Back to Journals » International Journal of Nanomedicine » Volume 13
Fabrication of a triptolide-loaded and poly-γ-glutamic acid-based amphiphilic nanoparticle for the treatment of rheumatoid arthritis
Authors Zhang L ![]() , Chang J, Zhao Y, Xu H, Wang T, Li Q, Xing L, Huang J, Wang Y, Liang Q
, Chang J, Zhao Y, Xu H, Wang T, Li Q, Xing L, Huang J, Wang Y, Liang Q
Received 8 September 2017
Accepted for publication 8 December 2017
Published 4 April 2018 Volume 2018:13 Pages 2051—2064
DOI https://doi.org/10.2147/IJN.S151233
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Li Zhang,1–3 Junli Chang,1–3 Yongjian Zhao,1–3 Hao Xu,1–3 Tengteng Wang,1–3 Qiang Li,1–3 Lianping Xing,4 Jing Huang,5 Yongjun Wang,1–3,6 Qianqian Liang1–3
1Department of Orthopaedics, Longhua Hospital, 2Institute of Spine, 3Key Laboratory of Theory and Therapy of Muscles and Bones, Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 4Department of Pathology and Laboratory Medicine, University of Rochester Medical Center, Rochester, NY, USA; 5School of Life Science, East China Normal University, 6School of Rehabilitation Science, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
Abstract: Triptolide (TP) exhibits immunosuppressive, cartilage-protective and anti-inflammatory effects in rheumatoid arthritis. However, the toxicity of TP limits its widespread use. To decrease the toxic effects, we developed a novel nano-drug carrier system containing TP using poly-γ-glutamic acid-grafted di-tert-butyl L-aspartate hydrochloride (PAT). PAT had an average diameter of 79±18 nm, a narrow polydispersity index (0.18), a strong zeta potential (-32 mV) and a high drug encapsulation efficiency (EE1=48.6%) and loading capacity (EE2=19.2%), and exhibited controlled release (t1/2=29 h). The MTT assay and flow cytometry results indicated that PAT could decrease toxicity and apoptosis induced by free TP on RAW264.7 cells. PAT decreased lipopolysaccharides/interferon γ-induced cytokines expression of macrophage (P<0.05). In vivo, PAT accumulated at inflammatory joints, improved the survival rate and had fewer side effects on tumor necrosis factor α transgenic mice, compared to TP. The blood biochemical indexes revealed that PAT did not cause much damage to the kidney (urea nitrogen and creatinine) and liver (alanine aminotransferase and aspartate aminotransferase). In addition, PAT reduced inflammatory synovial tissue area (P<0.05), cartilage loss (P<0.05), tartrate-resistant acid phosphatase-positive osteoclast area (P<0.05) and bone erosion (P<0.05) in both knee and ankle joints, and showed similar beneficial effect as free TP. In summary, our newly formed nanoparticle, PAT, can reduce the toxicity and guarantee the efficacy of TP, which represents an effective drug candidate for RA with low adverse side effect.
Keywords: triptolide, rheumatoid arthritis, γ-PGA, tumor necrosis factor α transgenic mice, drug carrier system
A corrigendum for this article has been published
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease that causes chronic inflammatory polyarthritis, which leads to pain and swelling, erosions of the cartilage and bone and even joint deformity. RA decreases motor function, leads to disability and diminishes quality of life.1,2 Currently, nonbiologic disease-modifying antirheumatic drugs (DMARDs) are the first-line treatment for RA. The patients will receive biologic DMARDs, such as tumor necrosis factor (TNF) inhibitors, if they did not respond to the nonbiologic DMARDs.3–9 Current treatments can improve symptoms and slow the progress of the disease, but only partial RA patients respond to DMARDs, and some biologics are contraindicated for others.10 Thus, there is an obvious need to develop novel effective drugs for RA due to the insufficient response, adverse effects and high cost of the present medicine treatments.11
Tripterygium wilfordii Hook F. (TWHF) is clinically used as a potential source of new drugs for RA. Several clinical trials proved that the extracts of TWHF achieve better effectiveness than DMARDs monotherapy.12,13 Triptolide (TP) is an active compound of TWHF, which exhibits immunosuppressive, cartilage-protective and anti-inflammatory effects on both humans and animals with RA.14–17 Unfortunately, the use of TP increases the risk for the occurrence of severe side effects, including nephrotoxicity, hepatic injury and bone marrow dysfunction.18–20 Development of self-assembled polymeric materials is a promising way to carry out drug delivery to achieve better therapeutic effect and minimize the side effects.21–23 These polymeric materials are prepared from a range of inorganic and organic compounds including polymers, lipids, dendrimers, carbon nanotubes and quantum dots.24,25 Among the polymers, poly-γ-glutamic acid (PGA) is stable with highly hydrophilic monoglutamate chain and can be synthesized cheaply by fermentation. Another advantage of this material is that it is nontoxic and fully biodegradable, and has been used clinically.26–29 PGA has been used as a carrier of various drugs and biomolecules, such as proteins,30 vaccine31 and genes,32 to treat liver tumor,33 glioma,34 bladder and lung cancer cells.35 However, whether PGA could be used as a carrier to treat RA was not known.
In this study, we investigated the use of PGA to wrap TP for RA to reduce the toxicity and guarantee the curative effect of TP. Previously, we used a PGA and L-phenylalanine ethylester complex to wrap TP, and found that the nanoparticle loaded with TP decreased TP toxicity in wild-type (WT) mice.36 In the current study, we conjugated PGA with di-tert-butyl L-aspartate hydrochloride (Asp), a hydrophobic structure that can allow the hydrophilic PGA-wrapped hydrophobic TP, to develop a new material, poly-γ-glutamic acid-grafted Asp (PA), which was utilized as a drug carrier to form a novel TP-loaded poly-γ-glutamic acid nanoparticle (PAT).
Materials and methods
Drugs and reagents
TP was purchased from Yuanye Biotech Company (Shanghai, People’s Republic of China). γ-PGA (molecular weight 300 kD) was purchased from Haining Zijingang Biotechnology Co. Ltd. Asp, penicillin–streptomycin sulfate, dimethyl sulfoxide (DMSO) and indocyanine green (ICG) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI) was purchased from Shanghai Medpep Company (Shanghai, People’s Republic of China). MTT and trypsin were from AMRESCO (Solon, OH, USA). 4′,6-Diamidino-2-phenylindole was purchased from Roche (Basel, Switzerland), DMEM, a cell culture medium, and fetal calf serum (FBS) were purchased from Hyclone (Logan, UT, USA). Apoptosis detection kit Annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) were purchased from BD (Franklin Lakes, NJ, USA).
Cells and animals
RAW264.7 cells were obtained from the cell bank of the Shanghai Institute of Biochemistry and Cell Research and were cultured in DMEM with 10% FBS and 1% penicillin–streptomycin solution at 37°C with 5% CO2. Tumor necrosis factor α transgenic (TNFα-Tg) mice in C57BL/6 background were generated by Dr George Kollias,37,38 which were bred as heterozygotes, and the WT littermates were used as control. The TNFα-Tg mice displayed mild bone erosion and joint inflammation from 2 to 2.5 months of age. Mice (2 months old) were reproduced in the East China Normal University animal center and were housed in specific pathogen-free room. All the mice studies were performed as per the Guiding Principles for the Care and Use of Laboratory Animals according to the Regulations of the People’s Republic of China for Administration of Laboratory Animals. This study was approved by the East China Normal University Ethics Committee on Animal Resources.
Preparation of PAT
The amphiphilic copolymers consisted of hydrophilic γ-PGA backbone and hydrophobic side chain Asp. The copolymer was synthesized by coupling Asp with γ-PGA.27 Briefly, Asp (10 mg) was conjugated to γ-PGA (38.4 mg) through amidation reaction under the catalyst EDCI (26 mg) for 24 h to form PA at room temperature. The unconjugated Asp and catalyst EDCI were removed by ether extraction. PAT complex was obtained by adding 5 mg TP to 20 mg PA in DMSO solution, and then the solution was dropped into a baker with ultrapure water under magnetic stirring at a speed of 120 rpm. After centrifugation (12,000 rpm) for 10 min, the free TP was removed, and dry PAT nanoparticle was obtained by vacuum freeze-drying equipment (Yibokang, Beijing, People’s Republic of China). Biopolymer dispersion samples (4 mg/mL) were stored at 4°C in ultrapure water.
