Back to Journals » Journal of Inflammation Research » Volume 19
Exploring the Potential Value of Inflammatory Response-Associated Genes in Chronic Rhinosinusitis with Nasal Polyps: An Integrated Bioinformatics and Experimental Validation Analysis
Authors Hossen MA, Liang S, Yan R, Zhu H, Yang S, Qin G
Received 13 January 2026
Accepted for publication 6 June 2026
Published 30 June 2026 Volume 2026:19 593214
DOI https://doi.org/10.2147/JIR.S593214
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
MD Arefin Hossen, Sai Liang, Runjie Yan, Han Zhu, Shishi Yang, Gang Qin
Department of Otolaryngology Head and Neck Surgery, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, 646000, People’s Republic of China
Correspondence: Gang Qin, Email [email protected]
Objective: Chronic rhinosinusitis with nasal polyps (CRSwNP) is a heterogeneous chronic inflammatory disease with unclear pathogenesis and a high recurrence rate. Inflammatory response-related genes (IRGs) play a key role in disease progression but remain to be further explored in CRSwNP.
Methods: CRSwNP-related transcriptome datasets and single-cell RNA sequencing (scRNA-seq) data were obtained from the Gene Expression Omnibus (GEO) database. IRGs were derived from the Molecular Signatures Database (MSigDB). Core genes were screened using weighted gene co-expression network analysis (WGCNA) and protein-protein interaction (PPI) network analysis, and the expression of the core genes was experimentally validated. The CIBERSORT algorithm was applied to analyze the correlation between core genes and immune cell infiltration. scRNA-seq data were used to explore the expression patterns of core genes in different cell subsets. Finally, Connectivity Map (CMap) and molecular docking were combined to predict potential therapeutic drugs for CRSwNP.
Results: A total of 96 key genes were identified through differential expression analysis and WGCNA, which were mainly enriched in the IL-17 signaling pathway, chemokine-cytokine interaction, and cytokine-cytokine receptor interaction pathways. Among them, FOXP3, C5AR1 and LIF can serve as effective prognostic predictors for CRSwNP. CIBERSORT analysis showed that activated CD4⁺ memory T cells and M2-type macrophages were significantly enriched in CRSwNP tissues. CMap analysis predicted that bicuculline had potential therapeutic value for CRSwNP.
Conclusion: FOXP3, C5AR1 and LIF could serve as diagnostic biomarkers for CRSwNP and may participate in the pathogenesis of CRSwNP by regulating inflammatory responses and immune cell infiltration, which provides novel insights into clarifying the etiology and pathogenesis of CRSwNP.
Keywords:
chronic rhinosinusitis with nasal polyps, inflammatory response, WGCNA, biomarkers, immune microenvironment
Introduction
Chronic rhinosinusitis (CRS) is characterized by persistent nasal congestion, rhinorrhea, and hyposmia, which significantly impairs patients’ quality of life.1 CRS exhibits substantial heterogeneity, with one distinct phenotype—chronic rhinosinusitis with nasal polyps (CRSwNP)—characterized by chronic inflammation and nasal polyp formation.2,3 The pathogenesis of CRSwNP remains incompletely understood, but it is broadly linked to defective sinus epithelial barriers, increased exposure to pathogenic microbes or bacterial colonization, and immune dysregulation.4,5 Clinically, CRSwNP often recurs despite aggressive treatments or exhibits incomplete responses, leading to frequent medical visits and prolonged disease courses.6 Although interventions such as intranasal glucocorticoids, endoscopic sinus surgery, and biologic agents have improved outcomes to some extent, the prognosis of CRSwNP patients remains suboptimal due to unclear molecular mechanisms and a lack of standardized treatment guidelines for diverse clinical scenarios.7
Inflammation represents the body’s immune response to pathogens, damaged cells, or toxic stimuli, which can induce acute or chronic reactions and subsequently cause tissue injury or disease.8 The pathogenesis of CRSwNP is closely linked to dysregulated Th2-type inflammation, characterized by eosinophil infiltration and tissue remodeling.9–11 Its core pathological features include disruption of the mucosal barrier, abnormal immune regulation, and persistent inflammatory infiltration.12,13 Current research indicates that inflammatory biomarkers can predict inflammatory diseases and correlate with their etiology and prognosis; stimuli activate inflammatory cells to induce the production of inflammatory cytokines (eg, IL-1β, IL-6, TNF-α), as well as inflammatory proteins and enzymes, which hold potential as biomarkers for disease diagnosis, prognosis, and therapeutic decision-making.14 Traditional cytokine markers are limited by their transient expression and inability to reflect complex regulatory networks. In contrast, Inflammatory Response-related Genes (IRGs) coordinately regulate inflammatory cascades.15 Expression profiling of IRGs can systematically reveal characteristics of the inflammatory microenvironment while simultaneously reflecting regulatory pathways and immune cell status. Furthermore, gene expression signatures demonstrate greater stability and reproducibility in tissue specimens. Therefore, systematically screening biomarkers based on IRGs holds promise for overcoming the limitations of traditional cytokine markers and elucidating the molecular heterogeneity of CRSwNP.
In this study, we integrated public transcriptomic and single-cell RNA sequencing (scRNA-seq) data from CRSwNP and healthy control samples. Weighted Gene Co-expression Network Analysis (WGCNA) and protein-protein interaction (PPI) network analysis were employed to screen for key genes associated with inflammatory responses.16,17 These genes were validated using independent datasets, and their diagnostic value was evaluated via receiver operating characteristic (ROC) curve analysis. Notably, the differential expression of the key genes was confirmed in clinical specimens obtained from our institution. Furthermore, we performed functional enrichment analysis, immune cell infiltration analysis, and single-cell sequencing analysis to elucidate the critical roles of the core genes in the pathogenesis of CRSwNP. This study aims to identify potential biomarkers and therapeutic targets for the diagnosis and prognosis of CRSwNP.
Materials and Methods
Data Collection and Preprocessing
All transcriptomic and scRNA-seq data sets that were used in this study were downloaded from the GEO database (GEO, http://www.ncbi.nlm.nih.gov/geo).18 The transcriptomic datasets comprised GSE136825, GSE179265, GSE23552, and GSE36830 (Table 1) and the scRNA-seq dataset was GSE196169. The Molecular Signatures Database (MSigDB, https://www.gsea-msigdb.org/gsea/msigdb) provided 1787 IRGs (Table S1). Data preprocessing was done with R software (version 4.2.2). In the case of transcriptomic data, batch effects were eliminated by using the sva package, and platform correction was done by the ComBat function; normalization was carried out with the limma package to normalize gene expression values.
 |
Table 1 Research Dataset and Sources of Sample Information |
Screening of Differentially Expressed Genes (DEGs)
The limma package was used to calculate the DEGs between the CRSwNP group and the control group, where the threshold is adj. P < 0.05 and |logFC| > 2.19 The intersection of DEGs and IRGs was analyzed to get inflammation-associated DEGs, which were visualized by Venn diagram.
Gene Set Enrichment Analysis (GSEA)
GSEA analysis was performed using the GSEA software (version 4.2.3) to explore key signaling pathways closely related to the progression of CRSwNP disease.20 Enrichment that was significant was taken as P < 0.05 and false discovery rate (FDR, q-value) < 0.05. The findings were represented in terms of normalized enrichment score (NES).
Weighted Gene Co-Expression Network Analysis (WGCNA) Identifies Characteristic Gene Sets
The WGCNA was carried out with the help of the package “WGCNA” in R software using the entire transcriptome dataset of the training set.21 To begin with, soft threshold was calculated following the scale-free topology criterion to make sure that the network constructed would be scale-free. The dynamic tree-cutting approach was used to determine gene modules and correlation coefficients between each module and the CRSwNP phenotype were computed to examine the relationship between the module and the phenotype. The key module was chosen as the one having the highest correlation with the CRSwNP phenotype. Candidate hub genes were identified by conducting intersection analysis of genes within this key module, IRGs, and DEGs.
Construction of Protein-Protein Interaction (PPI) Network for Key Genes
Using the STRING database (http://string-db.org), analyze the DEGs associated with the inflammatory response to construct a protein-protein interaction network.17,22 A medium confidence score (0.4) was selected as the minimum score required to screen key gene interactions. The Cytoscape software (version 3.9.1) was used to visualize the network. Betweenness Centrality, Degree, and Maximal Clique Centrality (MCC) algorithms in the CytoHubba plugin were used to identify hub genes. The overlap between genes found by the three algorithms was deemed candidate core genes.
GO/KEGG Enrichment Analysis
Enrichment analysis of candidate core genes was performed using the Metascape database and the “clusterProfiler” package in R software, including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. Enrichment was considered statistically significant at P < 0.05.23
Dataset Validation and ROC Curve Evaluation
The levels of candidate core genes were confirmed in the validation set. The ROC curves were drawn to assess their diagnostic performance, and AUC was applied to measure diagnostic accuracy.24,25 The nomogram model for predicting CRSwNP was built by using the “rms” package in R software. Its effectiveness was measured by calibration curves and concordance indices (C-index), and the Hosmer-Lemeshow test was used to assess the goodness of fit of the calibration model.
