")
Back to Journals » International Journal of Nanomedicine » Volume 17
Exploring a Novel Fasudil-Phospholipid Complex Formulated as Liposomal Thermosensitive in situ Gel for Glaucoma
Authors Khallaf AM , El-Moslemany RM, Ahmed MF, Morsi MH , Khalafallah NM
Received 15 October 2021
Accepted for publication 22 December 2021
Published 11 January 2022 Volume 2022:17 Pages 163—181
DOI https://doi.org/10.2147/IJN.S342975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Aya M Khallaf,1 Riham M El-Moslemany,1 Mahmoud F Ahmed,2 Mahmoud H Morsi,3 Nawal M Khalafallah1
1Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt; 2Managing Director at Ultimate Pharma Company, Alexandria, Egypt; 3Department of Ophthalmology, Faculty of Medicine, Alexandria University, Alexandria, Egypt
Correspondence: Riham M El-Moslemany
Department of Pharmaceutics, Faculty of Pharmacy, Alexandria University, 1, Khartoum Square, Azarita, Alexandria, 21521, Egypt
Tel +20 1006020405
Email [email protected]
Purpose: Fasudil hydrochloride (Fas), a rho-associated protein kinase inhibitor, proved to be promising for glaucoma management owing to its IOP lowering and antioxidant effects. However, its highly hydrophilic nature limits ocular permeation and bioavailability. Hence, the study objective was the development of Fas loaded vesicular system with high entrapment efficiency formulated as a thermosensitive gel for local administration aiming to enhance ocular retention and permeation and hence therapeutic efficacy.
Methods: Fasudil complex with phospholipid (Fas/PL) was prepared by solvent evaporation technique and characterized by Fourier transform infrared spectroscopy (FT-IR) and X-ray diffraction (XRD). Fas/PL was further formulated as liposomes by methanol injection method and characterized regarding colloidal properties, entrapment efficiency (EE%) and in vitro drug release. The prepared liposomes were incorporated into an optimized thermosensitive in situ gel (Fas/PL-LipoP407/HPMCgel) selected based on gelling time and temperature and rheological properties. The effect of incorporation into gel on the in vitro characteristics of liposomes was investigated. The in vitro mucoadhesive potential, ex vivo permeation, irritability and efficacy in a glaucoma rabbit model were also assessed.
Results: FT-IR and XRD suggested interactions between Fas and PL, proposing complexation. Fas/PL liposomal dispersions showed good colloidal properties (particle size: 132.5 ± 1.6 nm, zeta potential: − 21.6 ± 0.9 and %EE 78.6 ± 0.3%) with sustained drug release. In situ thermosensitive gel (20% poloxamer 407 and 0.5% HPMC) showed optimum gelling properties. The selected gel formulation reduced burst release of the drug, enhanced mucoadhesive properties and prolonged corneal permeation ex vivo. HET-CAM test confirmed that the prepared formulations were non-irritant. In vivo pharmacodynamic study indicated improved bioavailability and significantly lower intraocular pressure (IOP) of Fas/PL-LipoP407/HPMC gel compared to drug solution and liposomal dispersion.
Conclusion: The results present Fas/PL-LipoP407/HPMC gel as a potential platform for ophthalmic delivery of fasudil with improved pharmaceutical attributes and enhanced bioavailability and efficacy in glaucoma.
Keywords: fasudil hydrochloride, glaucoma, phospholipid-complex, liposomes, thermosensitive gel
Introduction
Glaucoma stays unequivocally a perplexing issue of high importance in ophthalmology as it remains asymptomatic until irreversible blindness may occur and the number of individuals affected by glaucoma is constantly increasing worldwide.1,2 Primary open-angle glaucoma (POAG) is the most predominant type of glaucoma commonly treated with eye drop medications intended to reduce the intra ocular pressure (IOP) which is a fundamental modifiable risk factor in the improvement of the disease.3–5 The main key issue is that most accessible medications try not to follow up on the pathological changes that are answerable for the expanded IOP in glaucoma.6 This reality plainly shows the requirement for new and causative treatment alternatives to improve the therapeutic outcome without causing permanent tissue damage.
With the accreditation of rho-associated protein kinase (ROCK) inhibitors Glanatec® (ripasudil) in 2014 and Rhopressa® (netarsudil) in 2017, a promising new class of medications has been presented for glaucoma.7,8 Their advantages compared to other medications are not limited to lowering IOP but also intensifying blood flow to ocular tissues and protecting nerve cells against various types of oxidative stress thus, promoting neuron survival and regeneration.9,10 Fasudil is a novel ROCK inhibitor previously proved its efficacy against glaucomatous eyes.11 Its hydrophilic nature (log P 0.16) is a major limitation resulting in low ocular bioavailability.12 Several strategies have been developed to deal with compounds’ high polarity. Among these, complexation of drugs with phospholipids was found to be useful in modifying pharmacokinetics and bioavailability of some therapeutics.13,14 To the best of our knowledge, complexation of fasudil with phospholipid has been reported for the first time in the presented work.
Although the advantages of topical drug delivery to the anterior segment of the eye are self-evident, only 5% of the applied drug reaches the aqueous humor as a result of several ocular barriers which require frequent instillation of medication leading to gradual deterioration of patient compliance.15,16 These barriers increment the interest for an ocular drug delivery system that can conquer the limits of traditional eye drops and furthermore can give a sustained release of the drug with higher therapeutic efficiency.
The use of liposomes in ocular drug delivery continues to demonstrate immense potential as they are totally biodegradable, generally nontoxic and have the ability to bypass several biological barriers.17–19 Yet, liposomes are still aqueous dispersion systems with low viscosity possessing poor maintenance capability on the ocular surface. To overcome this issue, liposomes can be incorporated into viscous systems (eg, hydrogels) to provide the thickness of the final formulations and extend their precorneal residence time.
Among these approaches, thermosensitive in situ-forming hydrogels which undergo fast sol–gel transition upon exposure to physiological temperature can provide sustained delivery of instilled drugs at the ocular tissues.20 Poloxamer 407 is a recognized thermo responsive polymer widely used for ocular administration.21 However, its poor mucoadhesive ability and concentration-dependent response require the use of high concentration of P407 for efficient gelation which may lead to hypertriglyceridemia in the eye.22,23 Hydroxypropyl methyl cellulose (HPMC) can be added to reduce the required P407 concentration and improve the mucoadhesive properties of the final preparation.21,24
The present work focused on developing a liposomal thermosensitive hydrogel loaded with fasudil-phospholipid complex, in the form of ophthalmic eye drops, aiming to tackle the root cause of glaucoma. Characterizing the optimized formulation in vitro, ex vivo, and investigating its safety and efficacy against glaucomatous rabbit eyes was carried out.
Materials and Methods
Materials
Fasudil HCl (Fas) (5-(1,4-diazepan-1-yl sulfonyl) isoquinoline hydrochloride), molecular formula C14H18ClN3O2S, molecular weight 327.83 g/mole, freely soluble in water (up to 200 mg/mL) with LogP 0.16 and pKa 8.046 was purchased from Henan Tianfu Co., Ltd., China. Lipoid S100 (PL, soybean phosphatidylcholine 100%) was a kind gift from Lipoid GmbH, Ludwigshafen, Germany. Poloxamer 407 (P), hydroxypropyl methylcellulose (HPMC K4M) and cholesterol were obtained from Sigma Aldrich chemical Co., Gillingham, UK. PEG-2000 and tween-80 were procured from Tianjin Guangfu Fine Chemical Research Institute, Tianjin, China. Porcine mucin (Sigma Aldrich, USA). All solvents were of analytical grade and obtained from commercial sources.
Preparation of Fasudil HCl Phospholipid Complex (Fas/PL)
Fasudil phospholipid complex (Fas/PL) was developed by solvent evaporation technique with slight modifications.25,26 Initially, Fas and PL were dispersed in dichloromethane at molar ratios 1:1 and 1:2 and shaken until complete dissolution was achieved. The resultant mixture was then stirred either at room temperature or under reflux at a temperature not exceeding 45 °C for 1–2 h to ensure maximal contact between the drug and the phospholipid. The solvent was then evaporated under vacuum using a rotary evaporator at 50 °C and the dried residue was collected and placed in a desiccator for further investigation.
Characterization of Fasudil-Phospholipid Complex (Fas/PL)
Fourier Transform Infrared (FT-IR) Spectroscopy
FT-IR spectra of pure Fas, PL, their corresponding physical mixture and Fas/PL were recorded in potassium bromide discs. The samples were scanned over the range 4000–400 cm−1 (mid-infrared region) at ambient temperature using FT-IR spectrophotometer (Shimadzu, Kyoto, Japan). Spectra obtained represent an average of 20 individual scans possessing a spectral resolution of 2 cm−1.
