Back to Journals » International Journal of General Medicine » Volume 14
Exploring the Potential Key IncRNAs with Endometriosis by Construction of a ceRNA Network
Authors Gu C, Meng Y ![]() , Meng Q, Fan W, Ye M, Zhang Q, Zhang N, Li L
, Meng Q, Fan W, Ye M, Zhang Q, Zhang N, Li L
Received 24 May 2021
Accepted for publication 16 July 2021
Published 5 August 2021 Volume 2021:14 Pages 4161—4170
DOI https://doi.org/10.2147/IJGM.S321648
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Chenglei Gu,1,* Yuanguang Meng,1,* Qingqing Meng,2 Wensheng Fan,1 Mingxia Ye,1 Qian Zhang,1 Nina Zhang,1 Lian Li1
1Department of Obstetrics and Gynecology, The Seventh Medical Center, Chinese PLA General Hospital, Beijing, People’s Republic of China; 2Health Care Office, Agency for Offices Administration of Central Military Commission of People’s Republic of China, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lian Li
Department of Obstetrics and Gynecology, The Seventh Medical Center, Chinese PLA General Hospital, Beijing, 100700, People’s Republic of China
Tel +86-10-6693-8244
Fax +86-10-6693-7516
Email [email protected]
Purpose: The etiology and pathophysiology of endometriosis remain unclear. Current research indicates long noncoding RNA (lncRNA) may play an important role in the pathogenesis and development of endometriosis. However, the molecular mechanism of lncRNA in endometriosis is far from clear.
Patients and Methods: The lncRNA and mRNA expression of 8 patients with ovarian endometriosis were determined by high-throughput RNA sequencing (8 ectopic endometria samples vs 8 eutopic endometria samples), and miRNA expression profiles were obtained from our previous study. Then a lncRNA-associated competing endogenous RNA (ceRNA) network was constructed by combining the regulatory interaction and negative co-expression interaction between the differentially expressed lncRNAs/mRNAs and miRNAs by different rules.
Results: The constructed lncRNA-related ceRNA network was composed of two separate networks, network 1 including 14,137 dysregulated lncRNA–mRNA interactions, referring to 242 lncRNAs, 55 miRNAs and 1600 mRNAs, network 2 including 4459 dysregulated lncRNA–mRNA interactions, referring to 111 lncRNAs, 39 miRNAs and 1151 mRNAs. The top six hub lncRNAs (LINC01140, MSC-AS1, HAGLR, CKMT2-AS1, JAKMIP2-AS1, AL365361.1) in the significant ternary relationship of mRNA–miRNA–lncRNA in network 1, and the top six hub lncRNAs (PAX8-AS1, MIR17HC, PART1, HOXA-AS3, PLAC4, LINC00511) in the significant ternary relationship of mRNA–miRNA–lncRNA in network 2 were selected. Functional enrichment analysis of these lncRNA-related mRNAs indicated that the lncRNAs in network 1 mainly take part in positive regulation of phagocytosis, myeloid leukocyte activation, and tissue remodeling, while the lncRNAs in network 2 mainly take part in negative regulation of cell proliferation, blood vessel development and regulation of epithelial cell differentiation, which is consistent with the results obtained from the different rules to construct the networks.
Conclusion: lncRNA-related ceRNA network analysis recognized key lncRNAs related to the development of endometriosis.
Keywords: endometriosis, endometrium, microRNAs, lncRNA, integrative analysis, ceRNA network
Introduction
Endometriosis is characterized by endometrial tissue (endometrial glandular and stromal) abnormally growing outside the uterine cavity.1 The incidence rate in fertile women is up to 10%, and may be up to 50% in women who are experiencing infertility and/or pelvic pain.2,3 Due to the high prevalence and the resulting physical and mental pain in fertile women, the etiology and pathophysiology of endometriosis needs further research.
Long non-coding RNA (lncRNA) is a non-coding RNA varying in length from 200 nucleotides to 100 kilobases, which has been involved in a wide range of biological processes from regulation transcription to immune responses.4–7 Many researchers have discovered that lncRNA may play an important role in regulated genes expression and the progression of endometriosis.8–10 Huang et al. reported that lncRNA UCA1 was downregulated in endometriosis,9 and Zhu et al. showed an effect of lncRNA BANCR on endometriosis through the ERK/MAPK pathway.11 Therefore, more attention should be paid to lncRNAs in the study of progression of endometriosis.