Characterization of PAT
The chemical structure of PAT was identified by 1H nuclear magnetic resonance spectroscopy (NMR) spectroscopy (Bruker Avance 500 Spectrometer; Billerica, MA, USA). Dynamic light scattering (DLS; Malvern Zetasizer Nano-ZS; Malvern, UK) was used to detect the mean particle size and zeta potential of PAT at 25°C in triplicates. The particle size distribution was detected by the mean diameter and polydispersity index (PDI). Transmission electron microscopy (TEM; JEM-2100; JEOL) was performed to study the morphological appearance of PAT.
Drug encapsulation efficiency
Agilent 1100 high-performance liquid chromatography (HPLC) system with a reverse-phase LiChrospher ODS C18 column was used to detect the TP encapsulation ratio. PAT (5 mg) was added in 1 mL DMSO to release TP under an oscillator for 5 min, and the solution was removed by freeze dryer (Yibokang), and then 2 mL methanol was added. The mobile phase was a mixture of acetonitrile–water (30/70, v/v), and the current speed was controlled at 1.0 mL/min. The column temperature was controlled at 30°C, and the detection wavelength was maintained at 204 nm by a variable wavelength detector. Drug encapsulation efficiency (EE1) was determined using the following equation: EE1 = M1/M2, where M1 represents the mass of TP in the PAT and M2 represents the mass of TP used in the equation. The drug loading capacity (EE2) was determined using the following equation: EE2 = W1/W2, where W1 represents the weight of TP in the PAT and W2 represents the total weight of PAT.
In vivo TP release profiles
The dialysis diffusion technique was used to study the TP release profiles. PAT (10 mg) was deposited in 2 mL phosphate buffer saline (PBS) and suspended in a dialysis bag (cut-off, 10 kD; Spectrum Laboratories Inc, Rancho Dominguez, CA, USA). Then, the dialysis bag was immersed into 50 mL PBS or saline with continuous magnetic shaking at a speed of 60 rpm at 37°C. At predetermined time point, 0.5 mL external dialysis solution was extracted for HPLC analysis, and same volume of PBS or saline was added simultaneously. A profile was plotted to show the accumulative release of TP.
In vitro cytotoxicity
RAW264.7 cells were grown in 96-well plates (4×103/well). TP or PAT was mixed in culture medium at the equivalent TP concentrations of 6.25, 12.5, 25, 50, 100 and 200 nM/L and cultured for another 24, 48 and 72 h. MTT was used to quantify the cell survival rate. Briefly, each well was added with 10 μL MTT (5 mg/mL) solution and incubated for 2 h. DMSO (200 μL) was added into each well after the removal of supernatant. To evaluate the cell viability, a microplate reader (Multiskan GO; Thermo Fisher Scientific, Waltham, MA, USA) was used to detect the optical density values of each well under the wavelength of 570 nm.
Flow cytometric analysis
RAW264.7 cells (5×105) were seeded in a 6 cm dish and treated with 100 nM TP or PAT (containing 100 nM TP) for 24 h. Then, double-staining using Annexin V-FITC/PI kit (BD) was performed to quantify apoptosis rate. Briefly, after incubation, cells were centrifuged (1,000 rpm) for 5 min and washed with PBS 3 times, and then incubated in 500 μL flow cytometric buffer added with 5 μL of PI and 5 μL of Annexin V-FITC for 30 min at 4°C. The apoptosis rate of the different groups was detected by BD FACSCalibur and analyzed with BD CellQuest Pro software.
Real-time polymerase chain reaction (PCR)
The expression of TNFα, IL-1β, IL-6, iNOS and β-actin (the primer sequences are listed in Table 1) genes in RAW246.7 cells was detected by quantitative real-time PCR (Q-PCR). Total RNA was isolated with the TRIzol reagent (Thermo Fisher Scientific) according to the product instruction. A NanoDrop instrument (Thermo Fisher Scientific) was used to quantify mRNA concentration extracted from the cells. Total RNA (1 μg) from each sample was reverse transcribed into cDNA using the SYBR Green I Kit (Takara, Kusatsu, Japan) according to the manual. The cDNA templates were diluted 5 times, and Q-PCR was conducted with SYBR Premix Ex Taq II (Takara) according to the instruction in a Bio-Rad CFX 96 (Hercules, CA, USA) real-time PCR system. Each experiment was performed in triplicates. The samples were heated at 94°C for 5 min, followed by 40 PCR cycles (94°C for 15 s; 56°C for 15 s; 72°C for 30 s). The gene of β-actin was used as internal control, and fold changes were counted with the 2−ΔΔCt method.
 | Table 1 Sequences of primers used in real-time PCR |
In vivo toxicity
In vivo toxicity of PAT was studied in WT and TNFα-Tg mice. Two-month-old TNFα-Tg mice were divided into 4 groups (n=10/group), and treated intravenously in tail with free TP (0.15 mg/kg), PAT (0.15 mg/kg TP), PA (100 μL), or PBS (100 μL), every 3 days for 30 days. WT mice (n=10) were treated intravenously in tail with 100 μL PBS. A continuous observation for 30 days was conducted to assess the survival rate after the first injection. As an index of systemic toxicity, body weight was measured every 5 days. On the last day, mice were sacrificed, and serum samples were collected. Aspartate aminotransferase (AST), serum alanine aminotransferase (ALT), creatinine (CRE) and blood urea nitrogen (BUN) were detected using an auto-analyzer (BM704; Hitachi) to evaluate hepatic function (ALT and AST) and renal function (CRE and BUN).
In vivo imaging of nanoparticles
ICG (1 mg) was incubated with PA (20 mg) or with 5 mg TP in DMSO solution at room temperature for 30 min. Then, the mix was dropped into ultrapure water under magnetic stirring at a speed of 120 rpm. The ICG-modified nanoparticles (PA-ICG and PAT-ICG) were harvested after centrifugation at 5,000 rpm for 10 min at 4°C, followed by lyophilization. One hundred microliters of PA-ICG (10 mg/mL) and 100 μL of PAT-ICG (10 mg/mL) in PBS (pH 7.4) were injected into mice by intravenous tail injection. The body images (from 0.5 to 80 h) were acquired after injection using the IVIS Spectrum in vivo imaging system (Caliper, Hopkinton, MA, USA).
Histological detection
The samples of the liver, kidney and spleen were collected for histopathological analysis from all groups at the end of treatment. The samples were treated with formalin, dehydrated, paraffin embedded and sliced into 5 μm sections for hematoxylin–eosin (HE) staining.
Microcomputed tomography (micro-CT)
The knee and ankle joints of each experimental mouse were scanned using micro-CT system (Scanco VIVA CT80; SCANCO Medical AG, Brüttisellen, Switzerland). Images were acquired at 55 kV, 72 μA and 300 ms/frame, with 360 views. The 3-dimensional structures of knee joint and ankle joint were reconstructed and evaluated by μCT Evaluation Program V6.6 (SCANCO Medical AG). Three-dimensional bone volume of patellas and astragalus was measured using Aquarius software (T Ray V4.04) to determine the volumetric change of arthritis joints, and a density threshold was set higher than 11,000 AU as “bone”. Analysis typically took around 15 min per joint for an experienced operator.
Pathological examination of ankle and knee
The ankle and knee joints were utilized for tissue fixation with 10% formalin, decalcification with 10% EDTA, and were paraffin embedded and cut into 4 μm sections. Alcian blue/orange G (ABOG)39 and tartrate-resistant acid phosphatase (TRAP) were used to stain the sections. In TRAP staining, nuclei were counterstained with hematoxylin. The osteoclast-like cells were defined as having more than 4 nuclei and positive to TRAP staining.40 The distal tibia and proximal tibia of metatarsal bone in joints were digitized by an Olympus microscope (BX50). Images were analyzed using ImageJ software. The area rate of cartilage, inflammatory synovial tissue and TRAP+ osteoclast was expressed as percentage in comparison to the total tissue area.
Statistical analysis
GraphPad Prism 5 software was used for statistical analyses. One-way analysis of variance test followed by a Bonferroni-corrected posttest was used for comparisons. P<0.05 was considered statistically significant.
Results
Characterization of PAT
The 5 characteristic peaks of PA in 1H NMR spectrum are shown in Figure 1: 1 was the symbolic peak of aspartic acid (d 1.44 ppm), and the remaining 4 were symbolic peaks of PGA (d 1.88, d 2.01, d 2.33 and d 4.27 ppm). The Asp loading rate for PGA could be calculated by comparing the intensities of the peaks with d 1.44 and d 4.27 ppm. A 25% Asp loading for PA (0.82 Asp per repeating units) (Figure 1A) was identified from the 1H NMR results.
