Back to Journals » Journal of Hepatocellular Carcinoma » Volume 12
Exploring Autophagy-Related Gene Expression in Hepatocellular Carcinoma via TCGA, GEPIA2, and HPA Databases: Implications for Prognosis
Authors Gu X, Chen Y ![]() , Hu X, Li Y, Zhu R, Li H
, Hu X, Li Y, Zhu R, Li H ![]() , Yuan Z
, Yuan Z ![]() , Wang Y
, Wang Y ![]()
Received 10 March 2025
Accepted for publication 21 June 2025
Published 22 July 2025 Volume 2025:12 Pages 1557—1586
DOI https://doi.org/10.2147/JHC.S520917
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Xiangran Gu,1,* Yuan Chen,2,* Xinyue Hu,1,* Yunhui Li,3 Renlong Zhu,2 Hongxu Li,1 Zhengrong Yuan,1 Yajie Wang2,3
1College of Biological Sciences and Technology, Beijing Forestry University, Beijing, People’s Republic of China; 2Department of Clinical Laboratory, Peking University Ditan Teaching Hospital, Beijing, People’s Republic of China; 3Department of Clinical Laboratory, Beijing Ditan Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhengrong Yuan, College of Biological Sciences and Technology, Beijing Forestry University, No. 35 Tsinghua East Road, Haidian District, Beijing, 100083, People’s Republic of China, Email [email protected] Yajie Wang, Department of Clinical Laboratory, Beijing Ditan Hospital, Capital Medical University, Beijing, People’s Republic of China, Email [email protected]
Objective: This study aimed to identify autophagy-related genes (ARGs) with prognostic significance in hepatocellular carcinoma (HCC) using bioinformatics and survival analysis.
Materials and Methods: ARGs were sourced from multiple references, including the Human Autophagy Database (HADb), relevant literatures, the Gene Set Enrichment Analysis (GSEA), and a final list was confirmed after eliminating duplicate entries. Differential expression analysis between normal and tumor tissues relied on data from The Cancer Genome Atlas (TCGA). Subsequently, the univariate and multivariate Cox regression analysis, along with the Kaplan-Meier survival analysis, were conducted to identify survival-associated genes. These findings were cross-validated using the Gene Expression Profiling Interactive Analysis 2 (GEPIA2) database and the Human Protein Atlas (HPA) database. Furthermore, expression levels of randomly selected ARGs were validated in HCC and normal cell lines using Real-time quantitative PCR (RT-qPCR), confirming bioinformatics findings.
Results: 41 ARGs were pinpointed. The bioinformatics analysis revealed elevated expression levels of these genes in HCC tissues compared to normal tissues. Notably, mRNA expression levels of ARGs were markedly higher in the tumor tissue samples than in the normal liver tissue samples. This observation was corroborated by data from the GEPIA2 and HPA databases, except for ATG4B and CAPN10. Results from the HPA database aligned with those from the TCGA analysis. GSEA uncovered potential signaling pathways associated with ARGs, including pathways relevant to cancer and autophagy. RT-qPCR analysis further confirmed significant upregulation of mRNA expression levels of randomly selected BAG3, EIF2AK2, KIF5B, and RAB24 in HCC cell lines, consistent with the bioinformatics analysis findings.
Conclusion: This study showed that the 41 obtained ARGs, such as ATG16L1, ATG4B, BAG3, KIF5B, MAPK1, RAB24, and SOGA1, these findings suggest that ARGs may serve as prognostic biomarkers for HCC, warranting further validation in clinical cohorts and functional studies.
Keywords: hepatocellular carcinoma, bioinformatics, autophagy, prognostic markers, TCGA
Introduction
Globally, cancer ranks as a primary cause of mortality and poses a formidable obstacle to enhancing life expectancy.1 Among liver cancers, hepatocellular carcinoma (HCC) stands out as the most prevalent form.2 Notably, in 2020, primary liver cancer emerged as the sixth most frequently diagnosed cancer and the third leading contributor to cancer-related fatalities worldwide,3 within these statistics, HCC accounts for 85% to 90% of cases, often presenting late-stage diagnoses, depriving patients of curative interventions, and resulting in bleak prognoses.4 The primary risk factors associated with HCC encompass aflatoxin B1 (AFB1) contamination in food, chronic infections of hepatitis B virus (HBV) or hepatitis C virus (HCV), excessive alcohol consumption, and metabolic disorders such as diabetes, obesity, and fatty liver disease. These factors collectively contribute to the induction of chronic inflammation in the liver.5 HCC has more than 840,000 new cases and more than 780,000 deaths each year.6 Transcatheter arterial chemoembolization (TACE) has become the preferred therapeutic approach for patients with HCC who cannot be resectable, but its long-term therapeutic effect is not ideal.4 Therefore, the pursuit of dependable prognostic biomarkers is imperative to address the challenge of poor prognosis and low survival rates in HCC. It is essential to elucidate the mechanisms underlying increased tumor burden, facilitating the refinement of prognosis prediction for HCC immunotherapy and the exploration of more efficacious treatment strategies.
Over four decades ago, Christian de Duve introduced the term “autophagy”, stemming from Greek origins denoting self-consumption. This concept emerged largely from observations of mitochondria and other cellular components undergoing degradation within lysosomes, particularly in rat liver perfused with glucagon, a pancreatic hormone. Autophagy presents itself in three primary forms: micro-autophagy, macro-autophagy, and chaperone-mediated autophagy. These pathways collectively converge to facilitate the proteolytic degradation of cytosolic components within lysosomes.7 Fundamental autophagy targets long-lived proteins and regulates organelle turnover. Notably, autophagy intensifies in response to starvation or cellular stress, serving dual roles in cellular dynamics. During starvation, autophagy facilitates the recycling of cellular constituents, ensuring a sustainable supply of macromolecules to sustain metabolic functions internally. In contrast, autophagy plays a vital role in averting the buildup of impaired proteins and organelles during stressful conditions, thus reducing potential harm. The inability to eliminate intracellular waste leaves cells vulnerable to demise, tissue damage, and persistent inflammation, all of which create an environment conducive to tumor formation. Consequently, autophagy serves to enhance cell survival and minimize harm. However, in the context of cancer, this phenomenon presents a complex dilemma. On the one hand, inhibiting autophagy in cancerous cells could confer benefits by disrupting their ability to withstand stress induced by cancer treatments. On the other hand, promoting autophagy might mitigate ongoing tissue damage and chronic inflammation, thereby preventing the development of genetic mutations that initiate and advance tumorigenesis. This perspective holds relevance for strategies aimed at cancer prevention.8 Autophagy exhibits a dual role in HCC: it acts as a tumor suppressor in early stages by maintaining cellular homeostasis and genome stability, yet facilitates tumor progression in advanced stages by supporting cancer cell survival under stress conditions.9
Autophagy’s significance in liver physiology and disease pathology is undeniable. The malfunction or imbalance in autophagy has been linked to various liver ailments, encompassing non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), alcohol-related liver disease (AALD), drug-induced liver injury (DILI), cholestasis, viral hepatitis, and HCC.10 The intricate involvement of autophagy in carcinogenesis reflects its dual nature. Autophagy exerts tumor-suppressive effects by eliminating dysfunctional organelles, enhancing protein degradation, fostering cell differentiation, and even triggering apoptosis in cancerous cells. However, autophagy can paradoxically bolster cancer progression by facilitating nutrient recycling and cellular energy generation. The intricate nature of autophagy’s involvement in both limiting tumor expansion and bolstering the resilience of cancer cells highlights the complexity of its function.11
Autophagy has been linked to tumor onset, progression, metastasis, therapeutic response, and treatment resistance. However, there is a lack of studies utilizing extensive clinical datasets to systematically screen for ARGs that could serve as prognostic biomarkers for HCC patients. In this study, a large of ARGs were obtained and more biomarkers with differential expression of liver cancer prognosis were screened, which could help to find new effective anti-cancer targets, and be used to predict clinical diagnosis and effectively evaluate the prognosis of HCC patients, provide new possibilities and theoretical support for emerging therapies for the treatment of liver cancer by autophagy.
Materials and Methods
Data Acquisition
ARGs were gathered from various databases including the Human Autophagy Database (HADb, http://www.autophagy.lu), paper citation,12 and the GSEA (Gene Set Enrichment Analysis) (https://www.gsea-msigdb.org) database. The final list of ARGs was obtained by removing duplicate genes. Data about the clinical profiles and gene expressions of HCC patients were retrieved from the TCGA database. This encompassed the primary transcriptomic analyses of tumor specimens as well as those from normal tissue samples. Patients with complete clinical data such as gender, age, tumor stage (T classification), distant metastasis (M classification), lymph node metastasis (N classification), and survival information were included. Perl (version 5.30.3) was utilized to organize the downloaded data for further analysis. This research was conducted using de-identified secondary datasets that lacked any personally identifiable details. As such, it was exempt from the requirement for ethical approval. Since the data were sourced from publicly accessible genomic repositories and no direct engagement with human participants or invasive techniques was involved, informed consent was not necessary. In line with applicable ethical standards and guidelines governing the use of public data, this study qualifies for exemption from both ethical review and consent procedures.
Differential Analysis and Survival Analysis
For the analysis of differential expression and survival, R software (version 4.3.2) was employed. Given the non-normal characteristics of the gene expression data and the limitation of sample size, the Wilcoxon signed-rank test was used for differential expression analysis, and the R-packages “limma” and “beeswarm” were employed for this purpose. This analysis compared gene expression disparities between tumor and normal samples, visually represented through scatter plots and pairing plots. For further analysis, ARGs with a P-value < 0.05 were specifically chosen. Utilizing the R-package “survival”, Kaplan-Meier survival curves were constructed, and the relationship between patient survival and the expression levels of candidate ARGs was assessed via Log rank tests. Genes exhibiting a P-value < 0.05 were deemed to possess statistical significance in the analysis. The collated gene expression data was uploaded to the website for heat mapping (https://www.bioinformatics.com.cn).
Given the non-normal characteristics of the gene expression data and the limitation of sample size, the Wilcoxon signed-rank test was used for differential expression analysis, and the R-packages “limma” and “beeswarm” were employed for this purpose.