Current reports have demonstrated an intrinsic interaction among diverse RNA, referring to protein-coding messenger RNAs and non-coding RNAs such as long non-coding RNAs.4 These RNA transcripts could behave as competing endogenous RNAs (ceRNAs) or natural microRNA sponges - they co-regulate each other by competing for binding to shared microRNAs, a family of small non-coding RNAs that are important post-transcriptional regulators of gene expression.4,12,13 By studying this novel RNA crosstalk, new insight about gene regulatory networks and the association with human disease will be clear. Besides, our previous study has identified other regulatory networks involved in microRNA, transcription factors, and genes in order to try to understand the abnormal moleculer changes in the occurrence and development of endometriosis.14 This study aims to construct the lncRNA-related ceRNA network and further survey the potential key lncRNAs for understanding the molecular mechanism of endometriosis by performing high-throughput sequencing techniques to detect lncRNA and mRNA expression profiling in eight paired ectopic endometria (EC) samples and eutopic endometria (EU) samples from eight patients with ovarian endometriosis.
Patients and Methods
Patient Samples
This study was approved by the Chinese PLA General Hospital (No. S2016-123-01), and was conducted in accordance with the Declaration of Helsinki. All patients were informed about the purpose of this study, and each patient signed informed consent. Eight paired EC and EU samples were selected and prepared for RNA sequencing. The detailed information about patients can be seen in our previous work,14 which were the same samples with miRNA sequencing. Endometrial tissue samples were obtained by pipelle sampling with inner membrane in the secretory phase including two endometrial gland cells and stromal cells.15
RNA Sequencing and Data Processing
All tissue samples were preserved in RNA-later solution (Sigma Aldrich, Poland) at 4°C for 24 h and then kept at −80 °C until utilized. RNA extraction, purification and RNA integrity analysis were performed as previously described in our previous work.14
Three μg of RNA in each sample was prepared for RNA sequencing. Library construction and rRNA removal were performed by the Ribo-Zero Gold Kit (Epicentre, USA) as well as NEB Next Ultra RNA Library Prep Kit (NEB, Ipswich, USA) according to the manufacturer’s protocols. Then, an Illumina HiSeq4000 system (Illumina, USA) was used to sequence paired-end 150-bp of the cDNAs conducted by Annoroad Genome (Beijing, China).
The raw data were processed to filter out adaptor-polluted reads, low-quality reads and reads with the number of N bases accounting for more than 5% with Perl scripts. Bowtie2 (v2.2.3)16 was used as gapped read aligner, and TopHat (v2.0.12)17 was used to align RNA-seq reads to the human genome (GRCh37) (http://grch37.ensembl.org/index.html). HTSeq (v0.6.0) was used to count the read counts of each gene,18 and genes expression in each sample were estimated by the reads per kilobase of one gene per million reads (RPKM).
Data Collection
The miRNA expression profiles were obtained from GSE105765, which involved eight paired ectopic endometria (EC) samples and eutopic endometria (EU) samples from eight patients with ovarian endometriosis in our previous study.14 miRNA-mRNA and miRNA-lncRNA interaction data were obtained from miRcode (291,978 miRNA-lncRNA interactions and 1,588,069 miRNA-mRNA interactions),19 miRDB (5332 miRNA-lncRNA interactions and 3,224,437 miRNA-mRNA interactions),20 miRTarBase (968 miRNA-lncRNA interactions and 477,771 miRNA-mRNA interactions),21 RNAInter (24,142 miRNA-lncRNA interactions and 2,123,235 miRNA-mRNA interactions)22 and StarBase (71,952 miRNA-lncRNA interactions and 1,075,989 miRNA-mRNA interactions).23 In sum, a total of 377,739 miRNA-lncRNA interactions and 4,757,629 miRNA-mRNA interactions were collected.