X-Ray Diffraction (XRD)
The crystallinity of Fas, PL, their physical mixture, and Fas/PL was assessed using XRD (XRD-7000 X-Ray diffractometer, Bruker D2-Phaser; Madison, WI, USA) employing Cu Kα1 radiation and step scan model of 30 kV and 30 mA. Samples were placed in the sample holder (20 mm × 15mm × 2mm) and then X-ray was passed through the sample. Diffractograms were plotted in the angular range of 1–100º with step size 0.02º.
Preparation of Blank and Fas/PL Loaded Liposomes
Liposomes were prepared by methanol injection method.27 For fasudil phospholipid complex-loaded liposome dispersion (Fas/PL-Lipodisp), Fas/PL, about 83 mg, was prepared as mentioned above, then 3 mL methanol containing 0.013 M cholesterol was added to the dried residue and mixed well until a clear solution was obtained. The organic mixture was subsequently added dropwise to 5 mL of deionized water containing 0.5% w/v PEG 2000 and 0.2% tween while stirring.28 The obtained dispersion was kept on the stirrer for 1 h to ensure homogeneous distribution and optimum size reduction. The organic solvent was then evaporated by the rotary evaporator at 50 °C. The resultant liposomes were kept overnight in a refrigerator at 2–8 °C before further investigation. Blank liposomes (B-Lipodisp) were prepared following the same technique except for the addition of Fas. Factors affecting the size distribution of liposomes were studied including the effect of cholesterol, solvent/non-solvent volume ratio and stirring rate.
Preparation of in situ Gels
Blank poloxamer 407 in situ gel (BP407 gel) was prepared using the cold method.29 Different concentrations of P407 ranging from 17.5 to 25% w/w were dissolved in cold deionized water (4–8 °C) with continuous stirring for 2 h, then refrigerated overnight to ensure complete dissolution. For blank poloxamer/HPMC K4M in situ gel (BP407/HPMC gel), HPMC K4M (0.5% w/v in the final preparation) was first dissolved in filtered deionized water before addition of poloxamer 407 powder.30 For Fas-containing in situ gels, Fas was dissolved in the cold in situ gels until a clear solution was obtained (FasP407 gel and FasP407/HPMC gel). To prepare fasudil complex-loaded liposome-P407 in situ gel (Fas/PL-LipoP407 gel), P407 (17.5 to 25%) was dispersed in cold liposomal dispersion and refrigerated overnight. For fasudil complex-loaded liposome-P407/HPMC in situ gel (Fas/PL-LipoP407/HPMC gel), 1% w/v HPMC solution was added to a double strength liposomal dispersion in the ratio 1:1 and stirred for 10 minutes to obtain a final liposomal dispersion with 1.5% w/v total lipids and 5mg/mL Fas. Different amounts of P407, to yield 17.5–25% w/v concentrations, were then dissolved in the previous dispersion on cold followed by overnight refrigeration. For all prepared formulations, Fas concentration was 0.5% w/v (5 mg/mL).
Characterization of Gel Formulations
Determination of Gelling Time and Temperature
The sol–gel transition temperature was determined by the tube inversion method.31 A 2 mL sample was heated in a water bath from 20 °C to 70 °C at a rate of 1 °C/min. The temperature at which the test solution changed to gel was recorded. Time for sol–gel transition was determined on 100 µL samples using an aluminum pan placed on a hot plate maintained at 35 °C. The pan was then tilted at 90º and the time at which the sol stopped to flow was recorded in seconds.31
Clarity and pH
The final appearance of the gel formulations including color and clarity was examined visually prior and after gelation. The pH of the optimized formulations was determined before gelation using a calibrated pH meter (Hanna, type 211, Romania). Readings were recorded in triplicates, and the average reading was computed.
Rheological Behavior of in situ Gel Formulations
Viscosity of gel formulations was estimated at 25 °C and 37 °C using a cone and plate viscometer (Brookfield, DV2T, USA) at 10 rpm. To study the possible thixotropic behavior of formulations, shear stress was assessed at different shear rates (1, 3, 5, 7 and 10 rpm) at 37 °C using spindle 40 for 60 s. Profiles were plotted using Rheocalc T software.
In vitro Characterization of Liposomal Formulations
Colloidal Properties
The particle size and polydispersity index (PdI) of B-Lipodisp and Fas/PL-Lipodisp were determined using dynamic light scattering (DLS), and zeta potential (ZP) was measured by Laser Doppler Velocimetry (LDV) using a Malvern Zetasizer (Zetasizer Nano ZS90, Malvern Instruments, UK) at a fixed angle 173 at 25 °C using a 4 mW He-Ne laser at 633 nm. The samples were appropriately diluted with filtered deionized water before measurement. The same procedure was applied for sizing liposomal gels. All measurements were performed in triplicates, and the mean values and standard deviations were estimated.
Morphological Examination
Transmission electron microscopy (TEM) (JEM-100S; JEOL Ltd., Japan) was used to detect the morphology of B-Lipodisp, Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel at the selected optimized poloxamer concentration. Samples were 5-fold diluted with deionized water, settled on copper grids and negatively stained with 2% w/v aqueous uranyl acetate solution. Photographs were taken at X25K magnification at an acceleration voltage of 80 kV.
Entrapment Efficiency (EE%) and Drug Loading Capacity (DL%)
The entrapment efficiency (EE%) of Fas in Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel was calculated by measuring the free (unentrapped) Fas (indirect method)32 separated from liposomal dispersion using an ultrafiltration/centrifugation technique for 30 min at 4000 rpm at 4°C (Vivaspin® 6, molecular weight cut-off 100,000). The concentration of free Fas in the filtrate was determined by UV spectrophotometry at λmax 320 nm.33 The method was linear in the concentration range 20–70 µg/mL with a determination coefficient 0.999 and limits of detection and quantitation 2.023 and 6.131 µg/mL, respectively. EE% and DL% were calculated using the following equations:


In vitro Fasudil Release
Fas release from Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel in comparison to Fas solution in normal saline was investigated using the dialysis bag method.36 A 1 mL volume of each formula equivalent to 5 mg Fas was placed in a dialysis bag (molecular weight cut-off 12,000–14,000 Da) and suspended in 10 mL of freshly prepared simulated tear fluid (STF) adjusted to pH 7.4 (0.678 g NaCl, 0.218 g NaHCO3, 0.138 g KCl, 0.0084 g CaCl2.2H2O and purified water to 100 mL37) and shaken (50 strokes per minute) at 35±1 °C. Samples were withdrawn at different time intervals (0.5,1,2,4,6,12 and 24 h) followed by compensation with fresh STF. Concentration of Fas in the dialysate was determined spectrophotometrically at λmax 320 nm, and the cumulative percentage Fas released was plotted versus time.
Stability Testing
Selected formulations (Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel were refrigerated for 3 months at 4 °C. Particle size, PdI, and ZP were assessed monthly. Changes in EE% as a measure of drug leakage were also monitored.
In vitro Mucoadhesion Study
The mucoadhesiveness of Fas/PL-Lipodisp, Fas/PL-LipoP407 gel, Fas/PL-LipoP407/HPMC gel and B-LipoP407/HPMC gel was indirectly assessed by measuring the changes in viscosity following incubation with mucin.38 Mucin 20% w/v was first hydrated with distilled water, then gently stirred at room temperature for complete dispersion. Formulations were incubated with hydrated mucin in a 2:1 ratio, under continuous magnetic stirring at room temperature for 15 minutes. Viscosity was measured in triplicate as previously described under rheological behavior of in situ gels and the mean values ± SD were calculated. Formulation controls were prepared by replacing mucin with distilled water, whereas mucin control sample was prepared similarly but with the formulations being replaced with distilled water. The possible rheological interaction was verified using the following formula:39
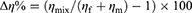
where Δη% is the rheological synergism on viscosity, ηmix is the apparent viscosity of the 2:1 formulation/mucin mixture, ηf is the apparent viscosity of the tested formulation similarly diluted with water (2:1), and ηm is the apparent viscosity of a 20% mucin dispersion (ie, having the same concentration as in the mixture).