Analysis of Immune Infiltration
The infiltration levels of 22 immune cell types in the CRSwNP and control groups were quantified using the CIBERSORT algorithm.26 The relative proportions of immune cells between the two groups were compared, and the correlations between immune cell infiltration and the expression of core genes was further calculated. The “corrplot” and “ggplot2” packages in R software were used to visualize the results.
Single-Cell RNA (scRNA-Seq) Data Analysis
The analysis of scRNA-seq data was carried out with the help of the packages “Seurat” (version 4.3.0) and “harmony” (version 0.1.1) packages in R software. The normalization of gene expression was done using the NormalizeData function and the default method of LogNormalize. The unsupervised clustering of cell subsets was done by the FindNeighbors, FindClusters (resolution = 0.5), and RunUMAP functions. Annotation of cell type was done through the use of the Human Primary Cell Atlas with the help of the SingleR package. Core genes expression patterns among various cell types were represented in UMAP plots and heatmaps.27
Screening of Therapeutic Drugs
The analysis of the Connectivity Map (CMap) was employed to forecast small-molecule compounds against inflammation subtypes of CRSwNP.28 Drug signatures were extracted from CMAP database (https://clue.io/about), and negative CMap scores were generated and selected by inputting the upregulated and downregulated core genes.
Molecular Docking Analysis
Obtain the protein structure files (in PDB format) of key proteins FOXP3, C5AR1, and LIF from the RCSB Protein Data Bank (https://www.rcsb.org/),29 and 3D molecular structures of small-molecule drugs were downloaded from the PubChem database, Molecular docking experiments were conducted with CB-Dock2 platform (https://cadd.labshare.cn/cb-dock2/),30 in which binding energy was used to measure ligand-receptor interactions: a binding energy ≤ −5 kcal/mol means stable binding and ≤ −7 kcal/mol means strong binding. The visual analysis of docking results was done using R software.
Clinical Samples and Experimental Validation
This study was approved by the Ethics Committee of The Affiliated Hospital of Southwest Medical University (Ethics No.: KY2025735) and conducted in strict accordance with the requirements of the Declaration of Helsinki. Written informed consent was obtained from all participants. Samples were collected from inpatients of The Affiliated Hospital of Southwest Medical University. The diagnosis of CRSwNP was based on the 2020 European Position Paper on Rhinosinusitis and Nasal Polyps (EPOS).31 Nasal polyp tissues were collected from 15 CRSwNP patients (8 males and 7 females, aged 28–49 years), and normal nasal mucosal tissues were collected from 15 control individuals who underwent septoplasty (8 males and 7 females, aged 24–45 years). Exclusion criteria include patients with concomitant respiratory diseases (including epidemic respiratory infectious diseases), hypertension, diabetes mellitus, congenital immunodeficiency, concomitant tumors, long-term use of immunosuppressive medications, mental disorders, and emotional or cognitive impairments.
RT-qPCR
Total RNA was extracted from tissue samples using TRIzol reagent (Invitrogen, USA), and complementary DNA (cDNA) was synthesized using a reverse transcription kit (Takara Bio Inc., Japan). Real-time PCR was performed using the SYBR-Green kit (Sichuan Acres Biotechnology Co., Ltd., China). The upstream and downstream primers used for qPCR are listed in Supplementary Table S2. All results were calculated using the 2-∆∆CT method after normalization to GAPDH.
Western Blotting
Total proteins were extracted from minced frozen tissue samples by lysis in RIPA lysis buffer (MCE, USA) supplemented with protease inhibitor (PMSF; Solarbio, China) and a phosphatase inhibitor cocktail (MCE, USA) at a volume ratio of 100:1:1. The protein concentration was determined using a BCA Protein Assay Kit (Solarbio, China). First, 10 μg of total protein per lane was separated by 10% SDS-PAGE electrophoresis (Servicebio, China) and then transferred to a polyvinylidene fluoride (PVDF) membrane (Merck KGaA, Germany) at a constant current of 300 mA. After blocking, the membrane was incubated overnight with the following primary antibodies: anti-FOXP3 antibody (Cusabio, 1:2000), anti-C5AR1 antibody (Cusabio, 1:1000), anti-LIF antibody (Cusabio, 1:1000), and anti-GAPDH antibody (Proteintech Group, 1:50,000). After washing the membrane three times with TBST buffer, the membrane was incubated with the corresponding HRP-conjugated secondary antibody at room temperature for 1 hour. Protein bands were detected by chemiluminescence using ECL reagents (Biosharp, China). Finally, the gray values of the bands were quantitatively analyzed using ImageJ software (version 1.8.0). All experiments were independently repeated three times.
Statistical Analysis
This study conducted statistical analysis using R software (version 4.3.1) and GraphPad Prism 9.0. The differences between groups were analyzed using Student’s t-test, and the correlation between variables was evaluated using Student’s t-test. Additionally, ROC curves and AUC values were combined to assess the diagnostic accuracy of core genes. In this study, a P-value < 0.05 was considered statistically significant. The study’s flow chart is depicted in Figure 1.
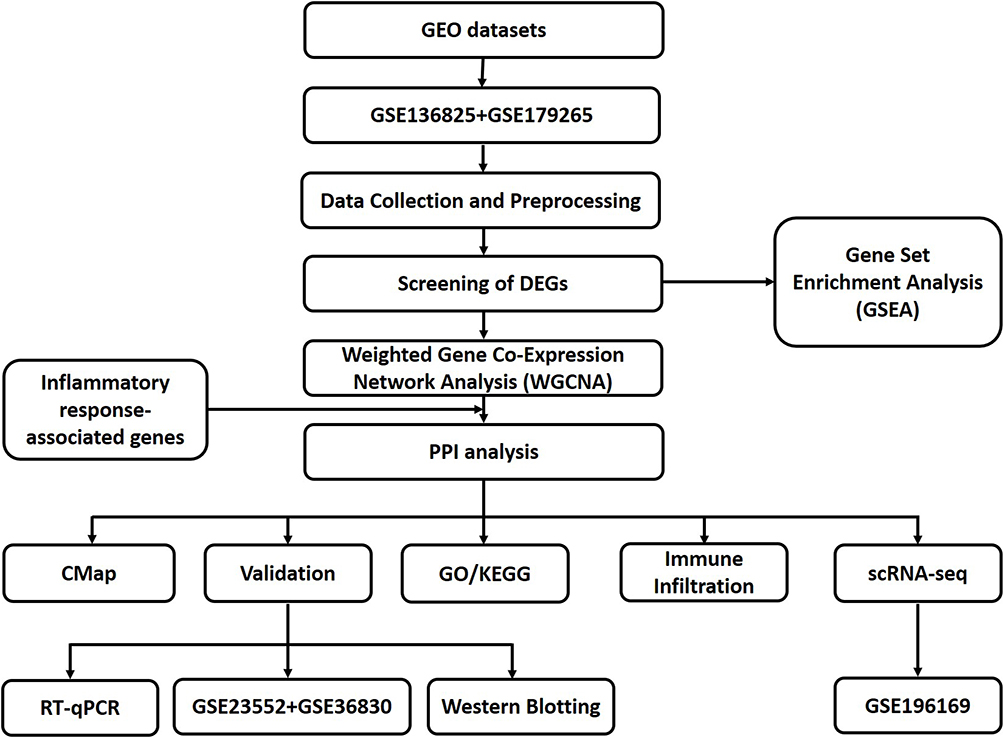 |
Figure 1 The workflow of the study. Abbreviations: DEGs, differentially expressed genes; PPI, Protein–protein interaction; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; CMap, Connectivity Map; RT-qPCR, reverse transcription quantitative PCR; scRNA-seq, Single-Cell RNA Sequencing. |
Results
Identification of Differentially Expressed Genes (DEGs)
After batch effect correction and normalization, differential expression analysis was performed on the training set (59 CRSwNP samples and 35 control samples). Compared with control samples, 975 genes were upregulated and 1805 genes were downregulated in CRSwNP samples (|logFC| > 2, P < 0.05) (Figure 2A). The heatmap shows the expression profiles of the top 50 upregulated and top 50 downregulated DEGs (Figure 2B).