Data Analysis
A total of 31,078 genes were involved in the study after experimental and data preprocessing, referring to 18,244 mRNAs, 11,325 lncRNAs, and 1509 miRNAs. DESeq224 was used to analyze differentially expressed genes (DEGs) with padj < 0.01 and abs(log2(foldChange)) > 1. Metascape (http://metascape.org/) was performed for GO and KEGG pathway enrichment analysis.25 Cytoscape software was used to visualize the lncRNA-related ceRNA networks and the lncRNA–mRNA interactions.26
Construction of the lncRNA-Associated ceRNA Network
First, collecting all the miRNA-lncRNA and miRNA-mRNA interactions from five databases mentioned in Data collection. Second, calculating the negative co-expression regulatory pattern of miRNA-lncRNA and miRNA-mRNA by differential expression miRNA, lncRNA, and mRNA with Spearman correlation coefficient < −0.5 and p-value < 0.01. Also, the network and metascape results of the co-dysregulated pairs whose CC were ranked in the top 0.05 and 0.1 percentile and p-value < 0.01 were calculated to compare. Third, intersecting the miRNA-lncRNA and miRNA-mRNA interactions mentioned in step one and two above, then all the interactions with common miRNA make up the lncRNA-associated ceRNA network.
Significant Ternary Relationship Analysis
We further selected the significant ternary relationship of lncRNA-miRNA-mRNA by calculating the score of any pair of the ith lncRNA and the jth mRNA,27 which was evaluated by the Jaccard similarity coefficient, the formula is below.

Where Li represents the number of miRNA which interactwith ith lncRNA and Mj represents the number of miRNA which interaction with the jth mRNA.
And p-value of any pair of the ith lncRNA and the jth mRNA calculating by hypergeometric test as follows:
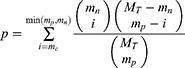
where MT represents all the number of miRNA, mp represents miRNA number which regulated the jth mRNA, mn represents miRNA number which regulated the ith lncRNA, mc represents miRNA number which regulated both the jth mRNA and the ith lncRNA.
And significant lncRNA-miRNA-mRNA ternary relationships were determined by score > 0.5 and P < 0.01.
The Flowchart of the whole work can be seen in Figure S1.
Results
Differential Expression Analysis
A total of 1437 differential expression lncRNAs (DElncRNAs), 4225 differential expression mRNAs (DEmRNAs), 207 differential expression miRNAs (DEmiRNAs) were obtained by DESeq2. In total 742 lncRNAs were up-regulated and 695 lncRNAs were down-regulated; 2345 mRNAs were up-regulated and 1880 mRNAs were down-regulated; and 105 miRNAs were up-regulated and 102 miRNAs were down-regulated (seeTables S1–S6). The volcano plot of DEG (lncRNA, miRNA, mRNA) is shown in Figures S2–S4. The heat map of DEG (lncRNA, miRNA, mRNA) to classify tissue and stage is shown in Figures S5–S7.
lncRNA-Associated ceRNA Network
A total of 56,935 DElncRNA-DEmiRNA pairs and 178,004 DEmRNA-DEmiRNA pairs were attained by calculating the negative co-expression regulatory patterns of these DElncRNA-DEmiRNA interactions and DEmRNA-DEmiRNA interactions. Then, a sum of 832 interactions involving 353 lncRNAs and 94 miRNAs, and 21,533 interactions involving 2912 mRNAs and 195 miRNAs were attained by intersecting DElncRNA-DEmiRNA and DEmRNA-DEmiRNA pairs from negative co-expression regulatory patterns with pairs from five databases introduced in ‘Data collection’ section. Finally, the interaction with 94 in common miRNA pairs of lncRNA and mRNA were retained to construct the lncRNA-associated ceRNA network, which included 2811 DEmRNAs, 353 DElncRNAs, and 94 DEmiRNAs.
As shown in Figure 1, the lncRNA-associated ceRNA network was composed of two separate networks naturally. Especially, network 1 including 14,137 dysregulated lncRNA–mRNA interactions, referred to 242 lncRNAs, 55 miRNA and 1600 mRNAs, and network 2 including 4459 dysregulated lncRNA–mRNA interactions, referred to 111 lncRNAs, 39 miRNA and 1151 mRNAs (seen in Table 1).
