Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 17
Exploring Antifibrotic Strategies for Interstitial Lung Disease in Rheumatoid Arthritis: A Narrative Review
Authors Farizani Gohari NS, Hoseinzadeh F, Khalaji A, Mirzaei S, Gholinataj jelodar M
Received 16 August 2025
Accepted for publication 28 November 2025
Published 12 December 2025 Volume 2025:17 Pages 233—250
DOI https://doi.org/10.2147/OARRR.S561159
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Narjes Sadat Farizani Gohari,1 Farahnaz Hoseinzadeh,2 Amirmohammad Khalaji,3 Samaneh Mirzaei,4,5 Mohsen Gholinataj jelodar6,7
1International Campus, Shahid Sadoughi University of Medical Science, Yazd, Iran; 2Pharmaceutical Sciences Research Center, School of Pharmacy, Student Research Committee, Shahid Sadoughi, University of Medical Sciences and Health Services, Yazd, Iran; 3School of Medicine, Tehran University of Medical Science, Tehran, Iran; 4Department of Health in Disaster and Emergencies, School of Public Health, Shahid Sadoughi University of Medical Sciences and Health Services, Yazd, Iran; 5Trauma Research Center, Shahid Sadoughi University of Medical Sciences, Yazd, Iran; 6Clinical Research Development Center, Shahid Rahnemoon Hospital, School of Medicine, Shahid Sadoughi University of Medical Sciences and Health Services, Yazd, Iran; 7Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, Shahid Sadoughi University of Medical Sciences and Health Services, Yazd, Iran
Correspondence: Mohsen Gholinataj jelodar, Division of pulmonary and Critical Care Medicine, Department of Internal Medicine, Shahid Sadoughi University of Medical Sciences and Health Services, Yazd, Iran, Tel +989113143911, Email [email protected]
Abstract: Rheumatoid arthritis (RA) is an autoimmune disease that primarily affects the joints, although extra-articular involvement, including interstitial lung disease (ILD), is also seen. Usual interstitial pneumonia and nonspecific interstitial pneumonia (NSIP) are the most common ILD in RA, which may be associated with the development of fibrotic changes in the lungs and a poorer prognosis for patients. However, the precise mechanism of ILD in RA remains unclear. A combination of environmental triggers, genetic predisposition, and enhanced immune system activity contributes to the formation of a chronic inflammatory process in the lungs, leading to uncontrolled fibroblast activity, which is ultimately associated with progressive fibrosis. Although currently available treatments for RA are effective for joint involvement, the efficacy of these anti-inflammatory treatments in progressive pulmonary fibrosis has not been encouraging. In recent years, the use of antifibrotic agents, which have been well-tested in the treatment of idiopathic pulmonary fibrosis (IPF), has been tested in the treatment of fibrotic and interstitial lung involvement in other diseases. In this study, we compared the pulmonary fibrotic developmental process of RA with IPF. Given the similarities in the pathogenesis and inflammatory pathways of these two entities, the use of antifibrotic drugs may offer a suitable and potentially promising strategy for treating fibrotic changes in RA. Currently, we face the challenge of a lack of sufficient studies with an appropriate sample size in this area. Therefore, the design and implementation of appropriate trials in the future should be considered a policy.
Keywords: rheumatoid arthritis, antifibrotic, interstitial lung disease
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disorder characterized by inflammation of the synovial membrane, which leads to joint destruction and systemic complications.1 This debilitating condition affects approximately 0.5–1% of the adult population, with an estimated 23.7 million individuals affected worldwide, in which women are three to four times more likely to develop RA than men.2,3 The prevalence of RA varies widely among different populations, with higher rates reported in developed countries.4 Moreover, RA typically affects individuals between the ages of 30 and 60, although it can occur at any age.5
The hallmark clinical features of RA include joint pain, swelling, stiffness, and functional impairment.6,7 The small joints of the hands and feet are typically affected symmetrically, although RA can involve any joint in the body.8,9
Extra-articular manifestations are common in RA and can involve multiple organ systems.10 Pulmonary complications are a significant cause of morbidity and mortality in patients with RA.10 Interstitial lung disease (ILD) is the most common pulmonary manifestation of RA, affecting approximately 10–20% of patients.11–14 Usual interstitial pneumonia and nonspecific interstitial pneumonia (NSIP) are the two most common histological patterns of ILD in patients with RA. However, other patterns, such as organizing pneumonia (OP) and diffuse alveolar damage, can also occur.15–19 Another pulmonary manifestation of RA is pleural disease, including pleurisy, pleural effusion, and rheumatoid nodules within the pleura.20
UIP is a histological pattern of interstitial pneumonia characterized by heterogeneous fibrosis with alternating areas of fibrosis and normal lung parenchyma.21 The hallmark histological features of UIP include temporal and spatial heterogeneity, honeycombing, and fibroblastic foci. Temporal heterogeneity refers to the presence of fibrosis of varying ages, while spatial heterogeneity refers to the presence of fibrosis adjacent to areas of normal lung parenchyma.22 Honeycombing is characterized by cystic airspaces lined by bronchiolar epithelium, while fibroblastic foci represent areas of active fibrosis with myofibroblast proliferation.23 On imaging, subpleural reticular opacities are usually seen primarily at the base, with honeycombing areas, irregular interpulmonary wall thickening, and traction bronchiectasis that predominate in the posterior regions of the lower lobes, especially in the cortical areas, with distinct boundaries between areas of fibrosis and normal lung parenchyma.24
NSIP is another histological pattern of interstitial pneumonia characterized by uniform interstitial inflammation and fibrosis.25 Unlike UIP, NSIP is characterized by more uniform fibrosis without the temporal and spatial heterogeneity seen in UIP.26 The prominent histological features of NSIP include interstitial inflammation, primarily consisting of lymphocytes and plasma cells, as well as fibrosis that is usually less severe than that of UIP.22 NSIP can be further classified into two subtypes based on the presence or absence of cellular inflammation: cellular NSIP and fibrotic NSIP.27 The NSIP pattern on high resolution computed tomography (HRCT) is characterized by bilateral, peripheral, and irregular ground-glass opacities, predominantly basal, usually with subpleural sparing, accompanied by septal thickening, reticular opacities, thickening of bronchovascular bundles, tractional bronchiectasis, and absence of honeycombing.24 The HRCT findings of RA-associated interstitial lung disease (RA-ILD), UIP and NSIP, are shown in Figure 1.
 |
Figure 1 High resolution computer tomography (HRCT) findings of the most common interstitial lung involvement in rheumatoid arthritis (RA); (A) NSIP pattern: Ground glass opacity with sub-pleural sparing and tractional bronchiectasis, (B) UIP pattern: Coarse reticulation with cystic lesion (Honey-combing), architectural distortion, and tractional bronchiectasis. |
The exact pathogenesis of pulmonary manifestations in RA remains poorly understood but is thought to involve a combination of genetic, environmental, and immunological factors.14 Chronic systemic inflammation in RA is believed to play a central role in the development of pulmonary complications by promoting aberrant immune responses and tissue remodeling in the lungs.28
The prognosis of pulmonary manifestations in RA varies depending on the underlying histological pattern and severity of lung involvement.29 Patients with RA-associated UIP tend to have a poorer prognosis compared to those with NSIP, with a median survival of 3–5 years from the time of diagnosis.30
The management of RA and its pulmonary manifestations is challenging and requires a multidisciplinary approach involving rheumatologists, pulmonologists, and radiologists.14 Current therapeutic strategies primarily focus on immunosuppression and anti-inflammatory agents, but the management of progressive pulmonary fibrosis remains a significant challenge.31 The efficacy of the two available antifibrotics in patients with idiopathic pulmonary fibrosis (IPF) has been demonstrated in several studies. Given the hypothesis of a common pathogenesis, recent evidence suggests that these agents may also be beneficial in pulmonary fibrosis beyond IPF.32 Pirfenidone, an antifibrotic agent, and Nintedanib, a small-molecule tyrosine kinase inhibitor,33 have shown promise in ameliorating pulmonary fibrosis associated with various interstitial lung diseases, prompting an investigation into their potential impact on pulmonary fibrotic changes related to RA.34
Currently, our understanding of the pathogenesis of pulmonary fibrosis and interstitial lung tissue involvement in RA patients is limited and imprecise. Due to the limitations of animal models, identifying the mechanism of RA-ILD has been challenging, the most important reason being the limitation of creating an animal model of RA that simultaneously demonstrates articular and extra-articular manifestations of the disease.35 Further research is needed to elucidate the optimal management strategies for pulmonary fibrotic changes related to RA and to determine the long-term safety and efficacy of Pirfenidone and Nintedanib in this patient population. In this review, we will discuss the potential mechanisms involved in the development of pulmonary fibrosis in RA patients, elucidate the differences and similarities between pulmonary fibrosis in RA and IPF, and clarify the potential role of antifibrotic medications in the management of RA-ILD.
Search Strategy
An electronic search was conducted to identify relevant studies for this review. The scientific literature available in databases, including Medline, PubMed, Web of Science, Scopus, and Google Scholar, was searched from inception to June 2025. Our search was limited to articles published in the English language. No restrictions were placed on the type of studies. Concisely, natural language terminology, Embase Subject Headings (Emtree), and Medical Subject Headings (MeSH) of interstitial lung disease, pulmonary fibrosis, rheumatoid arthritis, Pirfenidone, Nintedanib, and antifibrotics were used to identify articles in databases.
Two authors independently evaluated the titles and abstracts of the articles. In case of no initial concurrence, a final decision was reached through discussion or the opinion of a third author. In addition, the full text of eligible articles was reviewed, and their bibliographies were analyzed to identify further relevant studies.
