticles. Synth Met. 2007;157:454–459. | ||
Patil RS, Kokate MR, Jambhale CL, Pawar SM, Han SH, Kolekar SS. One-pot synthesis of PVA-capped silver nanoparticles their characterization and biomedical application. Adv Nat Sci Nanosci Nanotechnol. 2012;3:015013. | ||
Sonntag MD, Klingsporn JM, Zrimsek AB, Sharma B, Ruvuna LK, Van Duyne RP. Molecular plasmonics for nanoscale spectroscopy. Chemical Society Reviews. 2014;43:1230–1247. | ||
Zhu H, Du M, Zhang M, et al. Facile fabrication of AgNPs/(PVA/PEI) nanofibers: high electrochemical efficiency and durability for biosensors. Biosens Bioelectron. 2013;49:210–215. | ||
Obliosca JM, Liu C, Yeh HC. Fluorescent silver nanoclusters as DNA probes. Nanoscale. 2013;5:8443–8461. | ||
Moon KS, Dong H, Maric R, et al. Thermal behavior of silver nanoparticles for low-temperature interconnect applications. J Electron Mater. 2005;34:168–175. | ||
Yang C, Wong CP, Yuen MMF. Printed electrically conductive composites: conductive filler designs and surface engineering. Journal of Materials Chemistry C. 2013;1:4052–4069. | ||
McFarland AD, Van Duyne RP. Single silver nanoparticles as real-time optical sensors with zeptomole sensitivity. Nano Lett. 2003;3: 1057–1062. | ||
Lal S, Link S, Halas NJ. Nano-optics from sensing to waveguiding. Nat Photonics. 2007;1:641–648. | ||
El-Shamy AG, Attia W, Abd El-Kader KM. The optical and mechanical properties of PVA-Ag nanocomposites films. J Alloys Compd. 2014; 590:309–312. | ||
Jin M, Zhang X, Nishimoto S, et al. Large-scale fabrication of Ag nanoparticles in PVP nanofibres and net-like silver nanofibre films by electrospinning. Nanotechnology. 2007;18(7):075605. | ||
Lee HY, Park HK, Lee YM, Kim KM, Park SB. Mechanism of silver nanoparticles action on insect pigmentation reveals intervention of copper homeostasis. Chem Commun. 2007;28:2959–2961. | ||
Shanmugam S, Viswanathan B, Varadarajan TK. A novel single step chemical route for noble metal nanoparticles embedded organic–inorganic composite films. Mater Chem Phys. 2006;95:51–55. | ||
Shenoy S, Bates W, Frisch H, Wnek G. Role of chain entanglements on fiber formation during electrospinning of polymer solutions: good solvent, non-specific polymer–polymer interaction limit. Polymer. 2005;46:3372–3384. | ||
Allouni ZE, Cimpan MR, Høl PJ, Skodvin T, Gjerdet GR. Agglomeration and sedimentation of TiO2 nanoparticles in cell culture medium. Colloids Surf Biointerfaces. 2009;68:83–87. | ||
Ershov Bg, Janata E, Henglein A. Growth of silver particles in aqueous solution: long-lived “magic” clusters andionic strength effects. J Phys Chem. 1993;97:339–343. | ||
Banerjee M, Sharma S, Chattopadhyay A, Ghosh SS. Enhanced antibacterial activity of bimetallic gold-silver core-shell nanoparticles at low silver concentration. Nanoscale. 2011;3:5120–5125. | ||
Polte J, Tuaev X, Wuithschick M, et al. Formation mechanism of colloidal silver nanoparticles: analogies and differences to the growth of gold nanoparticles. ACS Nano. 2012;6(7):5791–5802. | ||
Park JH, Karim MH, Kim IK, et al. Electrospinning fabrication and characterization of poly(vinyl alcohol)/montmorillonite/silver hybrid nanofibers for antibacterial applications. Colloid Polym Sci. 2010;288:115–121. | ||
Park JH, Kim IK, Choi JY, et al. Electrospinning fabrication of poly(vinyl alcohol)/waterborne polyurethane/silver composite nanofibre mats in aqueous solution for anti-bacterial exploits. Polymer and Polymer Composites. 2011;19:753–761. | ||
National Institute of Standards and Technology (NIST). Technology Services. [Webpage on the Internet]. Available from http://srdata.nist.gov/xps/XPSDetailPage.aspx?AllDataNo=28020. Accessed May 4, 2014. | ||
Kim YW, Lee DK, Lee KJ, Min BR, Kim JH. In situ formation of silver nanoparticles within an amphiphilic graft copolymer film. J Polym Sci Part B Polym Phys. 2007;45:1283–1290. | ||
Shin HS, Choi HC, Jung Y, Bin Kim S, Song HJ, Shin HG. Chemical and size effects of nanocomposites of sliver and polyvinyl pyrrolidone determined by X-ray photoemission spectroscopy. Chem Phys Lett. 2004;383:418–422. | ||
Loher S, Schneider OD, Maienfisch T, Bokorny S, Stark WJ. Micro-organism-triggered release of silver nanoparticles from biodegradable oxide carriers allows preparation of self-sterilizing polymer surfaces. Small. 2008;4:824–832. | ||
Sambhy V, MacBride MM, Peterson BR, Sen A. Silver bromide nanoparticle/polymer composites: dual action tunable antimicrobial materials. J Am Chem Soc. 2006;128:9798–9808. | ||
Martínez-Castañón GA, Niño-Martínez N, Martínez-Gutierrez F, Martínez-Mendoza JR, Ruiz F. Synthesis and antibacterial activity of silver nanoparticles with different sizes. J Nanopar Res. 2008;10:1343–1348. | ||
Agnihotri S, Mukherji S, Mukherji S. Immobilized silver nanoparticles enchance contact killing and show highest efficacy: elucidation of the mechanism of bactericidal action of silver. Nanoscale. 2013;5:7328–7340. | ||
Lin S, Wang S, Qi J, et al. One-pot fabrication and antimicrobial properties of novel pet nonwoven fabrics. Biomedical materials. 2011;6:045009 and the reference therein. | ||
Chen CZ, Beck-Tan NC, Dhurjati P, van Dyk TK, LaRossa RA, Cooper SL. Quaternary ammonium functionalized poly(propylene imine) dendrimers as effective antimicrobials: structure-activity studies. Biomacromolecules. 2000;1:473–480. | ||
Lin S, Wu J, Jia H, Hao L, Wang R, Qi J. Facile preparation and antibacterial properties of cationic polymers derived from 2-(dimethylamino)ethyl methacrylate. RSC Adv. 2013;3:20758–20764. | ||
Zhou WY, Guo B, Liu M, Liao R, Rabie ABM, Jia D. Poly(vinyl alcohol)/halloysite nanotubes bionanocomposite films: properties and invitro osteoblasts and fibroblasts response. J Biomed Mater Res. 2010;93:1574–1587. | ||
Panácek A, Kolár M, Vecerová R, et al. Antifungal activity of silver nanoparticles against Candida spp. Biomaterials. 2009;30:6333–6340. | ||
Persano L, Camposeo A, Tekmen C, Pisignano D. Industrial upscaling of electrospinning and applications of polymer nanofibers: a review. Macromol Mater Eng. 2013;298:504–520. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.