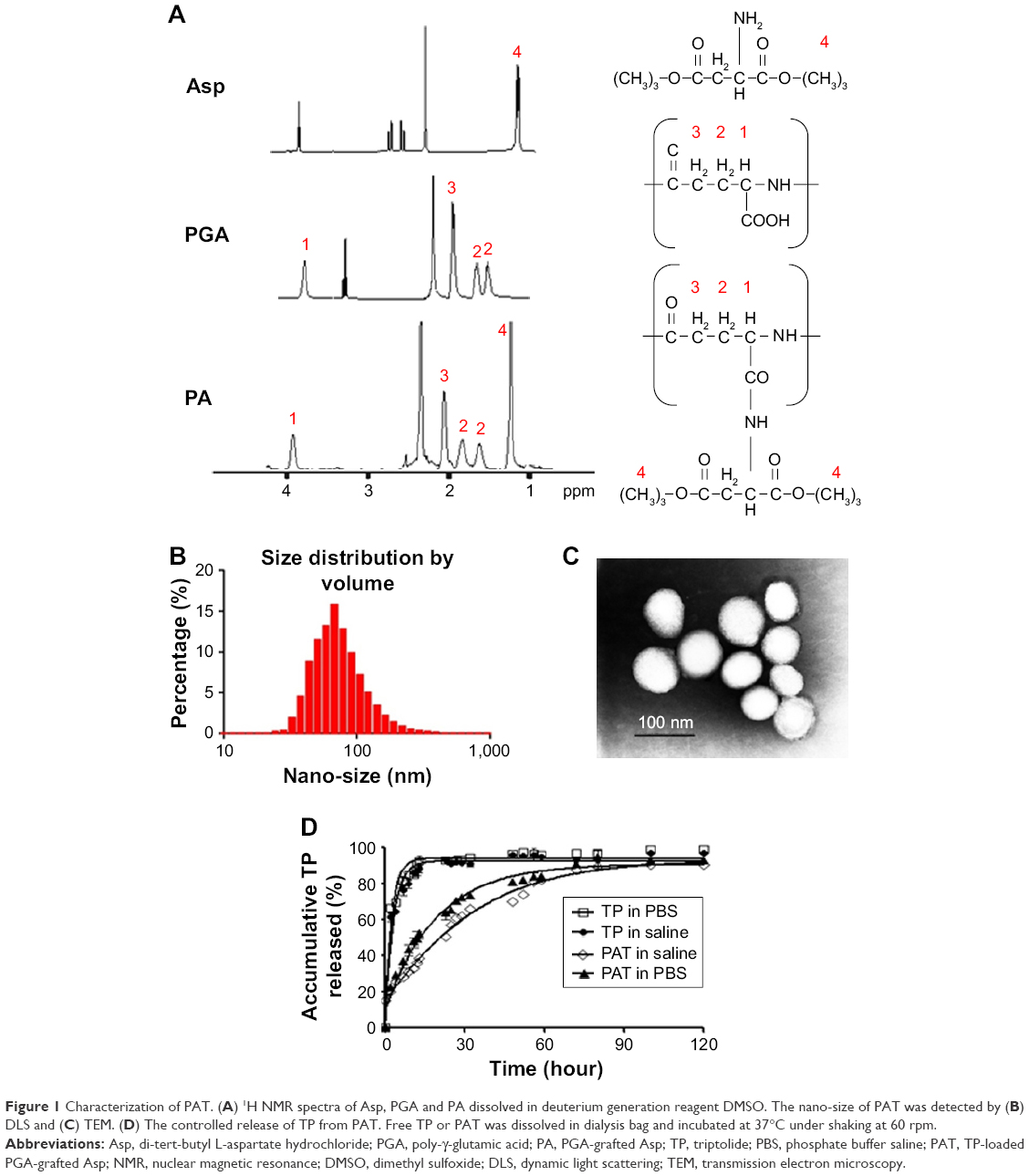 | Figure 1 Characterization of PAT. (A) 1H NMR spectra of Asp, PGA and PA dissolved in deuterium generation reagent DMSO. The nano-size of PAT was detected by (B) DLS and (C) TEM. (D) The controlled release of TP from PAT. Free TP or PAT was dissolved in dialysis bag and incubated at 37°C under shaking at 60 rpm. |
The mean particle dimension of PAT was 79±18 nm with a narrow size distribution (PDI =0.18; Figure 1B) as detected by DLS. Nanoparticles were round in shape with a diameter of 89±23 nm (Figure 1C) as detected by TEM. The particle size was in the range suitable for enhanced permeability and retention (EPR) effect in inflammatory tissue.41
Stronger zeta potentials will make the particles repel each other which can create a more stable dispersion system.42 In aqueous phase, the zeta potential border of stable particles is commonly considered as above +30 or below −30 mV.43 The zeta potential of the PAT developed in our study was −32 mV, which indicated that PAT was stable in aqueous phase due to strong mutual electrostatic repulsion.
In addition, the TP encapsulation rate EE1 and loading capacity EE2 were determined by HPLC to be 48.6% and 19.2%, respectively. The high EE1 and EE2 of PAT could avoid the poor aqueous solubility and allow rapid parenteral infusion without the risk of drug precipitation.44
In vitro release profile
The slow-release formulation can hold the effective medicine concentration in the blood for a long time, which can reduce the usage frequency and the drug dose, and increase the patient compliance. In our research, the median maximum release time (t1/2) of free TP was 1.5 h, but for PAT the t1/2 was about 29 h. There was 79% TP released, while only 36% TP was released by PAT at 6 h. At 120 h, 89% TP was released from PAT (Figure 1D). Compared with free TP, TP in PAT was released much more slowly. The release profile results indicated that the in vitro release of TP was burst-like, while TP release from PAT was sustainable.
PAT decreased TP-induced cytotoxic effect in the early stages and lipopolysaccharides/interferon γ (LPS/IFNγ)-induced cytokines expression of macrophage
To determine the cytotoxic effect of PAT in vitro, we applied MTT assay to detect the cell survival rate and flow cytometry method to detect cell apoptosis rate of macrophage (RAW264.7). MTT assay demonstrated that TP significantly decreased the cell survival rate of macrophage in a dose-dependent manner at different time points. Compared with TP-treated macrophage, PAT, containing same TP concentration, remarkably maintained the cell viability of macrophage from 24 to 48 h. The half-inhibition concentration of PAT was much higher than that of free TP at 24 (236±39.32 vs 47±12.34 nM) and 48 h (123±11.34 vs 21±4.68 nM), which could be accounted for by the slow release of TP from PAT. But with the treatment duration increase to 72 h, PAT caused similar decrease of the cell viability of macrophage as TP (Figure 2A).
 | Figure 2 PAT reduces the cytotoxicity of TP on RAW264.7 cells. (A) Growth curves of RAW264.7 cells were determined by MTT assay after incubation with TP (200, 100, 50, 25, 12.5 or 6.25 nM) or PAT (equal to TP concentration) for 24, 48 and 72 h. The values are the mean ± SD of 8 wells. (B) The apoptosis was assessed by FACS. Annexin V+/PI- cells represent early apoptotic cells, while Annexin V+/PI+ cells represent late apoptotic cells. The percentage of both early and late apoptotic cells is shown in the lower right corner. (C) The mRNA expression of TNFα, IL-1β, IL-6 and iNOS was analyzed by real-time PCR. The values are the mean ± SD of 3 wells. *P<0.05, compared with TP; **P<0.05, compared with PBS treated groups. |
We treated macrophage with PBS, PA, TP and PAT for 24 h and found no obvious apoptosis in the PBS- or PA-treated group. The apoptosis rate in free TP-treated group was 68.9%, while the apoptosis rate in PAT-treated group was 7.2% (Figure 2B). In addition, in PAT-treated group, there were 88% of live cells, which was higher than free TP-treated group (72%; Figure 2B). These results indicated that PAT treatment markedly improved the cell survival rate compared to free TP treatment. The result of Q-PCR indicated both TP and PAT dramatically decreased the mRNA level of TNFα (P<0.05), interleukin-1β (P<0.05), interleukin-6 (P<0.05) and inducible nitric oxide synthase (P<0.05) induced by LPS/IFNγ in RAW264.7 cells (Figure 2C).