Predisposing Factors Involved in the Pathogenesis of Rheumatoid Arthritis-Related Pulmonary Fibrosis
Rheumatoid arthritis-related pulmonary fibrosis (RA-PF) presents as progressive scarring and thickening of the lung tissue, leading to a decline in respiratory function, chronic cough, and dyspnea.31 The underlying mechanisms are complex and multifactorial, involving immune-mediated inflammation and aberrant tissue repair processes.31,36 The pathogenesis of RA-PF encompasses a combination of genetic predispositions, environmental triggers, and dysregulated immune responses that drive chronic inflammation and subsequent lung fibrosis.37 Meanwhile, the prominent role of factors such as unbridled fibroblast activity, oxidative stress, and tissue hypoxia is also emphasized. Figure 2 illustrates a brief overview of factors involved in the pathogenesis of RA-PF.
 |
Figure 2 The Pathogenesis of RA-Associated Pulmonary Fibrosis- A combination of genetic predispositions, environmental triggers, and dysregulated immune responses drives chronic inflammation and subsequent fibrosis in the lungs. Abbreviations: TGF, transforming growth factor; PDGF, platelet-derived growth factor; CTGF, connective tissue growth factor; FGF, fibroblast growth factor; EMT, epithelial-to-mesenchymal transition; End.MT, endothelial-to-mesenchymal transition. |
Genetic Predisposing Factors
In recent decades, there has been a growing interest in investigating genetic associations in interstitial lung and fibrotic diseases. MUC5B promoter variations are one of the most common genetic risk factors for primary and established pulmonary fibrosis.38 Juge et al identified the MUC5B promoter variant rs35705950 as a strong risk factor for developing ILD in RA patients. Their study found that this association was specific to patients with a UIP pattern of lung involvement and was not generalizable to other autoimmune lung involvements.39 A large cohort study found that the presence of the MUC5B variant in RA patients was associated with a tenfold increase in the risk of developing ILD. Based on the results obtained, it was concluded that increasing age also plays an important role, and this risk increases rapidly after the age of 65.40
In recent years, more attention has been paid to the potential role of shortened telomeres in contributing to pulmonary involvement in patients with RA. Telomeres are specialized regions of chromosomal DNA that contain the repeating sequence TTAGGG and play a crucial protective role. These regions shorten each time during cell division, eventually reaching a critical point that causes cell apoptosis.41 Doyle et al42 in a case-control study demonstrated an association between shortened telomeres in peripheral blood leukocytes and RA-ILD. Recently, Benedittis et al43 showed that, in addition to both groups of RA patients with and without ILD having significantly shorter telomeres compared to healthy individuals, RA-ILD patients were reported to have shorter telomeres than both healthy controls and RA patients without ILD.
The association between HLA-DRB1 and RA has been established. This association is related to a group of alleles called shared epitopes (SE) and appears to be strongly associated with anti-cyclic citrullinated peptide antibody (ACPA)-positive RA patients.44 Despite the strong association between SE and ACPA-positive RA, the frequency of these alleles in RA-ILD patients is relatively low.45 In a few studies, it has been found that HLA-DRB1*16 and DQB1*06 are risk factors for the development of interstitial lung involvement in RA patients, while HLA-DRB1*04 and DQB1*04 play a protective role against this issue.46
Environmental Triggers
The association between smoking and the development of ILD in RA patients has been demonstrated in various studies.47,48 Zhang et al conducted a meta-analysis of 22 studies to identify factors associated with the development of ILD in RA patients. Smoking (OR = 1.69, 95% CI: 1.30–2.18; P < 0.0001) was reported as a risk factor for developing RA-ILD. The precise mechanism by which smoking is associated with RA-ILD is still unclear. However, cigarette smoke components may induce an immune response, leading to the production of serum autoantibodies against multiple citrullinated proteins in the lungs, which in turn causes inflammation and epithelial cell damage, ultimately leading to ILD. Male gender, older age, longer duration of RA, and older age at onset of RA were also identified as other risk factors for developing interstitial lung disease in RA patients.49
The role of air pollution and occupational pollutants has been implicated as risk factors for incident RA and RA-ILD. The result of A large case-control study comparing 9701 incident RA, including 531 RA-ILD and 68852 matched controls, revealed that fire smoke PM2.5 exposure was not associated with RA (aOR 1.07, 95% CI 0.92–1.23) but was associated with RA-ILD (aOR 1.98, 95% CI 1.08–3.62, per 1 μg/m3). However, increased levels of nitrogen oxides (NOx) were associated with RA (aOR 1.16, 95% CI 1.06–1.27, highest vs lowest quartile). The authors concluded that air pollution may enhance the risk of RA and RA-ILD.50
Dysregulated Immune System Activity
Central to the development of RA-PF is the activation of the immune system.31 Autoantibodies, such as RF and ACPA, play a crucial role in initiating and perpetuating the inflammatory process.51 The presence of ACPAs in RA patients has been associated with a higher risk of developing pulmonary fibrosis.31,52
Inflammatory pathways in RA-PF are driven by both innate and adaptive immune responses.53,54
The Innate Immune Response
Alveolar macrophages, key players in the innate immune response, are activated and release a plethora of pro-inflammatory mediators, including TNF-α, IL-1β, and IL-6.55,56 These cytokines play a crucial role in recruiting and activating other immune cells, thereby amplifying the inflammatory response.55 TNF-α is particularly significant in RA-PF, as it promotes the recruitment of neutrophils and lymphocytes to the lungs, enhances the expression of adhesion molecules on endothelial cells, and stimulates fibroblast proliferation and activation.57 IL-1β is another pivotal cytokine that contributes to inflammation and fibrosis in RA-PF.55 It induces the production of other pro-inflammatory cytokines, promotes the expression of matrix metalloproteinases (MMPs), and facilitates the differentiation of fibroblasts into myofibroblasts.58 IL-6, a multifunctional cytokine, plays a dual role in RA-PF by contributing to both inflammation and fibrosis.55 It enhances the differentiation of T-helper (Th) 17 cells, a subset of Th cells that produce IL-17, a cytokine implicated in chronic inflammation and tissue remodeling.59
The Adaptive Immune Response
The adaptive immune response in RA-PF involves T cells and B cells.37,54 CD4+ T cells, particularly Th1 and Th17 subsets, are central to the inflammatory process.60 Th1 cells produce interferon (IFN)-γ, which activates macrophages and promotes the release of pro-inflammatory cytokines.61 Th17 cells, through the production of IL-17, contribute to chronic inflammation by recruiting neutrophils and stimulating fibroblasts.62 IL-17 also enhances the production of other pro-inflammatory cytokines and chemokines, perpetuating the inflammatory milieu in the lungs.63 B cells play a crucial role in RA-PF by producing autoantibodies, such as RF and ACPAs, which form immune complexes deposited in the lungs. These immune complexes activate complement pathways, leading to further recruitment of inflammatory cells and tissue damage.37
The Aberrant Activity of Fibroblasts
The chronic inflammatory environment in RA-PF leads to repeated injury and repair cycles in the lung tissue.64,65 This aberrant wound-healing process is characterized by the activation and proliferation of fibroblasts, which are key effector cells in the development of fibrosis.66 Fibroblasts in RA-PF can originate from various sources, including resident lung fibroblasts, epithelial-to-mesenchymal transition (EMT), endothelial-to-mesenchymal transition (EndMT), and the recruitment of circulating fibrocytes.67
EMT, a process in which epithelial cells lose their characteristics and acquire mesenchymal properties, is a significant source of fibroblasts in RA-PF.68 This transition is driven by cytokines such as TGF-β and IL-6 and is characterized by the loss of epithelial markers (eg, E-cadherin) and the acquisition of mesenchymal markers (eg, α-SMA).68,69 EndMT, similar to EMT, involves the transition of endothelial cells into mesenchymal cells, a process driven by TGF-β, which contributes to the pool of activated fibroblasts in the fibrotic lung.70
Furthermore, the activation of fibroblasts into myofibroblasts is a critical step in the pathogenesis of RA-PF.31,71 Myofibroblasts are characterized by the expression of α-SMA and the production of excessive amounts of extracellular matrix (ECM) proteins, such as collagen and fibronectin.72 TGF-β is the most potent pro-fibrotic cytokine, driving myofibroblast differentiation through the Smad pathway, which leads to the transcription of genes involved in ECM production and the suppression of fibroblast apoptosis.73,74
Besides TGF-β, other growth factors, such as platelet-derived growth factor (PDGF), connective tissue growth factor (CTGF), and fibroblast growth factor (FGF), play significant roles in fibroblast proliferation, migration, and ECM production in RA-PF.74,75 PDGF is a potent mitogen for fibroblasts, promoting their proliferation and migration. It also enhances the production of ECM components, contributing to tissue scarring and fibrosis.76 CTGF, also known as CCN2, is upregulated in response to TGF-β and acts synergistically with it to promote fibroblast proliferation, myofibroblast differentiation, and ECM production.77 FGF, particularly FGF-2, stimulates fibroblast proliferation and enhances the production of proteoglycans and collagen, further contributing to the fibrotic process.78
The Oxidative Stress
Oxidative stress also contributes to the pathogenesis of RA-PF.31 England et al79 demonstrated that patients with RA and ILD have significantly higher levels of serum IgA and IgM anti-Malondialdehyde-acetaldehyde antibody (MAA) concentrations compared to patients with RA and COPD, as well as RA without lung disease. A similar result was reported in a recent study by Aripova et al.80 Among 2739 participants with RA, they revealed that protein-specific anti-MMA antibodies to collagen, fibrinogen, and vimentin were related to the prevalence of RA-ILD. The results of these studies emphasize the potential role of MMA in the pathogenesis of RA-ILD. ROS, produced by inflammatory cells and damaged epithelial cells, induce cellular injury and activate pro-fibrotic signaling pathways. ROS can activate TGF-β and other pro-fibrotic cytokines, enhance fibroblast proliferation, and promote ECM production. The antioxidant defenses are often overwhelmed in RA-PF, exacerbating oxidative damage and fibrosis.81
Tissue Hypoxia and Fibrogenesis
Hypoxia, a common feature in fibrotic tissues, further drives fibrosis in RA-PF.31 Hypoxia-inducible factors (HIFs) are stabilized in low-oxygen conditions, leading to the transcription of genes involved in fibrosis and inflammation.82 Transient and mild hypoxia can enhance cellular repair mechanisms and reduce oxidative damage. However, excessive or prolonged activation of HIF abolishes this adaptive response, leading to fibrosis and inflammation.83 Increased expression of HIF-1 has been observed in the serum as well as in the synovial membrane of RA patients.84 HIFs enhance the expression of pro-fibrotic genes such as TGF-β, vascular endothelial growth factor (VEGF), and endothelin (ET)-1, promoting fibroblast activation and ECM production.85