Univariate and Multivariate Cox Regression Analysis
The univariate and the multivariate Cox regression analysis models were both built using R software. To ensure the integrity of the data, the missing information in the sorted clinical information data was first eliminated. The “survival” R package was utilized to perform univariate Cox regression analysis, investigating the correlations between gene expression and clinicopathological factors across different levels. The aim was to identify genes closely associated with HCC prognosis, setting the stage for subsequent multivariate Cox regression analysis. This analysis was carried out using the “survival” and “survminer” R packages, integrating patient prognostic data such as age, sex, clinical characteristics, and tumor stage. The objective was to assess whether the target gene could serve as an independent prognostic factor and to generate a forest plot accordingly. Typically, a Hazard Ratio value (HR) >1 signifies the gene’s role as a risk factor, while an HR < 1 denotes its function as a protective factor. Significance was attributed to P-values < 0.05 in statistical evaluation.
Verification of GEPIA2 and HPA
The verification of ARG expression between tumor and normal samples was conducted using the GEPIA2 (Gene Expression Profiling Interactive Analysis 2) (http://gepia2.cancer-pku.cn) database and the HPA (Human Protein Atlas) (https://www.proteinatlas.org/) database. Boxplots of gene expression differences, clinical stage maps, and Kaplan-Meier survival analysis maps in the GEPIA2 database validated the results of ARGs. In the analysis of GEPIA2, a gene symbol or collection ID was entered and LIHC (Liver Hepatocellular Carcinoma) was selected as the cancer name. When the box plot was drawn, the P-value cutoff was set to 0.05, “Multiple Datasets” and “Match TCGA normal and GTEx (Genotype-Tissue Expression) data” were selected, and log2(TPM(Transcripts Per Million)+1) was used as the logarithmic scale. The data in the GTEx database contains more data on normal liver samples. When mapping clinical staging stages, log2(TPM+1) was used as the logarithmic scale, and major stages were selected for mapping. For the survival analysis chart, select “Overall Survival” and “Median”, and leave the other options as default. Immunohistochemical and pathological staging profiles of tumor samples and normal samples stained with the same antibody were obtained from the HPA database.
The Related Signaling Pathway in GSEA
The genes retrieved from the TCGA database were subjected to analysis and enrichment using the GSEA database. The gene set databases chosen were “c2.cp.kegg.HS.symbols” and “c5.go.HS.symbols”. Enriched gene pathways were identified utilizing the “KEGG” package. A significance threshold of P-value < 0.05 was applied to select the enriched signal pathways.
Immune Cell Infiltration Analysis
Using the BEST (Biomarker Exploration for Solid Tumors) website (https://rookieutopia.hiplot.com.cn/app_direct/BEST/), the dataset was analyzed with eight popular algorithms to estimate immune cell infiltration in the tumor microenvironment (TME), including CIBERSORT, CIBERSORT ABS, EPIC, ESTIMATE, MCP-counter, Quantiseq, TIMER, and xCell.
Cell Culture
The human HCC cell lines HepG2 and Huh7, as well as the HCCLM3 cells, were purchased from the Cell Bank, Chinese Academy of Sciences (Beijing, China). The human normal hepatocyte line L02 was purchased from Wuhan Churuike Pharmaceutical Technology Co., Ltd. (CRK Pharma, Wuhan, China). Cell culture medium was prepared according to the proportions of DMEM (90 %), FBS (10 %), and P/S (1 %). HCC cell lines HepG2, Huh7, HCCLM3, and normal HCC cell line L02 were cultured and passed from one plate to three plates once every 2 days. If the cell density was not enough, the fluid was changed once every two days. The culture bottle was cultured in an incubator with 5 % CO2 and 37°C, and the cells in the logarithmic growth stage were selected for follow-up experiments.
Total RNA Extraction and RT-qPCR Analysis
Total intracellular RNA was isolated from cell lines utilizing the BioSci™ Animal Tissue/Total Cell RNA Extraction Kit (Dakewe, Shenzhen, China), following the instructions provided by the manufacturer. Subsequently, 1 μg of RNA was employed for cDNA synthesis using the PrimeScript RT reagent Kit with a gDNA Eraser (Takara, Dalian, China). The gene sequences of BAG3 (BAG Family Molecular Chaperone Regulator 3), EIF2AK2 (Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2), KIF5B (Kinesin Family Member 5B), RAB24 (RAB24, Member RAS Oncogene Family), and GAPDH (Glyceraldehyde-3-phosphate Dehydrogenase) were retrieved from the GenBank section of the NCBI database. Primer design was facilitated using Primer 5.0, and the NCBI online tool Primer-Blast was employed to align primers with gene sequences (Table 1). Primers were subsequently purified by Tsingke Biotech, Beijing, China. With GAPDH as the internal parameter, each group was provided with three multiple holes. GAPDH is a commonly used reference gene in molecular biology research, which is widely accepted due to its relatively stable expression in various tissues and conditions.13 RT-qPCR (Reverse Transcription Quantitative Polymerase Chain Reaction) was performed on a LightCycler 480 (Roche Life Sciences, Switzerland) with Rapid SYBR Green Mix (TransGen Biotech, Beijing, China), following the manufacturer’s protocol. The pre-denaturation was at 94°C for 30s, the denaturation was at 94°C for 5s per cycle, and the annealing was at 60°C for 15s per cycle, extending 72°C for 10s per cycle, a total of 40 cycles. The formula 2−ΔΔCt was used to calculate the relative expression level of the target gene. All experimental results were replicated at least 3 times, and the results were expressed as mean ± standard deviation. GraphPad 10.2.0 software was utilized for data visualization. Statistical comparisons between groups were conducted using independent sample t-tests, with significance set at P-vlaue < 0.05.
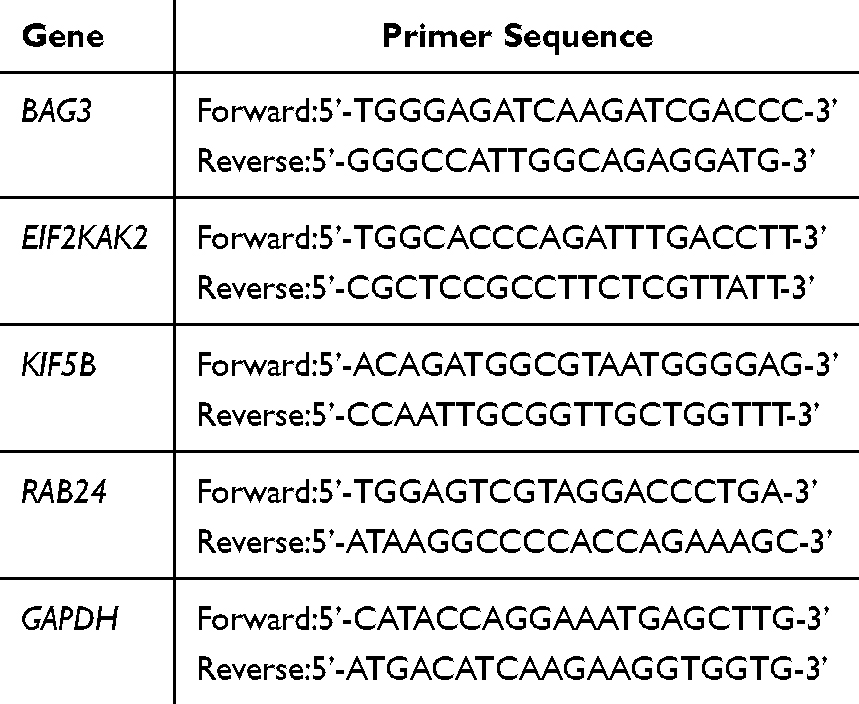 |
Table 1 The Primers Used for RT-qPCR |
Results
The Expression of Selected ARGs in HCC Tissues and Normal Tissues
A list of ARGs was obtained, including three parts: ARGs in the HADb database, ARGs cited in the literatures,14 and ARGs in the GSEA gene set database. After deleting duplicate genes, a total of 576 genes were obtained (Table S1). Genes that have been published in the field of liver cancer through TCGA bioinformatics were excluded, and 41 genes with different expressions in the HCC tissues and normal tissues were finally obtained by analysis (Table 2). From the TCGA database, 465 samples were gathered, comprising 407 tumor specimens and 58 samples of normal tissue. Autophagy Related 16 Like 1 (ATG16L1), Autophagy Related 4B Cysteine Peptidase (ATG4B), BAG3, BCL2 Interacting Protein 1 (BNIP1), Calcium-Activated Neutral Proteinase 10 (CAPN10), Casein Kinase 2 Alpha 1 (CSNK2A1), Death Associated Protein (DAP), EIF2AK2, G Protein Subunit Alpha I3 (GNAI3), KIF5B, Mitogen Activated Protein Kinase 1 (MAPK1), RAB24, Receptor Interacting Serine/Threonine Kinase 2 (RIPK2), Ras Related GTP Binding C (RRAGC), Suppressor of glucose, autophagy associated 1 (SOGA1), Sphingosine Kinase 1 (SPHK1), Serine Palmitoyltransferase Long Chain Base Subunit 1 (SPTLC1), Transmembrane Protein 39A (TMEM39A), Ubiquitin Specific Peptidase 13 (USP13), Von Hippel-Lindau Tumor Suppressor (VHL) were randomly selected to show in the text.