PAT targeted inflammatory joints of TNFα-Tg mice
Within the near-infrared range, ICG could penetrate deep tissue and could be used for fluorescence visualization.45–47 A mixture of amphiphilic ICG48,49 and PA can self-assemble in water. To detect the retention duration of PAT in vivo and to determine the frequency of drug administration, we conjugated PA or PAT with ICG by self-assembling. PA-ICG and PAT-ICG were injected into the tail vein of 2-month-old TNFα-Tg mice and WT littermates. The 2-month-old TNFα-Tg mice injected with PBS and free TP were utilized as negative controls, because no bioluminescence could be detected. Bioluminescent images were taken intermittently at 0.5, 1, 4, 24, 48 and 80 h. The ICG signal spread into the internal organs immediately after injection at tail vein of both WT and TNFα-Tg mice, accumulated at the knee joints of TNFα-Tg mice at 24 h, reduced but remained positive at the knee joints of TNFα-Tg mice at 48 h and was totally cleared in both WT and TNFα-Tg mice at 80 h. In WT mice, PA-ICG did not assemble at knee joints at any time point (Figure 3A and B). These results suggested that 1) the retention duration of PAT and PA in WT and TNFα-Tg mice was less than 80 h, and the frequency of drug administration should be once every 3 days, and 2) EPR effect made the nanoparticles, PA and PAT, accumulate in the inflammatory joints of TNFα-Tg mice.
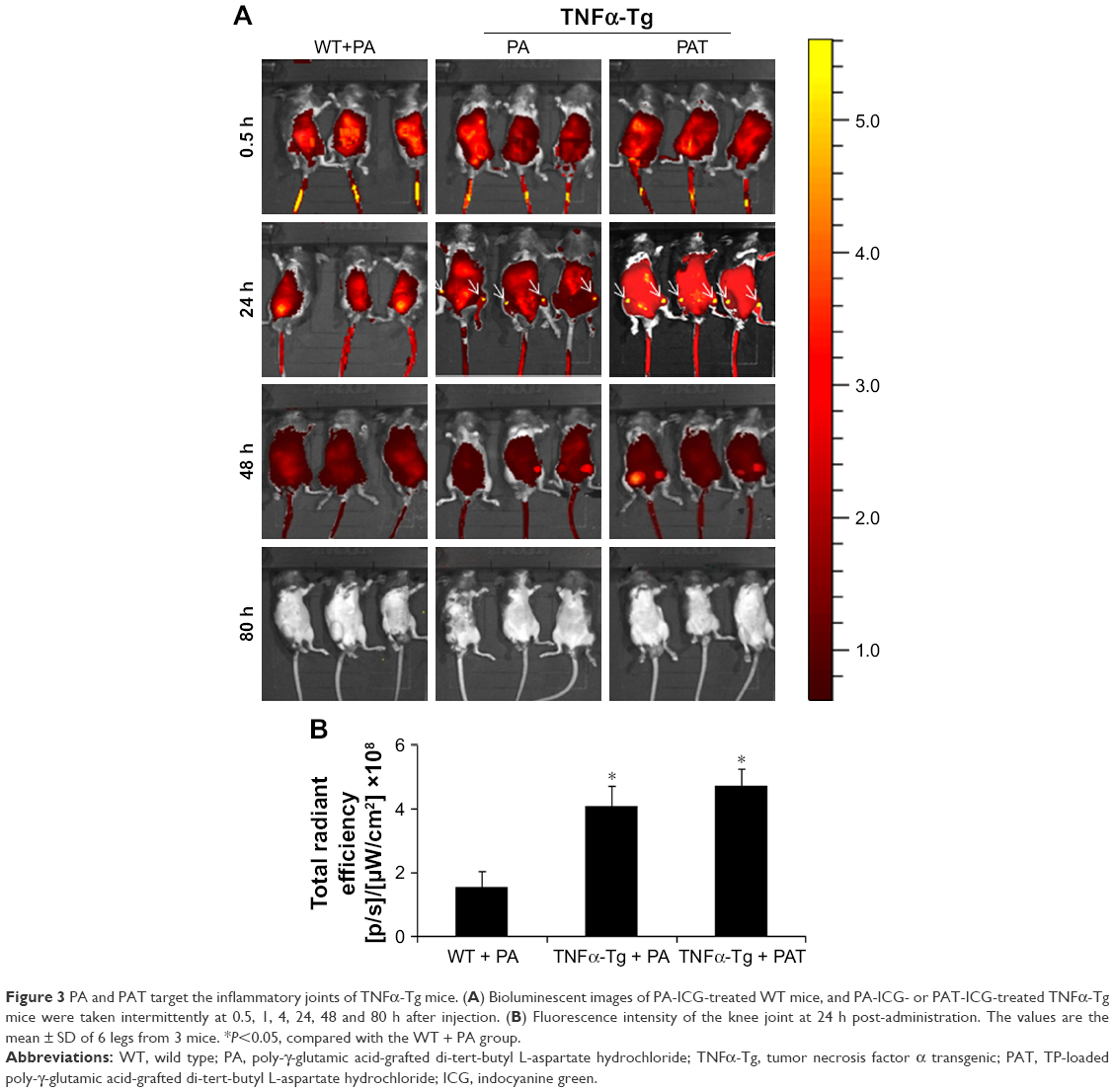 | Figure 3 PA and PAT target the inflammatory joints of TNFα-Tg mice. (A) Bioluminescent images of PA-ICG-treated WT mice, and PA-ICG- or PAT-ICG-treated TNFα-Tg mice were taken intermittently at 0.5, 1, 4, 24, 48 and 80 h after injection. (B) Fluorescence intensity of the knee joint at 24 h post-administration. The values are the mean ± SD of 6 legs from 3 mice. *P<0.05, compared with the WT + PA group. |
PAT decreased in vivo toxicity
To determine whether PAT can reduce the toxicity of TP on TNFα-Tg mice, we applied PA, TP and PAT in 2-month-old TNFα-Tg mice once in 3 days for 30 days, and found that the survival rate was 100% in mice treated with PBS, PA and PAT, whereas the survival rate was 70% in mice treated with 0.15 mg/kg TP (Figure 4A). Besides, 0.15 mg/kg TP brought about a 20% body weight loss in TNFα-Tg mice; however, the PAT, containing same concentration of TP, caused no obvious body weight changes (Figure 4B). Only TP treatment group showed kidney (BUN and CRE) and liver (ALT and AST) damage, and no significant differences between PAT and PBS groups were detected (Table 2). These results revealed that PAT exhibited lower toxicity.