 |
Table 1 Details of the lncRNA-Associated ceRNA Networks and Significant Ternary Relationships |
 |
Figure 1 Constructed lncRNA-associated ceRNA network. Orange represents lncRNA, green represents miRNA, blue represents mRNA. |
Functional enrichment analysis was performed on the mRNAs in these two networks. Notably, mRNAs in network 1 were enriched in biological processes including myeloid leukocyte activation, lymphocyte activation, positive regulation of cytokine production and activation of immune response (seen in Figure 2A), network 2 was mainly enriched in biological processes including tissue morphogenesis, embryonic organ development, epithelial cell development, and regulation of hormone levels (seen in Figure 2B). The network and functional analysis based on co-dysregulated pairs whose CC were ranked in the top 0.05 and 0.1 percentile and p-value < 0.01 is shown in Figures S8–S13. The functional enrichment analysis was consistent.
 |
Figure 2 Functional enrichment analysis of mRNA for two networks. (A) Functional enrichment analysis for network 1. (B) Functional enrichment analysis for network 2. |
Significant Ternary Relationship and Key lncRNAs
Based-on the lncRNA-associated ceRNA network, we further screened the significant ternary relationship (lncRNA-miRNA-mRNA), which also involved two independent modules (shown in Figure 3A and B, Table 1). As shown in Figure 3C and D, the degree of lncRNA is significantly larger than mRNA in two modules, which means lncRNA plays a major role in regulation expression of mRNA, so according to the degree of lncRNA in the significant ternary relationship, the top six lncRNAs were selected to research their function by these enriched lncRNA related mRNAs.
 |
Figure 3 Network visualization of significant ternary relationships and degree of lncRNA and mRNA in significant ternary relationships. (A) Network visualization of significant ternary relationship for module 1. (B) Network visualization of significant ternary relationship for module 2. (C) Degree of lncRNA and mRNA in significant ternary relationship for module 1. (D) Degree of lncRNA and mRNA in significant ternary relationship for module 2. |
The top six degrees of lncRNAs in module 1 were LINC01140, MSC-AS1, HAGLR, CKMT2-AS1, JAKMIP2-AS1, and AL365361.1 (shown in Figure 4A). Functional enrichment analysis of these lncRNA related mRNAs revealed that these lncRNAs take part in positive regulation of phagocytosis and myeloid leukocyte activation, cell adhesion mediator activity and tissue remodeling, etc. (shown in Figure 4B).
 |
Figure 4 Subnetwork of top six degrees of lncRNA in significant ternary relationships and functional enrichment analysis with top six degrees of lncRNA related mRNAs. (A) Subnetwork of top six degrees of lncRNA in significant ternary relationship for module 1 (B) Functional enrichment analysis with top six degrees of lncRNA related mRNAs for module 1. (C) Subnetwork of top six degrees of lncRNA in significant ternary relationship for module 2. (D) Functional enrichment analysis with top six degrees of lncRNA related mRNAs for module 2. |
As for module 2, the top six degrees of lncRNAs were PAX8-AS1, MIR17HC, PART1, HOXA-AS3, PLAC4, and LINC00511 (shown in Figure 4C). Functional enrichment analysis of these lncRNA related mRNAs revealed that these lncRNAs take part in negative regulation of cell proliferation, blood vessel development and regulation of epithelial cell differentiation, etc. This may account for the endometrium migration (Figure 4D).
Discussion
The competitive endogenous RNA (ceRNA) hypothesis proposes that RNA transcripts, including circRNAs, messenger RNAs (mRNAs), and long non-coding RNAs (lncRNAs), contain miRNA response elements (MREs) which compete among themselves for miRNA binding to regulate the expression of each other.12,28,29 Although there is still some debate about the clinical relevance of ceRNA,30 many studies have adopted the ceRNA mechanism to explain the action of lncRNA31,32 and their relationship with disease.33,34 Meanwhile, in this study, we tested the difference among different co-expression thresholds, and the functional enrichment results of the network obtained under different thresholds were very similar (see Figures S8–S13). This suggests that the analysis pipeline of this study is robust to a certain extent.