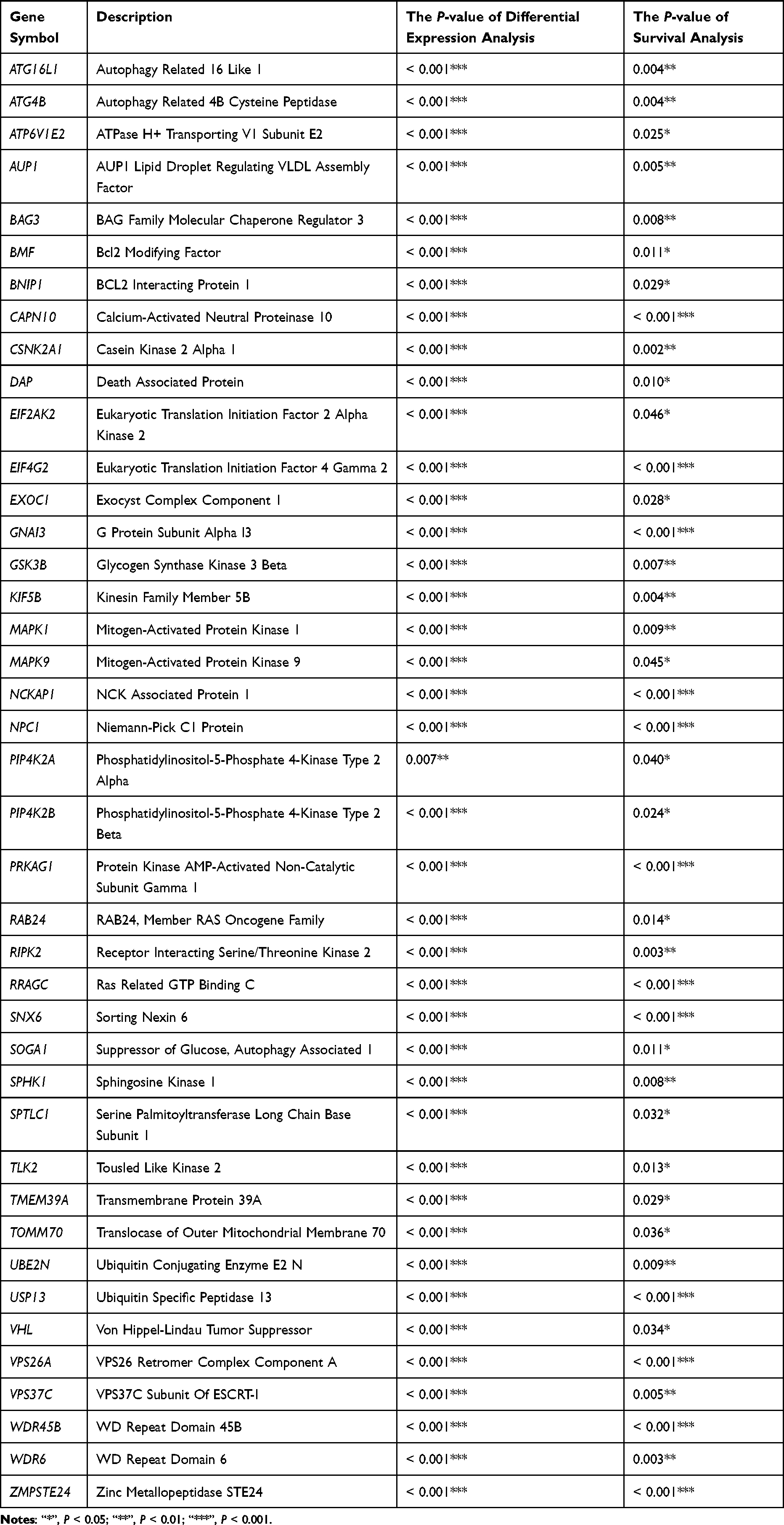 |
Table 2 The 41 Autophagy-Related Genes (ARGs) Associated with Survival |
The analysis of scatter differences revealed a notable increase in mRNA expression levels of 41 ARGs in HCC tissues when compared to normal liver tissues, exhibiting associated P-values < 0.001 (Table 2). Pairing analysis results demonstrated a significant upregulation in the mRNA expression levels of 41 ARGs within HCC tissues compared to normal liver tissues in 58 patient-matched pairs (Figure 1 and Figure S1). In both HCC tissues and their paired counterparts, the mRNA expression levels of the 20 randomly selected genes displayed a consistent trend of upregulation, with all associated P-values < 0.001 (Figure 1). Other ARGs were showed in Figure S1.
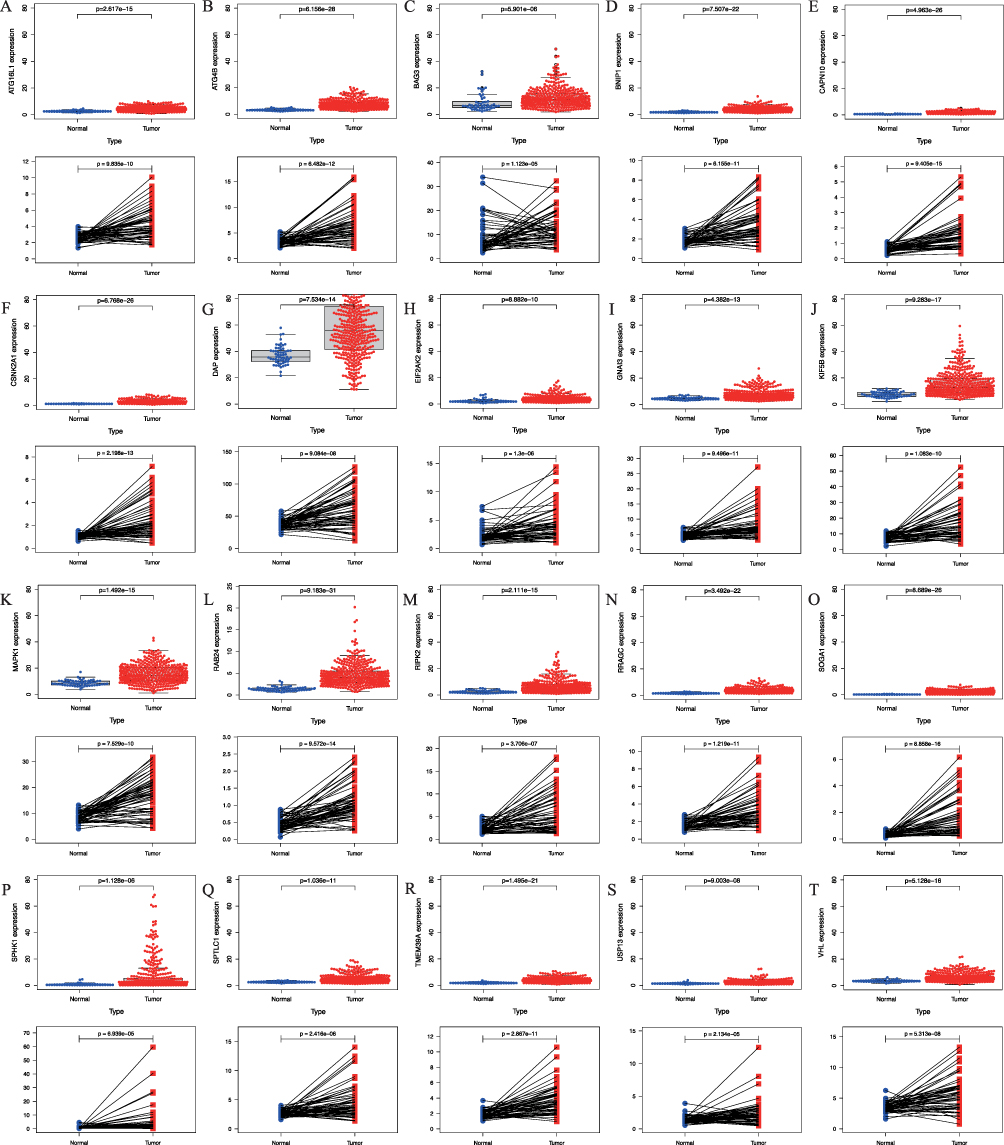 |
Figure 1 The mRNA expression of screened autophagy-related genes (ARGs) in adjacent normal and hepatocellular carcinoma (HCC) tissues, including (A) ATG16L1, (B) ATG4B, (C) BAG3, (D) BNIP1, (E) CAPN10, (F) CSNK2A1, (G) DAP, (H) EIF2AK2, (I) GNAI3, (J) KIF5B, (K) MAPK1, (L) RAB24, (M) RIPK2, (N) RRAGC, (O) SOGA1, (P) SPHK1, (Q) SPTLC1, (R) TMEM39A, (S) USP13, (T) VHL. Abbreviations: ATG16L1, Autophagy Related 16 Like 1; ATG4B, Autophagy Related 4B Cysteine Peptidase; BAG3, BAG Family Molecular Chaperone Regulator 3; BNIP1, BCL2 Interacting Protein 1; CAPN10, Calcium-Activated Neutral Proteinase 10; CSNK2A1, Casein Kinase 2 Alpha 1; DAP, Death Associated Protein; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; GNAI3, G Protein Subunit Alpha I3; KIF5B, Kinesin Family Member 5B; MAPK1, Mitogen-Activated Protein Kinase 1; RAB24, Ras-Related Protein Rab-24; RIPK2, Receptor Interacting Serine/Threonine Kinase 2; RRAGC, Ras Related GTP Binding C; SOGA1, Suppressor of Glucose, Autophagy Associated 1; SPHK1, Sphingosine Kinase 1; SPTLC1, Serine Palmitoyltransferase Long Chain Base Subunit 1; TMEM39A, Transmembrane Protein 39A; USP13, Ubiquitin Specific Peptidase 13; VHL, Von Hippel-Lindau Tumor Suppressor. |
Survival Analysis of ARGs
Based on the survival status and duration of patients, the Kaplan-Meier survival curve was constructed using clinical data from the TCGA database for HCC. This analysis shed light on the correlation between patient survival rates and the expression levels of differentially expressed ARGs, aiding in identifying potential associations between genes and survival outcomes. Generally, the mRNA expression levels of ARGs exhibited an elevation in tumor tissues when compared to normal liver tissues. High expression levels of ATG16L1 (P = 0.004), ATG4B (P = 0.004), BAG3 (P = 0.008), BNIP1 (P = 0.029), CAPN10 (P < 0.001), CSNK2A1 (P = 0.002), DAP (P = 0.01), EIF2AK2 (P = 0.046), GNAI3 (P < 0.001), KIF5B (P = 0.004), MAPK1 (P = 0.009), RAB24 (P = 0.014), RIPK2 (P = 0.003), RRAGC (P < 0.001), SOGA1 (P = 0.011), SPHK1 (P = 0.008), SPTLC1 (P = 0.032), TMEM39A (P = 0.029), USP13 (P < 0.001), and VHL (P = 0.034) genes were significantly associated with the decreased survival rates compared to those with low expression (Figure 2), aligning with the differential analysis results indicating the increased gene expression in HCC tissues relative to normal tissues. Additional ARGs were depicted in Figure S2. The heatmap was drawn for the selected ARGs, and the heatmap could represent the data more intuitively through color. The results of the heatmap were the same as those of differential expression analysis (Figure 3).