Ex vivo Corneal Permeation Test
Freshly excised corneas, from healthy adult white male New Zealand albino rabbits (2–2.5 kg), were used to assess ex vivo corneal permeation of fasudil formulations using modified Franz diffusion cells.40 Corneas along with 2–4 mm of surrounding sclera were mounted between donor and receiver compartments with the endothelial side facing the latter (at 35°C and shaking 50 strokes per minute). Fas solution, Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel formulations (0.5 mL) equivalent to 2.5 mg Fas were applied onto the mucosal surface of the cornea (permeation area 0.645 cm2). Receiver solution (freshly prepared degassed STF at pH 7.4) was selected to simulate in vivo conditions. At specified time intervals, 1, 2, 3, 4 and 6 h, aliquots from the receiver compartment were withdrawn and replaced with an equal volume of fresh medium. Concentration of drug in the samples was quantified spectrophotometrically at λmax 320 nm, and the cumulative amounts permeated were calculated in addition to flux values. To check for corneal integrity, corneas were separated from the scleral tissue at the end of the experiment, washed with normal saline and weighed (Ww), then dried in an oven at 90 °C and reweighed (Wd) for the calculation of corneal hydration level (HL%) using the following equation:41
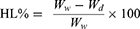
Ocular Tolerance Test (HET-CAM Test)
Ocular tolerability of the developed formulations was analyzed by Hen’s egg test-chorioallantoic membrane (HET-CAM) as reported.42 Fertilized hen’s eggs were incubated at 37 ± 0.5°C and 40% ± 5% relative humidity with manual rotation in a gentle manner every 12 h and exclusion of defective ones containing damaged CAM or showing no live embryo. Three eggs for each formulation weighing between 50 and 60 g were selected and incubated in an equatorial position for the development of chorioallantoic membrane (CAM) away from the shell. On the tenth day, a window (2 × 2 cm) was made on the equator of the eggs through which the tested formulation (0.5 mL) was instilled and left in direct contact with the CAM surface for 5 minutes. The membrane was then examined for any vascular damage, and the time taken for an injury to occur was recorded. Normal saline and 0.1 N sodium hydroxide were used as negative and positive controls, respectively. The scores were recorded according to the scoring scheme described by Gupta et al.43 Digital images of the CAM were taken and further analyzed using Image J software (version 1.49). Images were first converted to greyscale using Photoscape®. To quantify the extent of lysis, haemorrhage or coagulation, greyscale images were loaded into Image J and analyzed for the greyscale values of the pixels over a standardized square area of the membrane. An average of four squares was calculated and plotted for each of the tested formulations as described by McKenzie et al.44
In vivo Evaluation Studies
Animals
Healthy adult male New Zealand albino rabbits (2–2.5 kg) free from any signs of eye abnormalities were provided by the animal facility of the Faculty of Agriculture, Alexandria University. Rabbits were provided with free access to food and water and were kept under 12 h/12 h light/dark cycle in a temperature-controlled room (22–25°C). Experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee, Faculty of Pharmacy, Alexandria University (Approval number: AU-06201985157).
Intraocular Pressure Measurement
Ocular hypertension was induced by topical application of 1% cortisone 2 times daily over the course of one week45 with routine measurement of intraocular pressure (IOP) using a Schiotz tonometer (Riester GmbH & Co. KG, Germany). Animals were then randomly allocated into four groups corresponding to Fas 0.5% w/v solution in normal saline, Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel (n = 3). For all groups, the right eye received a single 50 μL dose of the tested formulation equivalent to 0.25 mg Fas in the conjunctival sac, while the left eye served as control and received 0.9% normal saline. IOP was measured directly prior to formulation administration (zero time) and at 1, 2, 3, 4, 5, 6, 8 and 24 h. All measurements were performed by the same operator and the same tonometer. At each time interval, an average of three readings was recorded; measurements were preceded by instillation of one drop of tetracaine hydrochloride (1% w/v), serving as a local anesthetic. No blepharostat was used. The tonometer readings were transformed into IOP using the conversion table 1955 provided with the instrument, and the percentage decrease in IOP for each time interval was calculated according to the following equation:46
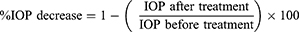
The percentage decrease in IOP was plotted against time. The therapeutic profiles obtained were used to determine the pharmacodynamic parameters including maximum percentage decrease in IOP, time required to attain this decrease (Tmax) and area under the curve (AUC0-24h).
Statistics
The data obtained were compared by one-way ANOVA test followed by pairwise (Tukey’s test) using SPSS 20.0 (SPSS Inc., Chicago, IL, USA). Differences at p values ≤0.05 were considered statistically significant.
Results and Discussion
Preparation of Fasudil HCl Phospholipid Complex (Fas/PL)
The solvent evaporation technique was used for preparation of Fas/PL due to its simplicity and practicability. Several formulation factors including the molar ratio of drug: phospholipid, reaction time, and the effect of reflux were optimized on the basis of complexation efficiency. As shown in Table 1, there is a statistically significant difference (p < 0.001) between the two tested ratios; however, 1:1 molar ratio of drug: phospholipid was selected as the optimum ratio to minimize total lipid concentration in the final formulation to be 1.5% instead of 3% w/v. Complex was better developed at 40°C and 2 h reaction time.
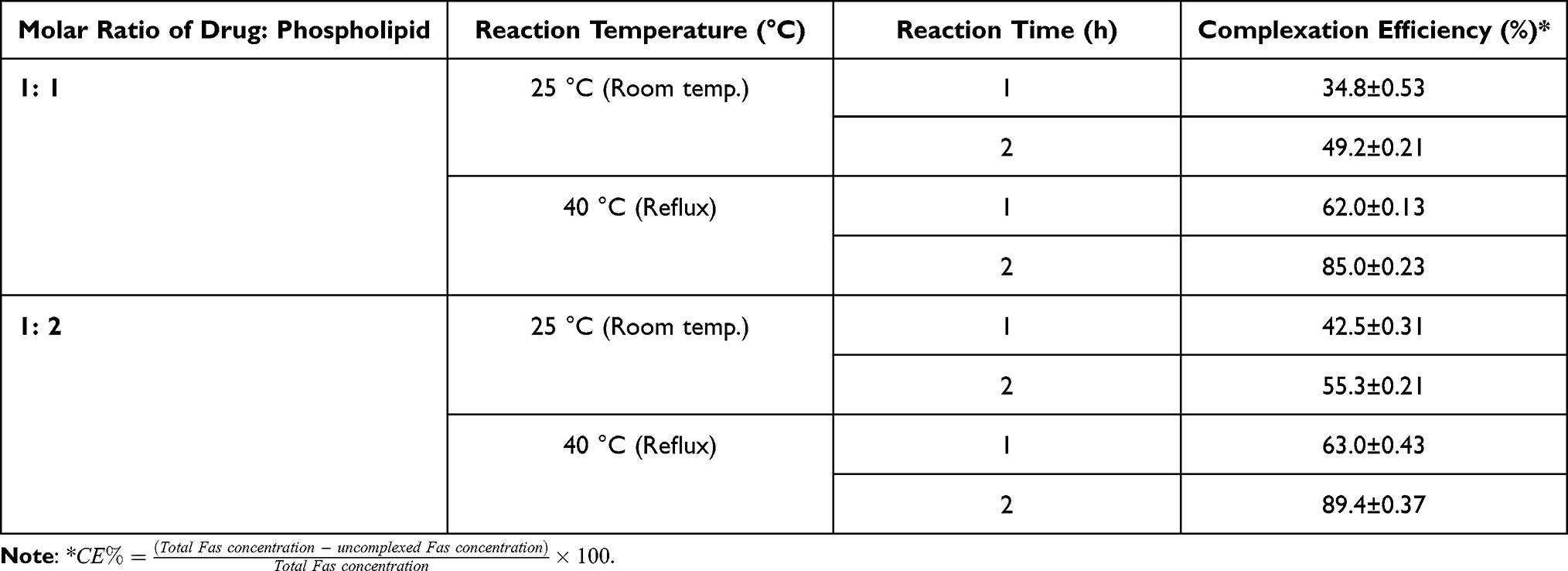 |
Table 1 Optimization Parameters for Preparation of Fasudil Phospholipid Complex (Data are Presented as Mean ± SD, n = 3) |
Physicochemical Characterization of Fas/PL
Fourier Transform Infrared (FT-IR) Spectroscopy
Figure 1A demonstrates the FTIR spectra of Fas, PL, their physical mixture and Fas/PL complex. Fas showed characteristic peaks at approximately 3400 corresponding to N–H stretching, at 1620 cm−1 due to the skeletal stretching vibration of the isoquinoline skeleton, a peak at approximately 1332 cm−1 due to the asymmetric stretching vibration of the sulfonamide group and a peak at approximately 1140 cm−1 due to the symmetric stretching vibration of the sulfonamide group. Phospholipid spectrum exhibited a band between 2925 and 2854 cm−1 corresponding to CH2, a band at 1736 cm−1 due to C=O stretching, P–O asymmetric and symmetric vibration bands at 1243 cm−1 and 1090 cm−1, respectively, and a band at 969 cm−1 corresponding to N+(CH3)3 stretching.47 Fas/Pl physical mixture showed comparative spectral analysis with no shift in the absorption peaks of either the drug or the phospholipid. On the other hand, the proposed complex FTIR chart showed significant changes in many peaks. The N–H peak of the drug and the P–O vibration peaks were obviously suppressed. These results set great evidence for the possible interaction between the two functional groups, suggesting the formation of drug-phospholipid complex.
