Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Fabrication of nanoadjuvant with poly-e-caprolactone (PCL) for developing a single-shot vaccine providing prolonged immunity
Authors Prashant C, Bhat M ![]() , Srivastava S, Saxena A, Kumar M, Singh A, Samim M
, Srivastava S, Saxena A, Kumar M, Singh A, Samim M ![]() , Ahmad FJ, Dinda AK
, Ahmad FJ, Dinda AK ![]()
Received 13 October 2013
Accepted for publication 6 December 2013
Published 12 February 2014 Volume 2014:9(1) Pages 937—950
DOI https://doi.org/10.2147/IJN.S55892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Professor Amit Kumar Dinda
Views: 785
Chandravilas Keshvan Prashant,1 Madhusudan Bhat,1 Sandeep Kumar Srivastava,1 Ankit Saxena,2 Manoj Kumar,3 Amar Singh,2 Mohammed Samim,4 Farhan Jalees Ahmad,5 Amit Kumar Dinda1
1Department of Pathology, 2Department of Transplant Immunology and Immunogenetics, All India Institute of Medical Sciences, 3Centre for Biomedical Engineering, Indian Institute of Technology, 4Department of Chemistry, Faculty of Sciences, 5Department of Pharmaceutics, Faculty of Pharmacy, Jamia Hamdard, New Delhi, India
Purpose: The aim of the study was to load a model antigen, tetanus toxoid (TT), in poly-ε-caprolactone nanoparticles (PCL NPs) of two size ranges, ie, mean 61.2 nm (small) and 467.6 nm (large), and study its effect on macrophage polarization as well as antigen presentation in human monocyte-derived macrophages in vitro, along with humoral and cell-mediated immune (CMI) response generated in Swiss albino mice following immunization with the TT-loaded NPs.
Materials and methods: PCL NPs were synthesized by solvent evaporation. The antigen-loaded PCL NPs were characterized for size, zeta potential, and protein-release kinetics. Swiss albino mice were immunized with the antigen-loaded PCL NPs. Flow cytometry was used to quantify interferon-γ- and interleukin-4-secreting cluster of differentiation (CD)4+ and CD8+ T cells in the spleen, and enzyme-linked immunosorbent assay was used to quantify anti-TT antibody levels in the serum of immunized mice.
Results: Small PCL NPs generated an M1/M2 type polarization of human blood monocyte-derived macrophages and T helper (Th)1/Th2 polarization of autologous CD4+ T cells. Efficient CD8+ T-cell responses were also elicited. Large PCL NPs failed to cause any type of macrophage polarization. They did not elicit efficient CD8+ T-cell responses.
Conclusion: TT-loaded small PCL NPs were able to generate persistent and strong CMI and humoral responses against TT 2 months after single injection in mice without booster dose. This biodegradable nanoadjuvant system may help to develop single-shot immunization for prolonged immunity without booster doses. The capability of enhanced CMI response may have high translational potential for immunization against intracellular infection.
Keywords: tetanus toxoid, antigen cross-presentation, poly-ε-caprolactone nanoparticles, vaccine, adjuvant
Corrigendum for this paper has been published
Introduction
Antigen-presenting cells are comprised of dendritic cells, macrophages, and B lymphocytes, and these are entrusted with the task of presenting both endogenous and exogenous antigens through major histocompatibility complex (MHC) class I and class II cell receptors, respectively, to cluster of differentiation (CD)8+ and CD4+ T lymphocytes. T lymphocytes are subdivided as CD4+ and CD8+ T cells depending on the expression of the coreceptors CD4 or CD8 on their cell membranes. CD4+ T cells are called T-helper (Th) cells, and CD8+ T cells are called cytotoxic T cells (CTLs). Th cells are further divided into Th1 and Th2 cells based on their cytokine-secreting pattern. This polarization of Th cell function was first shown in vitro in 1986 by Mosmann and Coffman.1 Th1 cells secrete interferon (IFN)-γ, promote cell-mediated immunity through CTLs, and activate macrophages, while Th2 cells produce interleukin (IL)-4 and promote humoral immunity through secretion of antibodies from B lymphocytes.2
Macrophages belong to the mononuclear phagocytic system, and function as important antigen-presenting cells. As they are prime targets for a variety of intracellular infections, targeting them with cell-based vaccination strategies using antigens entrapped in nanoparticles (NPs) may provide new tools to control and modulate antigen presentation with enhancement of efficiency of adaptive immune response.
As in the case of Th cells, macrophage response and function were proposed by Mills et al to be polarized, and were hence designated as M1 and M2 types in line with Th1/Th2 nomenclature.3 In general, M1 cells produce high amounts of IL-12 and low amounts of IL-10, efficiently generate reactive oxygen and nitrogen intermediates, and mediate resistance against intracellular infections and tumors, whereas the various forms of M2 macrophages share an IL-12-low, IL-10-high phenotype.4
There are several studies that have shown the efficacy of NPs for vaccine delivery.5–16 These studies have indicated their usefulness in eliciting good humoral, mucosal, and cell-mediated immune response depending on the mode of delivery. Antigens encapsulated in NPs are presented effectively on MHC class II molecules and may get cross-presented more efficiently on MHC class I molecules. The mechanism by which such effective cross-presentation is achieved through the use of NPs is not understood entirely, although recent studies have put forward some explanations for this phenomenon.17
Successful vaccination depends on the use of adjuvants.18 For almost 70 years, alum remained the only adjuvant approved for human use, but it has a major limitation of activating humoral responses with little activation of cell-mediated immunity (CMI). Therefore, a major issue today regarding the development of effective vaccines is the paucity of effective adjuvants that can elicit highly complex immune responses using all pathways of the immune system, especially CMI, for widespread and serious diseases like tuberculosis and human immunodeficiency virus that have escaped effective vaccination.
The aim of many vaccine-development programs has been to generate a strong T-cell response, which requires the presentation of pathogenic peptides on MHC molecules for T-cell stimulation. Therefore, the challenge for an effective vaccine is to induce long-lived central memory CD8+ T cells as well as CD4+ Tcells.19
The past decade has seen important developments in the use of nanotechnology in the field of vaccine adjuvants and carrier vehicles, especially polymeric materials in the nano- and microranges.20–35 These studies hold great promise to elicit complex immune responses from all branches of immunity for effective and safe vaccination against major infections that have escaped effective vaccination.