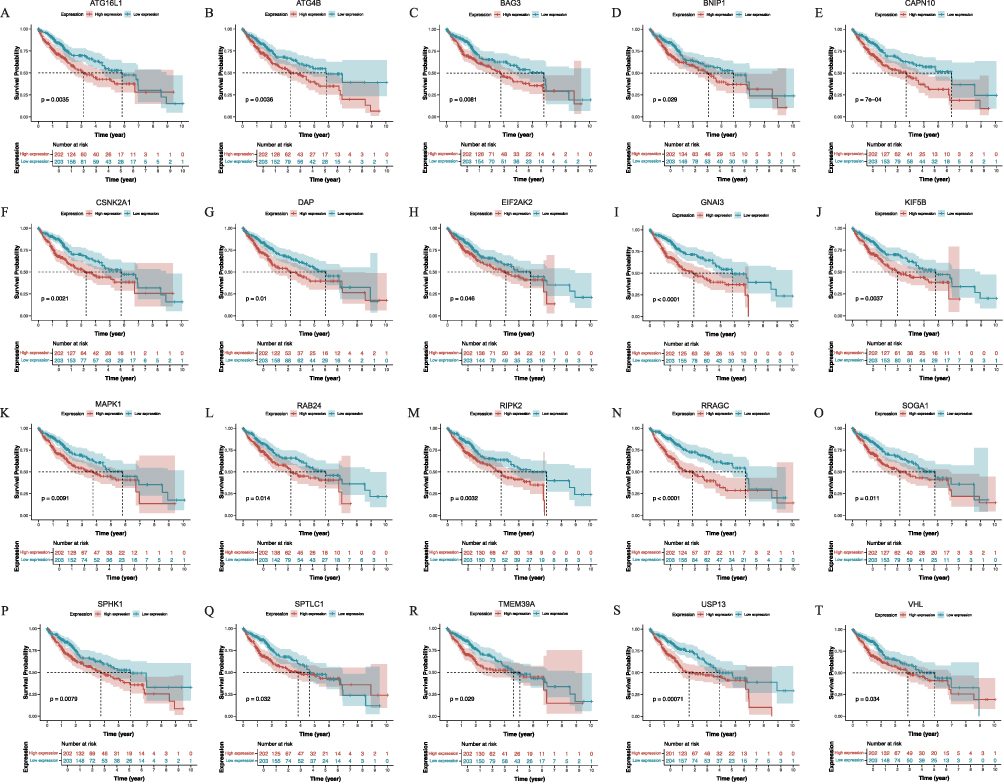 |
Figure 2 The Kaplan-Meier survival curves of screened autophagy related genes (ARGs) expression in hepatocellular carcinoma (HCC), including (A) ATG16L1, (B) ATG4B, (C) BAG3, (D) BNIP1, (E) CAPN10, (F) CSNK2A1, (G) DAP, (H) EIF2AK2, (I) GNAI3, (J) KIF5B, (K) MAPK1, (L) RAB24, (M) RIPK2, (N) RRAGC, (O) SOGA1, (P) SPHK1, (Q) SPTLC1, (R) TMEM39A, (S) USP13, (T) VHL. Abbreviations: ATG16L1, Autophagy Related 16 Like 1; ATG4B, Autophagy Related 4B Cysteine Peptidase; BAG3, BAG Family Molecular Chaperone Regulator 3; BNIP1, BCL2 Interacting Protein 1; CAPN10, Calcium-Activated Neutral Proteinase 10; CSNK2A1, Casein Kinase 2 Alpha 1; DAP, Death Associated Protein; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; GNAI3, G Protein Subunit Alpha I3; KIF5B, Kinesin Family Member 5B; MAPK1, Mitogen-Activated Protein Kinase 1; RAB24, Ras-Related Protein Rab-24; RIPK2, Receptor Interacting Serine/Threonine Kinase 2; RRAGC, Ras Related GTP Binding C; SOGA1, Suppressor of Glucose, Autophagy Associated 1; SPHK1, Sphingosine Kinase 1; SPTLC1, Serine Palmitoyltransferase Long Chain Base Subunit 1; TMEM39A, Transmembrane Protein 39A; USP13, Ubiquitin Specific Peptidase 13; VHL, Von Hippel-Lindau Tumor Suppressor. |
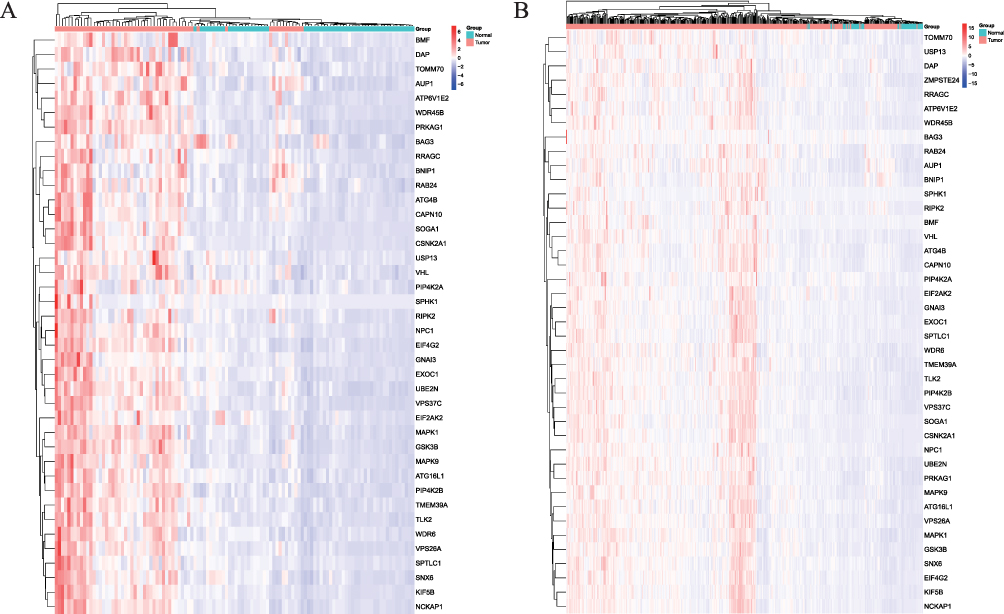 |
Figure 3 The Heatmap of screened autophagy-related genes (ARGs) expression in the adjacent normal and hepatocellular carcinoma (HCC) tissues. (A) The expression of 58 paired samples; (B) The expression of all 465 samples. |
Univariate and Multivariate Cox Regression Analysis
HR for 41 ARGs, obtained through univariate Cox regression analysis using clinical data, were greater than 1, suggesting their potential role as high-risk genes in HCC. Among them, the increased clinical stage, tumor stage (T), and high expression of CAPN10, CSNK2A1, GNAI3, MAPK1, RIPK2, SOGA1, SPTLC1, TMEM39A, and VHL in HCC patients were significantly linked to the increased survival risk (P < 0.001) (Tables 3 and 4). Poor survival in HCC patients was notably linked to the occurrence of distant metastasis (M) and the decreased expression of ATG16L1, ATG4B, BNIP1, DAP, EIF2AK2, KIF5B, RRAGC, SPHK1, and USP13 (P < 0.05) (Tables 3 and 4). However, survival in HCC patients was not correlated with Age, Gender, Histological grade (G), Lymph node metastasis (N), BAG3, and RAB24 (P > 0.05) (Table 3 and Figure 4). Other ARGs were displayed in Figure S3. Multivariate in multivariate Cox regression analysis, where a screening condition of P < 0.05 was applied, the expression of 41 ARGs emerged as an independent prognostic factor for the overall survival of HCC patients (P < 0.05) (Table 4). Figure 5 illustrated a random selection of 20 ARGs mentioned in the text. Figure S4 showed the other ARGs.