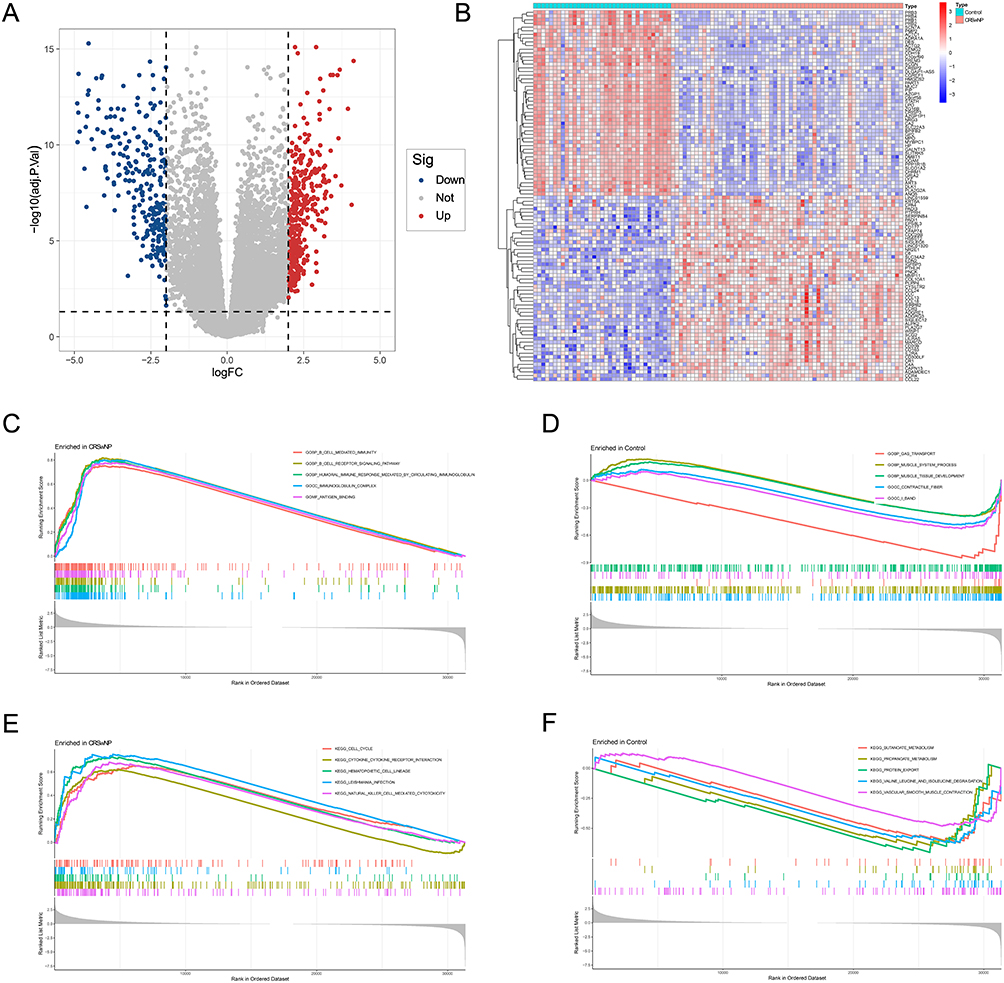 |
Figure 2 Identification of DEGs in the training set and function enrichment analysis of DEGs. (A and B) The volcano plot (A) and heatmap (B) of the DEGs. (C–F). GSEA enrichment results for CRSwNP and control groups. |
Gene Set Enrichment Analysis (GSEA)
GSEA results revealed that CRSwNP samples were significantly enriched in immune-related biological processes, including cell-mediated immunity, B-cell receptor signaling pathway, humoral immune response mediated by circulating immunoglobulins, and lymphocyte-mediated immunity (Figure 2C and D). In addition, inflammation-related pathways such as cytokine-cytokine receptor interaction, hematopoietic cell lineage, natural killer (NK) cell-mediated cytotoxicity, and cell cycle signaling pathway were also significantly enriched in CRSwNP samples (Figure 2E and F).
WGCNA for Identification of CRSwNP Co-Expression Modules
WGCNA was performed on the training set transcriptome data, and 12 gene modules with distinct expression patterns were identified using the dynamic tree-cutting method (Figure 3A and B). Module-trait correlation analysis showed that the ME-turquoise module had the strongest correlation with the CRSwNP phenotype (Figure 3C and D). Intersection analysis of genes in the ME-turquoise module, IRGs, and DEGs identified 96 overlapping genes, which were defined as candidate hub genes (Figure 3E).
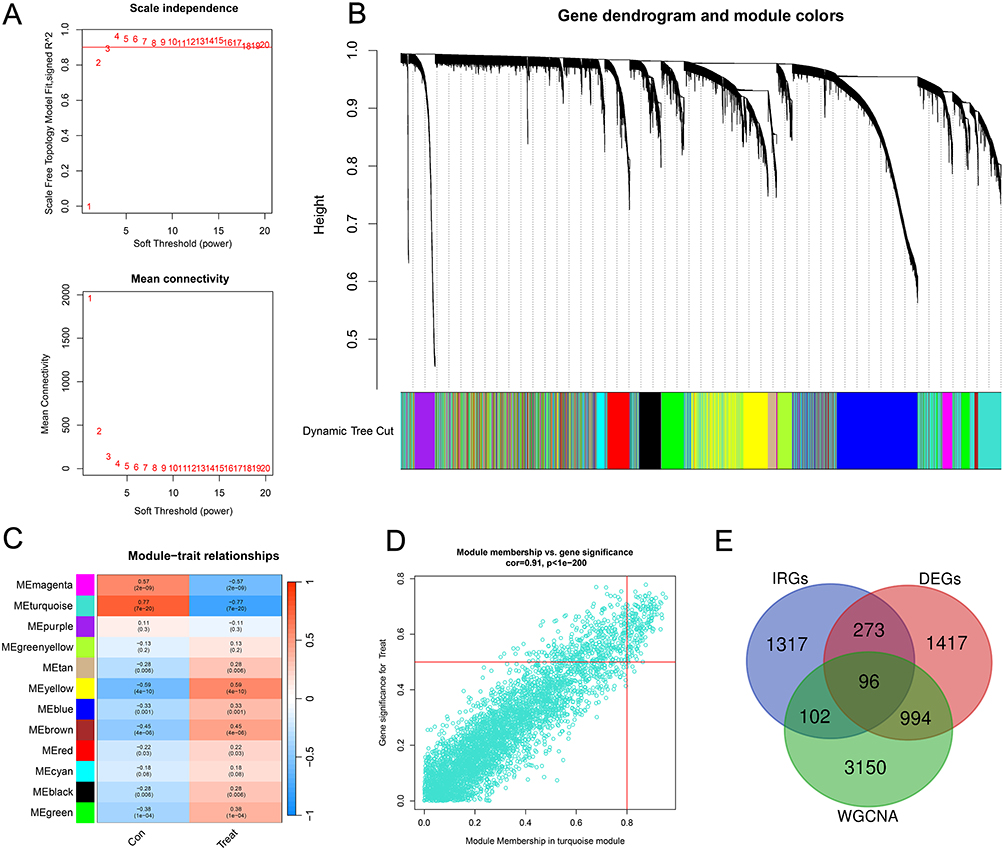 |
Figure 3 Identification of the key module in CRSwNP by WGCNA. (A) Scale-free exponent and average connectivity for each soft threshold. (B) Dendrogram of gene clusters, with different colors representing different modules. (C) Clustered tree plots of co-expression network modules. (D) Scatterplot of the strongest positive correlation of the turquoise module in the training set with the CRSwNP. (E) Venn diagram of the intersection of key module genes, IRGs, and DEGs. |
Functional Enrichment Analyses
To further explore the potential functions of these 96 candidate hub genes, GO and KEGG enrichment analyses were performed. GO enrichment analysis revealed that these genes are primarily involved in biological functions such as positive regulation of cytokine production, regulation of inflammatory response, secretory granule membrane, basal part of cell, cytokine activity and receptor ligand activity (Figure 4A). Similarly, KEGG enrichment analysis demonstrated that these hub genes are enriched in pathways including cytokine-cytokine receptor interaction, neuroactive ligand-receptor interaction, rheumatoid arthritis, viral protein interaction with cytokine and cytokine receptor, chemokine signaling pathway, and IL-17 signaling pathway (Figure 4B and C).