Various studies have investigated the effects of hypoxia in inducing and promoting the development of lung fibrosis. In an animal model of bleomycin-induced pulmonary fibrosis investigated by Guo et al. EMT was detected in the mice’s lung tissues. The study determined that hypoxia induced by fibrosis can promote the activation of EMT via the hypoxia-HIF-1α pathway.86 In another study, the effect of chronic intermittent hypoxia in an animal model of bleomycin-induced lung fibrosis was examined. Chronic intermittent hypoxia leads to the upregulation of inflammatory pathways, enhancing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation and expression of IL-17 mRNA and Col1α1 mRNA. The authors concluded that hypoxia may intensify collagen deposition and fibrosis development by inflammatory and oxidative pathways.79
Rheumatoid Arthritis-Related Pulmonary Fibrosis and Idiopathic Pulmonary Fibrosis: The Analogies and Discrepancies
Despite their different origins, RA-PF and IPF share several pathophysiological mechanisms, including chronic inflammation, fibroblast activation, and ECM deposition. In RA-PF, the initial trigger is autoimmune inflammation, where autoantibodies and immune complexes contribute to lung tissue damage.31 In IPF, the initial trigger remains unknown but is believed to involve repeated epithelial injury and abnormal wound healing.87
In RA-PF, the presence of autoantibodies such as RF and ACPAs is significant. These autoantibodies contribute to the autoimmune pathology of RA and its pulmonary manifestations.31 IPF, however, is not associated with autoantibodies, yet their presence can affect disease progression.51
The immune system plays a central role in RA-PF, with T- and B-cell-mediated responses contributing to lung inflammation and fibrosis.31 In IPF, the immune response is less well-defined, but it involves aberrant wound healing and fibroblast activation rather than a classical autoimmune process.54
Macrophages exhibit different functional states, or polarization, which influence fibrosis.88 In RA-PF, M1 (pro-inflammatory) and M2 (pro-fibrotic) macrophages contribute to disease pathology.89 In IPF, M2 macrophages are more prominent, driving fibrogenesis.90
Th17 cells, a subset of T helper cells, are involved in the autoimmune response in RA-PF, promoting inflammation and fibrosis. Their role in IPF is less clear, though they may contribute to chronic inflammation.31 Regulatory T Cells (Tregs), which suppress immune responses, are dysfunctional in RA-PF, leading to uncontrolled inflammation.37 In IPF, the function and number of Treg cells are also altered, contributing to an imbalance in immune regulation.54
B cells and autoantibodies play a critical role in RA-PF, driving chronic inflammation and fibrosis. In IPF, B cells are less central to pathogenesis, and autoantibodies are not a feature of the disease.31
Mast cells are involved in fibrosis through the release of pro-fibrotic mediators.91 Their role is more pronounced in RA-PF due to the autoimmune nature of the disease, while in IPF, they contribute to the fibrotic environment.31
Eosinophils, which are involved in allergic responses, can contribute to fibrosis in RA-PF by releasing pro-fibrotic cytokines. In IPF, eosinophils are less commonly implicated.92
In RA-PF, key inflammatory pathways include TNF-α, IL-1β, and IL-6, which are also central to the pathogenesis.37 In IPF, TGF-β is the predominant cytokine driving fibrosis, with less emphasis on classical inflammatory pathways seen in RA-PF.93 VEGF is involved in angiogenesis and fibrosis. Elevated levels of VEGF are seen in both RA-PF and IPF, contributing to abnormal blood vessel formation and fibrosis.94 Fibrogenic mediators, such as CTGF and PDGF, are elevated in both diseases and contribute to the proliferation of fibroblasts and the deposition of the extracellular matrix (ECM).75
Both RA-PF and IPF involve the activation and differentiation of fibroblasts into myofibroblasts, which produce ECM components, leading to fibrosis. In RA-PF, this process is driven by both inflammatory cytokines and autoimmune mechanisms.31 In IPF, fibroblast activation is primarily driven by TGF-β and mechanical stress.95 Fibroblastic Foci are a hallmark of UIP, seen in both RA-PF and IPF, representing areas of active fibrosis. The number and distribution of fibroblastic foci are similar in both conditions, although the surrounding inflammatory milieu is more prominent in RA-PF.31
ECM remodeling is a key feature of both diseases. In RA-PF, ECM deposition is influenced by chronic inflammation and autoimmunity.31 In IPF, ECM remodeling is driven by repeated epithelial injury and aberrant wound-healing processes.87 MMPs, enzymes that degrade ECM components, are involved in tissue remodeling in both diseases. Dysregulated MMP activity leads to excessive ECM deposition and fibrosis. TIMPs regulate MMP activity, and an imbalance between MMPs and TIMPs contributes to fibrosis in both RA-PF and IPF. Elevated TIMP levels are associated with disease severity.96,97
Injury and dysfunction of alveolar epithelial cells (AECs) are central to the pathogenesis of IPF, leading to aberrant repair and fibrosis.98 In RA-PF, AEC injury is secondary to chronic inflammation but still contributes to fibrotic processes.99 Furthermore, endothelial cell dysfunction contributes to the development of fibrosis in both diseases. In RA-PF, endothelial cells are damaged by chronic inflammation, while in IPF, they are affected by persistent epithelial injury and hypoxia.31,100
The mechanisms for resolving fibrosis are impaired in both diseases. In RA-PF, ongoing inflammation prevents resolution, while in IPF, persistent epithelial injury and fibroblast activation hinder resolution.31,101
The main challenge in RA-PF is the prevention of the progression of fibrosis and its treatment. Immunosuppressive therapies, including corticosteroids and DMARDs, are commonly used in RA-PF due to the underlying autoimmune nature. However, their use in IPF is controversial and generally not recommended due to a lack of efficacy and potential for harm.31,102 IPF treatment focuses on antifibrotic agents, such as Pirfenidone and Nintedanib, which slow disease progression.103 While antifibrotic therapies like Nintedanib and Pirfenidone are approved for IPF, their role in RA-PF is less clear. Ongoing studies are investigating their efficacy in RA-PF, given the similarities in fibrotic pathways.31,103
Pharmacology of Antifibrotic Agents: Pirfenidone and Nintedanib
Pirfenidone
Pirfenidone is an effective antifibrotic compound predominantly employed in the management of IPF. Its therapeutic benefits stem from its diverse pharmacodynamic characteristics, which encompass notable anti-inflammatory and antifibrotic actions. The pharmacokinetic and pharmacodynamic characteristics of Pirfenidone provide an essential understanding of its therapeutic capabilities and safety profile, emphasizing its significance in treating intricate fibrotic conditions.
Pharmacokinetics
Pirfenidone is rapidly absorbed, with a peak concentration reached within 1.8 to 2.2 hours and a short half-life of 2.1 to 2.4 hours, necessitating frequent dosing to maintain effectiveness.104 The drug is widely distributed in tissues, emphasizing the need for careful dosing strategies.105 Pirfenidone exhibits good oral bioavailability, which is crucial for its therapeutic effectiveness in treating conditions such as IPF.106
Primarily metabolized into 5-carboxy-pirfenidone, most of the drug is excreted in the urine. Monitoring renal function is crucial, as impaired kidney function can alter its pharmacokinetics and increase the risk of toxicity.104 Extended-release formulations offer more stable plasma concentrations, lower peak plasma concentrations, reduced fluctuations, and potentially minimized adverse effects, which may enhance treatment outcomes.107,108
Pharmacodynamics
The antifibrotic effects of Pirfenidone are characterized by its ability to inhibit fibroblast activation and the synthesis of extracellular matrix components, which are essential processes in the development of fibrosis.109 This has been demonstrated across various tissues, including the lungs, bladder, and pancreas.110 Additionally, Pirfenidone inhibits metabolic reprogramming associated with epithelial-mesenchymal transition in non-small cell lung cancer, showcasing its broad antifibrotic capabilities.111
Pirfenidone also exerts its effects by suppressing the TGF-β/Smad signaling pathway, a central regulator of fibrosis and inflammation, leading to reduced expression of fibrosis-related factors and enhanced tissue recovery.112,113 Molecular docking studies suggest that Pirfenidone binds to kinases such as p38 mitogen-activated protein kinase (MAPK), serine/threonine protein kinase B (AKT)-1, and extracellular signal-regulated kinase (ERK)-1/2, indicating a complex interaction that modulates inflammation and tissue remodeling.114 In nonalcoholic fatty liver disease models, Pirfenidone mitigates oxidative stress and inflammation through the activation of the nuclear factor-erythroid 2-related factor (Nrf)-2 signaling pathway,115 further extending its antifibrotic properties.
Clinical evidence supports the efficacy of Pirfenidone in managing IPF, where it significantly slows the decline in FVC and improves progression-free survival.116 Its protective effects against myocardial fibrosis have been observed in IPF patients, with a lower incidence of arrhythmic events.117
Generally well-tolerated, typical side effects include gastrointestinal symptoms and photosensitivity. However, the therapeutic efficacy of Pirfenidone may vary depending on the specific fibrotic condition and underlying pathophysiology, underscoring the need for individualized treatment strategies. While Pirfenidone is a promising therapeutic option for various fibrotic and inflammatory diseases, further research is essential to fully elucidate its long-term effects, optimize its clinical application, and explore its potential across a broader range of diseases.109,118
Nintedanib
Nintedanib is another oral antifibrotic agent approved for the treatment of IPF and is currently being investigated for the treatment of RA-associated interstitial lung disease (ILD).119 Nintedanib inhibits multiple receptor tyrosine kinases involved in the pathogenesis of pulmonary fibrosis, including the FGF receptor, PDGF receptor, and VEGF receptor.
Pharmacokinetics
Rapidly absorbed after oral administration, Nintedanib reaches maximum plasma concentrations (Cmax) within 2–4 hours and demonstrates a high volume of distribution (Vss = 1050 L), particularly in the lungs.120,121 Its extensive tissue partitioning in lung tissue is a key feature for treating pulmonary conditions.
The absorption rate constant (Ka) is about 0.0827 h−1, with a lag time of 25 minutes.122 The drug’s absorption, however, is limited by its poor solubility, which contributes to its low oral bioavailability of approximately 5% and first-pass metabolism.121 To address these limitations, advanced drug delivery systems have been explored. For instance, self-microemulsifying drug delivery systems (SMEDDS) have been shown to enhance Nintedanib’s permeability by 2.8- to 3.0-fold in vivo, significantly improving its bioavailability.123 Nanocrystal formulations, such as BIBF-NCs, enhance absorption by reducing particle size and maintaining drug supersaturation, resulting in a 2.58-fold increase in bioavailability compared to soft capsules.124 Moreover, nano-lipid carriers (NLCs) loaded with Nintedanib esylate demonstrated a 26.31-fold increase in bioavailability through enhanced lymphatic uptake.125 These formulation strategies highlight promising innovations to overcome the inherent absorption challenges of Nintedanib.