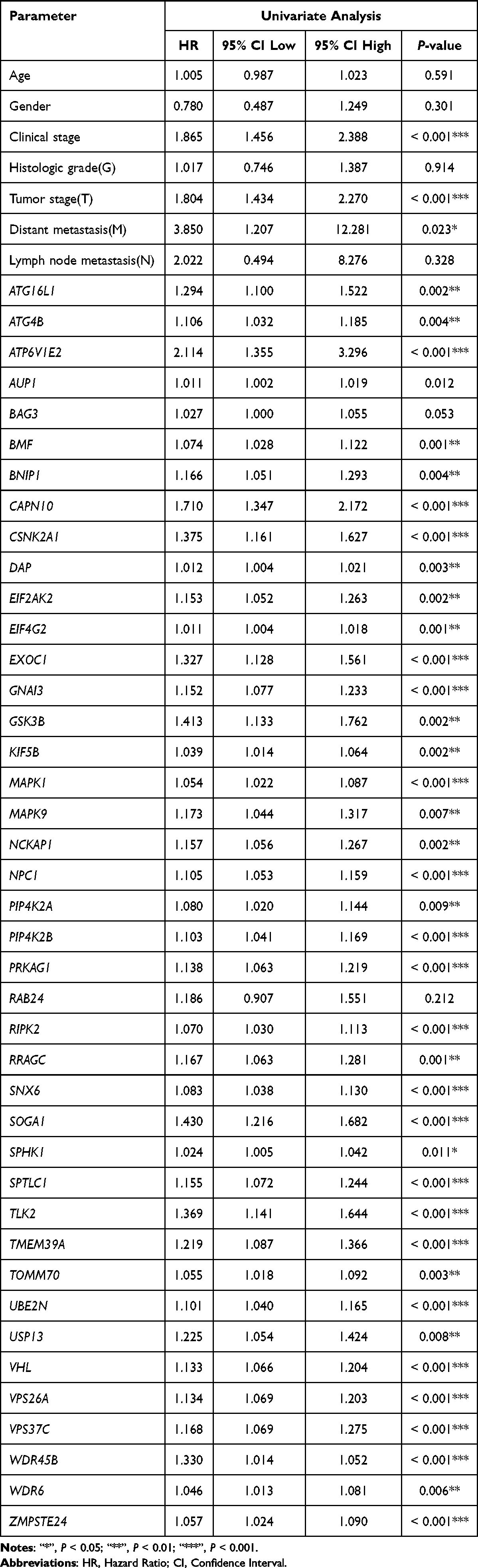 |
Table 3 The Univariate Cox Regression Analysis of Prognostic Risk Factors in Hepatocellular Carcinoma (HCC) Patients |
 |
Table 4 The Multivariate Cox Regression Analysis of Prognostic Risk Factors in Hepatocellular Carcinoma (HCC) Patients |
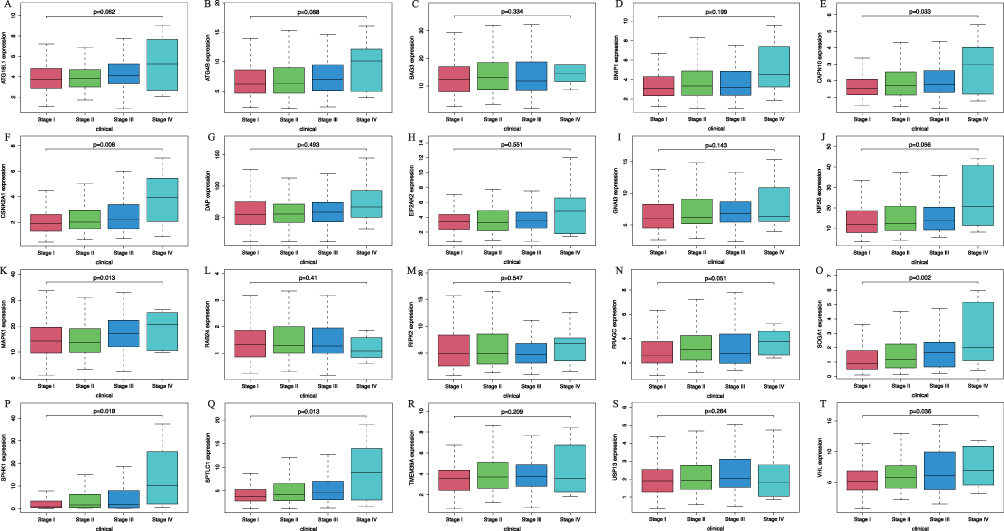 |
Figure 4 The result of the correlation between screened autophagy related genes (ARGs) expression levels and various clinicopathological features in hepatocellular carcinoma (HCC), including (A) ATG16L1, (B) ATG4B, (C) BAG3, (D) BNIP1, (E) CAPN10, (F) CSNK2A1, (G) DAP, (H) EIF2AK2, (I) GNAI3, (J) KIF5B, (K) MAPK1, (L) RAB24, (M) RIPK2, (N) RRAGC, (O) SOGA1, (P) SPHK1, (Q) SPTLC1, (R) TMEM39A, (S) USP13, (T) VHL. Abbreviations: ATG16L1, Autophagy Related 16 Like 1; ATG4B, Autophagy Related 4B Cysteine Peptidase; BAG3, BAG Family Molecular Chaperone Regulator 3; BNIP1, BCL2 Interacting Protein 1; CAPN10, Calcium-Activated Neutral Proteinase 10; CSNK2A1, Casein Kinase 2 Alpha 1; DAP, Death Associated Protein; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; GNAI3, G Protein Subunit Alpha I3; KIF5B, Kinesin Family Member 5B; MAPK1, Mitogen-Activated Protein Kinase 1; RAB24, Ras-Related Protein Rab-24; RIPK2, Receptor Interacting Serine/Threonine Kinase 2; RRAGC, Ras Related GTP Binding C; SOGA1, Suppressor of Glucose, Autophagy Associated 1; SPHK1, Sphingosine Kinase 1; SPTLC1, Serine Palmitoyltransferase Long Chain Base Subunit 1; TMEM39A, Transmembrane Protein 39A; USP13, Ubiquitin Specific Peptidase 13; VHL, Von Hippel-Lindau Tumor Suppressor. |
 |
Figure 5 The forest plots of multivariate Cox analysis about screened autophagy related genes (ARGs) expression levels among hepatocellular carcinoma(HCC), including (A) ATG16L1, (B) ATG4B, (C) BAG3, (D) BNIP1, (E) CAPN10, (F) CSNK2A1, (G) DAP, (H) EIF2AK2, (I) GNAI3, (J) KIF5B, (K) MAPK1, (L) RAB24, (M) RIPK2, (N) RRAGC, (O) SOGA1, (P) SPHK1, (Q) SPTLC1, (R) TMEM39A, (S) USP13, (T) VHL. Abbreviations: ATG16L1, Autophagy Related 16 Like 1; ATG4B, Autophagy Related 4B Cysteine Peptidase; BAG3, BAG Family Molecular Chaperone Regulator 3; BNIP1, BCL2 Interacting Protein 1; CAPN10, Calcium-Activated Neutral Proteinase 10; CSNK2A1, Casein Kinase 2 Alpha 1; DAP, Death Associated Protein; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; GNAI3, G Protein Subunit Alpha I3; KIF5B, Kinesin Family Member 5B; MAPK1, Mitogen-Activated Protein Kinase 1; RAB24, Ras-Related Protein Rab-24; RIPK2, Receptor Interacting Serine/Threonine Kinase 2; RRAGC, Ras Related GTP Binding C; SOGA1, Suppressor of Glucose, Autophagy Associated 1; SPHK1, Sphingosine Kinase 1; SPTLC1, Serine Palmitoyltransferase Long Chain Base Subunit 1; TMEM39A, Transmembrane Protein 39A; USP13, Ubiquitin Specific Peptidase 13; VHL, Von Hippel-Lindau Tumor Suppressor. |
Verification Results from GEPIA2 and HPA
Utilizing the GEPIA2 database, an analysis was conducted to investigate disparities in gene expression and survival risk between cancerous and normal tissues. The analysis revealed significant differences in the mRNA expression levels among most selected ARGs, indicating higher levels in HCC tissues compared to normal tissues (Figure S5–6). However, mRNA expression levels of ATG4B and CAPN10 in tumor tissues were found to be lower than in normal tissues (Figure S5B and E), contradicting the results of TCGA database analysis. Figure S5 showed the expression levels of 20 randomly selected genes. Other ARGs appeared in the Figure S6.
The HPA database provides a large number of tissue maps and pathological maps, from which immunohistochemical section verification results can be obtained, which can provide a reference for the bioinformatics analysis mentioned above in this study. The results of immunohistochemical sections verified that the expression levels of ARG proteins screened in this study were higher in HCC tissues than in normal tissues. There were differences in protein expression levels (Figure S7–8)
Signaling Pathways Associated with the ARGs in Enrichment Analysis
The GSEA (version 4.3.3) was employed to compare gene datasets with high and low expression, identifying potential signaling pathways significantly activated in HCC. The analysis aimed to detect the most enriched signaling pathways, applying a significance threshold of q-values of 0.05 or P-values of 0.05. In other words, when q < 0.05 was considered as the basis for being unable to screen out significantly enriched signaling pathways, P-values <0.05 were used as the basis for screening. The results of the “KEGG” signaling pathway indicated the differential expression of ARGs associated with survival and the high expression of HCC, Autophagy – other, Alcoholic liver disease, Autophagy – animal, Non-alcoholic fatty liver disease, Wnt signaling pathway, and other pathways were related to gene set enrichment. 20 randomly chosen genes were depicted in Figure 6. In Figure S9, additional ARGs were displayed.
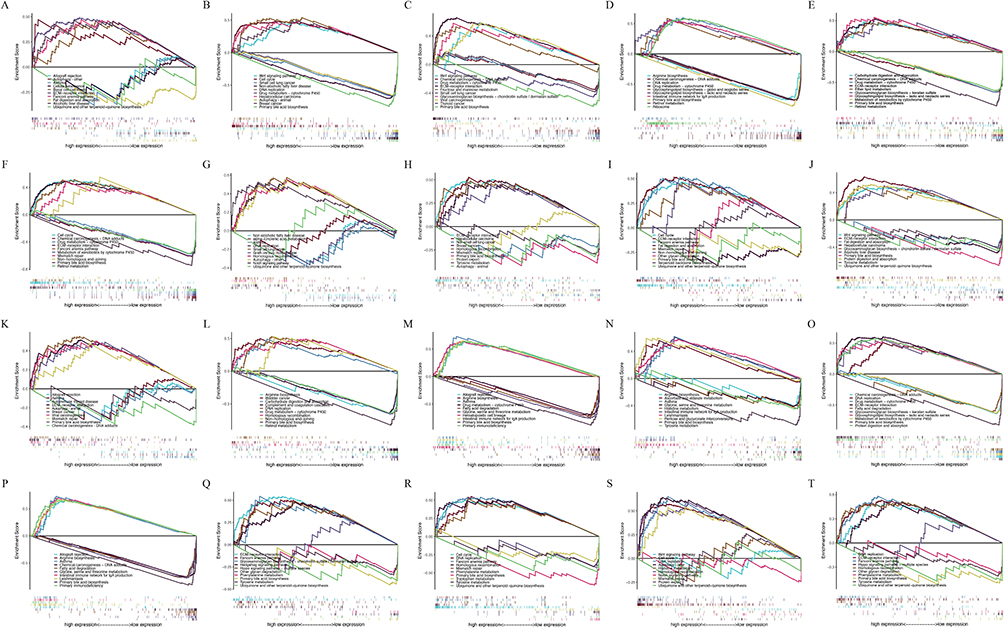 |
Figure 6 The enrichment plots of screened autophagy related genes (ARGs) expression levels in hepatocellular carcinoma (HCC) patients from Gene Set Enrichment Analysis (GSEA), including (A) ATG16L1, (B) ATG4B, (C) BAG3, (D) BNIP1, (E) CAPN10, (F) CSNK2A1, (G) DAP, (H) EIF2AK2, (I) GNAI3, (J) KIF5B, (K) MAPK1, (L) RAB24, (M) RIPK2, (N) RRAGC, (O) SOGA1, (P) SPHK1, (Q) SPTLC1, (R) TMEM39A, (S) USP13, (T) VHL. Abbreviations: ATG16L1, Autophagy Related 16 Like 1; ATG4B, Autophagy Related 4B Cysteine Peptidase; BAG3, BAG Family Molecular Chaperone Regulator 3; BNIP1, BCL2 Interacting Protein 1; CAPN10, Calcium-Activated Neutral Proteinase 10; CSNK2A1, Casein Kinase 2 Alpha 1; DAP, Death Associated Protein; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; GNAI3, G Protein Subunit Alpha I3; KIF5B, Kinesin Family Member 5B; MAPK1, Mitogen-Activated Protein Kinase 1; RAB24, Ras-Related Protein Rab-24; RIPK2, Receptor Interacting Serine/Threonine Kinase 2; RRAGC, Ras Related GTP Binding C; SOGA1, Suppressor of Glucose, Autophagy Associated 1; SPHK1, Sphingosine Kinase 1; SPTLC1, Serine Palmitoyltransferase Long Chain Base Subunit 1; TMEM39A, Transmembrane Protein 39A; USP13, Ubiquitin Specific Peptidase 13; VHL, Von Hippel-Lindau Tumor Suppressor. |
Verification Results of RT-qPCR
Compared with the L02 cell line, BAG3, EIF2AK2, KIF5B, and RAB24 were generally up-regulated in HCC cells (P < 0.05). The results were consistent with those of HCC samples detected in the TCGA database by bioinformatics analysis (Figure 7).