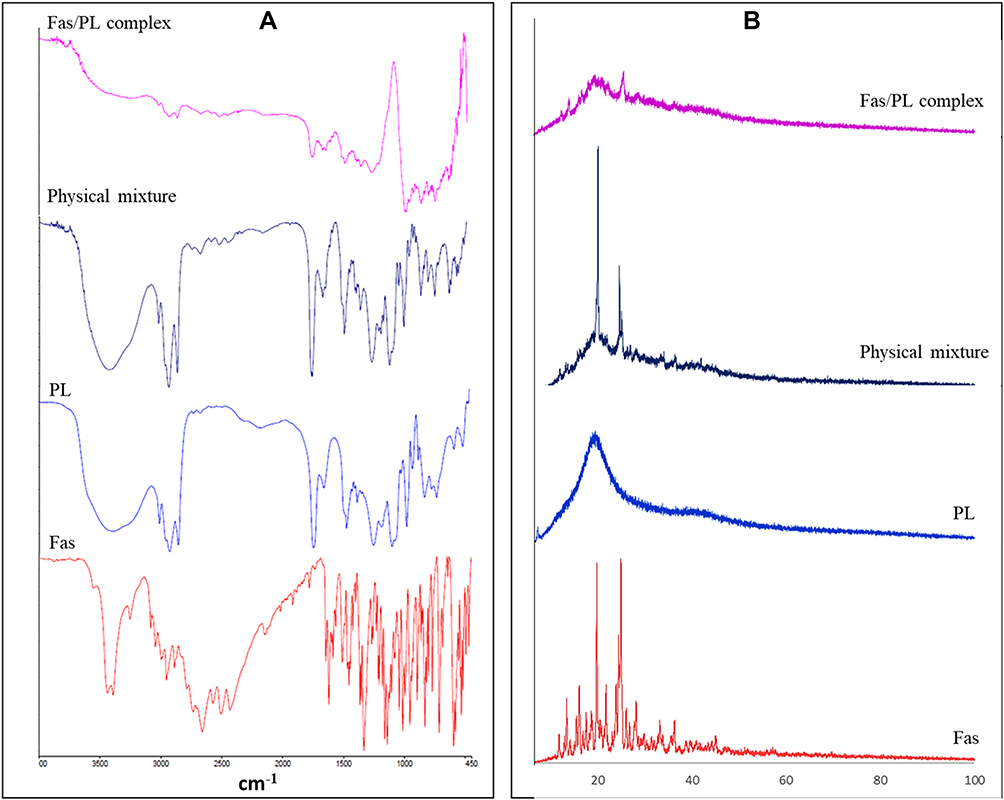 |
Figure 1 (A) FT-IR spectra and (B) XRD patterns of Fas, PL, physical mixture and Fas/PL complex. |
X-Ray Diffraction (XRD)
The XRD patterns of Fas, PL, their physical mixture and Fas/PL complex are shown in Figure 1B. Fas powder displayed sharp peaks at 2θ values approximately 13.4°, 16.2°, 19.9°, 21.8°, 25° and 28.2° with the 2 most characteristic intense peaks at 19.9° and 25° diffraction angles reflecting drug crystallinity. On the other hand, due to its amorphous nature, PL diffractogram showed a single broad peak at 20°.47 Whereas the diffraction pattern of the physical mixture retained both the characteristic crystalline peaks of Fas and the broad peaks of PL, the crystalline peaks disappeared in the case of Fas/PL complex indicating a change in the physicochemical properties of Fas suggesting successful complex formation with PL.
Preparation and Characterization of Liposomes
Liposomal dispersions prepared by methanol injection method were homogeneous in nature. For the optimization of drug-free liposomes’ size and PdI, several variables including the effect of cholesterol content, solvent/non-solvent volume ratio (S/NS) and stirring rate were studied. As shown in Table 2, the mean particle size of liposomes decreased by raising the concentration of cholesterol with respect to phospholipid at constant S/NS 0.6 and stirring rate 800 rpm. This is in line with previous findings that increasing cholesterol content has a negative effect on vesicle size of liposomes.48
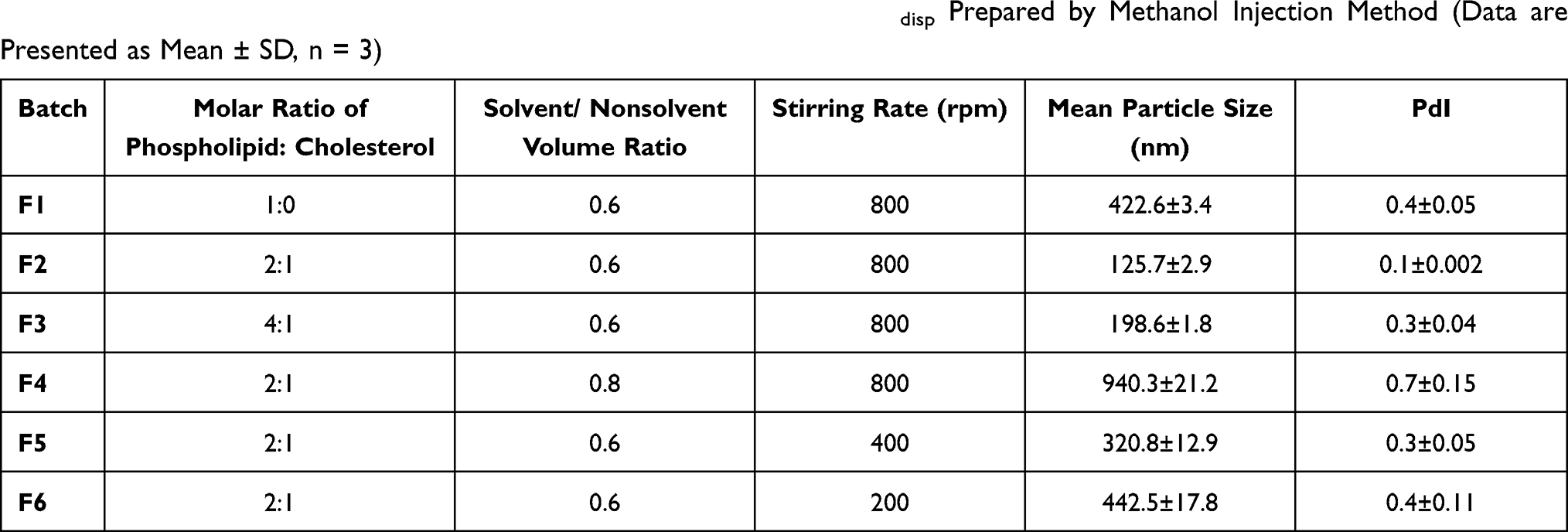 |
Table 2 Physicochemical Characterization of Different Batches of B-Lipodisp Prepared by Methanol Injection Method (Data are Presented as Mean ± SD, n = 3) |
Similarly, particle size reduction was achieved by increasing the stirring rate (Table 2). This could be explained by the intensification of micromixing between the two phases. This high micromixing efficiency may enhance mass transfer and rate of diffusion between the two phases leading to homogeneous supersaturation and self-arrangement of phospholipids, thus formation of smaller vesicles.49 To point out the impact of S/NS volume ratio on liposome formation, S/NS was varied from 0.6 to 1. It was observed that there is a statistically significant (p < 0.05) increase in liposomal particle size by increasing S/NS ratio. This may be attributed to increasing phospholipid solubility in the solvent/water mixture preventing liposome formation.49 Based on the previous results, F2 with smallest particle size and highest monodispersity (PS and PdI, 125.7 ± 2.9 nm and 0.1, respectively) was selected as the optimized liposomal formulation for Fas/PL loaded liposomes. Fas loading resulted in a slight increase in PS (132.5 ± 1.6 nm) while still being highly monodispersed (PdI, 0.18). Fas/PL-Lipodisp possessed a moderately negative ZP (−21.6 ± 0.9 mV).