In this study, we try to identify key lncRNAs and explore their potential function in endometriosis patients by construction of a ceRNA network. The endometrial tissue samples are inner membrane in the secretory phase including two endometrial gland cells and stromal cells. A large number of existing studies35,36 have shown that the occurrence and development of endometriosis is not only due to the role of glandular cells, but also the joint role of stromal cells, so we finally extracted glandular stromal cells, not specific to endometrial glandular cells to do further research. Firstly, lncRNA-associated ceRNA networks were constructed with combined DEmiRNAs, DEmRNAs and DElncRNAs with their interaction relationship of negative co-expression patterns and databases-sourced, which comprised two independent networks. Functional enrichment analysis found mRNAs in network 1 were enriched in immune-associated functions such as myeloid leukocyte activation, lymphocyte activation, and activation of immune response, and mRNAs in network 2 were enriched in development and differentiation associated functions such as gland development, cell-cell junction, cell division, and regulation of epithelial cell differentiation. Then, significant ternary relationships (DEmiRNA-DEmRNA-DElncRNA) were selected, and the top degrees of lncRNA in these were thought to be the key lncRNAs, and the mRNA interactions with these key lncRNAs were explored in depth. As a result, the top degrees of lncRNAs in the significant ternary relationship for network 1 were LINC01140, MSC-AS1, HAGLR, CKMT2-AS1, JAKMIP2-AS1 and AL365361.1, and for network 2 were PAX8-AS1, MIR17HC, PART1, HOXA-AS3, PLAC4 and LINC00511. Functional enrichment analysis showed the mRNAs’ interaction with these top degree lncRNAs were enriched in positive regulation of phagocytosis, myeloid leukocyte activation, cell differentiation and tissue remodeling for module 1, and enriched in negative regulation of cell proliferation, blood vessel development and regulation of epithelial cell differentiation for module 2.
Although lncRNA-associated research is limited, the key lncRNAs obtained above have been found to be related to some other diseases, for example, down-regulation of LINC01140 is associated with adverse features of breast cancer,37 and significantly correlates with overall survival in gastric cancer patients.38 High MSC-AS1 expression was correlated with poorer prognosis in patients with pancreatic ductal adenocarcinoma.39 HAGLR acted as a microRNA-143-5p sponge to regulate epithelial-mesenchymal transition and metastatic potential in esophageal cancer.40 Also, AL365361.1 was found to contribute to the modification of tumor immune microenvironment (TIME) in patients with head and neck squamous cell carcinoma.41 Han et al. found expression quantitative trait loci in long non-coding RNA PAX8-AS1 are associated with decreased risk of cervical cancer.42 Wang and Xu have found lncRNA PART1 promotes breast cancer cell progression by directly targeting miR-4516.43 Also, HOXA-AS3 were found to integrate NF-κB signaling to regulate endothelium inflammation.44
Especially, LINC00511 was found to play a major role in cancer in the reproductive system of women. LINC00511 accelerates proliferation and invasion in cervical cancer through targeting miR-324-5p/DRAM1 axis,45 and inhibition of LINC00511 via transcription factor RXRA-regulated PLD1 can promote cell autophagy and apoptosis in cervical cancer.46 LINC00511 can also mediate the effects of ESR1 on proliferation and invasion in ovarian cancer through miR-424-5p and miR-370-5p.47 This means intensive study may focus on LINC00511 to reveal the pathway of reproductive system cancer and endometriosis in women.
Previous studies have noticed some variation in endometriosis when researching the potential function of these key lncRNAs. Matthias and Menger have demonstrated the engraftment of endometriotic lesions is typically associated with angiogenesis, i.e. the formation of new blood vessels from pre-existing ones, and this angiogenic process underlies the complex regulation by angiogenic growth factors and hormones,48 which supports our findings of key lncRNAs function in blood vessel development and regulation of hormone levels. Moreover, 63-kDa membrane protein (p63), which plays an important role in regulating epithelial proliferation and differentiation, were differentially expressed in different endometriotic lesions,49 and Lin et al. explained integrin-mediated aberrant adhesion is also essential for establishment of endometriotic lesions,50 both of which support our findings of lncRNAs function associated with negative regulation of cell proliferation and cell adhesion mediator activity. Also, some immune-associated genes were elucidated as related to the progression of endometriosis. Sun et al. demonstrated that M-MDSCs (the predominant type of myeloid-derived suppressor cells) recruited and activated by CCR9/CCL25 play a crucial role in the pathogenic progression of endometriosis,51 which was consistent with our results about key lncRNA function of myeloid leukocyte activation.