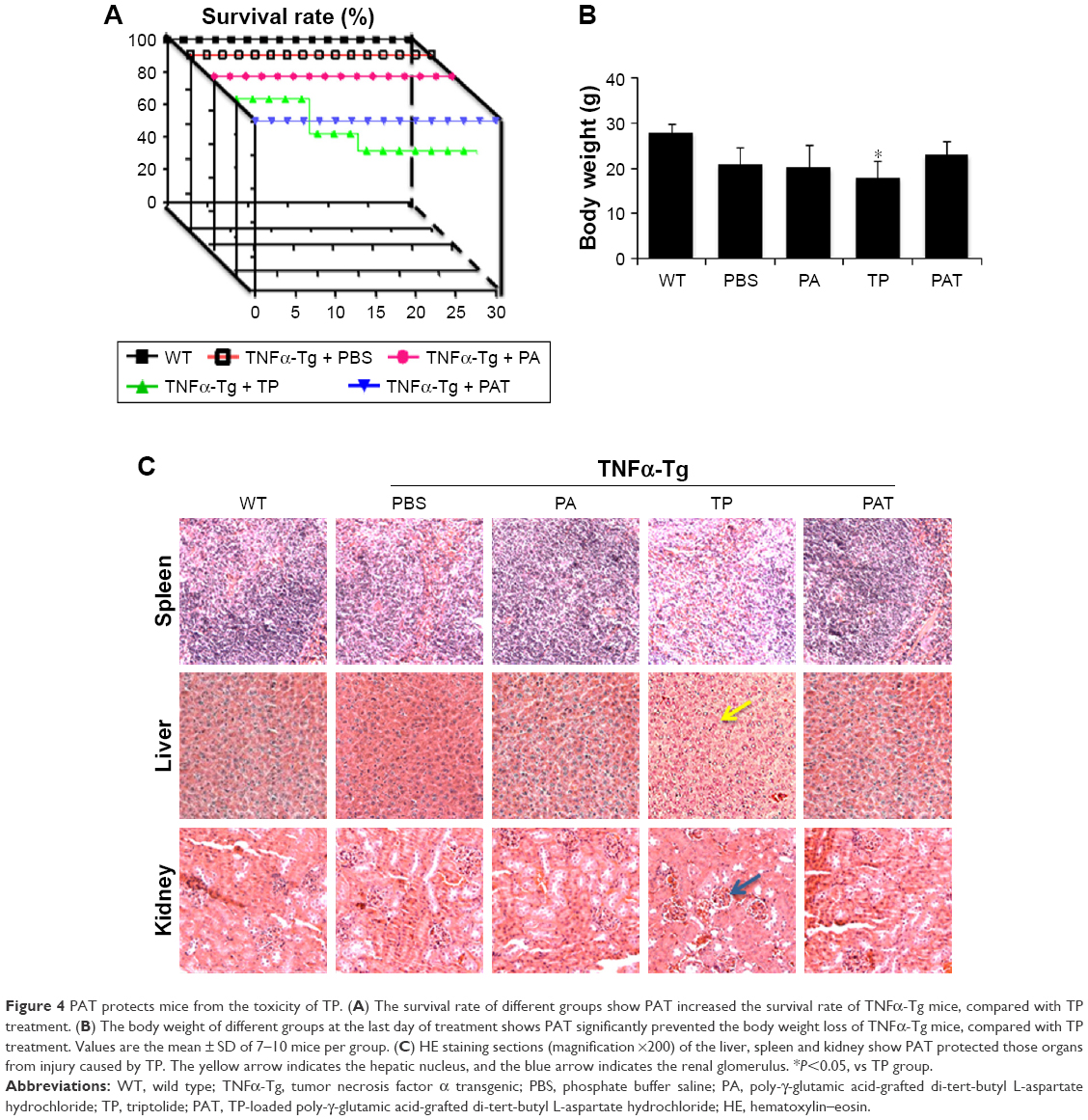 | Figure 4 PAT protects mice from the toxicity of TP. (A) The survival rate of different groups show PAT increased the survival rate of TNFα-Tg mice, compared with TP treatment. (B) The body weight of different groups at the last day of treatment shows PAT significantly prevented the body weight loss of TNFα-Tg mice, compared with TP treatment. Values are the mean ± SD of 7–10 mice per group. (C) HE staining sections (magnification ×200) of the liver, spleen and kidney show PAT protected those organs from injury caused by TP. The yellow arrow indicates the hepatic nucleus, and the blue arrow indicates the renal glomerulus. *P<0.05, vs TP group. |
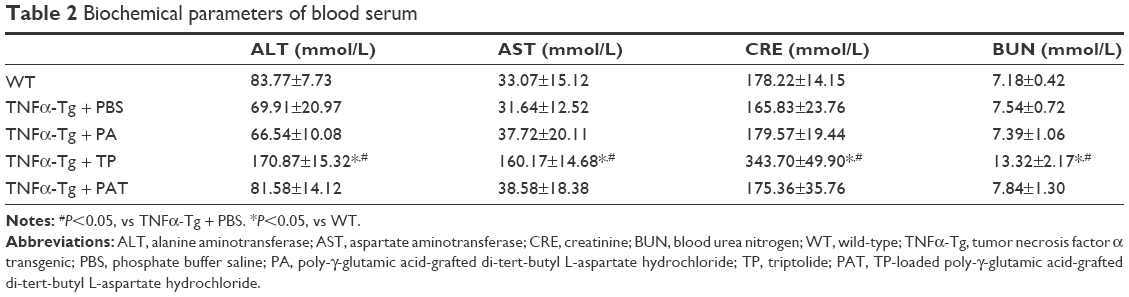 | Table 2 Biochemical parameters of blood serum |
Furthermore, the HE staining was performed to detect the side effects of PAT on the liver, kidney and spleen in TNFα-Tg mice, and found that the liver cells were intact in structure, and nucleus was arranged orderly without any degeneration or necrosis in WT and PA-treated TNFα-Tg mice. In free TP-treated group, the livers were mottled, fragile and hyperemic, and the nuclei became ruptured and pyknotic. However, the livers from PAT group exhibited normal structure, including clear cellular structures and orderly arranged cells. Similarly, free TP severely damaged the kidney, resulting in swelling, hyperemia, scattering and necrosis of renal glomerular cells and blurry borders of renal tubular epithelial cells and stenosis or atresia in lumens, while in PBS-, PA- and PAT-treated TNFα-Tg mice, the structures like renal glomerulus, interstitium and tubule were normal. Besides, in free TP-treated group, the spleen red pulp increased, whereas PAT treatment protected the spleen from the damaging effects of TP (Figure 4C). These results indicated that PAT protected the mice from the toxicity of TP.
PAT presented beneficial effect on both ankle and knee joints of TNFα-Tg mice
To determine if PAT had similar beneficial effect as TP on inflammatory arthritis, we injected PA, TP and PAT into tail veins of 2-month-old TNFα-Tg mice, because at this age, their ankle joints developed tenosynovitis.38,50 After 1-month treatment, we performed micro-CT scanning, and ABOG and TRAP staining on both ankle and knee joints of TNFα-Tg mice to investigate synovial inflammation, and bone and cartilage erosion at joints, which are the major characteristics of inflammatory arthritis.51
Micro-CT results indicated that mean values of bone volume of astragalus and patella significantly decreased in PBS- and PA-treated groups compared to WT mice (Figure 5A). In contrast, astragalus and patella bone volumes were significantly higher (P<0.05) in TP- and PAT-treated mice than in PBS-treated TNFα-Tg mice. However, the difference between TP- and PAT-treated groups was not significant (Figure 5B). These results suggested that PAT had similar beneficial effect as TP on inflammatory arthritis.
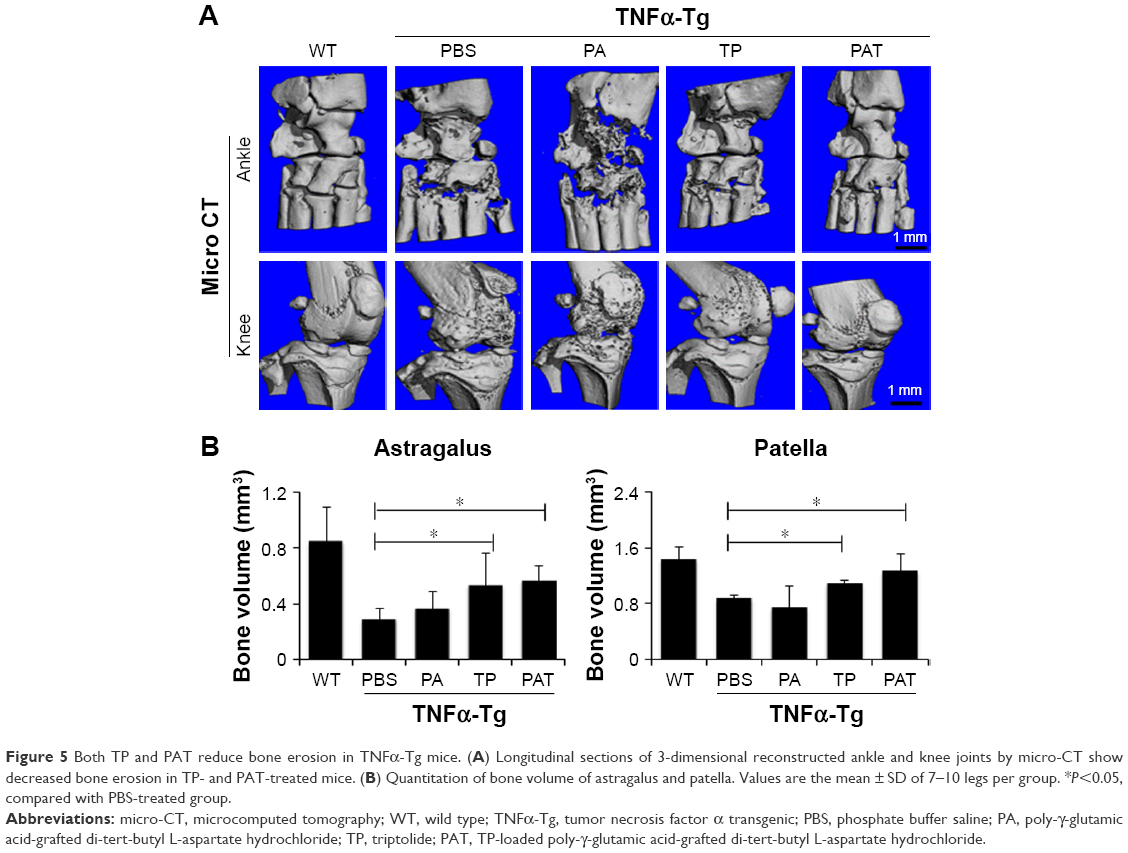 | Figure 5 Both TP and PAT reduce bone erosion in TNFα-Tg mice. (A) Longitudinal sections of 3-dimensional reconstructed ankle and knee joints by micro-CT show decreased bone erosion in TP- and PAT-treated mice. (B) Quantitation of bone volume of astragalus and patella. Values are the mean ± SD of 7–10 legs per group. *P<0.05, compared with PBS-treated group. |
Histological and histomorphometric analysis indicated that PA- and PBS-treated TNFα-Tg mice had conspicuous synovial inflammation, bone and cartilage destruction (Figure 6A and B) and numerous TRAP+ osteoclasts at ankle and knee joints (Figure 7A and B). All these symptoms were significantly less obvious in ankles and knee joints of PAT- and TP-injected mice.