The morphology of B-Lipodisp and Fas/PL-Lipodisp is illustrated in Figure 2. Liposomes were spherical with a double-layer structure suggesting the unilamellar vesicular nature of liposomes.50 It is worth noting that these spherical unilamellar liposomes can cross the biological barrier (cornea) easily and have better patient compliance as the human eyes could tolerate particles not more than 10 um.51
 |
Figure 2 TEM images of liposomal formulations at magnification x25,000. Scale bars represent 200 nm. |
Despite Fas high hydrophilicity, Fas/PL-Lipodisp showed a relatively high EE% of 78.6 ± 0.25% with %DL of 26.9 ± 0.25% further suggesting Fas/PL complexation.
Preparation and Characterization of in situ Gel Formulations
One of the main objectives of this work was the preparation of a thermo-responsive gel that is liquid at room temperature allowing for easier instillation then structures into gel rapidly upon exposure to the precorneal temperature (35°C) for prolonged ocular retention. Being liquid at room temperature also evades storage in the fridge, thus avoiding utilization of cold eye drops which might prompt eye irritation.22 P407 was selected as the in situ gelling agent due to its low critical temperature.52 This was combined with HPMC K4M for enhancing mucoadhesive properties and further promoting the gelling process.53 To distinguish the appropriate composition for use, systems containing various concentrations of P407 (17.5–25% w/w), with or without HPMC K4M (0.5% w/v), were prepared and evaluated for gelling time, gelling temperature and viscosity (Table 3).
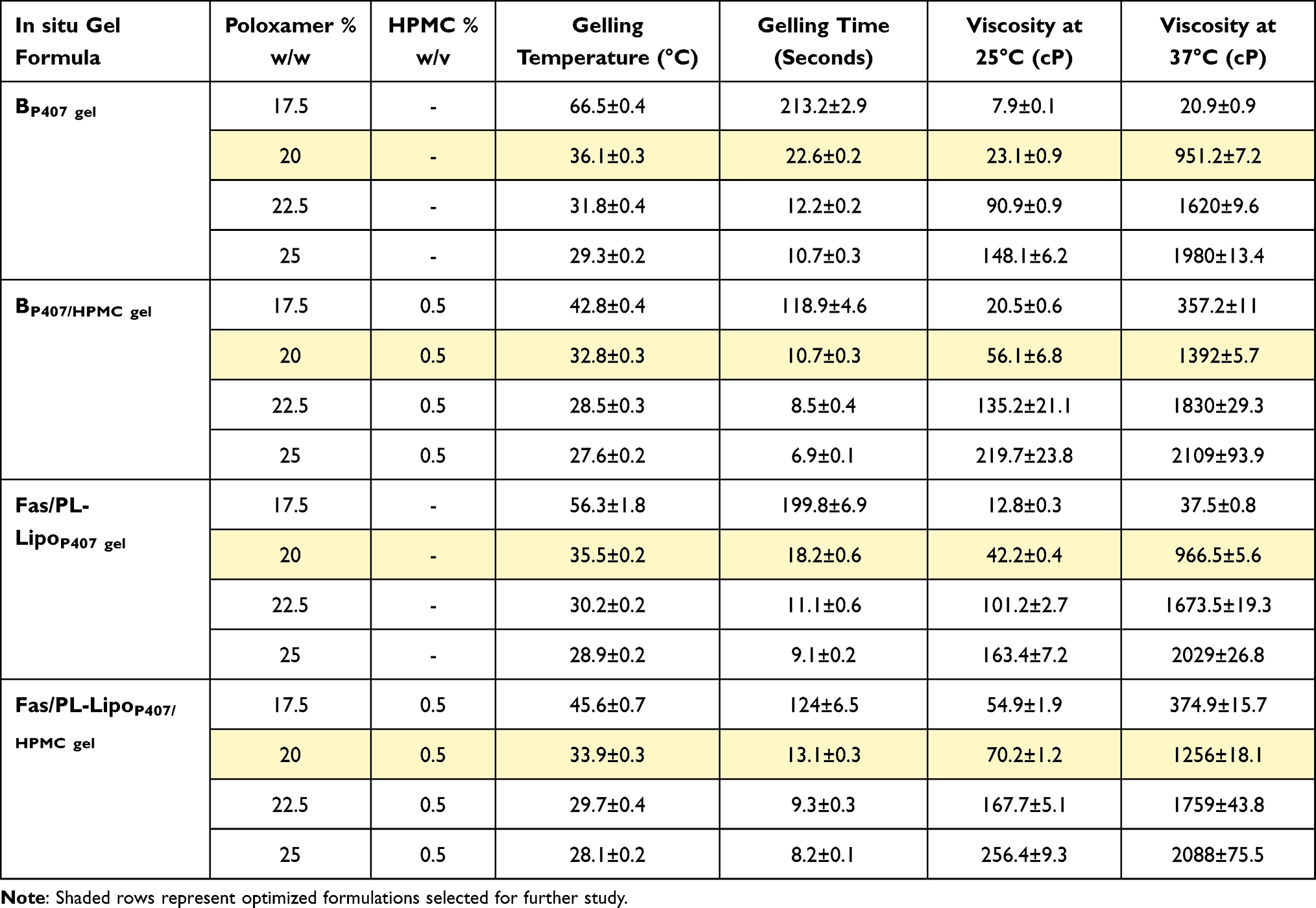 |
Table 3 Gelling Properties and Viscosity Data of P407 in situ Gels and P407/HPMC in situ Gels (Data are Presented as Mean ± SD, n = 3) |
Determination of Gelling Time and Temperature
Gelation temperature and time are those at which the formulation loses its fluidity and turns into gel. These factors are critical in ocular delivery to avoid draining by lacrimal fluid and ensure effective gel formation even when diluted by the small volume of tear fluid.54 The optimal P407 concentration was screened based on gelation temperature and time. Results are shown in (Table 3). By increasing P407 concentration, a shorter time for effective gelation was required indicating a reduction in sol–gel transition temperature.55 The addition of HPMC to P407 led to a significant decrease in gelling time and temperature as a result of the capacity of HPMC to attach to the polyoxyethylene chains present in the pluronic particles through hydrogen bonds which advances dehydration and increases entanglement of adjoining atoms resulting in increased viscosity and hence lower gelation temperature.30 Liposomal incorporation into gel formulations resulted in a slight change in gelling time and temperature.
Clarity and pH
Clarity is a profoundly beneficial characteristic of ocular formulations to avoid blurred vision and patient discomfort caused by non-transparent formulations.31 All formulations were found to be clear, homogeneous, and transparent in both liquid and gel state. Moreover, the pH of the optimized formulations (shaded in Table 3) was in the range 5.5–6, which is expected to be non-irritant as previous findings reported the suitability of pH 4–8 for ocular application.56
Viscosity Data
Viscosities of BP407 gel (Table 3) increased significantly upon increasing poloxamer concentrations from 17.5 to 25% at both room and physiological temperatures with higher values observed at physiological temperature. This may be attributed to the hydrated polypropylene oxide portion of the poloxamer chains at low temperature which undergoes dehydration by increasing the temperature resulting in chain entanglement.29 Similarly, BP407/HPMC gel exhibited similar characteristics to poloxamer alone concerning the change in viscosity with increasing P407 concentration, but with higher overall viscosity values due to the presence of HPMC. The increased viscosity in the presence of HPMC might be contributed to the methoxy group in HPMC molecule which binds to P407 chain and reduces the critical temperature to increase the viscosity as illustrated before.30 Incorporation of liposomes into P407-based gel showed a slight but significant increase in viscosity (p < 0.05) resulting from increased membrane fluidity as a result of interior thickening possibly attributed to the interference of P407 molecules with lipid bilayers of liposomes.57 This effect was reduced in gel formulations containing HPMC. Based on the results of in vitro gel characterization, formulations containing 20% P407 were considered optimum and were selected for further investigation.
Rheological Behavior of in situ Gel Formulations
All in situ gel formulations exhibited a pseudoplastic (shear thinning) behavior at physiological temperature (Figure 3A). The viscosity decreased significantly with increasing shear rate. This might be clarified by the way that any gradual increase in the shear rate brings about the arrangement of the polymer chains along one axis towards a stream, accordingly decreasing the internal resistance of the system to flow and lowering viscosity.31 These results are in a good agreement with those obtained by El-Kamel et al, who examined the properties of different P407 formulations comprising different cellulose derivatives.58 Figure 3B shows the thixotropic behavior of in situ gel formulations with the downward curves showing lower shear stress compared with the corresponding points on the upward ones. The inclusion of liposomes did not apparently affect the hysteresis loops obtained.