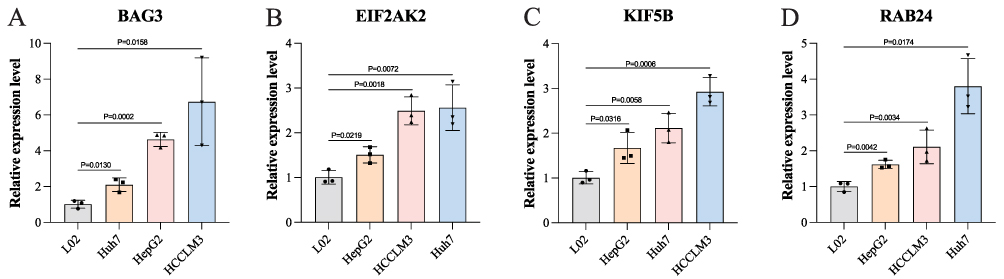 |
Figure 7 The validation of mRNA expressions of screened autophagy-related genes (ARGs) in the L02, HepG2, Huh7, and HCCLM3 cell lines by RT-qPCR analysis, including (A) BAG3, (B) EIF2AK2, (C) KIF5B, (D) RAB24. Abbreviations: BAG3, BAG Family Molecular Chaperone Regulator 3; EIF2AK2, Eukaryotic Translation Initiation Factor 2 Alpha Kinase 2; KIF5B, Kinesin Family Member 5B; RAB24, Ras-Related Protein Rab-24. |
Immune Infiltration Analysis of the BAG3, EIF2AK2, KIF5B, and RAB24
BAG3 expression is strongly positively correlated with stromal cells such as preadipocytes and smooth muscle cells, weakly positively correlated with M2 macrophages, but shows no significant correlation with anti-tumor immune cells such as CD8+ T cells. EIF2AK2 expression is strongly positively correlated with M0 and M2 macrophages, endothelial cells, and fibroblasts, and negatively correlated with CD8+ T cells and immune scores. KIF5B expression is strongly positively correlated with endothelial cells and fibroblasts, weakly positively correlated with M2 macrophages, and negatively correlated with M1 macrophages and CD8+ T cells, potentially promoting stromal support and an immunosuppressive microenvironment. RAB24 expression is strongly positively correlated with endothelial cells, smooth muscle cells, and fibroblasts, weakly positively correlated with M2 macrophages, and negatively correlated with CD8+ T cells and immune scores, potentially promoting stromal support and an immunosuppressive microenvironment (Figure S10).
Discussion
In this investigation, a comprehensive analysis was conducted on 576 instances of ARGs, encompassing difference analysis, survival analysis, as well as univariate and multivariate Cox regression analysis. The findings revealed significant disparities in mRNA expression levels among 41 ARGs between HCC tissue and normal liver tissue, concurrently indicating a pronounced correlation with the diminished survival rates associated with HCC. The results showed that the expression levels of these genes were markedly elevated in HCC tissues compared to their counterparts in normal tissues. Subsequent validation through GEPIA2 and HPA corroborated the findings, with 39 ARGs demonstrating consistency with the initial bioinformatics analysis results, albeit exceptions were noted for ATG4B and CAPN10. Further validation via RT-qPCR analysis confirmed alignment with the bioinformatics findings. The 41 ARGs obtained are expected to be tumor prognostic markers or potential targets for HCC therapy. At present, TACE is an important method for the treatment of unresectable HCC. According to the Guidelines for Diagnosis and Treatment of Primary Liver Cancer (2022 Edition), TACE can be used as the main treatment for stage II or III HCC in China.6 However, its long-term efficacy is not ideal, so there is a need to search for reliable prognostic biomarkers related to the control and prevention of poor prognosis and low survival rate of HCC.4
In recent years, autophagy, as an essential molecular process, has gradually attracted the attention of researchers. Although many reports have explored the role of individual ARGs in liver cancer, few studies have investigated the prognostic role of all ARGs in liver cancer. Therefore, we explored in depth the autophagy-associated risk genes that influence the prognosis of HCC patients by mining multiple public databases to guide the clinical evaluation of HCC patients.14
The functional role of ATG16L1 in pathophysiology may be governed by both its autophagy-dependent and independent functions. Initially, spotlighted by the identification of the Crohn’s disease-associated variant ATG16L1T300A, which correlates with residual beta-isomer levels.15 ATG16L1 polymorphisms have been observed in thyroid, gastric, and oral squamous cell carcinomas to be associated with cancer and to influence outcomes in colorectal and lung cancers.16 After accounting for variables such as age, sex, cirrhosis cause, and Patatin Like Phospholipase Domain Containing 3 (PNPLA3) genotype, the common genetic variant ATG16L1 p.T300A (rs2241880) showed a persistent association with HCC development in two distinct cohorts of cirrhotic patients. This link remained statistically significant, underscoring the potential relevance of this polymorphism in HCC susceptibility. Given its significance in cellular homeostasis and metabolic regulation, autophagy serves as a pivotal mechanism in bolstering the resilience of both healthy and cancerous cells against various stressors, including apoptotic, metabolic, and inflammatory challenges.16
ATG4 primarily catalyzes the conversion of inactive pro-LC3 to LC3-I and facilitates the expansion and closure of the autophagosome membrane.17 In HCC, ATG4B undergoes phosphorylation by AKT Serin/Threonine Kinase 1 (AKT1).18 Phosphorylation prediction analysis has identified a putative phosphorylation motif of AKT1 (31RKYS34) at the N-terminal of ATG4B, with Ser-34 being identified as a target in HCC cells. The study of long non-coding RNA CRNDE (Colorectal Neoplasia Differentially Expressed), which can regulate the stability of ATG4B mRNA, found that inhibition of ATG4B can enhance the sensitivity of cancer cells to sorafenib, a targeted drug for the treatment of HCC, which also indicates that ATG4B may be a new target for sensitizing HCC targeted drugs.19
Belonging to the BAG family, BAG3 interacts with the ATPase domain of the heat shock protein Hsp70 via the BAG domain.20 This interaction leads to BAG3’s competitive binding with BAG1 to the ATPase domain of HSP70, disrupting the delivery of client proteins for proteasomal degradation, which is typically HSP70/BAG1-dependent. In collaboration with HSP70 and LC3, BAG3 can also target the autophagy degradation of polyubiquitylation client proteins. Therefore, BAG3 extensively regulates major cellular protein degradation pathways, including proteasomal degradation and autophagy, playing a crucial role in cellular proteostasis.21 Through this mechanism of modulating the protein levels of diverse client proteins, BAG3 regulates various physiological processes such as apoptosis, development, and cytoskeletal dynamics/organization. In addition to its physiological functions, BAG3 has been implicated in various pathological conditions, including cardiomyopathy, age-related neurodegenerative diseases, and cancer. Particularly in malignancies, BAG3 exhibits a predominantly oncogenic role, influencing crucial aspects of cancer biology like cell survival, adhesion, metastasis, and angiogenesis.20 In human HCC, BAG3 is markedly overexpressed, promoting aggressive tumor behaviors such as invasiveness and angiogenesis.21
BNIP1, a member of the BH3 protein family, is primarily situated within the endoplasmic reticulum (ER), where it plays a crucial role in various cellular functions. These functions encompass apoptosis initiation, maintenance of ER network integrity, and modulation of mitochondrial dynamics. Insight into BNIP1’s multifaceted functions emerges from zebrafish research, which illustrates its involvement in detecting ER membrane vesicle fusion abnormalities, ultimately triggering apoptosis. Notably, studies suggest a potential interplay between RNF185 and BNIP1 in orchestrating autophagy and autophagosome formation, highlighting their significance in cellular homeostasis.22 BNIP1 is considered a marker for treatment in cancers such as esophageal cancer23 and cervical cancer,24 and BNIP1 may be used as a marker for HCC progression.
CAPN10 is a Ca2+-dependent cysteine protease, originally identified in the NIDDM1 (Non-Insulin-Dependent Diabetes Mellitus 1) locus region of chromosome 2 and associated with the risk of diabetes. CAPN10 has extensive effects on the human pancreas, fat, liver, and other tissues, and is involved in cell apoptosis, proliferation, and other processes in the body. The polymorphism of the CAPN10 gene is associated with the process of insulin processing, secretion, and function.25 Previous studies have shown that the regulation of the MIR-142-5P/CAPN10 axis can promote the development of ovarian cancer.26 However, current studies on CAPN10 mostly focus on the treatment of diseases highly associated with insulin secretion, and the role of CAPN10 in tumor progression is still unclear.