In our previous study, we identified global transcriptome abnormalities in eutopic endometria,52 and further integrated microRNA and mRNA paired expression profiling to attain deregulated microRNA-transcription factor gene regulatory networks to find the crucial miRNAs, TFs and genes in ovarian endometriosis.14 In the present study, key lncRNAs in endometriosis were surveyed by selecting top degrees of lncRNAs in a significant ternary relationship of the lncRNA-associated ceRNA network, and functional enrichment analysis was performed on the mRNAs interaction with these lncRNA to explore their functions. There is still some debate about the clinical relevance of ceRNA, which could be a potential limitation about the work. However, the present work still may provide novel and crucial insight for further study on molecular mechanisms involved in the development and proliferation of endometriosis.
Conclusion
In summary, we provide some potential key lncRNAs in endometriosis by constructing a lncRNA-associated ceRNA network, and further screened the significant ternary relationship (DEmiRNAs-DEmRNAs-DElncRNAs) to select the top degrees of lncRNAs. Functional enrichment analysis on these lncRNA related mRNAs indicated that these lncRNAs take part in positive regulation of epithelial cell differentiation, cell adhesion mediator activity, myeloid leukocyte activation, negative regulation of cell proliferation, and blood vessel development. While our previous research identified global transcriptome abnormalities in endometriosis and defined some crucial miRNAs, TFs and genes involved in the pathogenesis of endometriosis, the key lncRNAs identified in this work will provide a comprehensive insight in the study of the molecular mechanisms involved in the development and proliferation of endometriosis, which could also have implications in therapeutic strategies for endometriosis.
Acknowledgments
The authors thank all patients for their participation. The work was supported by a grant from National Natural Science Foundation of China: High-throughput sequencing identifying pathogenic genes of endometriosis (Grant No. 81571411).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kennedy S, Bergqvist A, Chapron C, et al. ESHRE guideline for the diagnosis and treatment of endometriosis. Hum Reprod. 2005;20(10):2698–2704. doi:10.1093/humrep/dei135
2. Stern RC, Dash R, Bentley RC, Snyder MJ, Haney AF, Robboy SJ. Malignancy in endometriosis: frequency and comparison of ovarian and extraovarian types. Int J Gynecol Pathol. 2001;20(2):133–139. doi:10.1097/00004347-200104000-00004
3. Meuleman C, Vandenabeele B, Fieuws S, Spiessens C, Timmerman D, D’Hooghe T. High prevalence of endometriosis in infertile women with normal ovulation and normospermic partners. Fertil Steril. 2009;92(1):68–74. doi:10.1016/j.fertnstert.2008.04.056
4. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi:10.1038/nature12986
5. Shi X, Sun M, Wu Y, et al. Post-transcriptional regulation of long noncoding RNAs in cancer. Tumour Biol. 2015;36(2):503–513. doi:10.1007/s13277-015-3106-y
6. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11(1):59. doi:10.1186/1741-7007-11-59
7. Huang Y, Wang J, Zhao Y, et al. cncRNAdb: a manually curated resource of experimentally supported RNAs with both protein-coding and noncoding function. Nucleic Acids Res. 2020;49(D1):D65–70.
8. Ahn SH, Singh V, Tayade C. Biomarkers in endometriosis: challenges and opportunities. Fertil Steril. 2017;107(3):523–532. doi:10.1016/j.fertnstert.2017.01.009
9. Huang H, Zhu Z, Song Y. Downregulation of lncrna uca1 as a diagnostic and prognostic biomarker for ovarian endometriosis. Rev Assoc Med Bras. 2019;65(3):336–341. doi:10.1590/1806-9282.65.3.336
10. Yu J, Chen LH, Zhang B, Zheng QM. The modulation of endometriosis by lncRNA MALAT1 via NF-κB/iNOS. Eur Rev Med Pharmacol Sci. 2019;23(10):4073–4080.
11. Zhu MB, Chen LP, Hu M, Shi Z, Liu YN. Effects of lncRNA BANCR on endometriosis through ERK/MAPK pathway. Eur Rev Med Pharmacol Sci. 2019;23(16):6806–6812.
12. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi:10.1016/j.cell.2011.07.014
13. Zhang Y, Liu T, Chen L, et al. RIscoper: a tool for RNA-RNA interaction extraction from the literature. Bioinformatics. 2019;35(17):3199–3202. doi:10.1093/bioinformatics/btz044
14. Zhao L, Gu C, Ye M, et al. Integration analysis of microRNA and mRNA paired expression profiling identifies deregulated microRNA-transcription factor-gene regulatory networks in ovarian endometriosis. Reprod Biol Endocrinol. 2018;16(1):4. doi:10.1186/s12958-017-0319-5
15. Dacco MD, Moustafa M, Papoutsis D, Georgantzis D, Halmos G, Magos A. An audit of using the H Pipelle for endometrial sampling at outpatient hysteroscopy and literature review comparison with the Pipelle de Cornier. Eur J Obstet Gynecol Reprod Biol. 2012;165(2):299–301. doi:10.1016/j.ejogrb.2012.07.021
16. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
17. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi:10.1093/bioinformatics/btp120
18. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi:10.1093/bioinformatics/btu638
19. Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28(15):2062–2063. doi:10.1093/bioinformatics/bts344
20. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–d131. doi:10.1093/nar/gkz757
21. Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–d302. doi:10.1093/nar/gkx1067
22. Lin Y, Liu T, Cui T, et al. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020;48(D1):D189–d197. doi:10.1093/nar/gkz804
23. Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92–97. doi:10.1093/nar/gkt1248
24. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
25. Zhou Y, Zhou B, Pache L, Chang M. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
26. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
27. Jiang W, Zhan H, Jiao Y, Li S, Gao W. A novel lncRNA-miRNA-mRNA network analysis identified the hub lncRNA RP11-159F24.1 in the pathogenesis of papillary thyroid cancer. Cancer Med. 2018;7(12):6290–6298. doi:10.1002/cam4.1900
28. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. doi:10.1038/nature11993
29. Zhang X, Chu H, Wen L, et al. Competing endogenous RNA network profiling reveals novel host dependency factors required for MERS-CoV propagation. Emerg Microbes Infect. 2020;9(1):733–746. doi:10.1080/22221751.2020.1738277
30. Mofidi M, Rahgozar S, Pouyanrad S. Increased level of long non coding RNA H19 is correlated with the downregulation of miR-326 and BCL-2 genes in pediatric acute lymphoblastic leukemia, a possible hallmark for leukemogenesis. Mol Biol Rep. 2021;48(2):1531–1538. doi:10.1007/s11033-021-06161-y
31. Lu W, Zhang H, Niu Y, et al. Long non-coding RNA linc00673 regulated non-small cell lung cancer proliferation, migration, invasion and epithelial mesenchymal transition by sponging miR-150-5p. Mol Cancer. 2017;16(1):118. doi:10.1186/s12943-017-0685-9
32. Nie L, Zhang P, Wang Q, Zhou X, Wang Q. lncRNA-triggered macrophage inflammaging deteriorates age-related diseases. Mediators Inflamm. 2019;2019:4260309. doi:10.1155/2019/4260309
33. Jiang J, Bi Y, Liu X-P, et al. To construct a ceRNA regulatory network as prognostic biomarkers for bladder cancer. J Cell Mol Med. 2020;24(9):5375–5386. doi:10.1111/jcmm.15193
34. Yao Y, Zhang T, Qi L, et al. Integrated analysis of co-expression and ceRNA network identifies five lncRNAs as prognostic markers for breast cancer. J Cell Mol Med. 2019;23(12):8410–8419. doi:10.1111/jcmm.14721
35. Wang Y, Nicholes K, Shih IM. The origin and pathogenesis of endometriosis. Annu Rev Pathol. 2020;15(1):71–95. doi:10.1146/annurev-pathmechdis-012419-032654
36. Tempest N, Maclean A, Hapangama DK. Endometrial stem cell markers: current concepts and unresolved questions. Int J Mol Sci. 2018;19(10):3240. doi:10.3390/ijms19103240
37. Li D, Li L, Cao Y, Chen X. Downregulation of LINC01140 is associated with adverse features of breast cancer. Oncol Lett. 2020;19(2):1157–1164.