Nintedanib undergoes complex hepatic metabolism, leading to the formation of inactive metabolites. Hydrolysis in the liver, mediated by carboxylesterase (CES)-1, converts the drug to a carboxylate derivative (BIBF1202), which is further glucuronidated by UGT1A1 and other UDP-glucuronosyltransferases (UGTs), such as UGT1A7, UGT1A8, and UGT1A10. This glucuronide metabolite can also be hydrolyzed back to BIBF1202, a reversible process that impacts its pharmacokinetics and excretion.120,126 The majority of Nintedanib is excreted via feces, with less than 1% eliminated unchanged through urine. Despite this complex metabolic profile, the drug exhibits a terminal elimination half-life of 10–15 hours, with minimal accumulation upon repeated dosing, making it manageable for long-term treatments.120
Hepatic impairment markedly affects Nintedanib pharmacokinetics, leading to increased drug exposure in these patients, which necessitates careful monitoring and potential dose adjustments.127 Nevertheless, despite these metabolic and pharmacokinetic challenges, Nintedanib is generally well-tolerated; however, adverse effects, such as gastrointestinal disturbances, can sometimes require dose reduction or treatment discontinuation.128 To mitigate systemic side effects, inhalation formulations of Nintedanib have been developed to improve localized pulmonary drug delivery, thereby enhancing therapeutic efficacy while reducing overall systemic exposure.129,130
Pharmacodynamics
Nintedanib, a potent tyrosine kinase inhibitor, is recognized for its significant pharmacodynamic properties, particularly its role in modulating angiogenesis and fibrosis, both critical processes in diseases like IPF and certain cancers. Its mechanism of action centers on inhibiting key receptor tyrosine kinases (RTKs), such as VEGF receptors, PDGF receptors, and FGF receptors, which are crucial in promoting angiogenesis, fibrosis, and tumor progression.120,131
Through the inhibition of the VEGF receptor, Nintedanib disrupts tumor vascularization, limiting the blood supply essential for tumor growth, while targeting the PDGF receptor interferes with cellular proliferation and survival mechanisms. FGF receptor inhibition further hampers cellular differentiation and growth, contributing to its broad antitumor and antifibrotic effects.120,131 This multi-target approach positions Nintedanib as a valuable agent across various conditions, including IPF and malignancies like prostate and gastric cancers.132–134
In IPF, Nintedanib has been shown to slow the decline in lung function by reducing the rate of FVC decline, thus prolonging patient survival and quality of life.132 It also exhibits significant antifibrotic effects by inhibiting fibroblast activity, a key driver in IPF, where fibroblast proliferation and extracellular matrix accumulation lead to progressive lung fibrosis.135 These effects extend to other fibrotic conditions, including autoimmune-related interstitial lung diseases (ILDs) and PPF, where it has consistently slowed disease progression across various underlying conditions.136,137
Nintedanib is actively explored for its role in inhibiting EMT, a process involved in fibrosis and cancer metastasis. In retinal pigment epithelial cells, for instance, Nintedanib reduced EMT, inhibiting cell migration and proliferation, which is significant in conditions like proliferative vitreoretinopathy.138
Moreover, its broad activity against multiple signaling pathways, including its anti-inflammatory effects, has been documented in various studies. For example, it suppresses the Janus kinase-2/signal transducer and activator of the transcription-3 (JAK2/STAT3) pathway, selectively inducing the death of senescent cells, which may have implications for age-related diseases and conditions associated with chronic inflammation.139
The Role of Pirfenidone and Nintedanib in RA-PF
In recent years, the repurposing of antifibrotic agents, initially approved for IPF, for the management of RA-PF has gained attention. However, the current clinical evidence on the efficacy and safety of these agents in RA patients with pulmonary fibrosis is inconclusive. Studies directly evaluating the effects of Pirfenidone and Nintedanib in patients with RA-PF offer valuable findings,140–144 although the evidence is minimal. Table 1 summarizes original research studies (randomized controlled trials and observational studies) investigating the effects of Pirfenidone and Nintedanib in patients with RA-PF.
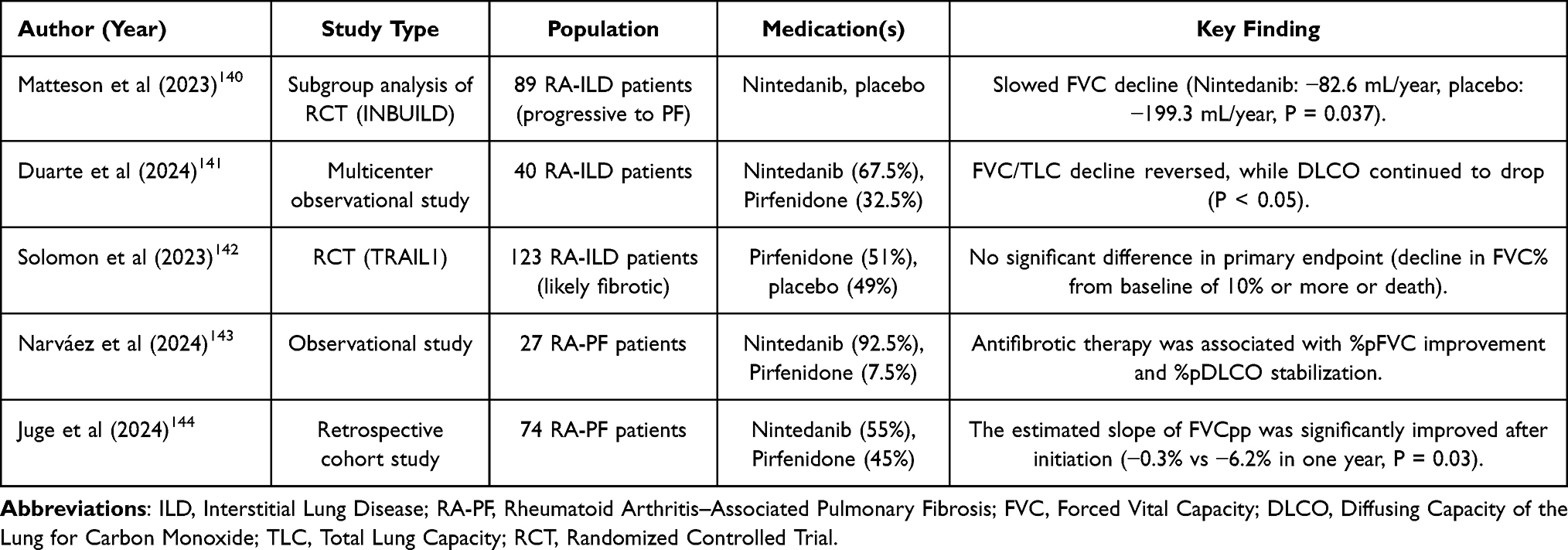 |
Table 1 Efficacy and Safety of Antifibrotic Agents in Patients with RA-PF |
The study by Matteson and colleagues utilized data from a subgroup of the INBUILD trial, explicitly focusing on RA-ILD patients,140 to evaluate the efficacy and safety of Nintedanib compared to placebo in patients with progressive RA-ILD who progressed to pulmonary fibrosis (PF). All the patients had shown progression to PF, despite clinical management, and were randomized to Nintedanib or placebo. In the 89 included patients, the FCV over 52 weeks declined in both groups, with a significantly higher decline in the placebo group, confirming the protective role of Nintedanib in the population (Nintedanib: −82.6 mL/year, placebo: −199.3 mL/year, P = 0.037). This study also demonstrated that patients treated with Nintedanib experienced fewer acute exacerbations of underlying lung disease and had a lower mortality rate compared to the placebo group. The most common adverse effect was diarrhea, which occurred more frequently in the Nintedanib group (61.9% vs 27.7%). It should be noted that the study design did not randomize patients based on treatments other than Nintedanib; however, among patients with RA-ILD, the effect of Nintedanib on reducing FVC decline in patients initially taking DMARDs and/or glucocorticoids was similar to that observed in all patients with RA-ILD.
Complementing the previous randomized controlled trial (RCT), Narváez et al evaluated the tolerability and effectiveness of Nintedanib and Pirfenidone in patients with progressive fibrosing RA-ILD in a longitudinal, retrospective observational study.143 They used data from 27 patients (25 using Nintedanib and 2 using Pirfenidone) with a median follow-up of 25 months. Among the total population, 18 patients completed one-year treatment and showed significant improvement in FVC% (+4.7%, P = 0.023), while nine patients completed two-year treatment with a higher increase in FVC% (+7.7%, P = 0.037). Moreover, the DLCO% remained unchanged during the one- and two-year treatment periods (one year: −3.8%, P = 0.175; two years: −2.2%, P = 0.621). The most common adverse events were gastrointestinal in nature. According to the reported results, adverse events led to permanent dose reductions in 40% of patients treated with Nintedanib and 14% of patients treated with pirfenidone, and resulted in treatment discontinuation in 18.5% of patients. The small sample size in this study is striking, and the lack of a control group somewhat overshadows the accuracy and precision of the results obtained. Additionally, due to the small sample size, it was not possible to perform any adjusted analyses to rule out possible confounding factors that might affect the association between antifibrotic drugs and outcomes. To conclude, antifibrotic therapy was associated with improvements in FVC% and stabilization of DLCO%. To conclude, antifibrotic therapy was associated with improvements in FVC% and stabilization of DLCO%.
Similar to the cohort studied by Narváez et al,143 the study conducted by Duarte et al evaluated the efficacy of antifibrotics in a real-world cohort of patients with RA-ILD.141 They used a mixed linear model with random intercept and random slope to compare the results of pulmonary function tests within 12 (±6) months before and 12 (±6) months after starting antifibrotic drugs. In this multicenter observational study, 40 RA-ILD patients were initially treated with either Nintedanib (27 patients) or Pirfenidone (13 patients). Using antifibrotics improved FVC one year after initiation, with a 200 mL improvement in the year after treatment compared to a 300 mL decline in the year before initiation (P = 0.336). Moreover, total lung capacity (TLC) improved by 600 mL after initiation compared to an 800 mL decline in the year before treatment (P = 0.147). Finally, DLCO remained in decline before and after the treatment (before: −3% vs after: −2.9%, P = 0.75).
The role of Pirfenidone in RA-PF remains less clear. In a randomized, double-blind, placebo-controlled RCT (TRIAL1), Solomon et al142 evaluated the safety, tolerability, and efficacy of Pirfenidone in RA-ILD patients. Of the total 123 patients, 63 initiated Pirfenidone, while 60 received a placebo. The trial was halted early due to the coronavirus disease-2019 (COVID-19) pandemic, which limited the scope of the findings. The primary outcome was a more than 10% decline in FVC% or death, which was not significantly different between the pirfenidone and placebo groups (Pirfenidone: 11% vs placebo: 15%, P = 0.48). However, patients receiving Pirfenidone showed a slower decline in absolute FVC (−66 vs −146, P = 0.0082) and FVC% (−1.02 vs −3.21, P = 0.0028). Although the effect of Pirfenidone in reducing the decline in FVC was promising, the primary endpoint showed no difference.
Finally, in a retrospective cohort study, Juge et al evaluated the effectiveness and tolerability of antifibrotics in RA-ILD patients receiving Nintedanib or Pirfenidone from 2014 to 2023.145 Among the 74 total patients, 40 initiated Nintedanib and 34 initiated Pirfenidone, with a median follow-up of 89 weeks. The estimated slope of FVCpp was significantly improved after initiation (−0.3%/year after initiation compared to −6.2%/year before treatment, P = 0.03). It should be noted that the small sample size of this study may have reduced the power to detect significant clinical and preclinical changes.