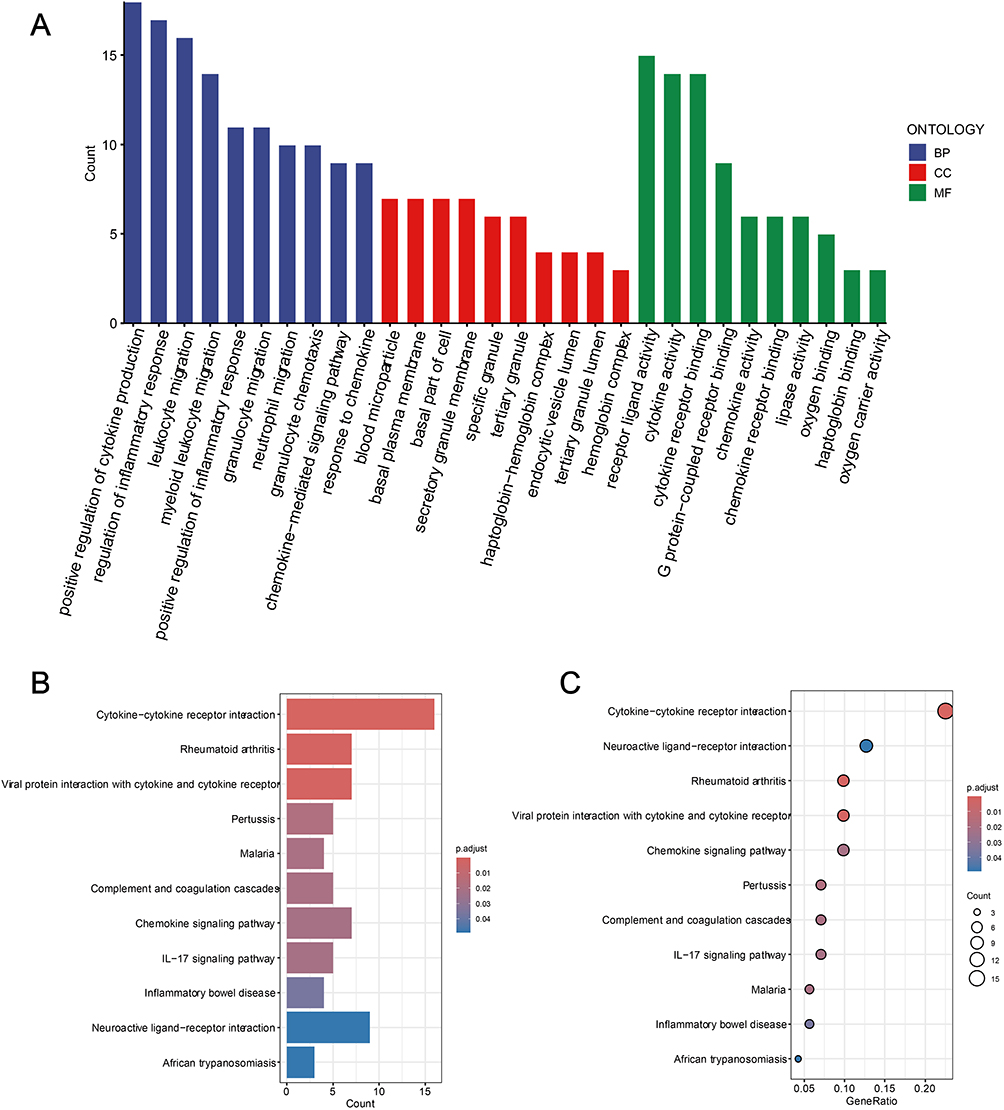 |
Figure 4 Enrichment analysis results of 96 candidate Hub genes. (A) GO analysis results of the first 10 enrichments. (B and C) KEGG pathway enrichment analysis of candidate Hub genes. Spot size indicates the number of genes and color indicates the P-value. Abbreviations: BP, Biological process; CC, Cellular component; MF, Molecular function. |
Screening of Core Genes via PPI Network Analysis
A PPI network was constructed for the 96 candidate hub genes using the STRING database (confidence score ≥ 0.4), and visualized with Cytoscape software. The top 15 genes identified by Betweenness Centrality (Figure 5A), Degree (Figure 5B), and MCC (Figure 5C) algorithms were selected (Table S3), and their intersection yielded 7 candidate core genes (Figure 5D and E).
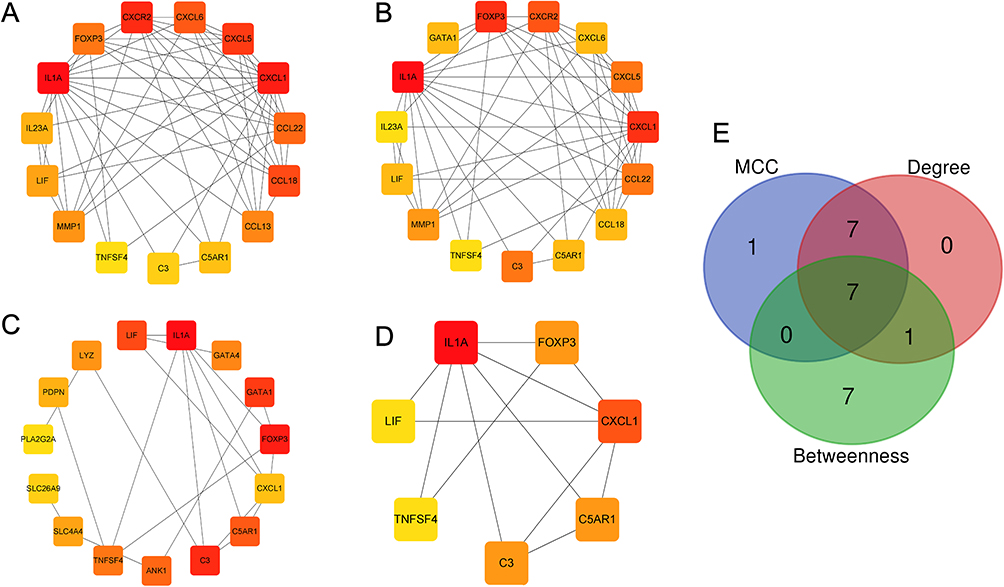 |
Figure 5 Screening of hub genes. (A–C) Algorithmic analyses of MCC (A), Degree (B), and Betweenness (C) in the PPI network; (D) Intersection genes of the 3 algorithms for PPI network graphs. (E) Venn diagram of the key hub genes of the 3 algorithms. |
Verification of Core Genes
Validation in the training and validation sets showed that FOXP3, C5AR1, and LIF exhibited significant differential expression between CRSwNP and control groups (P < 0.05) (Figure 6A and B), with AUC values exceeding 0.7 in both datasets (Figure 6C and D), indicating good diagnostic efficacy for CRSwNP. A nomogram model constructed based on FOXP3, C5AR1, and LIF showed high predictive accuracy for CRSwNP, as confirmed by calibration curves (Figure 7A and B). RT-qPCR and Western blot results demonstrated that the expression levels of FOXP3, C5AR1, and LIF in nasal polyp tissues were significantly higher than those in normal nasal mucosa tissues (P < 0.05) (Figure 7C–I).
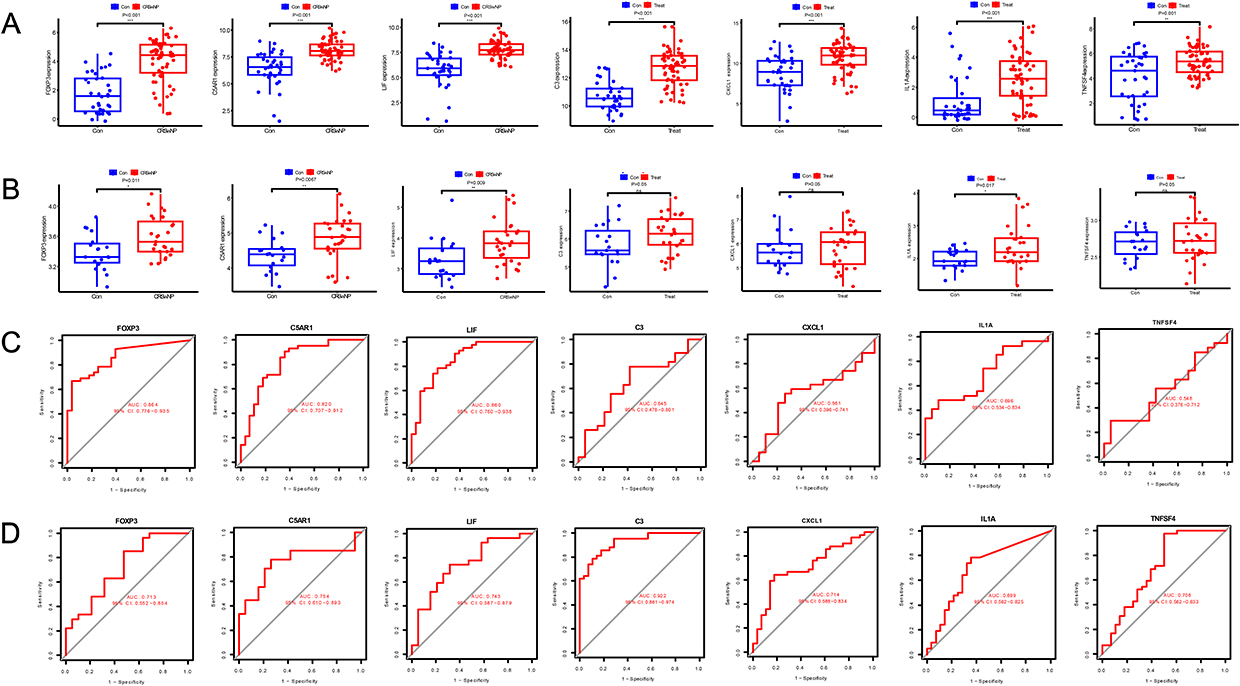 |
Figure 6 Expression validation of hub genes and Receiver Operating Characteristic (ROC) analysis. (A) Expression of Hub genes in CRSwNP and controls in the training set; (B) Expression of Hub genes in CRSwNP and controls in the validation set. (C) ROC curves of hub genes in the training set; (D) ROC curves of hub genes in the validation set; *P<0.05, **P<0.01, ***P<0.001. |
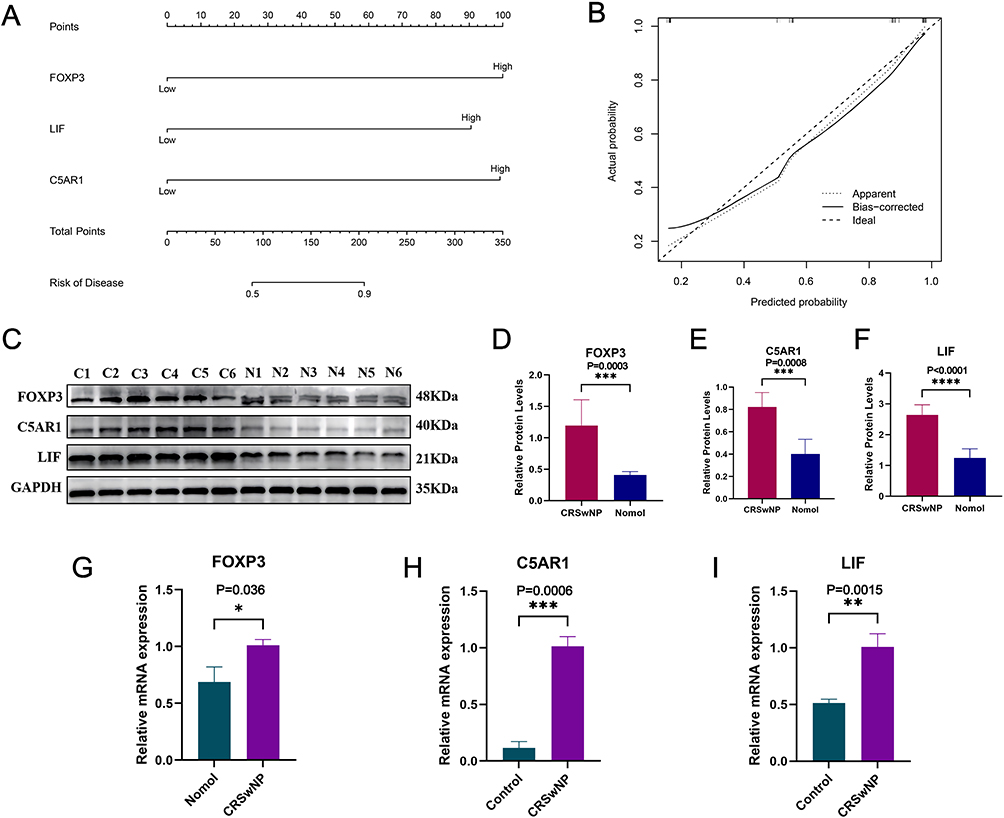 |