In the present study, we developed poly-ε-caprolactone (PCL) NPs in two size ranges, ie, (mean) 61.2 nm (small) and 467.6 nm (large) and loaded them with a model antigen – tetanus toxoid (TT). TT is an exogenous antigen that gets presented on MHC class II molecules generally, and humoral responses are sufficient to neutralize tetanus toxin. We aimed to study the effect of the size variation of PCL NPs on human monocyte-derived macrophage (hmoM) on antigen presentation to autologous CD4+ and CD8+ T cells, and study the differential immune responses elicited by the particles in vivo in Swiss albino mice immunized with the preparations. Our aim was to study the size-dependent modulation of immune response toward the entrapped antigen, and whether antigen cross-presentation depends on the NP size, along with efficient generation of Th1 responses. Cross-presentation is the process by which exogenous antigens are presented on MHC class I molecules. This is important to stimulate CD8+ T-cell response, which is essential for antitumor activity and for intracellular infections. Our objective was to use antigen-loaded PCL NPs and generate a strong humoral as well as T-cell response.
Materials and methods
Preparation of polycaprolactone nanoparticles
PCL (Mw 14 KD), Pluronic® F127, acetonitrile, polyvinyl alcohol, and dichloromethane were procured from Sigma-Aldrich (St Louis, MO, USA). PCL NPs were synthesized by double emulsion with Pluronic F127 and polyvinyl alcohol as surfactants for smaller and larger particles, respectively. Briefly, in one system (system A), varied concentrations of surfactant were added to 20 mL of water and stirred for 1 hour. In another system (system B), varied concentrations of PCL were dissolved in solvent (acetonitrile was used for smaller NPs, and dichloromethane for larger NPs) and stirred for 1 hour. TT was a gift from the Serum Institute of India. Pure TT (working concentration ~40 mg/mL) was added to system B under vigorous stirring with 5 seconds of ultrasonication. Immediately, polymer loaded with protein (system B) was added to system A using a 22-gauge syringe at a slow rate. The system was left under constant stirring overnight until the solvent evaporated. Void polylactic acid (PLA) NPs of a similar size range were prepared as above for comparison with PCL NPs to study their effects on macrophage polarization.
Characterization of PCL NPs
The size and morphology of the NPs were determined by transmission electron microscopy. One drop of the aqueous dispersion of PCL NPs was put on a Formvar-coated copper grid and then air-dried, followed by one drop of 1% phosphotungstic acid. The dried grid was examined under a Philips Morgagni 268 electron microscope (5600 MD; FEI, Hillsboro, OR, USA). The Malvern Zetasizer ZS-90 (Malvern Instruments, Malvern, UK) was used to determine the size distribution and zeta potential of PCL NPs at 25°C.
Cell culture
The human monocytic cell line THP1 was obtained from the National Centre For Cell Science, Pune, India, and used for PCL NP uptake and degradation studies, viability assay, and detection of reactive oxygen species (ROS) generation.
Cells were subcultured and maintained at 37°C in Roswell Park Memorial Institute (RPMI) medium (Sigma-Aldrich) under standard conditions. The medium was supplemented with 10% fetal calf serum (HyClone; Thermo Fisher Scientific, Waltham, MA, USA), and antibiotic containing 50 U/mL penicillin and 50 mg/mL streptomycin and actinomycin (Sigma-Aldrich).
MTT assay for cell viability
THP1 cells were seeded at 1×103 cells per well and grown in 96-well plates until subconfluent. A sterile aqueous solution of PCL NPs dissolved in phosphate-buffered saline (PBS; pH 7.2) was then added to the cells at a concentration of 500 μg/mL and incubated for 3, 6, 9, 12, 24, and 48 hours. After incubation, the medium was discarded and 90 μL fresh medium was added per well to the cells after thorough washing three times with sterile PBS (pH 7.2). MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; 10 μL) reagent (5 mg/mL stock) (Sigma-Aldrich) was then added to each well, and the plate was incubated for 6 hours in an incubator. After incubation, the medium was discarded from the wells, and dimethyl sulfoxide 100 μL was added to solubilize the formazan crystals formed. Readings were then taken in an enzyme-linked immunosorbent assay (ELISA) reader (Model 680 microplate reader, BIORAD, Hercules, California, USA) at 490 nm, with subtraction for plate absorbance at 650 nm. The percentage viability of the cells was calculated as the ratio of mean absorbance of triplicate readings of sample (Isample) with respect to mean absorbance of control wells (Icontrol):
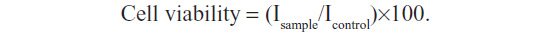
Assay for ROS generation
THP1 cells (2×104 cells per well) were grown on coverslips in six-well plates until subconfluent. Intracellular ROS was measured using a peroxide-sensitive fluorescent probe: carboxy-2′,7′-dichlorofluorescein diacetate (H2DCFDDA; Life Technologies, Carlsbad, CA, USA). The cells were loaded with 20 μM H2DCFDDA for 60 minutes at 37°C. After being loaded, the cells were thoroughly washed and fresh medium was added. The cells were incubated with the PCL NPs at 400 μg/mL and 500 μg/mL under standard conditions. After the defined time points, the cells were washed with sterile PBS and mounted with glycerol PBS (50% glycerol). The cells were observed under a fluorescent microscope (Eclipse 600; Nikon, Tokyo, Japan) with a green filter, and the images were captured with a DP-71 digital camera (Olympus, Tokyo, Japan). Cells incubated without PCL NPs were used as a negative control, and 100 μM H2O2 was used as a positive control.
Protein-release kinetics
Ten milligrams of TT-loaded PCL NPs (60 nm or 450 nm) was dissolved in 1 ml of either PBS (pH 7.2), PBS (pH 4.0), or double-distilled water (pH 6.0) and kept at 37°C for 7, 14, 21, 28, and 30 days. After the indicated time points, the Microfuge® (Beckman Coulter, Pasadena, CA, USA) tubes were centrifuged at 15,000 rpm, and the supernatants were assayed for protein content using a Bradford assay (Amresco, Solon, OH, USA). Values were obtained from the standard curve obtained with bovine serum albumin (BSA) as reference protein. Void PCL NPs were used as negative controls.