The protein EIF2AK2, also recognized as double-stranded RNA-activated protein kinase (PKR), is widely expressed ubiquitously in multiple cell types and may be activated in response to various types of cellular stress, such as viral infection, hypoxia, and nutrient depletion.27 While EIF2AK2 activation has demonstrated inhibitory effects on tumor proliferation, its expression and activity levels exhibit elevation in different cancer cell lines, including those associated with breast, lung, and colorectal cancers. EIF2AK2 is associated with the growth and migration of HCC and metastasis of gastric cancer.27
GNAI3 belongs to the G protein family, which not only participates in the regulation of various transmembrane signaling pathways, but also participates in the carcinogenic process. Previous studies have shown that GNAI3 can promote tumor cell metastasis through the activation of AKT,28 and can also significantly inhibit the migration and invasion of HCC cells through miR-222. Since the mRNA level of GNAI3 is up-regulated and the protein level down-regulated in HCC patients, there is post-transcriptional regulation, and miR-222 can directly bind to the 3’ end of GNAI3 and regulate the protein level of GNAI3 after transcription.29 The up-regulated mRNA level and down-regulated protein level of GNAI3 may be used as markers of HCC progression.
KIF5B is a component of the microtubule-associated motility protein complex, which mediates the transport of organelles in eukaryotic cells, plays a role in lysosomal localization, mediating natural killer cytotoxicity, and positive regulation of protein localization to the plasma membrane. Previous studies have found that KIF5B-EGFR (Epidermal Growth Factor Receptor) gene fusion and KIF5B-RET (Rearranged During Transfection) gene fusion driving non-small cell carcinoma appear in patients with lung adenoma.30 KIF5B protein expression shows a notable correlation with the advancement, recurrence, and outcome prediction in cases of oral squamous cell carcinoma,31 but the exact role of KIF5B in tumor development has not been determined and needs further study.
The MAPK1, a member of the MAP kinase family, participates in the MAPK pathway and can act as the integration point of various biochemical signals, participating in a series of cellular processes including proliferation, differentiation, transcriptional regulation, and development. The mutation of the MAPK pathway in somatic cells is related to the occurrence of solid tumors, and the dysfunction of the MAPK pathway is closely related to the growth of tumor cells, and the activation mutation of BRAF p.V600E, a major receptor cell in melanoma and thyroid cancer, is related to the drive of MAPK pathway.32 In addition, MAPK1 and MAPK14 signaling pathways are required for starvation or hypoxia to induce mitochondrial autophagy in mammalian cells, and when MAPK1 activity is inhibited, mitochondrial autophagy is effectively inhibited.32 The effect of long non-coding RNA CRNDE on HCC migration and invasion found that high expression of CRNDE is conducive to cell proliferation, migration, and invasion. MAPK1 plays the same role in HCC formation as CRNDE.33
Belonging to the RAB-associated protein subfamily, RAB24 is a small GTP (Guanosine Triphosphate) enzyme crucial for intracellular protein transport and implicated in immune response and protein metabolic pathways. Chen and colleagues, in their investigation of epigenetic modifications in HCC, noted the down-regulation of miR-615-5p, which correlates with HCC progression.34 Specifically, miR-615-5p down-regulates RAB24, while its diminished levels lead to increased RAB24 expression, thereby facilitating epithelial-mesenchymal transition, cell adhesion, and angiogenic mimicry in HCC. Consequently, RAB24 emerges as a direct target of miR-615-5p, promoting the malignant phenotype of HCC cells. However, the precise relationship between RAB24’s involvement in metastasis and cytokinesis and its functions in autophagy or endocytosis remains elusive.35
SOGA1, belonging to the autophagy-associated SOGA protein family, was found to be enriched in the CLASP2 (Cytoplasmic Linker Associated Protein 2) interaction group in 3T3-L1 mouse adipocytes and is proposed to contribute to a microtubule-dependent pathway regulating glucose metabolism, potentially via autophagy modulation. SOGA1 is expressed during mitosis and is regulated by CDK1 (Cyclin-Dependent Kinase 1)-dependent phosphorylation. It is a CLASP interacting protein required for faithful chromosome segregation in human cells.36 CDK1-mediated phosphorylation of SOGA1 promotes its direct binding to the polo box domain of PLK1 (Polo-like Kinase 1), thereby enhancing PLK1 activity and facilitating mitotic progression. Therefore, targeting the METTL16 (Methyltransferase-Like Protein 16)-SOGA1 pathway may provide a promising therapeutic strategy against colorectal cancer (CRC) by maintaining chromosome stability through accurate segregation during mitosis.37
USP13, a member of the deubiquitinase (DUB) superfamily, emerges as a pivotal player in tumorigenesis, offering diverse perspectives on its involvement. Notably, it stabilizes the tumor suppressors p53 and PTEN (Phosphatase and Tensin Homolog deleted on chromosome 10) through distinct mechanisms. By deubiquitinating USP10, USP13 enhances p53 stability, while directly binding to and deubiquitinating PTEN, it curbs PI3K/AKT signaling, impeding tumor growth across various cancer types. Moreover, the influence of USP13 extends to MITF (Microphthalmia-associated Transcription Factor), a pro-cancer protein implicated in melanoma development, where it promotes MITF deubiquitination. Despite extensive exploration of the biochemical and molecular functions of USP13 in human cancers like breast cancer, its precise role as a carcinogen or tumor suppressor remains contentious across different studies.38 In addition to the above-mentioned 41 ARGs, the other screened ARGs were also potentially associated with HCC.39–55
Utilizing gene expression and clinical data sourced from the TCGA database, GEPIA2 database, and HPA database through differential analysis, univariate and multivariate Cox regression models, Kaplan-Meier survival curve, and other analysis methods, the ARGs associated with prognostic risk were verified, and a total of 41 ARGs closely related to prognosis were obtained, describing the corresponding signaling pathways of gene set enrichment. They are expected to be utilized as tumor prognostic biomarkers and to play a potentially important role in cancer treatment and prognostic monitoring.
Conclusion
This study found that 41 ARGs, such as ATG16L1, ATG4B, BAG3, KIF5B, MAPK1, RAB24, SOGA1, were closely related to the prognosis of HCC. These genes may become markers and new targets for early diagnosis, accurate treatment, and prognosis evaluation of HCC. Further experiments are imperative to confirm this conclusion.
Ethical Statement
The data utilized in this study were obtained from publicly accessible, anonymized genomic datasets provided by the TCGA database. These datasets had undergone a rigorous three-tier de-identification procedure, including the removal of direct identifiers, masking of genomic linkage information, and aggregation of clinical features, ensuring that individuals could not be re-identified through any combination of data elements (refer to TCGA Data Use Policy, Section 4.2).
According to Article 32 of the Ethical Review Measures for Life Sciences and Medical Research Involving Humans (China, 2023) and Paragraph 32 of the Declaration of Helsinki (World Medical Association, 2013), secondary research involving completely anonymized data in non-interventional contexts does not require ethical review or informed consent.
This study involved no interaction with human participants, no biological sample collection, and no invasive procedures. Therefore, it meets the criteria for exemption from institutional ethical approval and informed consent under current regulatory guidelines.
Acknowledgments
We extend our heartfelt thanks to every author who devoted themselves earnestly to this study and the completion of this manuscript, as well as to the respected editors and reviewers for their invaluable contributions.
Funding
This study was supported by the Dengfeng Talent Support Program of Beijing Municipal Administration of Hospitals (DFL20241801).
Disclosure
The authors affirm that neither financial conflicts nor personal affiliations have influenced the reported findings within this paper.
References
1. Bray F, Laversanne M, Weiderpass E, Soerjomataram I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer. 2021;127(16):3029–3030. doi:10.1002/cncr.33587
2. Kim DW, Talati C, Kim R. Hepatocellular carcinoma (HCC): beyond sorafenib-chemotherapy. J Gastrointestinal Oncol. 2017;8(2):256–265. doi:10.21037/jgo.2016.09.07
3. Sung H, Ferlay J, Siegel RL et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clinicians. 2021;71(3):209–249. doi:10.3322/caac.21660
4. Tao J, Shi Z. Efficacy of hepatic arterial infusion chemotherapy combined with carrilizumab and lenvatinib versus hepatic arterial chemoembolization in the treatment of unresectable hepatocellular carcinoma. Chongqing Med. 2023;52:2758–2763.