Figure 7 Prognostic prediction and differential expression validation of hub genes. (A) Nomogram model for predicting overall survival, (B) Calibration curves for evaluating the accuracy of the nomogram model. (C–I) The expression levels of core genes FOXP3, C5AR1, and LIF were verified by PCR and Western blotting; *P<0.05, **P<0.01, ***P<0.001,****P<0.0001. |
Immune Cell Infiltration Analysis
CIBERSORT algorithm analysis showed that the infiltration levels of CD4⁺ memory activated T cells and M2 macrophages in CRSwNP tissues were significantly higher than those in control tissues (P < 0.05) (Figure 8A–C). Correlation analysis revealed that FOXP3 expression was significantly positively correlated with the infiltration of CD4⁺ memory-activated T cells and significantly negatively correlated with the infiltration of resting mast cells and activated NK cells (P < 0.05) (Figure 8D); C5AR1 expression was significantly positively correlated with the infiltration of monocytes, neutrophils, resting NK cells, and eosinophils, and significantly negatively correlated with the infiltration of resting mast cells, naive B cells, and M1-type macrophages (P < 0.05) (Figure 8E); LIF expression was significantly positively correlated with the infiltration of monocytes, activated dendritic cells, and CD4⁺ memory activated T cells (P < 0.05) (Figure 8F).
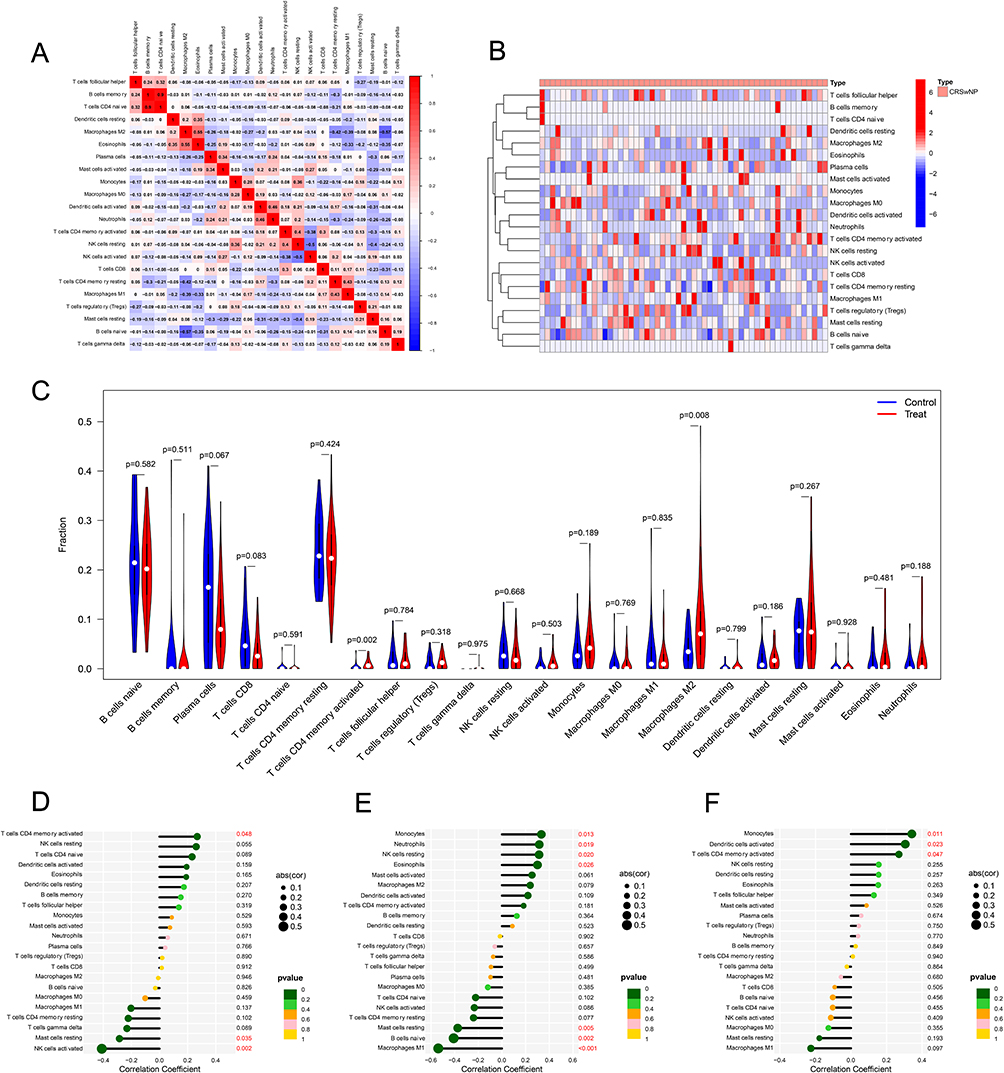 |
Figure 8 CIBERSORT algorithm analysis of the CRSwNP group versus the control group. (A) Immune cell correlation analysis, (B) Heatmap of the distribution of immune cell infiltration in the CRSwNP tissues. (C) Differential analysis of the infiltration of 22 immune cells in the CRSwNP group and the control group. (D–F) Correlation analysis between FOXP3 (D), C5AR1 (E), LIF (F) and immune cells. |
Single-Cell RNA Sequencing (scRNA-Seq) Analysis of Core Genes
Analysis of scRNA-seq data from GSE196169 identified 9 major cell types, including mast cells, T cells, plasma cells, NK cells, plasmacytoid dendritic cells, macrophages, epithelial cells, monocytes, and B cells (Figure 9A and B). Expression pattern analysis showed that FOXP3 was predominantly expressed in T cells (Figure 9C), C5AR1 was enriched in monocytes (Figure 9D), and LIF was mainly expressed in mast cells (Figure 9E). The heatmap of average expression levels further confirmed the cell-type-specific expression of these core genes (Figure 9F).