Loading efficiency
Ten milligrams of lyophilized PCL NPs was dissolved in a 1:1 ratio of dichloromethane:water. The sample was vortexed for 2 minutes and centrifuged at 8,000 rpm for 10 minutes. The supernatant was collected, and 10 μL of it was added to 90 μL of NaCl (0.15N) solution. The solution was brought up to 1 mL by adding 900 μL of Bradford solution. Optical density was read at 595 nm using a spectrophotometer (Shimadzu, Kyoto, Japan). Values were obtained from standard curves obtained with BSA as reference protein. Loading efficiency was calculated as:

where TTtotal was the protein added during synthesis of the PCL NPs, and TTsupernatant was the protein content in the supernatant.
Antigen-presentation assays
Ten milliliters of blood was drawn from healthy human volunteers, and mononuclear cells were separated over a Ficoll-Hypaque gradient (Sigma-Aldrich). The mononuclear cells were kept in petri dishes for 2 hours in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal calf serum (HyClone) and antibiotic (Sigma-Aldrich) containing 50 U/mL penicillin and 50 mg/mL streptomycin and actinomycin. After 2 hours, the medium was decanted to remove nonadherent cells, and fresh medium containing 100 μg/mL macrophage colony stimulating factor (M-CSF) was added. Cells were allowed to grow for 7 days in a 5% CO2 atmosphere, with a change in medium at day 4 with addition of M-CSF.
For isolation of autologous T cells, mononuclear cells were similarly isolated from the same individuals. CD4+ and CD8+ T cells were isolated by negative sorting using magnetic sorting as per the manufacturer’s instructions (Life Technologies).
For antigen-presentation assays, 2×103 hmoM per well was plated in 96-well plates in complete DMEM and allowed to adhere for 2 hours. TT-loaded PCL NPs were added at an antigen concentration of 10 μg/mL. Control wells had pure TT or alum-adjuvanted TT (Central Research Institute, Kasauli, India) at the same concentrations as above. The cells were then incubated for 24 hours in a CO2 incubator for antigen presentation to occur. Wells were then washed with PBS (pH 7.2) three times and once with serum-free DMEM. Fifty microliters of 1% (w/v) paraformaldehyde in PBS was then added for 15 minutes to fix the macrophages. Paraformaldehyde was then removed, and 50 μL of 0.1 M lysine was added for 15 minutes to quench the fixation reaction. The cells were then washed three times with complete DMEM and 100 μL of medium was then added. A total of 4×105/100 μL CD4+ or CD8+ autologous T cells obtained through magnetic sorting were then added to the macrophages and incubated for 72 hours at 37°C and 5% CO2 atmosphere. Culture supernatants were then collected for ELISA of IL-4, IL-10, IFN-γ and IL-12 for CD4+ T-cell response, while IL-2 was assayed for CD8+ T-cell response.
For studying the effect of the NPs alone on macrophage polarization, void PCL NPs or PLA NPs were added at 200 μg/mL to hmoM and incubated for 48 hours in a CO2 incubator. ELISA was performed with the culture supernatant for IL-10 and IL-12 response.
Enzyme-linked immunosorbent assay
ELISA was done for estimating the total immunoglobulin G titers of antibodies against TT in the serum of immunized animals. The microtiter plate provided in the ELISA kit (Cusabio, Wuhan, People’s Republic of China) had been precoated with specific antigen, ie, TT. Any antibodies specific for the antigen present in the serum of immunized mice will bind to the precoated antigen. Following a wash to remove any unbound reagent, a substrate solution was added to the wells, and color developed in proportion to the amount of mouse tetanus antibody bound in the initial step. The color development was stopped, and the intensity of the color was measured. Values were obtained as milli-international units per milliliter (mIU/mL) with reference to a standard curve.
ELISA was also used to measure the IFN-γ and IL-4 levels in the supernatant of cultured splenocytes. ELISA was done as per manufacturer’s instructions (PeproTech, Rocky Hill, NJ, USA). Experiments were done in triplicate, and an average of six different experiments were taken for analysis.
Animals
The animal study was approved by the Institute Animal Ethics Committee at the All India Institute of Medical Sciences, New Delhi. Female Swiss albino mice were taken for immunization. The mice were 6–8 weeks old and weighed 25–30 g. Three routes of immunization with TT-entrapped PCL NPs, ie, intramuscular (IM), subcutaneous (SC), and intravenous (IV), were taken. Alum-adjuvanted TT and pure TT antigen served as positive controls, the doses being administered through the conventional IM route. PBS served as negative control. A dosage of 10 μg/mL of TT antigen was taken for positive controls as well as for the TT-entrapped PCL NPs. No booster doses were given postimmunization with the first dose. Four mice per group of the immunization protocol were killed at the end of 2 months, blood was collected through cardiac puncture, and spleens were isolated and put in chilled PBS till isolation of splenocytes.
Isolation of splenocytes
Splenocytes were isolated from freshly removed spleens, as previously described.36 The spleens were cut into small pieces and teased apart. The pieces were then passed through a sterile 40 μm nylon filter mesh (Falcon™ cell strainer; BD Biosciences, San Jose, CA, USA), and splenocytes were collected in complete RPMI medium. Red blood cell lysis buffer was used to lyse the red blood cells.
Flow cytometry
Surface and intracellular staining and in vitro culture of cells
Isolated splenocytes were cultured in a 12-well plate (2×106 cells/well) and restimulated with TT-loaded PCL NPs, alum TT, or pure TT antigen, all at 10 μg/mL doses, for 72 hours. PBS served as negative control while phorbol myristate acetate (5 ng/mL)-stimulated cells served as positive control. The Golgi transport inhibitor, monensin was added to all wells at 1 μM concentration 6 hours prior to the termination of the culture. Intracellular staining was done as described earlier37 to assess IFN-γ and IL-4 production in CD4+ and CD8+ cells in the splenocytes of immunized mice. A total of 0.5×106 cells were taken in 200 μL medium for staining in a 96-well plate. Isolated cells were incubated directly with fluorescence-labeled monoclonal antibodies in staining buffer (PBS + BSA + azide) for 15 minutes on ice for surface staining of CD4+ and CD8+ cells. After being washed, fixated, and rewashed, for the staining of intracellular antigens (IL-4 and IFN-γ), cells were permeabilized by using permeabilization buffer (0.3% saponin) after fixation for 30 minutes at room temperature with 4% neutral buffered formalin. After being washed, cells were transferred in a fluorescence-activated cell-sorting (FACS) tube (Falcon; BD Biosciences) for data acquisition by a three/four-color flow cytometer (FACSCalibur; BD Biosciences). A total of 1×106 cells were acquired per sample.