5. Llovet JM, Kelley RK, Villanueva A et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
6. Yu H, Guo Y, Lie Z et al. Hepatic arterial infusion chemotherapy combined with Lenvatinib in the treatment of stage B or C hepatocellular carcinoma in Barcelona. China Interv Imag Therapeutics. 2024;21:70–74. doi:10.13929/j.issn.1672-8475.2024.02.002
7. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. doi:10.1002/path.2697
8. White E, Karp C, Strohecker AM et al. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22(2):212–217. doi:10.1016/j.ceb.2009.12.008
9. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–410. doi:10.1038/nrc3262
10. Qian H, Chao X, Williams J et al. Autophagy in liver diseases: a review. Mol Aspect Med. 2021;82:100973. doi:10.1016/j.mam.2021.100973
11. Wang K. Autophagy and apoptosis in liver injury. Cell Cycle. 2015;14(11):1631–1642. doi:10.1080/15384101.2015.1038685
12. Nan Z, Dou Y, Chen A et al. Identification and validation of a prognostic signature of autophagy, apoptosis and pyroptosis-related genes for head and neck squamous cell carcinoma: to imply therapeutic choices of HPV negative patients. Front Immunol. 2022;13:1100417. doi:10.3389/fimmu.2022.1100417
13. Barber RD, Harmer DW, Coleman RA et al. GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics. 2005;21(3):389–395. doi:10.1152/physiolgenomics.00025.2005
14. Zhang J, Zhang M, Huang J et al. Development and validation of an autophagy-related gene signature for predicting the prognosis of hepatocellular carcinoma. Biomed Res Int. 2021;2021(1):7771037. doi:10.1155/2021/7771037
15. Gammoh N. The multifaceted functions of ATG16L1 in autophagy and related processes. J Cell Sci. 2020;133(20). doi:10.1242/jcs.249227
16. Reuken PA, Lutz P, Casper M et al. The ATG16L1 gene variant rs2241880 (p.T300A) is associated with susceptibility to HCC in patients with cirrhosis. Liver Int. 2019;39(12):2360–2367. doi:10.1111/liv.14239
17. Xie Y, Fan S, Ni D et al. An ATG4B inhibitor blocks autophagy and sensitizes Sorafenib inhibition activities in HCC tumor cells. Bioorg Med Chem. 2023;84:117262. doi:10.1016/j.bmc.2023.117262
18. Park NY, Jo DS, Cho DH. Post-translational modifications of ATG4B in the regulation of autophagy. Cells. 2022;11(8):1330. doi:10.3390/cells11081330
19. Kunanopparat A, Kimkong I, Palaga T et al. Increased ATG5-ATG12 in hepatitis B virus-associated hepatocellular carcinoma and their role in apoptosis. World J Gastroenterol. 2016;22(37):8361–8374. doi:10.3748/wjg.v22.i37.8361
20. Rosati A, Graziano V, De Laurenzi V et al. BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis. 2011;2(4):e141. doi:10.1038/cddis.2011.24
21. Kögel D, Linder B, Brunschweiger A et al. At the crossroads of apoptosis and autophagy: multiple roles of the co-chaperone BAG3 in stress and therapy resistance of cancer. Cells. 2020;9(3):574. doi:10.3390/cells9030574
22. Holling T, Bhavani GS, von Elsner L et al. A homozygous hypomorphic BNIP1 variant causes an increase in autophagosomes and reduced autophagic flux and results in a spondylo-epiphyseal dysplasia. Human Mutation. 2022;43(5):625–642. doi:10.1002/humu.24368
23. Du H, Xie S, Guo W et al. Development and validation of an autophagy-related prognostic signature in esophageal cancer. Ann Translat Med. 2021;9(4):317. doi:10.21037/atm-20-4541
24. Li FH, Xiang L, Ran L et al. BNIP1 inhibits cell proliferation, migration and invasion, and promotes apoptosis by mTOR in cervical cancer cells. Eur Rev Med Pharmacol Sci. 2019;23(4):1397–1407. doi:10.26355/eurrev_201902_17096
25. Turner MD, Cassell PG, Hitman GA. Calpain-10: from genome search to function. Diabetes/Metab Res Rev. 2005;21(6):505–514. doi:10.1002/dmrr.578
26. Liu H, Chen R, Kang F et al. KCNQ1OT1 promotes ovarian cancer progression via modulating MIR-142-5p/CAPN10 axis. Mol Genetics Genomic Med. 2020;8(2):e1077. doi:10.1002/mgg3.1077
27. Kim TH, Cho SG. Kisspeptin inhibits cancer growth and metastasis via activation of EIF2AK2. Mol Med Reports. 2017;16(5):7585–7590. doi:10.3892/mmr.2017.7578
28. Ghosh P, Garcia-Marcos M, Bornheimer SJ et al. Activation of Galphai3 triggers cell migration via regulation of GIV. J Cell Biol. 2008;182(2):381–393. doi:10.1083/jcb.200712066
29. Zhang Y, Yao J, Huan L et al. GNAI3 inhibits tumor cell migration and invasion and is post-transcriptionally regulated by miR-222 in hepatocellular carcinoma. Cancer Lett. 2015;356(2):978–984. doi:10.1016/j.canlet.2014.11.013
30. Xu H, Shao C. KIF5B-EGFR fusion: a novel EGFR mutation in lung adenocarcinoma. Onco Targets Ther. 2020;13:8317–8321. doi:10.2147/ott.S263994
31. Luan YJ, Xu Y, Cai J et al. Expression of Kif5b protein is significantly associated with the progression, recurrence and prognosis of oral squamous cell carcinoma. Eur Rev Med Pharmacol Sci. 2018;22(14):4542–4550. doi:10.26355/eurrev_201807_15509
32. Ascierto PA, Kirkwood JM, Grob JJ et al . The role of BRAF V600 mutation in melanoma. J Transl Med. 2012;10(1):85. doi:10.1186/1479-5876-10-85
33. Wang H, Ke J, Guo Q et al. Long non-coding RNA CRNDE promotes the proliferation, migration and invasion of hepatocellular carcinoma cells through miR-217/MAPK1 axis. J Cell & Mol Med. 2018;22(12):5862–5876. doi:10.1111/jcmm.13856
34. Chen Z, Wang X, Liu R et al. KDM4B-mediated epigenetic silencing of miRNA-615-5p augments RAB24 to facilitate malignancy of hepatoma cells. Oncotarget. 2017;8(11):17712–17725. doi:10.18632/oncotarget.10832
35. Ylä-Anttila P, Eskelinen EL. Roles for RAB24 in autophagy and disease. Small GTPases. 2018;9(1–2):57–65. doi:10.1080/21541248.2017.1317699
36. Ferreira LT, Logarinho E, Macedo JC et al. SOGA1 and SOGA2/MTCL1 are CLASP-interacting proteins required for faithful chromosome segregation in human cells. Chromosome Res. 2021;29(2):159–173. doi:10.1007/s10577-021-09651-8
37. Li JM, Yang F, Wang ZY et al. METTL16-mediated N6-methyladenosine modification of Soga1 enables proper chromosome segregation and chromosomal stability in colorectal cancer. Cell Proliferation. 2023. doi:10.1111/cpr.13590
38. Zhao B, Huo W, Yu X et al. USP13 promotes breast cancer metastasis through FBXL14-induced Twist1 ubiquitination. Cellular Oncol. 2023;46(3):717–733. doi:10.1007/s13402-023-00779-9
39. Cohen O, Kimchi A. DAP-kinase: from functional gene cloning to establishment of its role in apoptosis and cancer. Cell Death Differ. 2001;8(1):6–15. doi:10.1038/sj.cdd.4400794
40. Jin L, Zhu J, Yao L et al. Targeting SphK1/2 by SKI-178 inhibits prostate cancer cell growth. Cell Death Dis. 2023;14(8):537. doi:10.1038/s41419-023-06023-4
41. Gramantieri L, Fornari F, Ferracin M et al. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin Cancer Res. 2009;15(16):5073–5081. doi:10.1158/1078-0432.Ccr-09-0092
42. He XX, Guo AY, Xu CR et al. Bioinformatics analysis identifies miR-221 as a core regulator in hepatocellular carcinoma and its silencing suppresses tumor properties. Oncol Rep. 2014;32(3):1200–1210. doi:10.3892/or.2014.3306
43. Nie D, Tang X, Deng H et al. Metabolic enzyme SLC27A5 regulates PIP4K2A pre-mRNA splicing as a noncanonical mechanism to suppress hepatocellular carcinoma metastasis. Adv Sci. 2024;11(5):e2305374. doi:10.1002/advs.202305374
44. Filali-Mouncef Y, Hunter C, Roccio F et al. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy. 2022;18(1):50–72. doi:10.1080/15548627.2021.1895658
45. Li S, Shao J, Lou G et al. MiR-144-3p-mediated dysregulation of EIF4G2 contributes to the development of hepatocellular carcinoma through the ERK pathway. J Experim Clin Cancer Res. 2021;40(1):53. doi:10.1186/s13046-021-01853-6
46. Li H, Li CW, Li X et al. MET inhibitors promote liver tumor evasion of the immune response by stabilizing PDL1. Gastroenterology. 2019;156(6):1849–1861.e1813. doi:10.1053/j.gastro.2019.01.252
47. Yang S, Peng LR, Yu AQ et al. CSNK2A2 promotes hepatocellular carcinoma progression through activation of NF-κB pathway. Ann Hepatol. 2023;28(5):101118. doi:10.1016/j.aohep.2023.101118
48. Cervello M, Augello G, Cusimano A et al. Pivotal roles of glycogen synthase-3 in hepatocellular carcinoma. Adv Bio Regul. 2017;65:59–76. doi:10.1016/j.jbior.2017.06.002
49. Wang H, Yang C, Jiang Y et al. A novel ferroptosis-related gene signature for clinically predicting recurrence after hepatectomy of hepatocellular carcinoma patients. American J Cancer Res. 2022;12(5):1995–2011.
50. Zhang E, Liu Q, Wang Y et al. MicroRNA miR-147b promotes tumor growth via targeting UBE2N in hepatocellular carcinoma. Oncotarget. 2017;8(69):114072–114080. doi:10.18632/oncotarget.23120
51. Wang X, Liao X, Huang K et al. Clustered microRNAs hsa-miR-221-3p/hsa-miR-222-3p and their targeted genes might be prognostic predictors for hepatocellular carcinoma. J Cancer. 2019;10(11):2520–2533. doi:10.7150/jca.29207
52. Zhang H, Chen G, Feng X et al. Targeting WDxR motif reprograms immune microenvironment and inhibits hepatocellular carcinoma progression. EMBO Mol Med. 2023;15(5):e15924. doi:10.15252/emmm.202215924
53. Cao Y, Zhang J, Xiong D et al. Hsa-miR-331-3p inhibits VHL expression by directly targeting its mRNA 3’-UTR in HCC cell lines. Acta Biochim Pol. 2015;62(1):77–82. doi:10.18388/abp.2014_779
54. Liu XT, Huang Y, Liu D et al. Targeting the SphK1/S1P/PFKFB3 axis suppresses hepatocellular carcinoma progression by disrupting glycolytic energy supply that drives tumor angiogenesis. J Transl Med. 2024;22(1):43. doi:10.1186/s12967-023-04830-z
55. Satyananda V, Oshi M, Tokumaru Y et al . Sphingosine 1-phosphate (S1P) produced by sphingosine kinase 1 (SphK1) and exported via ABCC1 is related to hepatocellular carcinoma (HCC) progression. American J Cancer Res. 2021;11(9):4394–4407 doi:.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.