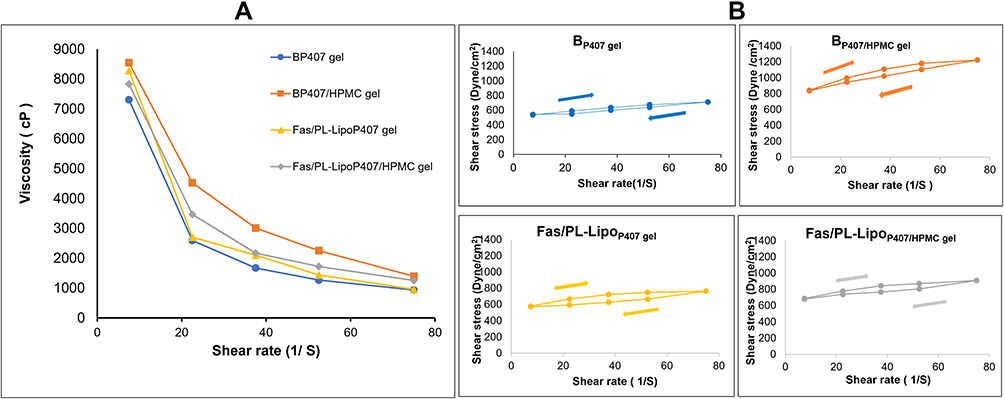 |
Figure 3 (A) Rheogram and (B) Thixotropic behavior of in situ gel formulations at physiological temperature. |
Effect of Gel Formulation on Liposomal Properties
A significant increase in PS (p < 0.05) was observed following incorporation of Fas/PL-Lipodisp into P407/HPMC gel (132.5 ± 1.6 nm and 238.7 ± 3.8 nm for Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel, respectively) with a slight change in PdI (0.18 ± 0.01 and 0.21 ± 0.06, respectively). Also, ZP changed from moderately negative (−21.6 ± 0.93 mV) to positive (27.2 ± 1.5mV). The increase in the average liposomal size recorded with P407/HPMC gel formulation might be due to increased viscosity of the outer phase caused by in situ gel ingredients59 and the formation of a polymer sheath as suggested by the change in ZP.
As shown in Figure 2, the morphology of liposomes did not change after incorporation into a temperature-sensitive gel. Liposomes appeared as vesicular structures surrounded by a polymer sheath.
Regarding EE%, no significant change was observed after incorporation into gel (78.6 ± 0.3 and 77.9 ± 0.5% for Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel, respectively). This insignificant change in encapsulation efficiency recorded for the temperature-sensitive in situ gel suggests liposomal integrity.
Storage Stability
Following 3 months of storage at 4°C, Fas/PL-Lipodisp showed no significant change in particle size or PdI (p > 0.05) indicating stability of the selected optimized formulation (Table 4). On the other hand, Fas/PL-LipoP407/HPMC gel showed a slightly significant apparent increase in particle size and PdI by the end of the study period (p < 0.05). Similar observations were reported for cyclosporine lipid nanocapsules and were attributed to the progressive hydration of the surrounding polymer sheath. This postulation is further supported by the insignificant change in liposomal PS upon storage.29 EE% was measured as an indicator of drug leakage which is a major drawback for liposomal delivery systems.60 The results (Table 4) showed a minor statistically insignificant reduction in EE% (p > 0.05) for both dispersion and gel formulations upon storage (78.6 ± 0.3 and 77.9 ± 0.5 at zero time compared to 75.3 ± 0.4 and 74.8 ± 0.3% after 3 months for Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel, respectively). This reflects the success of drug-phospholipid complexation in improving hydrophilic drug loading into liposomes.
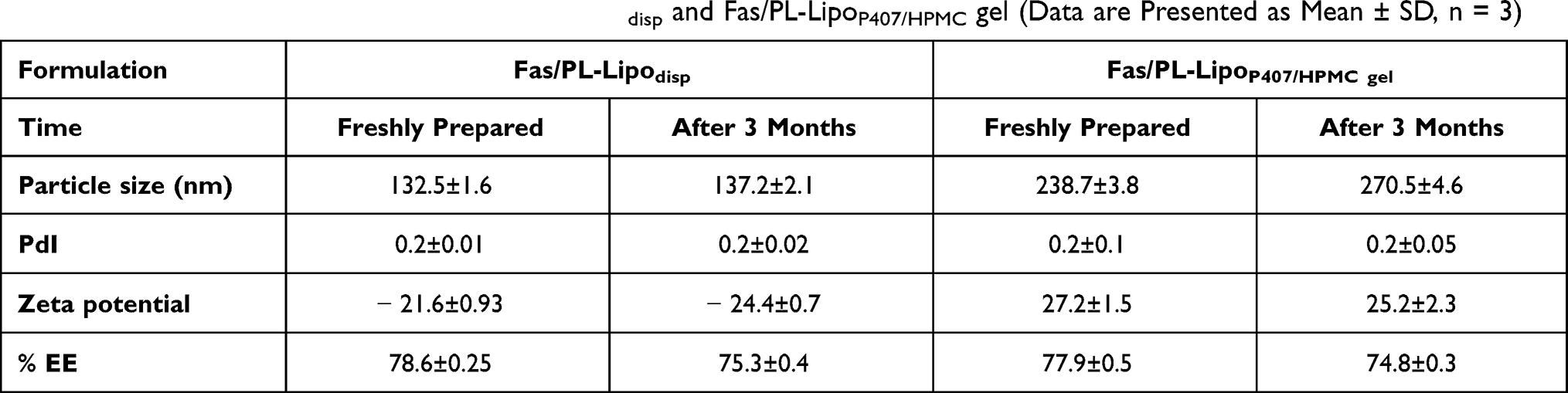 |
Table 4 Storage Stability Data at 4°C of Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel (Data are Presented as Mean ± SD, n = 3) |
In vitro Fasudil Release
In vitro release of Fas from optimized formulations was studied to test whether these formulations control the release of encapsulated drug compared to drug solution. As shown in Figures 4, 100% of Fas placed in dialysis bag as solution in normal saline, appeared in dialysate within 2 h indicating that the dialysis membrane was not a rate-restricting variable in Fas diffusion out of the bag when in the molecular state. A striking difference was noted in the release profiles of liposome dispersion Fas/PL-Lipodisp and Fas solution, particularly after half an hour (Figure 4). Both profiles showed nearly 50% release in the first half an hour, beyond which the liposome dispersion showed controlled release. The initial burst release from the liposome dispersion could be attributed to the rapid release of drug molecules attached to liposomal surface followed by slower release of the encapsulated ones.61 The drug release profile of liposome dispersion suggested that the release mechanism was mainly by diffusion of drug through the lipid bilayer rather than disruption of liposomes.33 This explanation was further supported by kinetic analysis, which indicated that Fas release from liposomes followed Fickian diffusion mechanism; the best fit kinetic model was Korsmeyer-Peppas model (n = 0.021).62
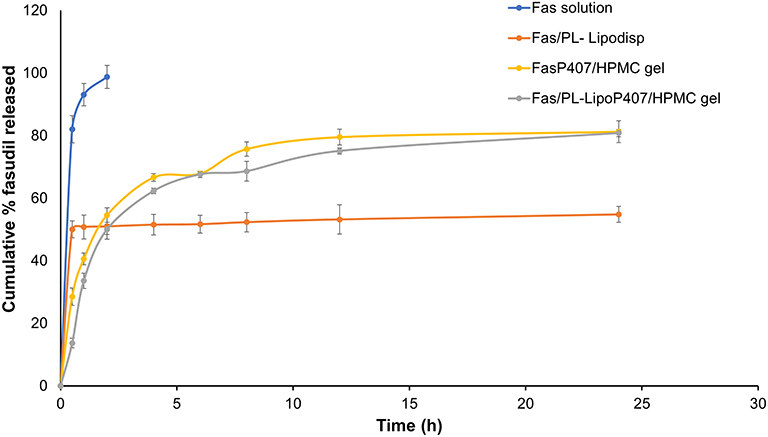 |
Figure 4 Release profiles of tested formulations over a 24 h period in STF pH 7.4 at 50 rpm and 35 °C (n=3). |
In order to reduce the excessive burst release of Fas from liposomes, P407-based liposomal gels (FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel) were prepared and assessed, which showed almost superimposable release profiles with disappearance of initial burst release. The kinetic analysis of the release profiles of both gels fitted the Weibull model with β values equal 0.428 and 0.515, respectively.