Discussion
Despite appropriate treatment options, DMARDs, in the management of joint disease in RA patients, the effectiveness of these treatments in pulmonary involvement has been inconsistent and challenging.146 While DMARDs may respond to some instances of RA-ILD with an inflammatory pattern, fibrotic RA-ILD, particularly with a UIP pattern, tends to be unresponsive to DMARDs.147 It is worth noting that there is conflicting data, leading to concerns about pulmonary toxicity from the use of immunomodulators for the treatment of RA.148 In recent years, due to the inadequacy of existing treatments and the progressive course of fibrotic changes in RA patients, which is associated with a poorer prognosis in patients, efforts have been made to use previously known antifibrotic drugs that have proven successful in studies on IPF patients. The positive effects of antifibrotic drugs, Pirfenidone and Nintedanib, in reducing the deterioration of lung function, as well as improving life expectancy in IPF patients, have been demonstrated in numerous studies.149,150 The similarities in the pathogenesis of IPF and RA-PF patients make the use of antifibrotic drugs in this group of patients rational. Currently, we are faced with limitations in the available studies investigating the role of these drugs in RA patients. The current results are based on small randomized clinical trials and small sample sizes, which cannot be relied upon as a sound scientific basis.
A decrease in FVC in patients with interstitial lung disease indicates disease progression and is used as a predictive factor for prognosis and mortality.151,152 It has been observed that DLCO is also reduced due to the diminished diffusion surface and impaired gas exchange caused by increased alveolar-capillary membrane thickness resulting from lung fibrosis in ILD. It is of great importance to note that while lung volumes decrease in ILD patients in more advanced cases, changes in DLCO have helped diagnose mild and early cases of the disease.153
Based on the results of the few available studies, the effect of antifibrotic drugs on pulmonary function has been variable, ranging from no change to delayed decline in FVC reported in different studies. Recently, in a meta-analysis study, Jang and colleagues examined the effects of antifibrotic drugs in RA-ILD. The results of the meta-analysis revealed a significant reduction in FVC decline in patients with RA-ILD treated with antifibrotic agents compared with placebo (mean difference, 88.30; 95% CI, 37.10–139.50).119 They include only two randomized clinical trials, and the small number of participants is undoubtedly a significant limitation in accepting the meta-analysis findings as reliable and substantial evidence in defense of the clinical use of these drugs in RA patients.
An essential point to consider when using antifibrotic drugs in RA patients is that the effectiveness of these drugs should not be focused solely on pulmonary function. The clinical effects of these drugs in improving the quality of life of patients and reducing mortality should be considered. These issues cannot be accurately assessed with our current data.
It is essential to note that in every treatment strategy, the discussion of cost-effectiveness in various forms should be carefully evaluated. In a study conducted in France on IPF patients, it was concluded that Pirfenidone is likely to be a cost-effective strategy compared to best supportive care and appears to be more efficient and less expensive than Nintedanib for treating patients.154 A study conducted in Belgium on IPF patients yielded a different conclusion from the previous study. A cost-effectiveness analysis revealed that the use of Nintedanib was superior to Pirfenidone in these patients.155 Indeed, our information on this issue in RA patients is minimal; however, regional conditions should be considered in terms of cost-effectiveness when recommending the initiation of treatment. In our observational experience in Iran on IPF patients, Limited access to these drugs, as well as their high prices in the country’s pharmaceutical market, have been common problems in treating patients.
We identified some limitations in the previous research conducted on the efficacy of antifibrotic agents in patients with RA-ILD. As mentioned earlier, the small number of studies conducted in this field has led to a scientific gap in answering the important clinical question of whether antifibrotic drugs can play a significant role in the treatment of RA-ILD. There is a lack of well-designed clinical trials with adequate sample sizes. Our review found only two RCTs, and the others were observational and mostly retrospective. The lack of randomization of study participants based on their receipt of medications other than antifibrotic drugs is an important consideration when interpreting the results obtained. The TRAIL1 study, designed to investigate the efficacy of pirfenidone in patients with RA-ILD, was terminated early due to the onset of the COVID-19 pandemic, resulting in an underpowered analysis of the primary endpoint. The small sample sizes of studies conducted in this area have limited the ability of these studies to detect significant changes in key outcomes. Our data from many trials are based on a 52-week treatment period, which may not fully reflect the results of treatment in diseases such as RA-ILD, which have a long-term clinical course. Heterogeneity is also observed in study design. Inclusion criteria for participants in studies varied and included different thresholds for spirometric and imaging indices. In some studies, the extent and type of interstitial lung involvement were not specified. The outcomes reported as endpoints in the studies have varied. Some studies have focused solely on spirometric measures and lung function tests, while others have also examined clinical outcomes, including mortality, exacerbations, and drug side effects. Overall, it appears that we currently have significantly less information about pirfenidone than Nintedanib in this regard. Even in studies that have examined both drugs in patients, the number of patients treated with pirfenidone has been substantially lower.
Given the currently available evidence and the scientific gap in this field, some directions are recommended for future studies on the efficacy of antifibrotic drugs in RA-ILD. Designing and implementing randomized controlled studies with careful participant selection and minimal confounding factors will greatly help identify the effects of these treatments in patients. Subgroup analysis is useful in evaluating the effectiveness of treatment in various patient groups. For example, it is suggested that this analysis be performed in patients with varying degrees of interstitial lung involvement on imaging (UIP or NSIP). On this basis, future studies can provide information on which groups of patients are likely to respond better to these treatments and derive greater benefits. Trial designs comparing pirfenidone and Nintedanib with each other in RA-ILD patients, and even their combination in the treatment of this disease, are recommended for future implementation. Considering the endpoints of future studies on patient-centered outcomes, such as cough, activity-related shortness of breath, and quality of life, is another important point that should be strongly considered in the design of future studies.
Conclusion
Given the common pathogenic process in the development of pulmonary fibrosis in RA and IPF patients, the use of antifibrotic drugs may be effective in reducing the decline in pulmonary function in patients with RA. Despite the evidence of a potential role in the pathogenesis of the disease, there is currently no strong clinical evidence to recommend the use of these drugs in this clinical setting. The need for further randomized clinical trials to evaluate the potential efficacy of these drugs in RA-ILD patients in the future is strongly felt.
Data Sharing Statement
The authors confirm that the data supporting the findings of this study are available within the article.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare no conflict of interest.
References
1. Wu D, Luo Y, Li T, et al. Systemic complications of rheumatoid arthritis: focus on pathogenesis and treatment. Front Immunol. 2022;13:1051082. doi:10.3389/fimmu.2022.1051082
2. Mian A. The impact of moderate disease activity rheumatoid arthritis and evaluating the management of this cohort. 2023.
3. Ohno T, Aune D, Heath AK. Adiposity and the risk of rheumatoid arthritis: a systematic review and meta-analysis of cohort studies. Sci Rep. 2020;10(1):16006. doi:10.1038/s41598-020-71676-6
4. Almutairi KB, Nossent JC, Preen DB, Keen HI, Inderjeeth CA. The prevalence of rheumatoid arthritis: a systematic review of population-based studies. J Rheumatol. 2021;48(5):669–676. doi:10.3899/jrheum.200367
5. Angum F, Khan T, Kaler J, Siddiqui L, Hussain A. The prevalence of autoimmune disorders in women: a narrative review. Cureus. 2020;12(5). doi:10.7759/cureus.8094
6. Mease PJ, Bhutani MK, Hass S, Yi E, Hur P, Kim N. Comparison of clinical manifestations in rheumatoid arthritis vs. spondyloarthritis: a systematic literature review. Rheumatol ther. 2022;1–48.
7. Saalfeld W, Mixon AM, Zelie J, Lydon EJ. Differentiating psoriatic arthritis from osteoarthritis and rheumatoid arthritis: a narrative review and guide for advanced practice providers. Rheumatol Ther. 2021;8(4):1493–1517. doi:10.1007/s40744-021-00365-1
8. Colquhoun M, Gulati M, Farah Z, Mouyis M. Clinical features of rheumatoid arthritis. Medicine. 2022;50(3):138–142. doi:10.1016/j.mpmed.2021.12.002
9. Khidir SJ, van Dijk BT, Krijbolder DI, Verstappen M, van Mulligen E, van der Helm-van AH. Joint involvement in RA starts predominantly in the hands: functional, clinical and imaging studies in clinically suspect arthralgia and during progression to RA. RMD Open. 2023;9(2):e003107. doi:10.1136/rmdopen-2023-003107
10. Figus FA, Piga M, Azzolin I, McConnell R, Iagnocco A. Rheumatoid arthritis: extra-articular manifestations and comorbidities. Autoimmunity Rev. 2021;20(4):102776. doi:10.1016/j.autrev.2021.102776
11. Laria A, Lurati AM, Zizzo G, et al. Interstitial lung disease in rheumatoid arthritis: a practical review. Front Med. 2022;9:837133. doi:10.3389/fmed.2022.837133
12. Akiyama M, Kaneko Y. Pathogenesis, clinical features, and treatment strategy for rheumatoid arthritis-associated interstitial lung disease. Autoimmunity Rev. 2022;21(5):103056. doi:10.1016/j.autrev.2022.103056
13. Cassone G, Manfredi A, Vacchi C, et al. Treatment of rheumatoid arthritis-associated interstitial lung disease: lights and shadows. J Clin Med. 2020;9(4):1082. doi:10.3390/jcm9041082
14. Kadura S, Raghu G. Rheumatoid arthritis-interstitial lung disease: manifestations and current concepts in pathogenesis and management. Eur Respir Rev. 2021;30(160):210011. doi:10.1183/16000617.0011-2021
15. Rzepka-Wrona P, Miądlikowska E, Skoczyński S, Barczyk A, Piotrowski W. Patterns of lung fibrosis in patients with interstitial pneumonia with autoimmune features and connective tissue diseases-associated interstitial lung disease-a narrative review. Ann Palliat Med. 2022;11(6):2110–2130. doi:10.21037/apm-21-3974
16. Samy N, Salah H, Hammoda RM. Rheumatoid arthritis patients with interstitial lung disease: clinical, radiological and laboratory characteristics. Egypt Rheumatologist. 2021;43(1):29–34. doi:10.1016/j.ejr.2020.08.004
17. Teoh AK, Corte TJ. editors. Nonspecific Interstitial Pneumonia. Seminars in Respiratory and Critical Care Medicine. Thieme Medical Publishers; 2020.
18. Yamakawa H, Ogura T, Kameda H, et al. Decision-making strategy for the treatment of rheumatoid arthritis-associated interstitial lung disease (RA-ILD). J Clin Med. 2021;10(17):3806. doi:10.3390/jcm10173806
19. Shao T, Shi X, Yang S, et al. Interstitial lung disease in connective tissue disease: a common lesion with heterogeneous mechanisms and treatment considerations. Front Immunol. 2021;12:684699. doi:10.3389/fimmu.2021.684699
20. Guo H, Iqbal B, Rahman NM. editors. Pleural Diseases in Connective Tissue Diseases. Seminars in Respiratory and Critical Care Medicine. Thieme Medical Publishers, Inc; 2024.