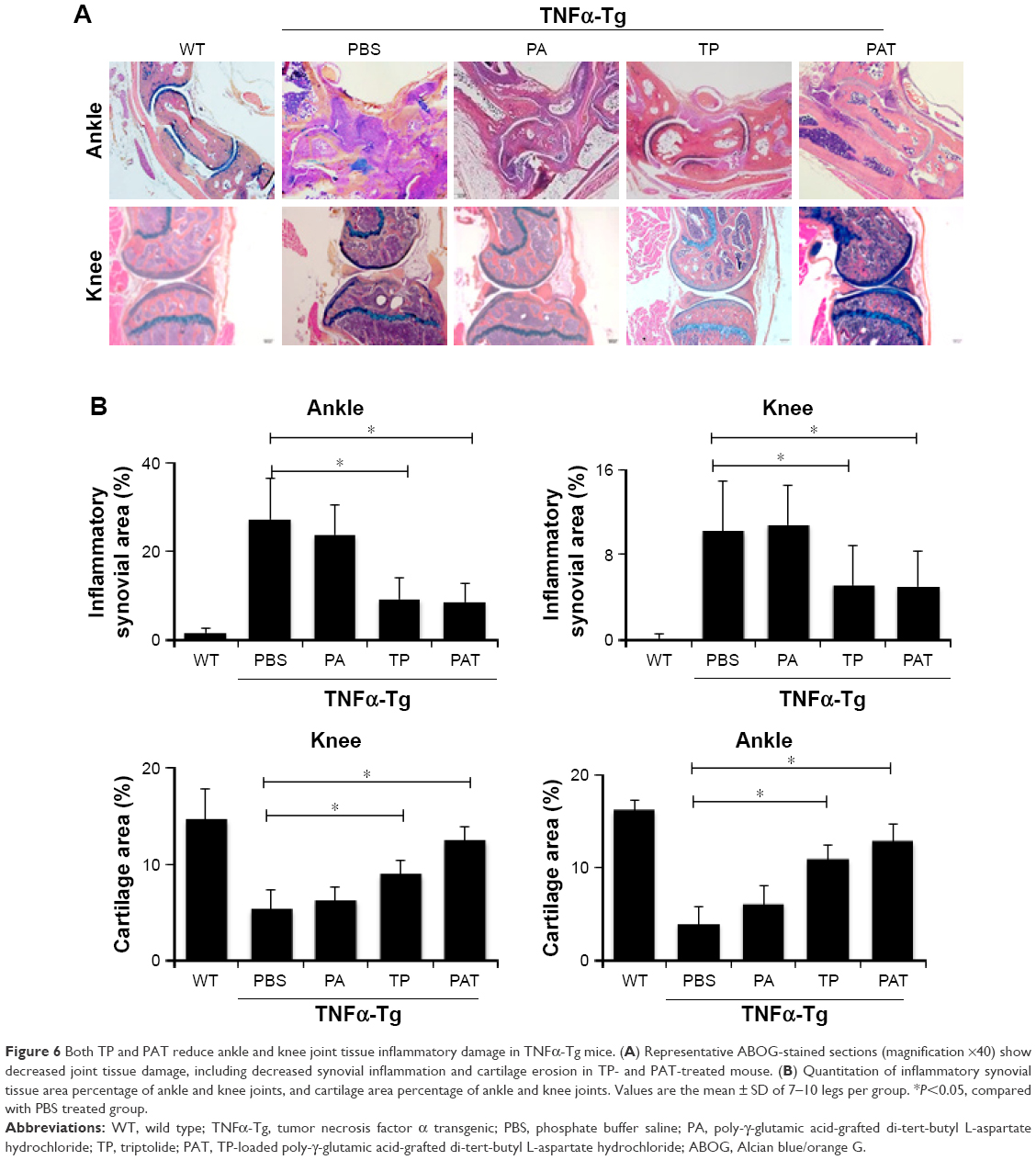 | Figure 6 Both TP and PAT reduce ankle and knee joint tissue inflammatory damage in TNFα-Tg mice. (A) Representative ABOG-stained sections (magnification ×40) show decreased joint tissue damage, including decreased synovial inflammation and cartilage erosion in TP- and PAT-treated mouse. (B) Quantitation of inflammatory synovial tissue area percentage of ankle and knee joints, and cartilage area percentage of ankle and knee joints. Values are the mean ± SD of 7–10 legs per group. *P<0.05, compared with PBS treated group. |
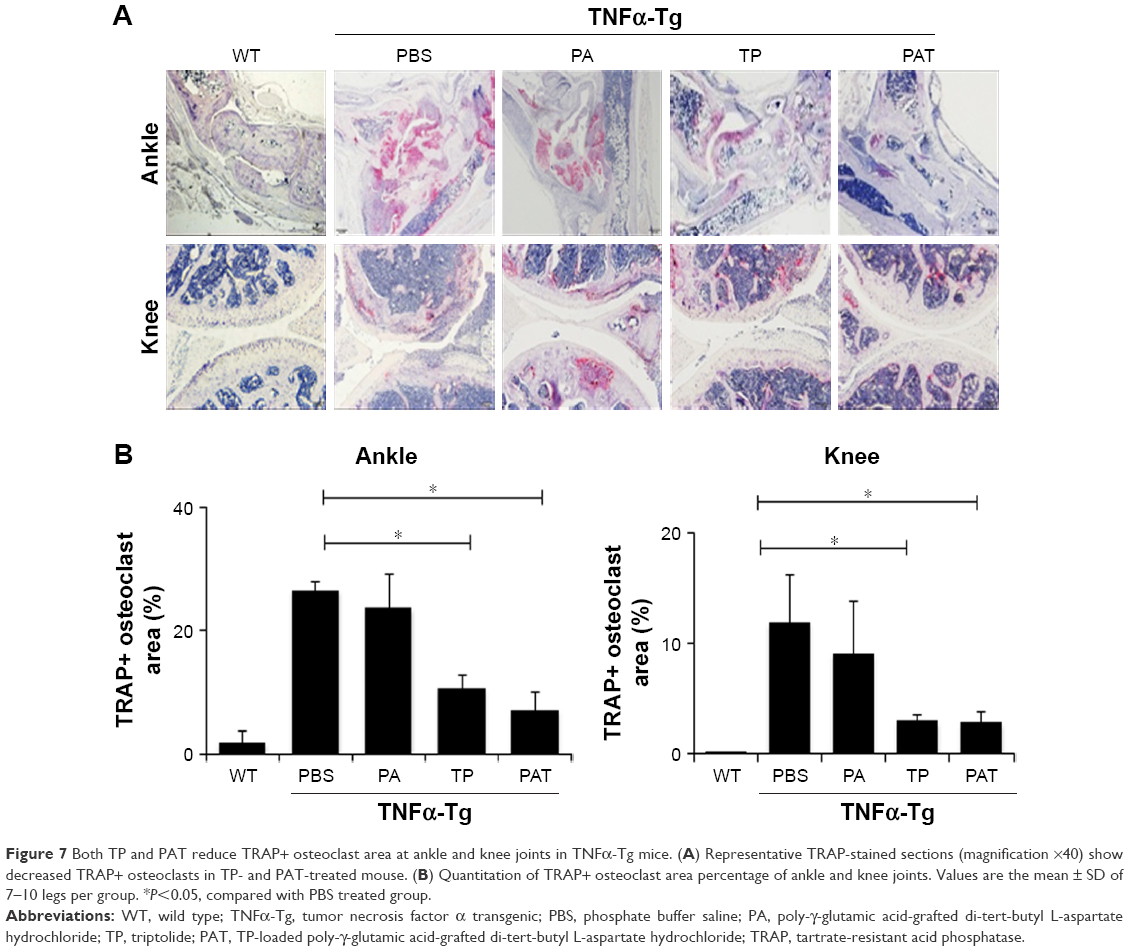 | Figure 7 Both TP and PAT reduce TRAP+ osteoclast area at ankle and knee joints in TNFα-Tg mice. (A) Representative TRAP-stained sections (magnification ×40) show decreased TRAP+ osteoclasts in TP- and PAT-treated mouse. (B) Quantitation of TRAP+ osteoclast area percentage of ankle and knee joints. Values are the mean ± SD of 7–10 legs per group. *P<0.05, compared with PBS treated group. |
Discussion
A variety of new strategies have been developed for the treatment of RA if the RA patients do not respond to the medications. Biologic agents and small-molecule drugs have been developed. Small potent molecules such as sulfasalazine, leflunomide, methotrexate, hydroxychloroquine, cyclosporine and azathioprine are widely used in the clinic but can cause unpleasant side effects. Some biologic agents including rituximab, abatacept, etanercept, infliximab, certolizumab, golimumab, adalimumab and tocilizumab are contraindicated in some patients and estimated to be cost-effective.52 Nano-drug is a good option to achieve cure and reduce toxicity and is a complementary therapeutic agent for RA.