Beyond 2 h, both gels showed faster release compared to the liposome dispersion which was not expected (Figure 4). Theoretically, a combination of transport resistance due to liposome bilayer and gel network are expected to result in overall slower release from liposomal gel compared to liposome dispersion.63 This did not happen in the present study. A possible explanation is that when the dialysis bags were placed in the release medium at 35 °C, the thermoresponsive gelling agent (Poloxamer) caused gelation of the sample in the bag (gelling temperatures 33.9 and 35.5 °C for both gels) (Table 3). Release medium diffused slowly into the bag causing swelling of the gel, but also causing dilution of the gelling agent and consequently, a rise in gelling temperature coupled with breakdown of gel structure. Data in Table 3 indicated that a decrease in Poloxamer concentration, for example, from 20 to 17.5% w/v, raised the gelling temperature beyond 45°C for the two drug-loaded gels. This, together with the surface activity of Poloxamer inside the bag, may have contributed to the higher percent release from both gels exceeding that of the liposome dispersion beyond 2 h.
In vitro Mucoadhesion Study
Good mucoadhesive properties of ocular formulations are important to improve their retention time in the eye. As a measurement of mucoadhesive potential, the interaction of liposomal formulations with 20% mucin was determined by analyzing the changes in viscosity following incubation. Enhanced interaction with mucin is reflected by higher elevation in viscosity.64 As shown in (Figure 5A), the viscosity of the four tested formulation/mucin mixtures at a 2:1 volume ratio was significantly higher (p˂ 0.05) than the theoretical sum of individual viscosities of the two components at similar dilution. Such a rheological synergy implies an intimate interaction with mucin proposing formulation capability of mucoadhesion to ocular tissues.39 The present results demonstrated that although a positive rheological synergism occurred for all tested formulations, the highest Δη % was observed with Fas/PL-LipoP407/HPMC gel (Figure 5B). The enhanced mucoadhesion observed for Fas/PL-LipoP407/HPMC gel compared to Fas/PL-LipoP407 gel indicates synergistic effect of the combination of P407 and HPMC on extending precorneal residence time which may be contributed to the weak chemical bonds formed between wetted and swelled mucoadhesive HPMC chains and mucin particles introducing prolonged adhesion of the hydrogel at the mucosal surface.22
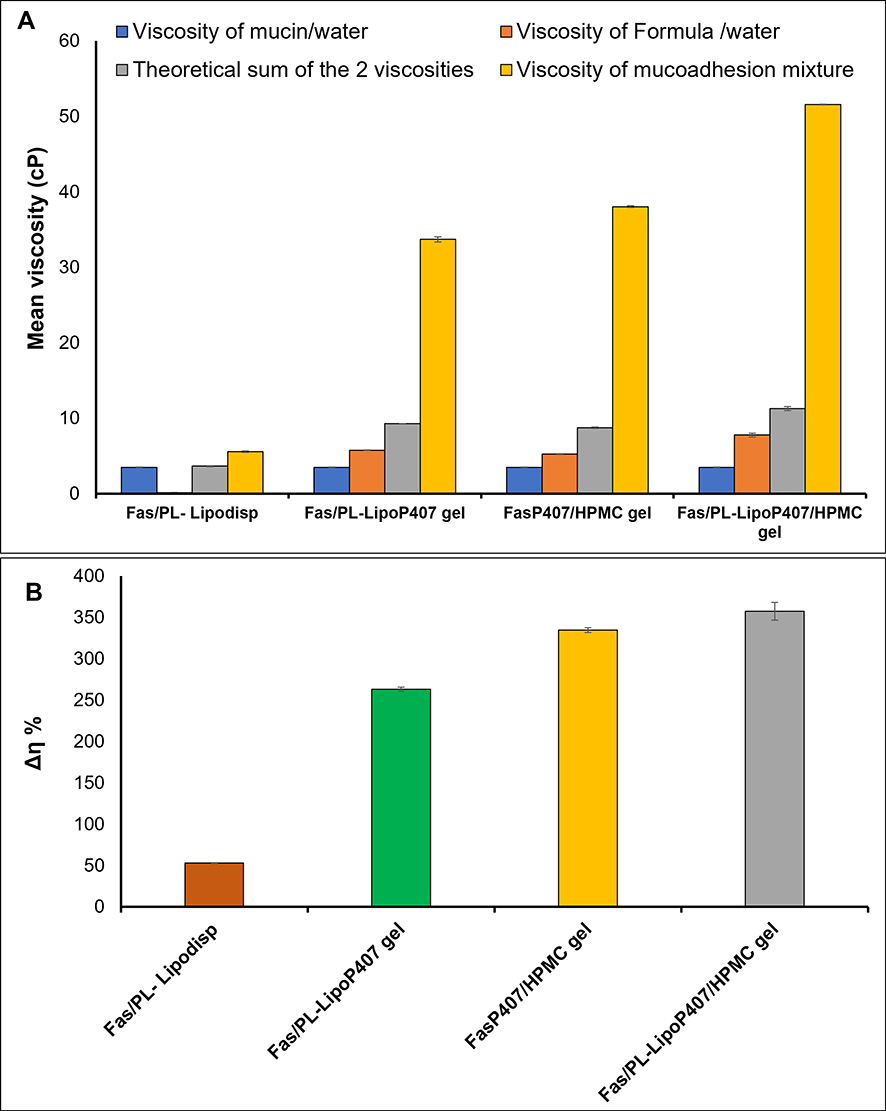 |
Figure 5 (A) Viscosity values of mucin dispersion in water (20% w/v), formulations, theoretical sum of formulation and mucin viscosities and the viscosity of formulation/mucin 2:1 mixtures (B) Rheological synergism of the four tested formulations upon mixing with mucin at a 2:1 ratio represented by Δη % (mean ± SD, n=3). |
Ex vivo Corneal Permeation Test
Corneal drug permeation profiles (Figure 6A) and calculated flux values (Table 5 and Figure 6B) provided ranking of the formulations tested, in terms of rate and extent of permeation over 6 h. Flux values represent slopes of the linear plots describing amounts permeated from 1–3h and from 3–6h. The corneal hydration level remained in the normal range of 76–80% (Table 5), indicating that the formulations and study conditions did not cause damage to any of the corneal layers.30
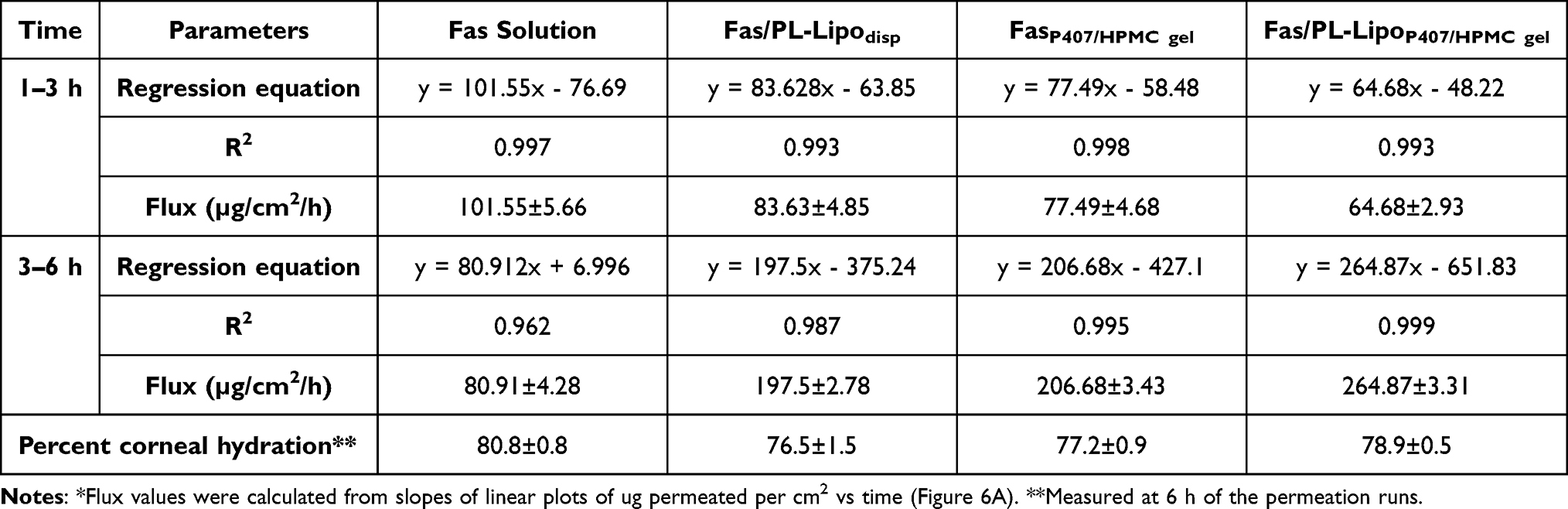 |
Table 5 Ex vivo Corneal Permeation Flux* Values |
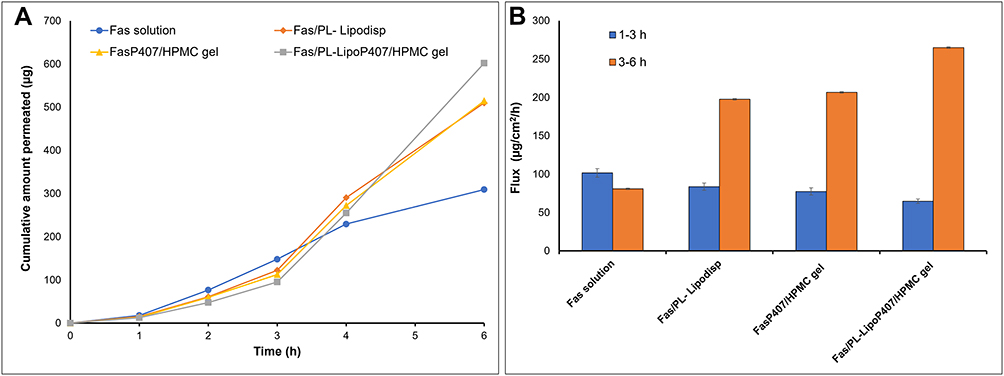 |
Figure 6 (A) Ex vivo corneal permeation profile of fasudil from Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel compared to Fas solution (mean ± SD, n=3) (B) Ex vivo corneal permeation flux values. |
Figure 6A shows the cumulative amount of Fas permeated into the receiver medium over 6 h. Compared to Fas solution, the cumulative permeation in the case of Fas/PL-Lipodisp was lower at 0–3 h, but increased from 3–6h. A similar pattern was observed for the two gel formulations tested (FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel) and was reflected in the flux values (Table 5 and Figure 6B) with maximum flux3-6h shown by Fas/PL-LipoP407/HPMC gel. The low flux1-3 h calculated for liposomal dispersion and gels suggested a lag time during which drug accumulates in the corneal tissues with minimum permeation, followed by high permeation driven by a concentration gradient (cornea to receiver). This permeation pattern could be attributed to the biocompatibility between the lipid vesicles and epithelial cells of the cornea leading to accumulation of drug-loaded liposomes as a depot within the cornea from which the drug is slowly released then transported through the cornea.51,65
Ocular Tolerance Test (HET-CAM Test)