21. Smith ML. The histologic diagnosis of usual interstitial pneumonia of idiopathic pulmonary fibrosis. Where we are and where we need to go. Mod Pathol. 2022;35:8–14. doi:10.1038/s41379-021-00889-5
22. Lucà S, Pagliuca F, Perrotta F, et al. Multidisciplinary approach to the diagnosis of idiopathic interstitial pneumonias: focus on the pathologist’s key role. Int J Mol Sci. 2024;25(7):3618. doi:10.3390/ijms25073618
23. Tomac IL. Idiopathic Lung Fibrosis: University of Zagreb. School of Medicine. Department of Internal Medicine; 2020.
24. de Souza SMP, Alves TSGN, da Silva AFSF, et al. Lung involvement in rheumatoid arthritis: what do we know? Discover Medicine. 2024;1(1):137. doi:10.1007/s44337-024-00165-w
25. Hino T, Lee KS, Yoo H, Han J, Franks TJ, Hatabu H. Interstitial lung abnormality (ILA) and nonspecific interstitial pneumonia (NSIP). Eur. J. Radiol. Open. 2021;8:100336. doi:10.1016/j.ejro.2021.100336
26. Lerma LA, Chandler CM, Xu H. Usual Interstitial Pneumonia Versus Nonspecific Interstitial Pneumonia. Practical Lung Pathology: Frequently Asked. Questions: Springer; 2022:233–238.
27. Ntiamoah P, Purpura R, Vehar S, et al. Nonspecific, Unclassifiable, and Rare Idiopathic Interstitial Pneumonia: Acute Interstitial Pneumonia, Respiratory Bronchiolitis Interstitial Pneumonia, Desquamative Interstitial Pneumonia, Nonspecific Interstitial Pneumonia. Orphan Lung Diseases: A Clinical Guide to Rare Lung. Disease: Springer; 2023:589–603.
28. De Zorzi E, Spagnolo P, Cocconcelli E, et al. Thoracic involvement in systemic autoimmune rheumatic diseases: pathogenesis and management. Clin Rev Allergy Immunol. 2022;63(3):472–489. doi:10.1007/s12016-022-08926-0
29. Conforti A, Di Cola I, Pavlych V, et al. Beyond the joints, the extra-articular manifestations in rheumatoid arthritis. Autoimmunity Rev. 2021;20(2):102735. doi:10.1016/j.autrev.2020.102735
30. Fui A, Bergantini L, Selvi E, et al. Rituximab therapy in interstitial lung disease associated with rheumatoid arthritis. Internal Med J. 2020;50(3):330–336. doi:10.1111/imj.14306
31. Diesler R, Cottin V. Pulmonary fibrosis associated with rheumatoid arthritis: from pathophysiology to treatment strategies. Expert Rev Respir Med. 2022;16(5):541–553. doi:10.1080/17476348.2022.2089116
32. Finnerty JP, Ponnuswamy A, Dutta P, Abdelaziz A, Kamil H. Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: a systematic review and meta-analysis. BMC Pulm Med. 2021;21(1):411. doi:10.1186/s12890-021-01783-1
33. Patel H, Perry S, Badu E, et al. A scoping review of interprofessional education in healthcare: evaluating competency development, educational outcomes and challenges. BMC Med Educ. 2025;25(1):409. doi:10.1186/s12909-025-06969-3
34. Yang M, Wu Y-Q, Liu X-M, et al. Efficacy and safety of antifibrotic agents in the treatment of CTD-ILD and RA-ILD: a systematic review and meta-analysis. Respir Med;2023. 107329. doi:10.1016/j.rmed.2023.107329
35. Kim Y, Yang H-I, Kim KS. Etiology and pathogenesis of rheumatoid arthritis-interstitial lung disease. Int J Mol Sci. 2023;24(19):14509. doi:10.3390/ijms241914509
36. Jönsson E, Ljung L, Norrman E, et al. Pulmonary fibrosis in relation to genetic loci in an inception cohort of patients with early rheumatoid arthritis from northern Sweden. Rheumatology. 2022;61(3):943–952. doi:10.1093/rheumatology/keab441
37. Gao Y, Zhang Y, Liu X. Rheumatoid arthritis: pathogenesis and therapeutic advances. MedComm. 2024;5(3):e509. doi:10.1002/mco2.509
38. Zhao R, Zhang Y-W, Guo J-C, et al. Genetic evidence reveals a causal relationship between rheumatoid arthritis and interstitial lung disease. Front Genetics. 2024;15:1395315. doi:10.3389/fgene.2024.1395315
39. Juge P-A, Lee JS, Ebstein E, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. 2018;379(23):2209–2219. doi:10.1056/NEJMoa1801562
40. Palomäki A, Palotie A, Koskela J, et al. Lifetime risk of rheumatoid arthritis-associated interstitial lung disease in MUC5B mutation carriers. Ann Rheumatic Dis. 2021;80(12):1530–1536. doi:10.1136/annrheumdis-2021-220698
41. Svyryd Y, Pascual-Ramos V, Contreras-Yañez I, et al. Telomeres length variations in a rheumatoid arthritis patients cohort at early disease onset and after follow-up. Revista de investigación clínica. 2022;74(4):202–211. doi:10.24875/RIC.22000048
42. Doyle TJ, Juge P-A, Peljto AL, et al. Short peripheral blood leukocyte telomere length in rheumatoid arthritis-interstitial lung disease. Thorax. 2024;79(2):182–185. doi:10.1136/thorax-2023-220022
43. De Benedittis G, Latini A, Morgante C, et al. Alteration of telomere length and mtDNA copy number in interstitial lung disease associated with rheumatoid arthritis. Autoimmunity. 2025;58(1):2473741. doi:10.1080/08916934.2025.2473741
44. Liu B, Shao Y, Fu R. Current research status of HLA in immune‐related diseases. Immun. inflamm. dis. 2021;9(2):340–350. doi:10.1002/iid3.416
45. Furukawa H, Oka S, Higuchi T, et al. Biomarkers for interstitial lung disease and acute-onset diffuse interstitial lung disease in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2021;13:1759720X211022506. doi:10.1177/1759720X211022506
46. Bernardinello N, Zen M, Castelli G, et al. Navigating interstitial lung disease associated with rheumatoid arthritis (RA-ILD): from genetics to clinical landscape. Front Med. 2025;12:1542400. doi:10.3389/fmed.2025.1542400
47. Kelly CA, Saravanan V, Nisar M, et al. Rheumatoid arthritis-related interstitial lung disease: associations, prognostic factors and physiological and radiological characteristics—a large multicentre UK study. Rheumatology. 2014;53(9):1676–1682. doi:10.1093/rheumatology/keu165
48. Kiely P, Busby A, Nikiphorou E, et al. Is incident rheumatoid arthritis interstitial lung disease associated with methotrexate treatment? Results from a multivariate analysis in the ERAS and ERAN inception cohorts. BMJ open. 2019;9(5):e028466. doi:10.1136/bmjopen-2018-028466
49. Zhang M, Yin J, Zhang X. Factors associated with interstitial lung disease in patients with rheumatoid arthritis: a systematic review and meta-analysis. PLoS One. 2023;18(6):e0286191. doi:10.1371/journal.pone.0286191
50. Kronzer VL, Yang Y, Roul P, et al. Associations of fire smoke and other pollutants with incident rheumatoid arthritis and rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheumatol. 2025;77:808–816. doi:10.1002/art.43113
51. Koether K, Besnard V, Sandig H, et al. Autoantibodies are associated with disease progression in idiopathic pulmonary fibrosis. Eur Respir J. 2023;61(5):2102381. doi:10.1183/13993003.02381-2021
52. Joshua V, Hensvold AH, Reynisdottir G, et al. Association between number and type of different ACPA fine specificities with lung abnormalities in early, untreated rheumatoid arthritis. RMD Open. 2020;6(2):e001278. doi:10.1136/rmdopen-2020-001278
53. Richards CD. Innate immune cytokines, fibroblast phenotypes, and regulation of extracellular matrix in lung. J Interferon Cytokine Res. 2017;37(2):52–61. doi:10.1089/jir.2016.0112
54. Spagnolo P, Tonelli R, Samarelli AV, et al. The role of immune response in the pathogenesis of idiopathic pulmonary fibrosis: far beyond the Th1/Th2 imbalance. Expert Opin Ther Targets. 2022;26(7):617–631. doi:10.1080/14728222.2022.2114897
55. Ross EA, Devitt A, Johnson JR. Macrophages: the good, the bad, and the gluttony. Front Immunol. 2021;12:708186. doi:10.3389/fimmu.2021.708186
56. Carroll TP, Greene CM, Taggart CC, McElvaney NG, O’Neill SJ. Interleukin-1, neutrophil elastase, and lipopolysaccharide: key pro-inflammatory stimuli regulating inflammation in cystic fibrosis. Curr Respir Med Rev. 2005;1(1):43–67. doi:10.2174/1573398052953640
57. Wan R, Jiang J, Hu C, et al. Neutrophil extracellular traps amplify neutrophil recruitment and inflammation in neutrophilic asthma by stimulating the airway epithelial cells to activate the TLR4/NF-κB pathway and secrete chemokines. Aging. 2020;12(17):16820. doi:10.18632/aging.103479
58. Robert S, Gicquel T, Victoni T, et al. Involvement of matrix metalloproteinases (MMPs) and inflammasome pathway in molecular mechanisms of fibrosis. Biosci. Rep. 2016;36(4):e00360. doi:10.1042/BSR20160107
59. Akhter S, Tasnin FM, Islam MN, et al. Role of Th17 and IL-17 cytokines on inflammatory and auto-immune diseases. Curr. Pharm. Des. 2023;29(26):2078–2090. doi:10.2174/1381612829666230904150808
60. Gomez-Bris R, Saez A, Herrero-Fernandez B, Rius C, Sanchez-Martinez H, Gonzalez-Granado JM. CD4 T-cell subsets and the pathophysiology of inflammatory bowel disease. Int J Mol Sci. 2023;24(3):2696. doi:10.3390/ijms24032696
61. Todorović-Raković N. The role of cytokines in the evolution of cancer: IFN-γ paradigm. Cytokine. 2022;151:155442. doi:10.1016/j.cyto.2021.155442
62. Ritzmann F, Lunding LP, Bals R, Wegmann M, Beisswenger C. IL-17 cytokines and chronic lung diseases. Cells. 2022;11(14):2132. doi:10.3390/cells11142132
63. Schinocca C, Rizzo C, Fasano S, et al. Role of the IL-23/IL-17 pathway in rheumatic diseases: an overview. Front Immunol. 2021;12:637829. doi:10.3389/fimmu.2021.637829
64. Bezerra FS, Lanzetti M, Nesi RT, et al. Oxidative stress and inflammation in acute and chronic lung injuries. Antioxidants. 2023;12(3):548. doi:10.3390/antiox12030548
65. Barnes PJ, Anderson GP, Fagerås M, Belvisi MG. Chronic lung diseases: prospects for regeneration and repair. Eur Respir Rev. 2021;30(159):200213. doi:10.1183/16000617.0213-2020
66. Roman J. Fibroblasts—warriors at the intersection of wound healing and disrepair. Biomolecules. 2023;13(6):945. doi:10.3390/biom13060945
67. LeBleu VS, Neilson EG. Origin and functional heterogeneity of fibroblasts. THE FASEB Journal. 2020;34(3):3519–3536. doi:10.1096/fj.201903188R
68. Jonckheere S, Adams J, De Groote D, Campbell K, Berx G, Goossens S. Epithelial-mesenchymal transition (EMT) as a therapeutic target. Cells Tissues Organs. 2022;211(2):157–182. doi:10.1159/000512218
69. Ray I, Michael A, Meira LB, Ellis PE. The role of cytokines in epithelial–mesenchymal transition in gynaecological cancers: a systematic review. Cells. 2023;12(3):416. doi:10.3390/cells12030416
70. Ma J, Sanchez-Duffhues G, Goumans M-J, Ten Dijke P. TGF-β-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Develop Biol. 2020;8:260. doi:10.3389/fcell.2020.00260
71. Wei Y, Wang D, Wu J, Zhang J. JAK2 inhibitors improve RA combined with pulmonary fibrosis in rats by downregulating SMAD3 phosphorylation. Int. J. Rheum. Dis. 2024;27(5):e15164. doi:10.1111/1756-185X.15164
72. Grigorieva O, Vigovskiy M, Dyachkova U, et al. Mechanisms of endothelial-to-mesenchymal transition induction by extracellular matrix components in pulmonary fibrosis. Bull. Exp. Biol. Med. 2021;171:523–531. doi:10.1007/s10517-021-05264-7
73. Budi EH, Schaub JR, Decaris M, Turner S, Derynck R. TGF‐β as a driver of fibrosis: physiological roles and therapeutic opportunities. J Pathol. 2021;254(4):358–373. doi:10.1002/path.5680
74. Ma H, Liu S, Li S, Xia Y. Targeting growth factor and cytokine pathways to treat idiopathic pulmonary fibrosis. Front Pharmacol. 2022;13:918771. doi:10.3389/fphar.2022.918771
75. Isshiki T, Naiel S, Vierhout M, et al. Therapeutic strategies to target connective tissue growth factor in fibrotic lung diseases. Pharmacol Ther. 2023;108578.