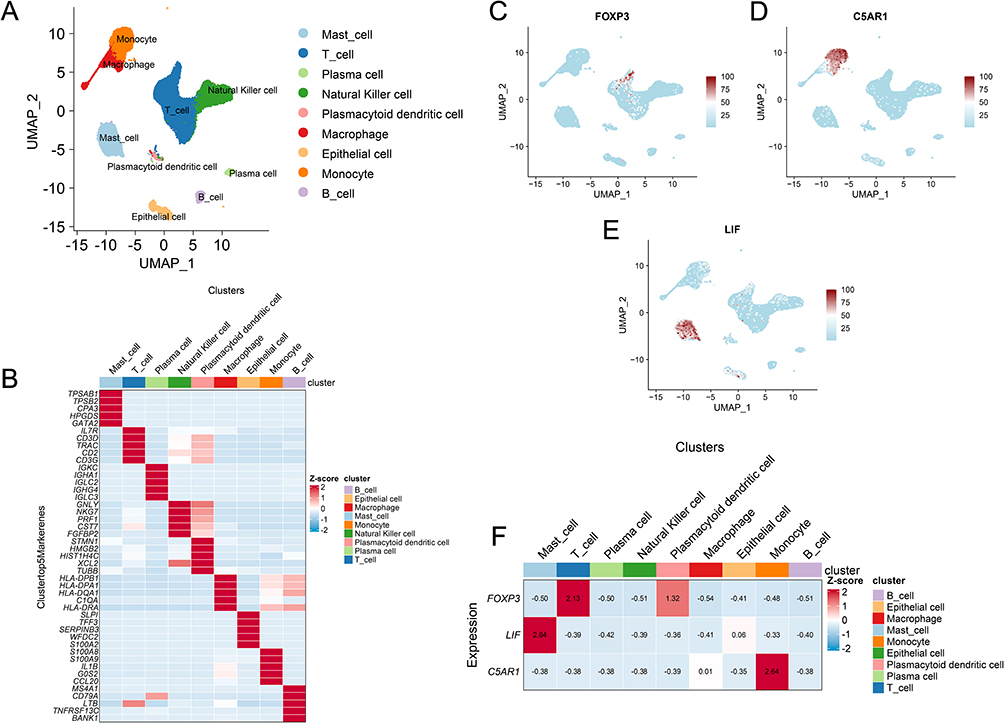 |
Figure 9 Analysis of single-cell data (GSE196169) to identify different immune subtypes. (A) Nine cell types were identified based on cell type-specific markers. (B) Heat map of characteristic genes of immune cell clusters. Each cell cluster is represented by 5 specifically expressed genes. (C–E) UMAP plots showing the spatial distribution of FOXP3 (C), C5AR1 (D), and LIF (E) expression across all cell types; (F) Heatmap of the relative expression levels of FOXP3, C5AR1, and LIF across different cell subtypes. |
Prediction of Therapeutic Targets
To identify potential therapeutic agents for CRSwNP, we performed CMap analysis. Compounds with negative connectivity scores are considered to have the potential to reverse disease-associated gene expression signatures. Among all compounds tested, bicuculline exhibited the lowest CMap score (Table S4), suggesting its strong therapeutic potential. Bicuculline is classically known as a competitive antagonist of GABA receptors, with emerging roles in immune modulation. We then performed molecular docking to evaluate its binding affinity to the three core proteins identified in this study. Molecular docking revealed that bicuculline binds with high affinity to FOXP3 (−7.3 kcal/mol) (Figure 10A and B), C5AR1 (−9.5 kcal/mol) (Figure 10C and D), and LIF (−10.7 kcal/mol) (Figure 10E and F), all below the threshold for strong binding. These results suggest that bicuculline may exert multi-target effects on key inflammatory regulators in CRSwNP.
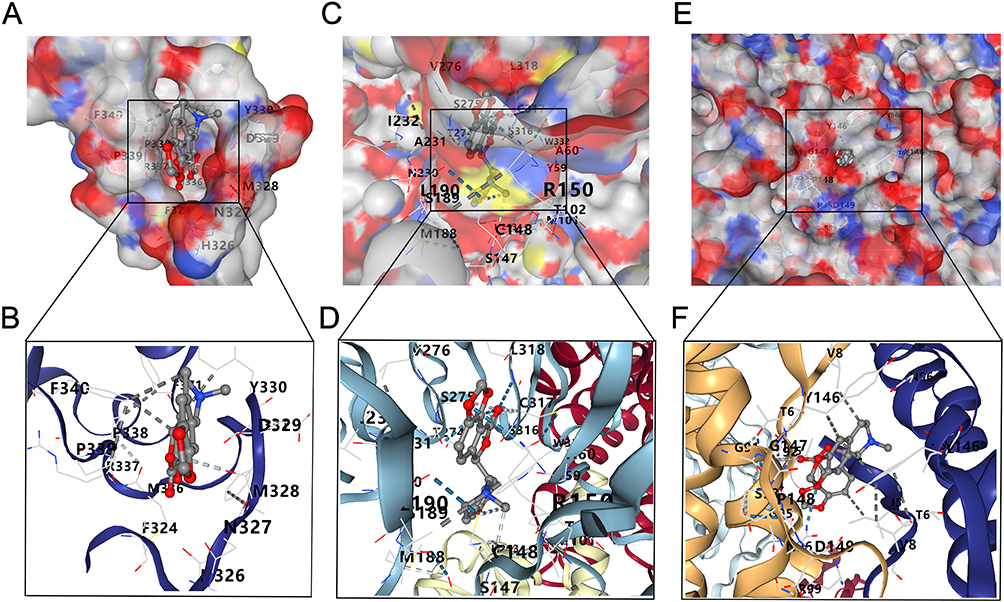 |
Figure 10 The best docked position of Bicuculline inside FOXP3 (A and B), C5AR1 (C and D) and LIF protein (E and F). |
Discussion
CRSwNP is a chronic inflammatory disease affecting the nasal cavity and paranasal sinuses, characterized by a complex pathogenesis. Despite the availability of diverse treatment modalities, its recurrence rate remains persistently high, severely impairing patients’ quality of life.32 Accumulating evidence indicates that immune dysregulation and persistent inflammatory responses are core drivers of CRSwNP progression;7 however, the specific molecular mechanisms and key regulatory targets still require further elucidation. By integrating multi-dimensional bioinformatics analyses with experimental validation, this study systematically investigated the role of IRGs in CRSwNP, supplementing and deepening the current understanding of this disease.
In this study, we integrated four publicly available RNA sequencing datasets to perform comprehensive bioinformatics analysis. Through application of the WGCNA algorithm, we identified distinct gene modules associated with CRSwNP. GSEA revealed significant enrichment of inflammation-related pathways in CRSwNP patients compared with controls, particularly cytokine-cytokine receptor interactions and NK cell-mediated cytotoxicity pathways. Notably, immune-related biological processes—including B cell-mediated immune response, B cell receptor signaling pathway, circulating immunoglobulin-mediated humoral immune response, and lymphocyte-mediated immune response—were prominently enriched. These findings collectively suggest that dysregulated immune responses play a pivotal role in the inflammatory progression of CRSwNP. Further KEGG enrichment analysis of 96 core IRGs demonstrated significant pathway enrichment in cytokine-cytokine receptor interactions, chemokine signaling, and IL-17 signaling pathways. This is consistent with previous reports showing that: (1) cytokine-cytokine receptor interaction pathways are characteristically enriched in CRSwNP pathogenesis;33,34 (2) IL-17A disrupts nasal mucosal epithelial barrier function via activation of the ERK/STAT3 pathway;35 and (3) IL-17A-induced cellular pyroptosis through ERK signaling contributes to steroid resistance in CRSwNP.36 Our findings thus corroborate existing evidence highlighting inflammatory mechanisms as critical drivers of CRSwNP development and progression. Notably, pathway analysis revealed concurrent enrichment of inflammatory response-associated disease pathways, including those involved in pertussis, inflammatory bowel disease, malaria, and African trypanosomiasis. This pattern suggests potential shared inflammatory mechanisms across these clinically distinct conditions, though further investigation is required to elucidate specific pathogenic overlaps.
We further screened CRSwNP-related inflammatory response genes using PPI networks and integrated results from three independent algorithmic approaches, ultimately identifying FOXP3, C5AR1, and LIF as core hub genes in CRSwNP. Compared with healthy controls, these genes exhibited significantly upregulated expression in nasal polyp tissues of CRSwNP patients and demonstrated remarkable diagnostic utility, with AUC values exceeding 0.7. Furthermore, protein expression levels of C5AR1, FOXP3, and LIF were significantly elevated in CRSwNP tissues relative to control tissues. FOXP3 is recognized as a specific marker for regulatory T (Treg) cells, which are crucial for maintaining immune homeostasis by mitigating excessive immune responses. In autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus, FOXP3 expression may be altered due to deregulation of Treg cell functionality. Study results have indicated that CD8⁺CD25⁺Foxp3⁺ Tregs regulate neutrophilic inflammation, whereas budesonide nasal spray therapy does not ameliorate inflammation in neutrophil-dominated CRSwNP.37 Furthermore, CD4⁺Foxp3⁺ Tregs have been shown to modulate the inflammatory milieu in CRSwNP.38 FOXP3 directly governs a core set of Treg cell signature genes essential for Treg cell homeostasis and sustains its own transcription through autoregulatory loops. As a hallmark transcription factor of regulatory T cells (Tregs), dysregulation of FOXP3 expression or function is a common feature of various autoimmune diseases (such as rheumatoid arthritis and systemic lupus erythematosus) and allergic disorders.39,40 In this study, FOXP3 was predominantly expressed in T cells, and its expression was positively correlated with activated CD4⁺ memory T cell infiltration. This suggests that FOXP3 may be involved in the regulation of T cell subset differentiation in CRSwNP, leading to the accumulation of activated CD4⁺ memory T cells and promoting persistent inflammation.
C5AR1 serves as the receptor for C5a, a key effector molecule of the complement system, and plays a central role in innate immune defense. Previous studies have demonstrated that aberrant activation of the C5AR1 signaling pathway contributes to the pathogenesis of various inflammatory diseases, including sepsis, asthma, and acute respiratory distress syndrome.41 In this study, C5AR1 was enriched in monocytes, and its expression was positively correlated with the infiltration of monocytes, neutrophils, and eosinophils. Monocytes can differentiate into macrophages, and neutrophils and eosinophils are key inflammatory cells in CRSwNP, participating in tissue damage and inflammatory response amplification.42 This suggests that C5AR1 may promote the recruitment and activation of inflammatory cells through the complement system, aggravating mucosal inflammation and tissue remodeling in CRSwNP. The complement and coagulation cascade pathways are markedly enriched in CRSwNP,43 which further supports the involvement of C5AR1 in the complement-mediated inflammatory response of CRSwNP.