In the current study, we firstly developed a novel nano-drug carrier system containing TP using γ-PGA-grafted L-Asp-OtBu, which can reduce the toxicity and guarantee the curative effect of TP for RA. Previously, PGA was conjugated with L-phenylalanine,53 leucine, tryptophan54 or carboxymethyl chitosan55 to form nanoparticles. The use of aspartic acid conjugated with PGA to form amphiphilic nanoparticles has not been reported. PA as a drug carrier was fabricated using mono-amino acid chain which helped to achieve a solid structure and reduced the leakage of TP. The TEM images indicated that the particle size of our newly formed PAT was in the range suitable for EPR effect in inflammatory tissue, and our experiments with PAT-ICG suggested that PAT was efficacious in targeting inflammatory joints. The zeta potential of PAT demonstrated that our nanoparticle was stable in the aqueous phase. The TP encapsulation ratio of PAT reached 48.6%, which exhibited the high encapsulation efficiency of PAT.
As a small molecule, TP could rapidly perform its therapeutic action, but it is hard to maintain its concentration in blood due to glomerular filtration and various forms of protein hydrolysis.56 TP release from PAT was in a sustainable manner. MTT studies suggested that PAT reduced the cytotoxicity of TP in the early stages and maintained its efficacy for a long time, which confirmed the slow-release effect of PAT. A slow-release drug formation could maintain a good drug concentration in blood and prolong the drug’s efficacy. These results suggest that PAT is stable, exhibits slow release and is suitable for EPR effect and targets inflammatory joints. Two anti-TNF monoclonal antibodies, etanercept and infliximab, have been approved by the US Food and Drug Administration and the European Medicines Evaluation Agency for RA.57 It was reported the TNFα-Tg mice developed an erosive arthritis with bone and cartilage destruction, and in the treatment of inflammatory bone loss disease, the administration of monoclonal antibodies against human TNF-α significantly decreased synovitis (~5-fold; P<0.05 vs placebo) and potentially suppressed the pathological development.58,59 In the present study, PAT decreased LPS/IFNγ-induced cytokines expression of macrophage and showed beneficial effect on TNFα-Tg mice, as PAT reduced inflammatory area, bone erosion, cartilage loss and TRAP+ osteoclast area in knee and ankle joints. The curative effect of TP was not bothered by the carrier system, γ-PGA-grafted L-Asp. These results suggested that our PAT exhibited anti-inflammatory effect, similar to anti-TNF therapy.
Despite the promising bioactivities, the toxicity of TP limits its widespread clinical usage. In the current study, PAT decreased the death rate and toxicity at the liver and spleen induced by TP in mice, which suggests that PAT is safe for in vivo usage.
The emulsification of TP in PAT resulted in favorable characteristics such as high encapsulation efficiency, low energy consumption and mild preparation conditions, which are promising for more hydrophobic substances. Therefore, PAT represents a very effective drug candidate for RA with low adverse side effect.
Acknowledgments
This work was sponsored by research grants from National Natural Science Foundation (81330085, 81673990 and 81220108027), Longyi innovation team program (LYCX-01), the program for innovative research team of Ministry of Science and Technology of the People’s Republic of China (2015RA4002), Changning District Guanghua Excellent PI Project (No 2016-01), Scientific Research Project of Shanghai Municipal Commission of Health and Family Planning (20174Y0108) and Yumiao project of Longhua Hospital (LYTD-42). The authors also thank Dr Qi Shi for his instruction on chemical synthesis and his generous permission to use his laboratory instrument.
Disclosure
The authors report no conflicts of interest in this work.
References
Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14(1):397–440. | ||
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–2038. | ||
Mok CC, Tsai WC, Chen DY, Wei JC. Immunogenicity of anti-TNF biologic agents in the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2016;16(2):201–211. | ||
Edginton S, Hitchon C, Froese W, El-Gabalawy H. Effects of rituximab and infliximab treatment on carboxypeptidase b and its substrates in RA synovium. J Rheumatol. 2016;43(5):846–854. | ||
Herenius MM, Oliveira AS, Wijbrandts CA, Gerlag DM, Tak PP, Lebre MC. Anti-TNF therapy reduces serum levels of chemerin in rheumatoid arthritis: a new mechanism by which anti-TNF might reduce inflammation. PLoS One. 2013;8(2):e57802. | ||
Fanet-Goguet M, Martin S, Fernandez C, Fautrel B, Bourgeois P. Le point sur les biothérapies dans la polyarthrite rhumatoïde: principes actifs disponibles, stratégie thérapeutique [Focus on biological agents in rheumatoid arthritis: newer treatments and therapeutic strategies]. Therapie. 2004;59(4):451–461. French [with English abstract]. | ||
Gravallese EM, Walsh NC. Rheumatoid arthritis: repair of erosion in RA – shifting the balance to formation. Nat Rev Rheumatol. 2011;7(11):626–628. | ||
Goldbach-Mansky R, Wilson M, Fleischmann R, et al. Comparison of Tripterygium wilfordii Hook F. versus sulfasalazine in the treatment of rheumatoid arthritis: a randomized trial. Ann Intern Med. 2009;151(4):229–240, W49–W51. | ||
O’Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med. 2004;350(25):2591–2602. | ||
Kalden JR. Emerging therapies for rheumatoid arthritis. Rheumatol Ther. 2016;3(1):31–42. | ||
Landewe RB, van der Heijde D. Comment on: ‘comparison of Tripterygium wilfordii Hook F with methotrexate in the treatment of active rheumatoid arthritis (TRIFRA): a randomised, controlled clinical trial’ by Qian-wen et al. Ann Rheum Dis. 2014;73(10):e62. | ||
Graziose R, Lila MA, Raskin I. Merging traditional Chinese medicine with modern drug discovery technologies to find novel drugs and functional foods. Curr Drug Discov Technol. 2010;7(1):2–12. | ||
Cameron M, Gagnier JJ, Little CV, Parsons TJ, Blumle A, Chrubasik S. Evidence of effectiveness of herbal medicinal products in the treatment of arthritis. Part 2: rheumatoid arthritis. Phytother Res. 2009;23(12):1647–1662. | ||
Kupchan SM, Court WA, Dailey RG, Gilmore CJ, Bryan RF. Triptolide and tripdiolide, novel antileukemic diterpenoid triepoxides from Tripterygium wilfordii. J Am Chem Soc. 1972;94(20):7194–7195. | ||
Lu Y, Zhang Y, Li L, et al. TAB1: a target of triptolide in macrophages. Chem Biol. 2014;21(2):246–256. | ||
Matta R, Wang X, Ge H, Ray W, Nelin LD, Liu Y. Triptolide induces anti-inflammatory cellular responses. Am J Transl Res. 2009;1(3):267–282. | ||
Zhang Y, Ma X. Triptolide inhibits IL-12/IL-23 expression in APCs via CCAAT/enhancer-binding protein alpha. J Immunol. 2010;184(7):3866–3877. | ||
Li J, Shen F, Guan C, et al. Activation of Nrf2 protects against triptolide-induced hepatotoxicity. PLoS One. 2014;9(7):e100685. | ||
Quan L, Zhang Y, Crielaard BJ, et al. Nanomedicines for inflammatory arthritis: head-to-head comparison of glucocorticoid-containing polymers, micelles, and liposomes. ACS Nano. 2014;8(1):458–466. | ||