PCL NP drainage to lymph nodes
Rhodamine-B labeled small PCL NP were injected intramuscularly in the thigh of mice. After 3 and 7 days, inguinal lymph nodes were isolated and put in chilled PBS. Frozen sections were immediately cut with a cryotome and observed under a fluorescence microscope (Eclipse 600).
Immunofluorescence staining
Isolated inguinal lymph nodes from immunized mice were formalin-fixed and paraffin-embedded, and 2–5 μm sections were cut. The sections were dual stained with polyclonal rabbit primary antibodies against CD8, IFN-γ, or IL-4 (Biorbyt, Cambridge, UK). Biotinylated secondary antibody (Vectastain® Universal Quick Kit; Vector Labs, Burlingame, CA, USA) raised in horse against the primary antibodies was used after serum blocking. Avidin-conjugated fluorescein and avidin-conjugated phycoerythrin were used to detect the biotinylated secondary antibody and visualize CD8 and IFN-γ or IL-4, respectively.
Statistical analysis
Statistical analysis was done using Prism version 5.04 for Windows, (GraphPad Software, La Jolla, CA, USA). A two-tailed unpaired t-test was used with Welch’s correction for comparing cytokine levels. P<0.05 was considered statistically significant.
Results
Characterization of PCL NPs
PCL NPs were successfully synthesized in the two size ranges (mean ± standard deviation [SD]) of 61.2±20 nm (small) and 467.6±120 nm (large), and sizes were determined using transmission electron microscopy (Figure 1A and B) and dynamic light scattering (Figure 1E and F). The zeta potential of 60 nm PCL NPs was −3±3.2 mV, and that of 450 nm PCL NPs was −14±4.8 mV. They were entrapped with TT antigen. For a comparative study, void PLA NPs were synthesized in the same size range, with average size (mean ± SD) of 48±32 nm and 485±127 nm (Figure 1C and D). The zeta potential of 50 nm PLA NPs was −20.7±8.84 mV, and that of 480 nm PLA NPs was −16.7±6.66 mV.
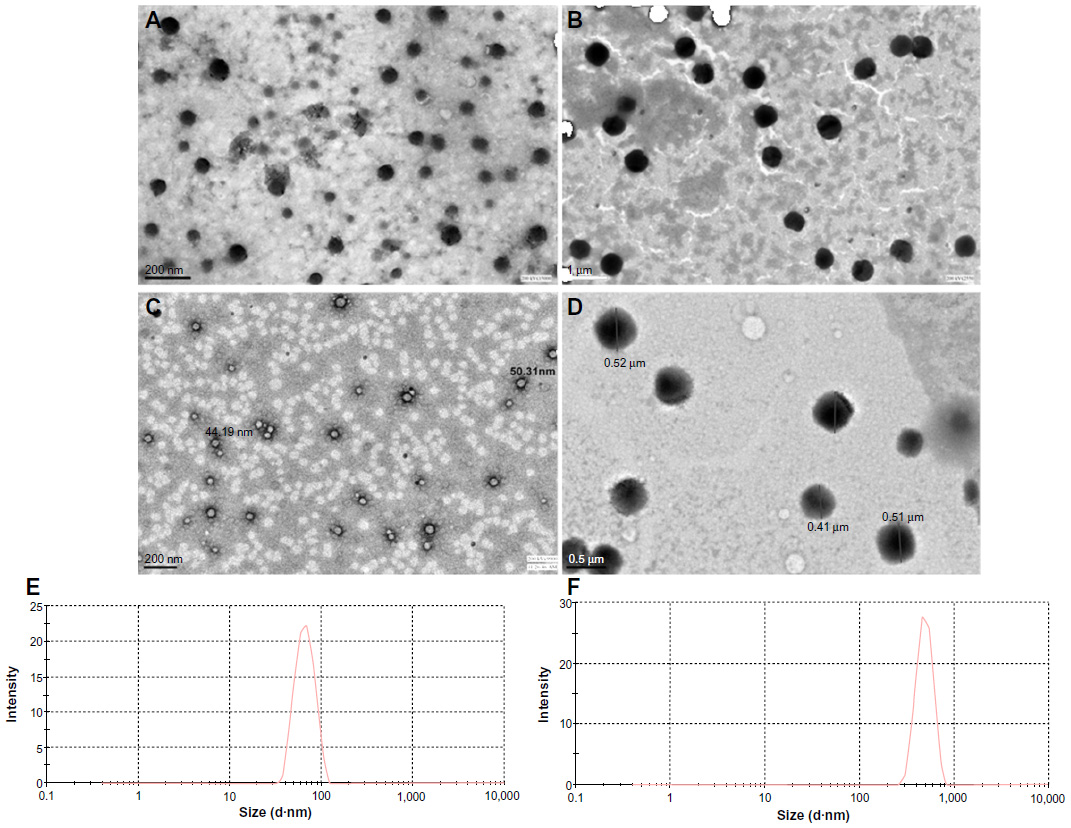 | Figure 1 Transmission electron microscopy images of poly-ε-caprolactone nanoparticles (PCL NPs). Small (A), large (B), Polylactic acid NP 50 nm (C) and 500 nm (D) NPs. Dynamic light-scattering data of small PCL NPs (E) and large PCL NPs (F). |
Loading efficiency, degradation, and protein-release kinetics
Small PCL NPs had an antigen-loading efficiency of 61.66%±16.96%, while large PCL NPs had an average loading efficiency of 49.5%±5.45%. Degradation kinetics of PCL NPs was studied with scanning electron microscopy, and is indicated in Figure S1. Scanning electron microscopy images clearly show that small PCL NPs retained their spherical morphology with some swelling without appreciable degradation even at 30 days, while large PCL NPs lost their spherical morphology with degradation after 30 days of incubation. Protein-release kinetics from the particles is shown in Figure S2. Small PCL NPs showed slow sustained-release profiles, whereas large NPs showed a burst release of almost 50% on day 1.