38. Song P, Jiang B, Liu Z, Ding J, Liu S, Guan W. A three-lncRNA expression signature associated with the prognosis of gastric cancer patients. Cancer Med. 2017;6(6):1154–1164. doi:10.1002/cam4.1047
39. Sun Y, Wang P, Yang W, Shan Y, Zhang Q, Wu H. The role of lncRNA MSC-AS1/miR-29b-3p axis-mediated CDK14 modulation in pancreatic cancer proliferation and Gemcitabine-induced apoptosis. Cancer Biol Ther. 2019;20(6):729–739. doi:10.1080/15384047.2018.1529121
40. Yang C, Shen S, Zheng X, et al. Long noncoding RNA HAGLR acts as a microRNA-143-5p sponge to regulate epithelial-mesenchymal transition and metastatic potential in esophageal cancer by regulating LAMP3. FASEB J. 2019;33(9):10490–10504. doi:10.1096/fj.201802543RR
41. Zhong Z, Hong M, Chen X, et al. Transcriptome analysis reveals the link between lncRNA-mRNA co-expression network and tumor immune microenvironment and overall survival in head and neck squamous cell carcinoma. BMC Med Genomics. 2020;13(1):57. doi:10.1186/s12920-020-0707-0
42. Han J, Zhou W, Jia M, et al. Expression quantitative trait loci in long non-coding RNA PAX8-AS1 are associated with decreased risk of cervical cancer. Mol Genet Genom. 2016;291(4):1743–1748. doi:10.1007/s00438-016-1217-9
43. Wang Z, Xu R. lncRNA PART1 promotes breast cancer cell progression by directly targeting miR-4516. Cancer Manag Res. 2020;12:7753–7760. doi:10.2147/CMAR.S249296
44. Zhu X, Chen D, Liu Y, et al. Long noncoding RNA HOXA-AS3 integrates NF-κB signaling to regulate endothelium inflammation. Mol Cell Biol. 2019;39(19):e00139–19. doi:10.1128/MCB.00139-19
45. Zhang X, Wang Y, Zhao A, Kong F, Jiang L, Wang J. Long non-coding RNA LINC00511 accelerates proliferation and invasion in cervical cancer through targeting miR-324-5p/DRAM1 axis. Onco Targets Ther. 2020;13:10245–10256. doi:10.2147/OTT.S255067
46. Shi Y, Liu M, Huang Y, Zhang J, Yin L. Promotion of cell autophagy and apoptosis in cervical cancer by inhibition of long noncoding RNA LINC00511 via transcription factor RXRA-regulated PLD1. J Cell Physiol. 2020;235(10):6592–6604. doi:10.1002/jcp.29529
47. Wang K, Zhu G, Bao S, Chen S. Long non-coding RNA LINC00511 mediates the effects of ESR1 on proliferation and invasion of ovarian cancer through miR-424-5p and miR-370-5p. Cancer Manag Res. 2019;11:10807–10819. doi:10.2147/CMAR.S232140
48. Matthias LW, Menger MD. Basic mechanisms of vascularization in endometriosis and their clinical implications. Hum Reprod Update. 2018;24(2):207–224.
49. Poli Neto OB, Ferreira HM, Ramalho LN, Rosa e Silva JC, Candido Dos Reis FJ, Nogueira AA. Expression of p63 differs in peritoneal endometriosis, endometriomas, adenomyosis, rectovaginal septum endometriosis, and abdominal wall endometriosis. Arch Pathol Lab Med. 2007;131(7):1099–1102. doi:10.5858/2007-131-1099-EOPDIP
50. Lin X, Dai Y, Xu W, et al. Hypoxia promotes ectopic adhesion ability of endometrial stromal cells via TGF-β1/Smad signaling in endometriosis. Endocrinology. 2018;159(4):1630–1641. doi:10.1210/en.2017-03227
51. Sun Y, Shao J, Jiang F, et al. CD33(+) CD14(+) CD11b(+) HLA-DR(-) monocytic myeloid-derived suppressor cells recruited and activated by CCR9/CCL25 are crucial for the pathogenic progression of endometriosis. Am J Reprod Immunol. 2019;81(1):e13067. doi:10.1111/aji.13067
52. Zhao L, Gu C, Ye M, et al. Identification of global transcriptome abnormalities and potential biomarkers in eutopic endometria of women with endometriosis: a Preliminary Study. Biomed Rep. 2017;6(6):654–662. doi:10.3892/br.2017.902
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.