76. Zhao F-Y, Xu S-L, Zhang C-F, et al. PDGF mediates pulmonary arterial smooth muscle cell proliferation and migration by regulating NFATc2. Mol. Med. Rep. 2021;23(1). doi:10.3892/mmr.2021.11833
77. Zaykov V, Chaqour B. The CCN2/CTGF interactome: an approach to understanding the versatility of CCN2/CTGF molecular activities. J Cell Commun Signal. 2021;15(4):567–580. doi:10.1007/s12079-021-00650-2
78. Farooq M, Khan AW, Kim MS, Choi S. The role of fibroblast growth factor (FGF) signaling in tissue repair and regeneration. Cells. 2021;10(11):3242. doi:10.3390/cells10113242
79. England BR, Duryee MJ, Roul P, et al. Malondialdehyde-acetaldehyde adducts and antibody responses in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2019;71(9):1483–1493. doi:10.1002/art.40900
80. Aripova N, Thiele GM, Duryee MJ, et al. Antibodies to malondialdehyde‐acetaldehyde adduct are associated with prevalent and incident rheumatoid arthritis–associated interstitial lung disease in us veterans. Arthritis Rheumatol. 2024;76(9):1353–1363. doi:10.1002/art.42916
81. Antar SA, Ashour NA, Marawan ME, Al-Karmalawy AA. Fibrosis: types, effects, markers, mechanisms for disease progression, and its relation with oxidative stress, immunity, and inflammation. Int J Mol Sci. 2023;24(4):4004. doi:10.3390/ijms24044004
82. Löfstedt T, Fredlund E, Holmquist-Mengelbier L, et al. Hypoxia inducible factor-2alpha in cancer. Cell Cycle. 2007;6(8):919–926. doi:10.4161/cc.6.8.4133
83. Thapa R, Marianesan AB, Rekha A, et al. Hypoxia-inducible factor and cellular senescence in pulmonary aging and disease. Biogerontology. 2025;26(2):64. doi:10.1007/s10522-025-10208-z
84. Tang YY, Wang DC, Wang YQ, Huang A-F, Xu W-D. Emerging role of hypoxia-inducible factor-1α in inflammatory autoimmune diseases: a comprehensive review. Front Immunol. 2023;13:1073971. doi:10.3389/fimmu.2022.1073971
85. Duric LF. It’s time for new insights into renovascular hypertension at the molecular level. J Biosci Med. 2024;12(2):180–201. doi:10.4236/jbm.2024.122014
86. Guo L, Xu J, Liu L, Liu S, Zhu R. Hypoxia‐induced epithelial‐mesenchymal transition is involved in bleomycin‐induced lung fibrosis. Biomed Res. Int. 2015;2015(1):232791. doi:10.1155/2015/232791
87. Confalonieri P, Volpe MC, Jacob J, et al. Regeneration or repair? The role of alveolar epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis (IPF). Cells. 2022;11(13):2095. doi:10.3390/cells11132095
88. Kishore A, Petrek M. Roles of macrophage polarization and macrophage-derived miRNAs in pulmonary fibrosis. Front Immunol. 2021;12:678457. doi:10.3389/fimmu.2021.678457
89. Yang S, Zhao M, Jia S. Macrophage: key player in the pathogenesis of autoimmune diseases. Front Immunol. 2023;14:1080310. doi:10.3389/fimmu.2023.1080310
90. Ge Z, Chen Y, Ma L, Hu F, Xie L. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front Immunol. 2024;15:1444964. doi:10.3389/fimmu.2024.1444964
91. Overed-Sayer C, Miranda E, Dunmore R, et al. Inhibition of mast cells: a novel mechanism by which nintedanib may elicit anti-fibrotic effects. Thorax. 2020;75(9):754–763. doi:10.1136/thoraxjnl-2019-214000
92. Xin Q, Yuan R, Shi W, Zhu Z, Wang Y, Cong W. A review for the anti-inflammatory effects of paeoniflorin in inflammatory disorders. Life Sci. 2019;237:116925. doi:10.1016/j.lfs.2019.116925
93. Epstein Shochet G, Brook E, Bardenstein-Wald B, Shitrit D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir Res. 2020;21:1–10. doi:10.1186/s12931-020-1319-0
94. Miao C, Zhu X, Wei X, et al. Pro-and anti-fibrotic effects of vascular endothelial growth factor in chronic kidney diseases. Renal Failure. 2022;44(1):881–892. doi:10.1080/0886022X.2022.2079528
95. Freeberg MA, Perelas A, Rebman JK, Phipps RP, Thatcher TH, Sime PJ. Mechanical feed-forward loops contribute to idiopathic pulmonary fibrosis. Am J Pathol. 2021;191(1):18–25. doi:10.1016/j.ajpath.2020.09.008
96. Cabral-Pacheco GA, Garza-Veloz I, Castruita-De la Rosa C, et al. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int J Mol Sci. 2020;21(24):9739. doi:10.3390/ijms21249739
97. Liu G, Philp AM, Corte T, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. 2021;225:107839.
98. Planté-Bordeneuve T, Pilette C, Froidure A. The epithelial-immune crosstalk in pulmonary fibrosis. Front Immunol. 2021;12:631235. doi:10.3389/fimmu.2021.631235
99. McCartney-Francis NL, Chan J, Wahl SM. Inflammatory joint disease: clinical, histological, and molecular parameters of acute and chronic inflammation and tissue destruction. Inflammation Protocols. 2003;147–159.
100. May J, Mitchell JA, Jenkins RG. Beyond epithelial damage: vascular and endothelial contributions to idiopathic pulmonary fibrosis. J Clin Investig. 2023;133(18):e172058. doi:10.1172/JCI172058
101. Ptasinski VA, Stegmayr J, Belvisi MG, Wagner DE, Murray LA. Targeting alveolar repair in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2021;65(4):347–365. doi:10.1165/rcmb.2020-0476TR
102. Trachalaki A, Irfan M, Wells AU. Pharmacological management of idiopathic pulmonary fibrosis: current and emerging options. Expert Opin Pharmacother. 2021;22(2):191–204. doi:10.1080/14656566.2020.1822326
103. Amati F, Stainer A, Polelli V, et al. Efficacy of pirfenidone and nintedanib in interstitial lung diseases other than idiopathic pulmonary fibrosis: a systematic review. Int J Mol Sci. 2023;24(9):7849. doi:10.3390/ijms24097849
104. Huang N-Y, Ding L, Wang J, et al. Pharmacokinetics, safety and tolerability of pirfenidone and its major metabolite after single and multiple oral doses in healthy Chinese subjects under fed conditions. Drug Res. 2013;63(08):388–395. doi:10.1055/s-0033-1341478
105. Sun G, Lin X, Zhong H, et al. Pharmacokinetics of pirfenidone after topical administration in rabbit eye. Mol Vision. 2011;17:2191.
106. Bai X, Nie P, Lou Y, et al. Pirfenidone is a renal protective drug: mechanisms, signalling pathways, and preclinical evidence. Eur. J. Pharmacol. 2021;911:174503. doi:10.1016/j.ejphar.2021.174503
107. Barranco-Garduño LM, Buendía-Roldan I, Rodriguez JJ, et al. Pharmacokinetic evaluation of two pirfenidone formulations in patients with idiopathic pulmonary fibrosis and chronic hypersensitivity pneumonitis. Heliyon. 2020;6(10):e05279. doi:10.1016/j.heliyon.2020.e05279
108. Poo JL, Aguilar JR, Bernal-Reyes R, et al. Prolonged release pirfenidone pharmacokinetics is modified in cirrhosis GENESIS study. Biomed. Pharmacother. 2023;168:115712. doi:10.1016/j.biopha.2023.115712
109. Torre A, Martínez‐Sánchez FD, Narvaez‐Chávez SM, Herrera‐Islas MA, Aguilar‐Salinas CA, Córdova‐Gallardo J. Pirfenidone use in fibrotic diseases: what do we know so far? Immun. inflamm. dis. 2024;12(7):e1335. doi:10.1002/iid3.1335
110. Ko I-G, Hwang L, Jin -J-J, et al. Pirfenidone improves voiding function by suppressing bladder fibrosis in underactive bladder rats. Eur. J. Pharmacol. 2024;977:176721. doi:10.1016/j.ejphar.2024.176721
111. Zhang S, Wang Y, Luo D, et al. Pirfenidone inhibits TGF‐β1‐induced metabolic reprogramming during epithelial‐mesenchymal transition in non‐small cell lung cancer. J Cell & Mol Med. 2024;28(3):e18059. doi:10.1111/jcmm.18059
112. Paik SS, Lee JM, Ko I-G, et al. Pirfenidone alleviates inflammation and fibrosis of acute respiratory distress syndrome by modulating the transforming growth factor-β/smad signaling pathway. Int J Mol Sci. 2024;25(15):8014. doi:10.3390/ijms25158014
113. Guo H-L, Liang X-S, Zeng X-P, et al. Pirfenidone alleviates chronic pancreatitis via suppressing the activation of pancreatic stellate cells and the M1 polarization of macrophages. Int Immunopharmacol. 2024;130:111691. doi:10.1016/j.intimp.2024.111691
114. Wijewardhane PR, Wells A, Muhoberac M, Leung KP, Chopra G. Modeling molecular mechanisms of pirfenidone interaction with kinases. bioRxiv. 2024.