MTT assay and ROS assay
A cell-viability assay done on the THP1 cell line with MTT showed that the synthesized PCL NPs of both sizes were nontoxic at the dose tested (Figure 2A), with viability of 99.34%±0.66% for small and 99.55%±0.15% for large PCL NPs. There was no significant ROS generation, as determined by H2DCFDDA assay (Figure 2B and C). Figure S3 shows THP1 cells with internalized rhodamine-labeled small PCL NPs at 6 hours of incubation.
 | Figure 2 (A) Cell viability measured using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay on THP1 cells with small and large poly-ε-caprolactone nanoparticles (PCL NPs). (B) Study of intracellular reactive oxygen species (ROS) generation in THP1 cells incubated with small PCL NPs showed no detectable green fluorescence of the carboxy-2′,7′-dichlorofluorescein diacetate dye, indicating no ROS generation. (C) Positive control. |
Antigen-presentation assay
The results of in vitro antigen-presentation assays using hmoM indicated that small PCL NPs caused M1/M2 polarization of the macrophages and generated a Th1/Th2-type immune response. Large PCL NPs, on the other hand, failed to generate any kind of response. This is clearly indicated by the ELISA results. Figure 3 shows the comparison of IL-12 and IL-10 cytokine production, by macrophages incubated with either small or large PCL NPs. When void small PCL NPs were incubated with hmoM for 72 hours, we found significantly higher concentrations (P<0.0001) of IL-12 and IL-10 (P=0.0007) in comparison to macrophages incubated with large PCL NPs. No such polarization of macrophage function was observed when PLA NPs of a similar size range were incubated with the macrophages.
 | Figure 3 (A and B) Polarization of macrophages after internalizing void small poly-ε-caprolactone nanoparticles (PCL NPs). Significant increase in secretion of interleukin (IL)-12 (A) and IL-10 (B) by human monocyte-derived macrophage (hmoM). Thus, void small PCL NPs generate both M1 and M2 responses in hmoM, whereas such response is not observed with void large PCL NPs or large and small polylactic acid (PLA) NPs. |
When TT-entrapped small PCL NPs were used in the antigen-presentation assays, CD4+ T cells produced IFN-γ in significantly higher amounts compared to the positive control, alum TT, while IL-4 and IL-10 secretion was significantly less than the positive control (Figure 4A–C). When large TT-entrapped PCL NPs were used, CD4+ T cells did not produce significant amounts of IFN-γ or IL-4. CD8+ T cells produced significant amounts of IL-2 when incubated with hmoM that had internalized TT-loaded small PCL NPs (Figure 4D). There was no detectable IL-2 production by CD8+ T cells when incubated with hmoM that had internalized TT-loaded large PCL NPs.
 | Figure 4 Enzyme-linked immunosorbent assay for measuring cytokine production from CD4+ T cells cocultured with nanoadjuvant-primed human monocyte-derived macrophage (hmoM): (A) interferon (IFN)-γ, (B) interleukin (IL)-4, (C) IL-10. Increased IL-2 production by CD8+ T cells was observed when cocultured with nanoadjuvant primed hmoM (D). |
Flow cytometry and ELISA
The flow-cytometry data of isolated splenocytes indicated that antigen-loaded small NPs delivered through the IM and SC routes elicited robust IFN-γ response in CD4+ T cells (2.655%±0.225%, P=0.0179; 2.205%±0.165%, respectively (percentage of positive cells expressed as mean ± standard error of mean), as a recall-memory response against the immunized antigen, TT (Figure 5A). The IV route did not elicit significant IFN-γ secretion in CD4+ cells. IL-4 production in CD4+ cells was significant in all three routes of immunization (Figure 5B). CD8+ cells did not produce significant IFN-γ or IL-4 compared to the positive control alum TT through any of the immunization routes, but cytokine secretion was significant compared to the negative control (Figure 6). TT-loaded large PCL NPs did not elicit significant cytokine responses in either CD4+ or CD8+ T cells. ELISA for detecting IFN-γ and IL-4 levels in the supernatant of the cultured splenocytes revealed that IFN-γ and IL-4 were produced in significant amounts in cases where immunization with small PCL NPs was done through all the three routes (Figure 7). Thus, the cytokine milieu in the spleen is overall a Th1/Th2 type promoting both CMI and humoral responses if immunized with TT-loaded small PCL NPs.