LIF is a pleiotropic cytokine whose functional receptor is expressed in multiple organs, such as the liver, bone, uterus, kidney, and central nervous system. It is essential for both the induction and maintenance phases of iPSCs derived from mature somatic cells.43 Recent studies have also revealed the potential role of LIF in airway inflammation and remodeling in asthma.44 In this study, LIF was mainly expressed in mast cells, and its expression was positively correlated with the infiltration of monocytes, activated dendritic cells, and CD4⁺ memory activated T cells. Mast cells are important in the pathogenesis of allergic diseases, and their degranulation can release a variety of inflammatory mediators, promoting Th2-type inflammation.45 Activated dendritic cells can present antigens and promote T cell activation, while CD4⁺ memory activated T cells can secrete inflammatory cytokines, sustaining the inflammatory response. This suggests that LIF may regulate the function of multiple immune cells, participating in the formation of the inflammatory microenvironment in CRSwNP.
We evaluated the infiltration abundance of 22 immune cell subsets in CRSwNP tissues and controls based on the expression of hub genes. Notably, the levels of activated CD4⁺ memory T cells and M2 macrophages were elevated in CRSwNP tissues compared with controls. Previous research has indicated that M2 macrophage infiltration is substantially increased in CRSwNP nasal polyps, and their quantity positively correlates with type 2 mediators such as IL-5, eosinophil cationic protein (ECP), and IgE.42 M2-type macrophages are critical in the pathogenesis of CRSwNP, where their increased presence aligns with enhanced levels of type 2 inflammatory mediators and disease severity.46 These cells are activated by various factors that elicit anti-inflammatory responses, facilitate tissue remodeling, and mediate Th2 immune responses. In CRSwNP, M2-type macrophages contribute to tissue remodeling by promoting fibroblast proliferation and disrupting the extracellular matrix via secretion of growth factors. Additionally, they regulate anti-inflammatory responses through molecules such as CD163 and interact with the complement system, with complement and coagulation cascade pathways being markedly enriched in CRSwNP.43 To more precisely dissect the functional contexts of these genes, we analyzed scRNA-seq data. The results revealed highly cell-type-specific expression patterns: FOXP3 was enriched in T cell subsets, C5AR1 was highly expressed in mast cells, and LIF was predominantly produced by the monocyte/macrophage lineage. This differential expression pattern strongly suggests that these genes act synergistically through distinct immune cell subsets in CRSwNP, collectively forming a complex inflammatory microenvironment network.45 Elucidating the cell-type-specific functions of these genes will lay a solid foundation for the development of cell-targeted therapies.
Based on CMap analysis, we predicted that bicuculline may have potential therapeutic value for CRSwNP. Molecular docking results indicated that bicuculline exhibits strong binding affinities with FOXP3, C5AR1, and LIF. As a γ-aminobutyric acid (GABA) receptor antagonist, bicuculline may exert immunomodulatory effects through both direct target binding and GABA-mediated immune regulation, given that GABA receptors are expressed on immune cells, including T cells, macrophages, and mast cells.47,48 Importantly, the target binding of bicuculline aligns with the pathogenesis of CRSwNP, potentially through modulation of FOXP3-mediated regulatory T cell function, C5AR1-driven complement activation, and LIF-related mast cell interactions. However, these computational predictions require experimental validation to confirm binding and assess therapeutic efficacy. In addition to CRSwNP, the core genes identified in this study may also be relevant to other inflammatory airway diseases, particularly local allergic rhinitis (LAR). LAR shares key pathological features with CRSwNP, including local type 2 inflammation, mast cell and eosinophil activation, and immune cell infiltration, as well as common inflammatory pathways such as cytokine-cytokine receptor interactions and chemokine signaling. FOXP3, as a master regulator of Treg cells, may contribute to the local immune imbalance characteristic of LAR by modulating T cell differentiation and function. C5AR1, which we found to be correlated with eosinophil and monocyte infiltration in CRSwNP, may similarly drive complement-mediated inflammatory cell recruitment in LAR. These shared mechanisms suggest that the molecular insights gained from this study may extend beyond CRSwNP and inform research on LAR and other localized inflammatory diseases of the upper airways.49
This study has several limitations. First, the sample size for clinical validation was relatively small, and larger-scale, multi-center studies are needed to confirm the diagnostic value of FOXP3, C5AR1, and LIF. Second, and more importantly, our study design only compared CRSwNP samples with healthy control tissues. While this approach effectively identifies genes associated with CRSwNP inflammation, it cannot distinguish whether these genes are specific to CRSwNP or represent inflammatory features shared with other inflammatory airway diseases. Future studies should incorporate multiple disease control groups to rigorously assess the disease specificity of potential biomarkers. Third, the functional mechanisms of these core genes in CRSwNP were not fully explored, and in vitro and in vivo experiments are required to elucidate their specific roles. Fourth, the therapeutic potential of bicuculline was only predicted through bioinformatics analysis and molecular docking, and further preclinical and clinical studies are needed to verify its efficacy and safety in CRSwNP treatment. Fifth, the bioinformatics analysis relies on public datasets, which may have potential selection biases; future studies can collect more diverse samples for verification. Sixth, the regulatory relationships between core genes (FOXP3, C5AR1, LIF) were not explored, and whether there is mutual regulation between them needs to be further studied.
Conclusions
In summary, this study identified three pivotal IRGs (FOXP3, C5AR1, and LIF) in CRSwNP. These genes are specifically expressed in particular immune cell subsets, where they regulate the recruitment and function of local immune cells, thereby influencing the inflammatory microenvironment and contributing to the pathogenesis of CRSwNP. Bicuculline is a promising candidate for targeted therapy of CRSwNP by binding to these core genes. These findings provide new insights into the etiology and pathogenesis of CRSwNP, and offer potential biomarkers and therapeutic targets for clinical practice. However, to translate these findings into clinical practice, further investigation in larger and more diverse populations is essential.
Abbreviations
CRSwNP, Chronic rhinosinusitis with nasal polyps; CRS, Chronic rhinosinusitis; IRGs, inflammatory response-associated genes; DEGs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; PPI, protein-protein interaction; GSEA, gene set enrichment analysis; KEGG, Kyoto Encyclopedia of Genes and Genomes; GO, Gene Ontology; MSigDB, Molecular Signatures Database; scRNA-seq, Single-Cell RNA Sequencing; CMap, Connectivity Map; ROC, Receiver Operating Characteristic; AUC, area under the curve; RT-qPCR, reverse transcription quantitative PCR; EMT, epithelial-mesenchymal transition; UMAP, Uniform Manifold Approximation and Projection; NK, natural killer; iPSCs, induced pluripotent stem cells; MCC, Maximal Clique Centrality; ECP, eosinophil cationic protein; GABA, γ-aminobutyric acid.
Data Sharing Statement
The datasets analyzed in this study are available in the GEO (http://www.ncbi.nlm.nih.gov/geo/) and MSigDB (https://www.gsea-msigdb.org/gsea/msigdb) databases. Datasets used in this study can be obtained from the corresponding author upon request.
Ethics Approval and Informed Consent
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the Affiliated Hospital of Southwest Medical University (KY2025735) on December 22, 2025. Informed consent for participation was obtained from all subjects involved in the study.
Author Contributions
MD Arefin Hossen: Conceptualization, Methodology, Visualization, Writing-Original Draft, Writing-Review & Editing, Supervision. Sai Liang: Conceptualization, Methodology, Data Curation, Writing-Original Draft, Writing-Review & Editing. Runjie Yan: Data Curation, Methodology, Formal Analysis, Writing-Original Draft. Han Zhu: Formal Analysis, Investigation, Writing-Original Draft. Shishi Yang: Data Curation, Formal Analysis, Writing-Review & Editing. Gang Qin: Conceptualization, Formal Analysis, Investigation, Writing-Review & Editing, Supervision. All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors did not receive support from any organization for the submitted work.
Disclosure
The authors declare that they have no competing interests in the reporting of this work.