Mei Z, Li X, Wu Q, Hu S, Yang X. The research on the anti-inflammatory activity and hepatotoxicity of triptolide-loaded solid lipid nanoparticle. Pharmacol Res. 2005;51(4):345–351. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. | ||
Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48:131–143. | ||
Sundaramoorthy P, Ramasamy T, Mishra SK, et al. Engineering of caveolae-specific self-micellizing anticancer lipid nanoparticles to enhance the chemotherapeutic efficacy of oxaliplatin in colorectal cancer cells. Acta Biomater. 2016;42:220–231. | ||
Ramasamy T, Ruttala HB, Choi JY, et al. Engineering of a lipid-polymer nanoarchitectural platform for highly effective combination therapy of doxorubicin and irinotecan. Chem Commun. 2015;51(26):5758–5761. | ||
Feng Z, Lai Y, Ye H, Huang J, Xi XG, Wu Z. Poly (gamma, L-glutamic acid)-cisplatin bioconjugate exhibits potent antitumor activity with low toxicity: a comparative study with clinically used platinum derivatives. Cancer Sci. 2010;101(11):2476–2482. | ||
Geng X, Ye H, Feng Z, et al. Synthesis and characterization of cisplatin-loaded, EGFR-targeted biopolymer and in vitro evaluation for targeted delivery. J Biomed Mater Res Part A. 2012;100(10):2839–2848. | ||
Xiong Y, Jiang W, Shen Y, et al. A poly(γ, l-glutamic acid)-citric acid based nanoconjugate for cisplatin delivery. Biomaterials. 2012;33(29):7182–7193. | ||
Zhang L, Geng X, Zhou J, et al. Fabrication of poly(gamma-glutamic acid)-based biopolymer as the targeted drug delivery system with enhanced cytotoxicity to APN/CD13 over-expressed cells. J Drug Target. 2015;23(5):453–461. | ||
Matsuo K, Koizumi H, Akashi M, et al. Intranasal immunization with poly(gamma-glutamic acid) nanoparticles entrapping antigenic proteins can induce potent tumor immunity. J Control Release. 2011;152(2):310–316. | ||
Yoshikawa T, Okada N, Oda A, et al. Nanoparticles built by self-assembly of amphiphilic gamma-PGA can deliver antigens to antigen-presenting cells with high efficiency: a new tumor-vaccine carrier for eliciting effector T cells. Vaccine. 2008;26(10):1303–1313. | ||
Geng Y, Lin D, Shao L, Yan F, Ju H. Cellular delivery of quantum dot-bound hybridization probe for detection of intracellular pre-microRNA using chitosan/poly(gamma-glutamic acid) complex as a carrier. PLoS One. 2013;8(6):e65540. | ||
Yamaguchi S, Tatsumi T, Takehara T, et al. EphA2-derived peptide vaccine with amphiphilic poly(gamma-glutamic acid) nanoparticles elicits an anti-tumor effect against mouse liver tumor. Cancer Immunol Immunother. 2010;59(5):759–767. | ||
Kulhari H, Telukutla SR, Pooja D, et al. Peptide grafted and self-assembled poly(gamma-glutamic acid)-phenylalanine nanoparticles targeting camptothecin to glioma. Nanomedicine (Lond). 2017;12(14):1661–1674. | ||
Sun C, Xu S, Guo J, Xu K, Wang C. The inhibitory and apoptotic effects of docetaxel-loaded mesoporous magnetic colloidal nanocrystal clusters on bladder cancer T24 cells in vitro. J Biomed Nanotechnol. 2014;10(3):455–462. | ||
Zhang L, Wang T, Li Q, et al. Fabrication of novel vesicles of triptolide for antirheumatoid activity with reduced toxicity in vitro and in vivo. Int J Nanomedicine. 2016;11:2663–2673. | ||
Keffer J, Probert L, Cazlaris H, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025–4031. | ||
Li P, Schwarz EM. The TNF-alpha transgenic mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25(1):19–33. | ||
Liang Q, Ju Y, Chen Y, et al. Lymphatic endothelial cells efferent to inflamed joints produce iNOS and inhibit lymphatic vessel contraction and drainage in TNF-induced arthritis in mice. Arthritis Res Ther. 2016;18:62. | ||
Kim T, Lee W, Baek SH, et al. Effects of alveolar bone displacement with segmental osteotomy: micro-CT and histomorphometric analysis in rats. Braz Oral Research. 2016;30(1):e132. | ||
Arfors KE, Rutili G, Svensjo E. Microvascular transport of macromolecules in normal and inflammatory conditions. Acta Physiol Scand Suppl. 1979;463:93–103. | ||
Mishra PR, Al Shaal L, Muller RH, Keck CM. Production and characterization of Hesperetin nanosuspensions for dermal delivery. Int J Pharm. 2009;371(1–2):182–189. | ||
Bhattacharjee S. DLS and zeta potential – what they are and what they are not? J Control Release. 2016;235:337–351. | ||
Patil S, Lis LG, Schumacher RJ, et al. Phosphonooxymethyl prodrug of triptolide: synthesis, physicochemical characterization, and efficacy in human colon adenocarcinoma and ovarian cancer xenografts. J Med Chem. 2015;58(23):9334–9344. | ||
Sharma R, Wendt JA, Rasmussen JC, Adams KE, Marshall MV, Sevick-Muraca EM. New horizons for imaging lymphatic function. Ann N Y Acad Sci. 2008;1131:13–36. | ||
Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19(4):316–317. | ||
Sevick-Muraca EM, Sharma R, Rasmussen JC, et al. Imaging of lymph flow in breast cancer patients after microdose administration of a near-infrared fluorophore: feasibility study. Radiology. 2008;246(3):734–741. | ||
Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17(5):545–580. | ||
Sano K, Nakajima T, Miyazaki K, et al. Short PEG-linkers improve the performance of targeted, activatable monoclonal antibody-indocyanine green optical imaging probes. Bioconjug Chem. 2013;24(5):811–816. | ||
Hayer S, Redlich K, Korb A, Hermann S, Smolen J, Schett G. Tenosynovitis and osteoclast formation as the initial preclinical changes in a murine model of inflammatory arthritis. Arthritis Rheum. 2007;56(1):79–88. | ||
Mcinnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442. | ||
Diamantopoulos A, Finckh A, Huizinga T, et al. Tocilizumab in the treatment of rheumatoid arthritis: a cost-effectiveness analysis in the UK. Pharmacoeconomics. 2014;32(8):775–787. | ||
Chen C, Yu CH, Cheng YC, Yu PHF, Cheung MK. Preparation and characterization of biodegradable nanoparticles based on amphiphilic poly(3-hydroxybutyrate)–poly(ethylene glycol)–poly(3-hydroxybutyrate) triblock copolymer. Eur Polym J. 2006;42(10):2211–2220. | ||
Shima F, Akagi T, Akashi M. Effect of hydrophobic side chains in the induction of immune responses by nanoparticle adjuvants consisting of amphiphilic poly(gamma-glutamic acid). Bioconjug Chem. 2015;26(5):890–898. | ||
Yan X, Tong Z, Chen Y, et al. Bioresponsive materials for drug delivery based on carboxymethyl chitosan/poly(gamma-glutamic acid) composite microparticles. Mar Drugs. 2017;15(5):pii: E127. | ||
Gong X, Chen Y, Wu Y. Absorption and metabolism characteristics of triptolide as determined by a sensitive and reliable LC-MS/MS method. Molecules. 2015;20(5):8928–8940. | ||
Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344(12):907–916. | ||
Alexopoulou L, Pasparakis M, Kollias G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol. 1997;27(10):2588–2592. | ||
Bouta EM, Kuzin I, de Mesy Bentley K, et al. Brief report: treatment of tumor necrosis factor-transgenic mice with anti-tumor necrosis factor restores lymphatic contractions, repairs lymphatic vessels, and may increase monocyte/macrophage egress. Arthritis Rheumatol. 2017;69(6):1187–1193. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.