 | Figure 5 Flow-cytometry data of percentage of (A) interferon (IFN)-γ-positive CD4+ T cells and (B) interleukin (IL)-4-positive CD4+ T cells in spleens of immunized mice. Gated on total lymphocytes. |
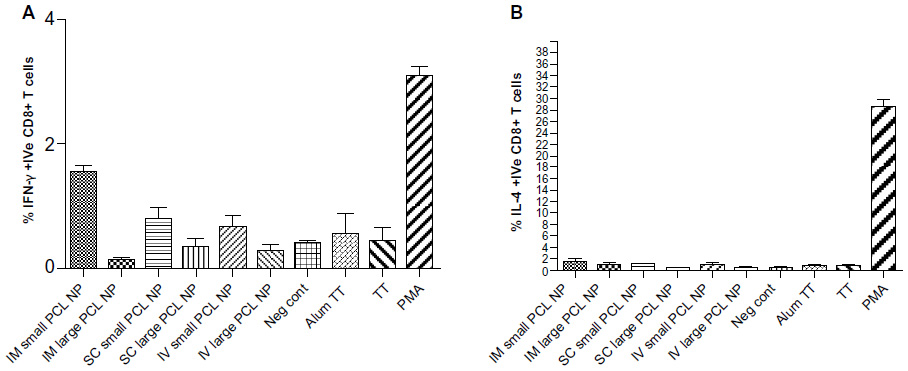 | Figure 6 Flow-cytometry data of percentage of (A) interferon (IFN)-γ-positive CD8+ T cells and (B) interleukin (IL)-4-positive CD8+ T cells in spleens of immunized mice. Gated on total lymphocytes. There was no significant difference between groups immunized with alum tetanus toxoid (TT) and those immunized with TT-loaded small poly-ε-caprolactone nanoparticles (PCL NPs). |
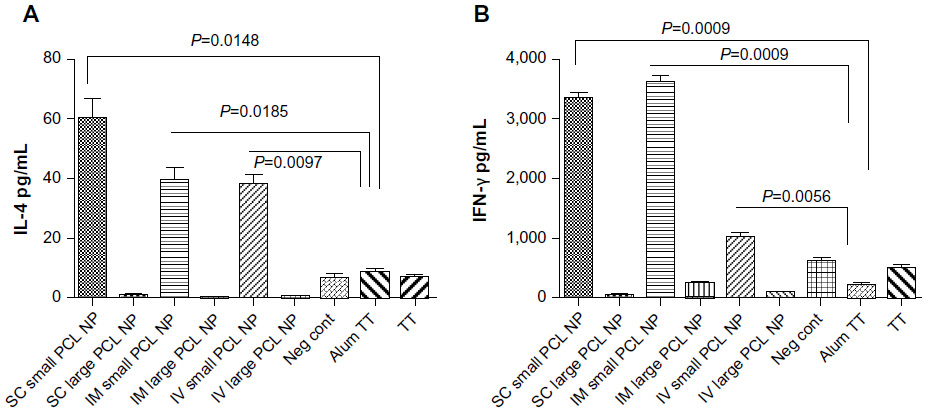 | Figure 7 (A and B) Interferon (IFN)-γ and interleukin (IL)-4 levels in supernatant of cultured splenocytes. Significant secretion of IL-4 (A) and IFN-γ (B) is observed from total splenocytes from mice immunized with small poly-ε-caprolactone nanoparticles (PCL NPs) through all three routes. |
ELISA for measuring anti-TT antibody titers 2 months after single injection in mice indicated that only small PCL NPs elicited a persistent and prolonged humoral response (Figure 8).These NPs delivered through the SC route elicited 96±5.2 mIU/mL immunoglobulin G titer, while the IM route elicited 74±11.3 mIU/mL anti-TT antibody titer. However, the IV route of administration of small PCL NPs resulted in the lowest antibody titer – 20±3.1 mIU/mL.
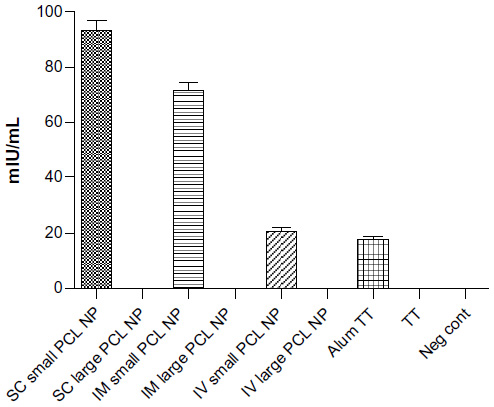 | Figure 8 Anti-tetanus toxoid antibody titers in serum of immunized mice 2 months after single injection without booster dose. Only mice immunized with TT-loaded small poly-ε-caprolactone nanoparticles (PCL NPs) show high antibody titer. |
PCL NP drainage to lymph nodes
Rhodamine B-labeled small PCL NPs injected IM in the thighs of mice were found to drain into the inguinal lymph nodes after 3 and 7 days of injection (Figure 9). Immunofluorescence staining done on lymph-node sections isolated from mice immunized with TT-loaded non-rhodamine B-labeled small PCL NPs showed that CD8+ T cells produced significantly more IFN-γ and IL-4 after 2 months of immunization compared to the negative control (Figure 10).
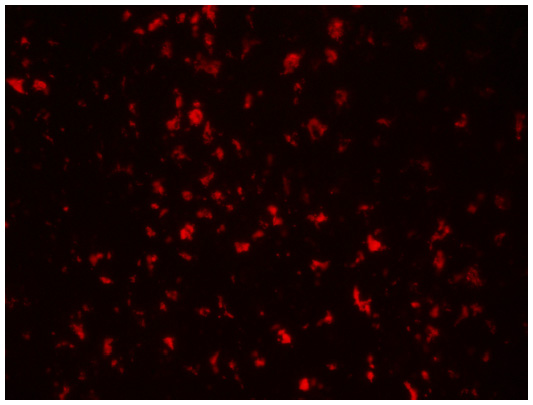 | Figure 9 Rhodamine B-labeled small poly-ε-caprolactone nanoparticles draining into inguinal lymph nodes after 7 days of intramuscular injection in thighs of mice. Magnification 200×. |
 | Figure 10 Immunofluorescence images of inguinal lymph nodes of (A) interferon (IFN)-γ-positive CD8+ T cells in mice immunized with small tetanus toxoid-loaded poly-ε-caprolactone nanoparticles and (B) interleukin (IL)-4-positive CD8+ T cells. (C and D) Negative controls. CD8 is tagged with fluorescein (green), IFN-γ and IL-4 with phycoerythrin (red). Magnification 200×. |
Discussion
In the present study, we used the US Food and Drug administration-approved biocompatible polymer PCL to develop NPs with an average diameter of 60 nm and loaded a model antigen, TT, in them. PCL degrades at much slower rates than other popular polymers like PLA/poly(lactic-co-glycolic) acid, making it ideal for slow and prolonged release of antigens. Microspheres were not preferred, as PCL microspheres are known to cause neutrophil activation and localized inflammatory responses.38
The study revealed that void PCL NPs by themselves can skew the response of macrophages. Small void PCL NPs cause significant secretion of IL-12 and IL-10 when macrophages internalize them. The small PCL NPs initially caused an M1/M2-type polarization of the macrophages. Large void PCL NPs do not cause polarization of the macrophages. PLA NPs of similar size ranges as PCL NPs did not cause macrophage polarization when incubated with them. It is likely that this initial skewing of macrophage response by small PCL NPs enhances T-cell response for efficient protective immunity.