References
1. Hopkins C. Chronic rhinosinusitis with nasal polyps. N Engl J Med. 2019;381(1):55–19. doi:10.1056/NEJMcp1800215
2. Cho SH, Hamilos DL, Han DH, Laidlaw TM. Phenotypes of chronic rhinosinusitis. J Allergy Clin Immunol Pract. 2020;8(5):1505–1511. doi:10.1016/j.jaip.2019.12.021
3. Orlandi RR, Kingdom TT, Smith TL, et al. International consensus statement on allergy and rhinology: rhinosinusitis 2021. Int Forum Allergy Rhinol. 2021;11(3):213–739. doi:10.1002/alr.22741
4. Yan B, Lan F, Li J, Wang C, Zhang L. The mucosal concept in chronic rhinosinusitis: focus on the epithelial barrier. J Allergy Clin Immunol. 2024;153(5):1206–1214. doi:10.1016/j.jaci.2024.01.015
5. Asamori T, Katoh H, Takata M, et al. Molecular mimicry-driven autoimmunity in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2025;155(5):1521–1535. doi:10.1016/j.jaci.2025.02.014
6. Fokkens WJ, De Corso E, Backer V, et al. EPOS2020/EUFOREA expert opinion on defining disease states and therapeutic goals in CRSwNP. Rhinology. 2024;62(3):287–298. doi:10.4193/Rhin23.415
7. Kratchmarov R, Dharia T, Buchheit K. Clinical efficacy and mechanisms of biologics for chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2025;155(5):1401–1410. doi:10.1016/j.jaci.2025.03.011
8. Deng Z, Liu S. Inflammation-responsive delivery systems for the treatment of chronic inflammatory diseases. Drug Deliv Transl Res. 2021;11(4):1475–1497. doi:10.1007/s13346-021-00977-8
9. Tokunaga T, Sakashita M, Haruna T, et al. Novel scoring system and algorithm for classifying chronic rhinosinusitis: the JESREC study. Allergy. 2015;70(8):995–1003. doi:10.1111/all.12644
10. Bachert C, Hicks A, Gane S, et al. The interleukin-4/interleukin-13 pathway in type 2 inflammation in chronic rhinosinusitis with nasal polyps. Front Immunol. 2024;15:1356298. doi:10.3389/fimmu.2024.1356298
11. AlBloushi S, Al-Ahmad M. Exploring the immunopathology of type 2 inflammatory airway diseases. Front Immunol. 2024;15:1285598. doi:10.3389/fimmu.2024.1285598
12. Baird AM, Masliah J, Filip P, et al. Histopathologic features of biologic therapy nonresponders in chronic rhinosinusitis with nasal polyposis. Int Forum Allergy Rhinol. 2024;14(5):939–949. doi:10.1002/alr.23283
13. Chen SY, Liu PQ, Qin DX, Lv H, Zhou HQ, Xu Y. E3ubiquitin ligase NEDD4L inhibits epithelial-mesenchymal transition by suppressing the β-catenin/HIF-1α positive feedback loop in chronic rhinosinusitis with nasal polyps. Acta Pharmacol Sin. 2024;45(4):831–843. doi:10.1038/s41401-023-01190-8
14. Schleimer RP. Immunopathogenesis of chronic rhinosinusitis and nasal polyposis. Annu Rev Pathol. 2017;12(1):331–357. doi:10.1146/annurev-pathol-052016-100401
15. Yang P, Miao Y, Wang T, Sun J. Identification of diagnostic markers related to inflammatory response and cellular senescence in endometriosis using machine learning and in vitro experiment. Inflamm Res. 2024;73(7):1107–1122. doi:10.1007/s00011-024-01886-5
16. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9(1):559. doi:10.1186/1471-2105-9-559
17. Chen YC, Chen YH, Wright JD, Lim C. PPI-hotspot(DB): database of protein-protein interaction hot spots. J Chem Inf Model. 2022;62(4):1052–1060. doi:10.1021/acs.jcim.2c00025
18. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
19. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
20. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
21. Pei G, Chen L, Zhang W. WGCNA application to proteomic and metabolomic data analysis. Methods Enzymol. 2017;585:135–158.
22. Romero-Molina S, Ruiz-Blanco YB, Harms M, Münch J, Sanchez-Garcia E. PPI-detect: a support vector machine model for sequence-based prediction of protein-protein interactions. J Comput Chem. 2019;40(11):1233–1242. doi:10.1002/jcc.25780
23. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
24. Obuchowski NA, Bullen JA. Receiver operating characteristic (ROC) curves: review of methods with applications in diagnostic medicine. Phys Med Biol. 2018;63(7):07tr01. doi:10.1088/1361-6560/aab4b1
25. Namdar K, Haider MA, Khalvati F. A modified AUC for training convolutional neural networks: taking confidence into account. Front Artif Intell. 2021;4:582928. doi:10.3389/frai.2021.582928
26. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–259.
27. Shen Y, Leng L, Hu Y. Exploring core genes associated with sepsis and systemic inflammatory response syndrome using single-cell sequencing technology. J Inflamm Res. 2025;18:1815–1838. doi:10.2147/JIR.S448900
28. Musa A, Ghoraie LS, Zhang SD, et al. A review of connectivity map and computational approaches in pharmacogenomics. Brief Bioinform. 2018;19(3):506–523. doi:10.1093/bib/bbw112
29. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
30. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
31. Fokkens WJ, Lund VJ, Hopkins C, et al. European position paper on rhinosinusitis and nasal polyps 2020. Rhinology. 2020;58(Suppl S29):1–464. doi:10.4193/Rhin20.401
32. Min HK, Lee S, Kim S, et al. Global incidence and prevalence of chronic rhinosinusitis: a systematic review. Clin Exp Allergy. 2025;55(1):52–66. doi:10.1111/cea.14592
33. Li K, Liu FF. Analysis of competing endogenous RNA (ceRNA) crosstalk in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2022;12(12):1468–1479. doi:10.1002/alr.23008
34. Ishino T, Takeno S, Takemoto K, et al. Distinct gene set enrichment profiles in eosinophilic and non-eosinophilic chronic rhinosinusitis with nasal polyps by bulk RNA barcoding and sequencing. Int J Mol Sci. 2022;23(10):5653. doi:10.3390/ijms23105653
35. Wu H, Li Y, Li X, et al. IL-17A disrupts the nasal mucosal epithelial barrier in patients with chronic rhinosinusitis by activating the ERK/STAT3 pathway. Rhinology. 2024;62(6):726–738. doi:10.4193/Rhin24.127
36. Li Y, Chang LH, Huang WQ, et al. IL-17A mediates pyroptosis via the ERK pathway and contributes to steroid resistance in CRSwNP. J Allergy Clin Immunol. 2022;150(2):337–351. doi:10.1016/j.jaci.2022.02.031
37. Lin L, Lan J, Dai F, Wei J, Chen Z. Effect of local corticosteroid administration on CD8+CD25+Foxp3+ tregs in neutrophilic CRSwNP. ORL J Otorhinolaryngol Relat Spec. 2022;84(5):396–405. doi:10.1159/000524385
38. Yusoff NN, Ahmad S, Wan Abdul Rahman WF, et al. CD4+ Foxp3+ regulatory T-cells in modulating inflammatory microenvironment in chronic rhinosinusitis with nasal polyps: progress and future prospect. Cytokine. 2024;178:156557. doi:10.1016/j.cyto.2024.156557
39. Caso F, Saviano A, Tasso M, et al. Analysis of rheumatoid- vs psoriatic arthritis synovial fluid reveals differential macrophage (CCR2) and T helper subsets (STAT3/4 and FOXP3) activation. Autoimmun Rev. 2022;21(12):103207. doi:10.1016/j.autrev.2022.103207
40. Sumida TS, Cheru NT, Hafler DA. The regulation and differentiation of regulatory T cells and their dysfunction in autoimmune diseases. Nat Rev Immunol. 2024;24(7):503–517. doi:10.1038/s41577-024-00994-x
41. Wang Y, Liu W, Xu Y, et al. Revealing the signaling of complement receptors C3aR and C5aR1 by anaphylatoxins. Nat Chem Biol. 2023;19(11):1351–1360. doi:10.1038/s41589-023-01339-w
42. Banks CA, Schlosser RJ, Wang EW, Casey SE, Mulligan RM, Mulligan JK. Macrophage infiltrate is elevated in CRSwNP sinonasal tissue regardless of atopic status. Otolaryngol Head Neck Surg. 2014;151(2):215–220. doi:10.1177/0194599814528672
43. Wang ZC, Yao Y, Wang N, et al. Deficiency in interleukin-10 production by M2 macrophages in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2018;8(11):1323–1333. doi:10.1002/alr.22218
44. Xin Y, Wen R, Song D, et al. Emu-miR-10a-5p in Echinococcus multilocularis-derived-extracellular vesicles alleviates airway inflammation in mice with allergic asthma by inhibiting macrophage M2a polarization through LIF-mediated JAK1-STAT3 signaling. Front Immunol. 2025;16:1577349. doi:10.3389/fimmu.2025.1577349
45. Kim DH, Lim JY, Jang JY, et al. Distinct subsets of innate lymphoid cells in nasal polyp. Allergol Int. 2023;72(1):151–160. doi:10.1016/j.alit.2022.06.007
46. Krysko O, Holtappels G, Zhang N, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy. 2011;66(3):396–403. doi:10.1111/j.1398-9995.2010.02498.x
47. Vezzani A, Moneta D, Richichi C, et al. Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia. 2002;43 Suppl 5(s5):30–35. doi:10.1046/j.1528-1157.43.s.5.14.x
48. Mousavi Majd A, Ebrahim Tabar F, Afghani A, et al. Inhibition of GABA A receptor improved spatial memory impairment in the local model of demyelination in rat hippocampus. Behav Brain Res. 2018;336:111–121. doi:10.1016/j.bbr.2017.08.046
49. Berghi O, Dumitru M, Cergan R, Musat G, Serboiu C, Vrinceanu D. Local allergic rhinitis-A challenge for allergology and otorhinolaryngology cooperation (scoping review). Life. 2024;14(8):965.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.