115. Xie R, Chen S, Li F, Yang L, Yu B. Pirfenidone attenuates nonalcoholic fatty liver disease through activation of the nuclear factor erythroid 2‐related factor 2 (Nrf2) signaling pathway. J Biochem Mol Toxicol. 2023;37(2):e23251. doi:10.1002/jbt.23251
116. Khokhlov A, Rybachkova J. Efficacy and safety of pirfenidone according to clinical trials. Patient-Oriented Med Pharmacy. 2024;2:6–14. doi:10.37489/2949-1924-0036
117. Im S. Impact of Pirfenidone on arrhythmic and clinical outcomes in patients with idiopathic pulmonary fibrosis. Europace. 2024;26(Supplement_1). doi:10.1093/europace/euae102.828
118. Lehmann M, Kolb M. Another piece in the pirfenidone puzzle. Eur Respir Soc. 2023;61:2300240. doi:10.1183/13993003.00240-2023
119. Jang JH, Ko J, Jung SY, et al. Antifibrotic agents in rheumatoid arthritis-associated interstitial lung disease: a systematic review and meta-analysis. Life. 2023;13(12):2318. doi:10.3390/life13122318
120. Wind S, Schmid U, Freiwald M, et al. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin. Pharmacokinet. 2019;58:1131–1147. doi:10.1007/s40262-019-00766-0
121. Dallinger C, Trommeshauser D, Marzin K, Liesener A, Kaiser R, Stopfer P. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J Clin Pharmacol. 2016;56(11):1387–1394. doi:10.1002/jcph.752
122. Schmid U, Liesenfeld K-H, Fleury A, Dallinger C, Freiwald M. Population pharmacokinetics of nintedanib, an inhibitor of tyrosine kinases, in patients with non-small cell lung cancer or idiopathic pulmonary fibrosis. Cancer Chemother Pharmacol. 2018;81:89–101. doi:10.1007/s00280-017-3452-0
123. Liu H, Mei J, Xu Y, et al. Improving the oral absorption of nintedanib by a self-microemulsion drug delivery system: preparation and in vitro/in vivo evaluation. Int J Nanomed. 2019;Volume 14:8739–8751. doi:10.2147/IJN.S224044
124. Che J, Fu Y, Li Y, et al. Eudragit L100-coated nintedanib nanocrystals improve oral bioavailability by reducing drug particle size and maintaining drug supersaturation. Int J Pharm. 2024;658:124196. doi:10.1016/j.ijpharm.2024.124196
125. Patel P, Patel M. Enhanced oral bioavailability of nintedanib esylate with nanostructured lipid carriers by lymphatic targeting: in vitro, cell line and in vivo evaluation. Eur. J. Pharm. Sci. 2021;159:105715. doi:10.1016/j.ejps.2021.105715
126. Nakashima S, Sato R, Fukami T, et al. Characterization of enzymes involved in nintedanib metabolism in humans. Drug Metab. Dispos. 2023;51(6):733–742. doi:10.1124/dmd.122.001113
127. Marzin K, Kretschmar G, Luedtke D, et al. Pharmacokinetics of nintedanib in subjects with hepatic impairment. J Clin Pharmacol. 2018;58(3):357–363. doi:10.1002/jcph.1025
128. Ferreira C, Costa E, Pereira P, et al. Ab1442 predictive factors for dose reduction and discontinuation of nintedanib in fibrotic interstitial lung diseases: a real-life. Ann Rheumatic Dis. 2024;83:2077–2078. doi:10.1136/annrheumdis-2024-eular.5399
129. Ruggiero V, Aquino G, Del Gaudio P, Mencherini T, Tedesco C, Russo P. Development and characterization of nintedanib inhalable powders as a potential pulmonary fibrosis treatment. J Drug Delivery Sci Technol. 2024;92:105340. doi:10.1016/j.jddst.2024.105340
130. Surber MW, Beck S, Pham S, et al. Inhaled nintedanib is well-tolerated and delivers key pharmacokinetic parameters required to treat bleomycin-induced pulmonary fibrosis. Pulm Pharmacol Ther. 2020;63:101938. doi:10.1016/j.pupt.2020.101938
131. Auberle C, Gao F, Sloan M, et al. A pilot study of nintedanib in molecularly selected patients with advanced non-small cell lung cancer. J Thoracic Dis. 2024;16(6):3782. doi:10.21037/jtd-23-1717
132. Mazzei ME, Richeldi L, Collard HR. Nintedanib in the treatment of idiopathic pulmonary fibrosis. Therapeutic advances in respiratory disease. Therap Adv Respir Dis. 2015;9(3):121–129. doi:10.1177/1753465815579365
133. da Silva RF, Dhar D, Raina K, et al. Nintedanib inhibits growth of human prostate carcinoma cells by modulating both cell cycle and angiogenesis regulators. Sci Rep. 2018;8(1):9540. doi:10.1038/s41598-018-27831-1
134. Dong X, Wang L, Wang D, Yu M, Jun Yang X, Cai H. Proteomic study on nintedanib in gastric cancer cells. PeerJ. 2024;12:e16771. doi:10.7717/peerj.16771
135. Thakkar D, Singh S, Wairkar S. Advanced delivery strategies of nintedanib for lung disorders and beyond: a comprehensive review. AAPS Pharm Sci Tech. 2024;25(6):150. doi:10.1208/s12249-024-02869-9
136. Ardan AR, Nurwidya F. The role of nintedanib in the treatment of progressive pulmonary fibrosis of autoimmune-related interstitial lung disease. J. Respir. 2023;3(4):200–207. doi:10.3390/jor3040019
137. Inoue Y, Kitamura H, Okamoto M, et al. The effect of nintedanib on health-related quality of life in Japanese patients with progressive fibrosing interstitial lung diseases: a subset analysis of the INBUILD trial. Respir. Investig. 2024;62(4):589–596. doi:10.1016/j.resinv.2024.04.008
138. Yin Y, Liu S, Liu H, Wu W. Nintedanib inhibits normal human vitreous-induced epithelial-mesenchymal transition in human retinal pigment epithelial cells. Biomed. Pharmacother. 2023;166:115403. doi:10.1016/j.biopha.2023.115403
139. Cho H-J, Hwang J-A, Yang EJ, et al. Nintedanib induces senolytic effect via STAT3 inhibition. Cell Death Dis. 2022;13(9):760. doi:10.1038/s41419-022-05207-8
140. Matteson EL, Aringer M, Burmester GR, Mueller H, Moros L, Kolb M. Effect of nintedanib in patients with progressive pulmonary fibrosis associated with rheumatoid arthritis: data from the INBUILD trial. Clin Rheumatol. 2023;42(9):2311–2319. doi:10.1007/s10067-023-06623-7
141. Duarte AC, Marques Gomes C, Correia M, et al. Antifibrotics in rheumatoid arthritis-associated interstitial lung disease - real-world data from a nationwide cohort. ARP Rheumatol. 2024;3(3):182–188. doi:10.63032/POPM9413
142. Solomon JJ, Danoff SK, Woodhead FA, et al. Safety, tolerability, and efficacy of pirfenidone in patients with rheumatoid arthritis-associated interstitial lung disease: a randomised, double-blind, placebo-controlled, Phase 2 study. Lancet Respir Med. 2023;11(1):87–96. doi:10.1016/S2213-2600(22)00260-0
143. Narváez J, Aguilar-Coll M, Vicens-Zygmunt V, Alegre JJ, Bermudo G, Molina-Molina M. Real-world clinical effectiveness and safety of antifibrotics in progressive pulmonary fibrosis associated with rheumatoid arthritis. J Clin Med. 2024;13(23):7074. doi:10.3390/jcm13237074
144. Juge P-A, Hayashi K, McDermott GC, et al. Effectiveness and tolerability of antifibrotics in rheumatoid arthritis-associated interstitial lung disease. In: Seminars in Arthritis and Rheumatism. Elsevier; 2024.
145. Juge PA, Hayashi K, McDermott GC, et al. Effectiveness and tolerability of antifibrotics in rheumatoid arthritis-associated interstitial lung disease. Semin Arthritis Rheum. 2024;64:152312. doi:10.1016/j.semarthrit.2023.152312
146. Yang M, Tan Y, Yang T, Xu D, Chen M, Chen L. Efficacy and safety of antifibrotic drugs for interstitial lung diseases other than IPF: a systematic review, meta-analysis and trial sequential analysis. PLoS One. 2025;20(2):e0318877. doi:10.1371/journal.pone.0318877
147. Song J, Lee H, Lee C, et al. Clinical course and outcome of rheumatoid arthritis-related usual interstitial pneumonia. Sarcoidosis, vasculitis, and diffuse lung diseases. Official J WASOG. 2013;30(2):103–112.
148. Roubille C, Haraoui B. editors. Interstitial lung diseases induced or exacerbated by DMARDS and biologic agents in rheumatoid arthritis: a systematic literature review. In: Seminars in Arthritis and Rheumatism. Elsevier; 2014.
149. Fisher M, Nathan SD, Hill C, et al. Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J Manag Care Spec Pharm. 2017;23(3–b Suppl):S17–S24. doi:10.18553/jmcp.2017.23.3-b.s17
150. Collard HR, Richeldi L, Kim DS, et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur Respir J. 2017;49(5):1601339. doi:10.1183/13993003.01339-2016
151. Solomon JJ, Chung JH, Cosgrove GP, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2016;47(2):588–596. doi:10.1183/13993003.00357-2015
152. Brown KK, Martinez FJ, Walsh SLF, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020;55(6):2000085. doi:10.1183/13993003.00085-2020
153. Kumar DP. Assessment and follow-up of interstitial lung disease. Indian J Rheumatol. 2021;16(Suppl 1):S69–S78. doi:10.4103/0973-3698.332980
154. Clay E, Cristeau O, Chafaie R, Pinta A, Mazaleyrat B, Cottin V. Cost-effectiveness of pirfenidone compared to all available strategies for the treatment of idiopathic pulmonary fibrosis in France. J Mark Access Health Policy. 2019;7(1):1626171. doi:10.1080/20016689.2019.1626171
155. Rinciog C, Diamantopoulos A, Gentilini A, et al. Cost-effectiveness analysis of nintedanib versus pirfenidone in idiopathic pulmonary fibrosis in belgium. Pharmacoecon Open. 2020;4(3):449–458. doi:10.1007/s41669-019-00191-w
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.