We studied the effect of antigen-loaded PCL NPs on hmoM in vitro along with in vivo study of antibody-mediated immunity AMI and CMI response in Swiss albino mice immunized through different routes. Following a single dose of IM or SC immunization, the mice produced robust AMI and CMI against TT for more than 2 months. A single IM injection of commercially available alum-based TT vaccine failed to produce significantly detectable AMI and CMI after 2 months.
The fact that the NPs injected IM are able to reach the local lymph nodes means that antigen-loaded small PCL NPs have access to antigen-presenting cells in the local lymph nodes, resulting in efficient direct delivery of antigen to T and B cells. Immunofluorescence staining in lymph nodes revealed that mice immunized with TT-loaded PCL NPs showed higher numbers of CD8+ cells that were positive for IFN-γ and IL-4 compared to the negative control 2 months postimmunization.
Yue et al showed that chitosan NPs elicit differential cytokine responses with the J774 A.1 murine macrophage cell line.39 They reported that nanosized chitosan NPs of about 430 nm diameter promoted the secretion of Th1-specific cytokines, while NPs of diameters 1.9 μm and 4.8 μm produced Th2-specific cytokines. Lucarelli et al showed that SiO2 nanoparticles, when incubated with phorbol myristate acetate, differentiated human macrophages obtained from the myelomonocytic cell line U937 caused strong M1 polarization of the macrophages, as indicated by cytokine secretion. Other ceramic NPs had little or no effect. They observed that the inflammatory and defense capability of macrophages can be modulated differently by different types of NPs.40
There is much controversy as to the size of NPs used and subsequent immune responses generated when antigens are entrapped in them, with literature citing both small and big NPs to be as efficient at cross-presentation.41 Xiang et al42 reported that 20–200 nm NPs are taken up by endocytosis and are more likely to elicit cellular-based immune response, whereas NPs with size of 500 nm to 5 μm are taken up by phagocytosis/macropinocytosis and are more likely to promote humoral immune response. However, the nature of material used is the most important criteria for the eventual immune responses elicited by NPs, as this would determine whether macrophages get activated or not after internalizing the material.
The results of the in vivo experiments indicated that not only is there a size-dependent immunomodulation toward the antigen of interest but also that these responses vary significantly with the route of administration of the antigen-entrapped PCL NPs. Route of administration is known to affect the quality of immune response generated with particulate vaccines.43 The rate of release of antigen is known to affect the efficacy of the vaccine, with slow release of antigen giving better vaccines compared to fast release of antigens. This may have to do with the persistence of the antigen for a longer time in circulation. Consequently, efficient CTL responses are generated by particulate antigen-delivery systems compared to the loading of soluble antigens.44
The study conclusively shows the potential of PCL NPs in the 60 nm range to effectively induce prolonged protective immunity from both humoral and cell-mediated branches of immunity, which is difficult to achieve with traditional adjuvants like aluminum hydroxide and aluminum phosphate.
This preclinical proof-of-concept study in a mouse model shows the potential of a PCL-based nanoadjuvant that can be loaded with antigen for developing single-shot booster-free vaccine.
Conclusion
The present study indicates the importance of size and composition of NPs in the modulation of immune responses in the context of polarization of macrophages and subsequent T-cell responses. The study shows that void small PCL NPs themselves are capable of macrophage polarization.
There is an NP size-dependent immunomodulation of antigenic responses toward TT, which also depends on the route of immunization. Small PCL NPs entrapped with TT are able to generate long-lasting humoral and CMI responses toward TT when mice are immunized through IM and SC routes by single injection. PCL NPs are slow to degrade and biocompatible, which can be useful for prolonged efficient antigen delivery. Thus, this may have good translational potential for the development of vaccines with less or no requirement for booster dose.
Acknowledgments
We acknowledge the Indian Council of Medical Research for financially supporting a research fellow for the work. We thank the Electron Microscopy Facility at the All India Institute of Medical Sciences. We acknowledge the Department of Biotechnology, Government Of India (DBT) and the All India Institute of Medical Sciences for financial help to file an Indian patent (application #1862/DEL/2013).
Disclosure
The authors report no conflicts of interest in this work.
References
Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. | |
Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557–1569. | |
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. | |
Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005:23(4):344–346. | |
Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc Natl Acad Sci U S A. 2012;109(4):1080–1085. | |
Li N, Peng LH, Chen X, Nakagawa S, Gao JQ. Effective transcutaneous immunization by antigen-loaded flexible liposome in vivo. Int J Nanomedicine. 2011;6:3241–3250. | |
Yue H, Wei W, Fan B, et al. The orchestration of cellular and humoral responses is facilitated by divergent intracellular antigen trafficking in nanoparticle-based therapeutic vaccine. Pharmacol Res. 2012;65(2):189–197. | |
Gupta NK, Tomar P, Sharma V, Dixit VK. Development and characterization of chitosan coated poly-(ε-caprolactone) nanoparticulate system for effective immunization against influenza. Vaccine. 2011;29(48):9026–9037. | |
Jewell CM, López SC, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2011;108(38):15745–15750. | |
Petersen LK, Ramer-Tait AE, Broderick SR, et al. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32(28):6815–6822. | |
Uto T, Akagi T, Yoshinaga K, Toyama M, Akashi M, Baba M. The induction of innate and adaptive immunity by biodegradable poly(γ-glutamic acid) nanoparticles via a TLR4 and MyD88 signaling pathway. Biomaterials. 2011;32(22):5206–5212. | |
Han R, Zhu J, Yang X, Xu H. Surface modification of poly(D,L-lactic-co-glycolic acid) nanoparticles with protamine enhanced cross-presentation of encapsulated ovalbumin by bone marrow-derived dendritic cells. J Biomed Mater Res A. 2011;96(1):142–149. | |
Da Costa Martins R, Gamazo C, Sánchez-Martínez M, Barberán M, Peñuelas I, Irache JM. Conjunctival vaccination against Brucella ovis in mice with mannosylated nanoparticles. J Control Release. 2012;162(3):553–560. | |
Wang T, Jiang H, Zhao Q, Wang S, Zou M, Cheng G. Enhanced mucosal and systemic immune responses obtained by porous silica nanoparticles used as an oral vaccine adjuvant: effect of silica architecture on immunological properties. Int J Pharm. 2012;436(1–2):351–358. | |
Klippstein R, Pozo D. Nanotechnology-based manipulation of dendritic cells for enhanced immunotherapy strategies. Nanomedicine. 2010;6(4):523–529. | |
Caputo A, Castaldello A, Brocca-Cofano E, et al. Induction of humoral and enhanced cellular immune responses by novel core-shell nanosphere- and microsphere-based vaccine formulations following systemic and mucosal administration. Vaccine. 2009;27(27):3605–3615. | |
Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20(1):89–95. | |
Stills HF Jr. Adjuvants and antibody production: dispelling the myths associated with Freund’s complete and other adjuvants. ILAR J. 2005;46(3):280–293. | |
Berzofsky JA, Ahlers JD, Janik J, et al. Progress on new vaccine strategies against chronic viral infections. J Clin Invest. 2004;114(4):450–462. | |
Ma W, Chen M, Kaushal S, et al. PLGA nanoparticle-mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine. 2012;7:1475–1487. | |
Keijzer C, Slütter B, van der Zee R, Jiskoot W, van Eden W, Broere F. PLGA, PLGA-TMC and TMC-TPP nanoparticles differentially modulate the outcome of nasal vaccination by inducing tolerance or enhancing humoral immunity. PLoS One. 2011;6(11):e26684. | |
Sneh-Edri H, Likhtenshtein D, Stepensky D. Intracellular targeting of PLGA nanoparticles encapsulating antigenic peptide to the endoplasmic reticulum of dendritic cells and its effect on antigen cross-presentation in vitro. Mol Pharm. 2011;8(4):1266–1275. | |
Chen J, Li Z, Huang H, et al. Improved antigen cross-presentation by polyethyleneimine-based nanoparticles. Int J Nanomedicine. 2011;6:77–84. | |
Rice-Ficht AC, Arenas-Gamboa AM, Kahl-McDonagh MM, Ficht TA. Polymeric particles in vaccine delivery. Curr Opin Microbiol. 2010;13(1):106–112. | |
Gutierro I, Hernández RM, Igartua M, Gascón AR, Pedraz JL. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine. 2002;21(1–2):67–77. | |
Cohen JA, Beaudette TT, Tseng WW, et al. T-cell activation by antigen-loaded pH-sensitive hydrogel particles in vivo: the effect of particle size. Bioconjug Chem. 2009;20(1):111–119. | |
Cruz LJ, Tacken PJ, Fokkink R, et al. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release. 2010;144(2):118–126. | |
Schliehe C, Redaelli C, Engelhardt S, et al. CD8– dendritic cells and macrophages cross-present poly(D,L-lactate-co-glycolate) acid microsphere-encapsulated antigen in vivo. J Immunol. 2011;187(5):2112–2121. | |
Wilson JT, Keller S, Manganiello MJ, et al. pH-responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7(5):3912–3925. | |
Patterson DP, Rynda-Apple A, Harmsen AL, Harmsen AG, Douglas T. Biomimetic antigenic nanoparticles elicit controlled protective immune response to influenza. ACS Nano. 2013;7(4):3036–3044. | |
Gamvrellis A, Gloster S, Jefferies M, et al. Characterisation of local immune responses induced by a novel nano-particle based carrier-adjuvant in sheep. Vet Immunol Immunopathol. 2013;155(1–2):21–29. | |
Lugade AA, Bharali DJ, Pradhan V, Elkin G, Mousa SA, Thanavala Y. Single low-dose un-adjuvanted HBsAg nanoparticle vaccine elicits robust, durable immunity. Nanomedicine. 2013;9(7):923–934. | |
Mahony D, Cavallaro AS, Stahr F, Mahony TJ, Qiao SZ, Mitter N. Mesoporous silica nanoparticles act as a self-adjuvant for ovalbumin model antigen in mice. Small. 2013;9(18):3138–3146. | |
Manish M, Rahi A, Kaur M, Bhatnagar R, Singh S. A single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challenge. PLoS One. 2013;8(4):e61885. | |
Li N, Peng LH, Chen X, et al. Antigen-loaded nanocarriers enhance the migration of stimulated Langerhans cells to draining lymph nodes and induce effective transcutaneous immunization. Nanomedicine. Epub June 7, 2013. | |
Herrera OB, Golshayan D, Tibbott R, et al. A novel pathway of alloantigen presentation by dendritic cells. J Immunol. 2004;173(8):4828–4837. | |
Rai AK, Thakur CP, Singh A, et al. Regulatory T cells suppress T cell activation at the pathologic site of human visceral leishmaniasis. PLoS One. 2012;7(2):e31551. | |
Woodruff MA, Hutmacher DW. The return of a forgotten polymer: polycaprolactone in the 21st century. Prog Polym Sci. 2010;35(10):1217–1256. | |
Yue H, Wei W, Yue Z, et al. Particle size affects the cellular response in macrophages. Eur J Pharm Sci. 2010;41(5):650–657. | |
Lucarelli M, Gatti AM, Savarino G, et al. Innate defence functions of macrophages can be biased by nano-sized ceramic and metallic particles. Eur Cytokine Netw. 2004;15(4):339–346. | |
Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vaccines. 2010;9(9):1095–1107. | |
Xiang SD, Scholzen A, Minigo G, et al. Pathogen recognition and development of particulate vaccines: does size matter? Methods. 2006:40(1):1 –9. | |
Mohanan D, Slütter B, Henriksen-Lacey M, et al. Administration routes affect the quality of immune responses: a cross-sectional evaluation of particulate antigen-delivery systems. J Control Release. 2010;147(3):342–349. | |
Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. |
Supplementary materials
 | Figure S1 (A and B) Scanning electron microscopy images of poly-ε-caprolactone nanoparticles (PCL NPs), showing degradation kinetics after 30 days in phosphate-buffered saline (pH 7.2). (A) Small PCL NPs; (B) large PCL NPs. |
 | Figure S2 Protein-release kinetics of (A) small poly-ε-caprolactone nanoparticles (PCL NPs) and (B) large PCL NPs (pH 7.2). |
 | Figure S3 Confocal image of THP1 cells showing rhodamine-labeled small poly-ε-caprolactone nanoparticles internalized by the cells after 6 hours of incubation. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.