This test is considered a qualitative appliance for estimating irritability of chemicals assessed in terms of adverse changes that occur in the chorioallantoic membrane (CAM) of the egg upon exposure to test chemicals.66 This test was chosen as it is a sensitive, rapid and inexpensive test which does not present any conflict with ethical legal obligations. Also, the CAM of the chick embryo represents a complete tissue containing blood vessels and reacts to injury with a total fiery cycle like that actuated in the conjunctival tissue of rabbit eyes.42 As shown in Figure 7A, the four optimized formulations were evaluated and results were compared with normal saline as a negative control claimed non-irritant and 0.1 N NaOH as a positive control known to be irritant. Based on the scoring system suggested by Gupta et al,43 Fas solution and Fas/PL-Lipodisp exhibited a score 0 and so were considered non-irritant. On the other hand, gel formulations were considered to be mildly irritant with a score of 1. Considering the subjectivity of this assay, and the high probability of the observer not perceiving mild changes in blood vessels, the images were analyzed quantitatively using Image J software. The data were analyzed and processed as “grey values”, where lower values correlate to more hemorrhage and hyperemia.44 As shown in Figure 7B, NaOH showed a significantly lower grey value compared to saline and to all test formulations (p < 0.05). On the other hand, a non-significant difference (p > 0.05) was observed for Fas solution and Fas/PL-Lipodisp compared to saline with a slightly significant difference observed for gel formulations. This reflects the relatively non-irritant nature and hence tolerability of the tested formulations.
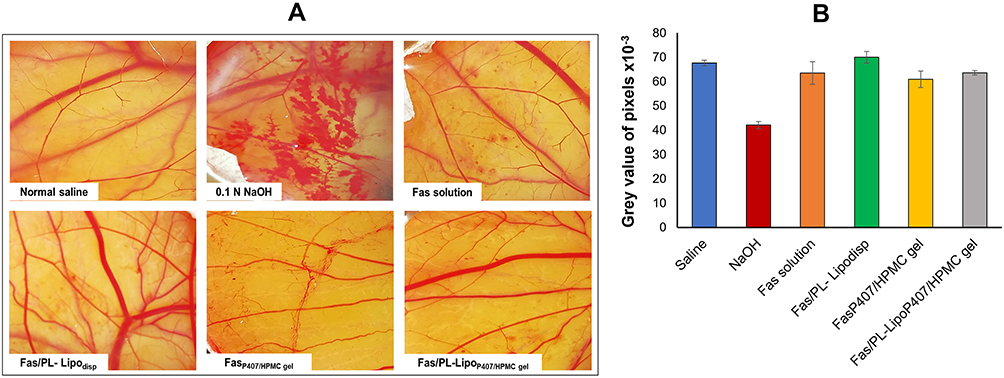 |
Figure 7 (A) Photographs of hen’s egg test-chorioallantoic membrane (HET-CAM) after treatment at room temperature to predict ophthalmic irritation potential and (B) Average grey value of pixels of test formulations analyzed using Image J software as an indicator of ocular irritation (n=4). |
In vivo Evaluation Studies
Considering multiple anatomical and physiological ocular resemblances to humans, rabbits are used as a standard model for eye investigations.67 At the end of 7 days induction by twice daily 1% cortisone eye drops in a rabbit high IOP model, glaucomatous rabbits (n = 15) showed IOP of 31.15 ± 1.36 mmHg which was significantly (p < 0.05) higher than the initial IOP (14.12 ± 0.51mmHg) indicating successful development of ocular hypertension.
The results shown in Figure 8 revealed the effect of a single instillation of the developed formulations (Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel) on the IOP of glaucomatous New Zealand male albino rabbits compared to 0.5% Fas solution in normal saline.
 |
Figure 8 Percentage decrease in IOP after ocular application of 0.5% Fas solution, Fas/PL-Lipodisp, FasP407/HPMC gel and Fas/PL-LipoP407/HPMC gel. Data expressed as mean ± SD (n = 3). |
The positive control group received 0.9% normal saline representing corticosteroid withdrawal pattern from ocular tissues. In the case of 0.5% Fas solution, the IOP decreased sharply reaching a maximum after 2 h. This comes in accordance with Pakravan et al who reported that a single topical dose of 0.5% fasudil can decrease IOP within 2 h.11 Unlike Fas solution, Fas/PL-Lipodisp and FasP407/HPMC gel exhibited a slower decrease with maximum peak after 4 h. On the other hand, the IOP continued to drop till 6 h in case of Fas/PL-LipoP407/HPMC gel. This decrease in IOP caused by all tested formulations was followed by a gradual increase over the rest of the time intervals.
Table 6 represents the calculated pharmacodynamic parameters, namely, Tmax, maximum percent decrease in IOP and AUC0–24h. The delay in Tmax (from 2 to 4 and 6 h) for Fas solution, Fas/PL-Lipodisp and Fas/PL-LipoP407/HPMC gel, respectively, clearly indicated the sustained release characteristics of the developed formulations. AUC0–24h of the temporal profile of IOP was used to characterize the pharmacodynamic effect of each treatment and was determined by the trapezoidal rule. This pharmacodynamic parameter is a measure of the strength, duration of treatment of ocular hypertension and the ocular bioavailability of the drug.68 A significant increase (p < 0.05) in the AUC0–24h values was clearly shown (Table 6), which was in the following order: Fas/PL-LipoP407/HPMC gel > FasP407/HPMC gel > Fas/PL-Lipodisp > Fas solution. Fas/PL-LipoP407/HPMC gel exhibited the highest bioavailability revealing an AUC0–24h value of 761.4 ± 27.7 (%·h). This enhanced ocular bioavailability may be attributed to the electrostatic interaction between positively charged gel molecules and negatively charged mucin particles, and to the proposed bioadhesion of P407, thus prolonging Fas corneal retention time.69 The results demonstrated that a drop of developed Fas/PL-LipoP407/HPMC gel succeeded in sustaining the release rate of Fas through the corneal tissue and improving the ocular drug bioavailability significantly.
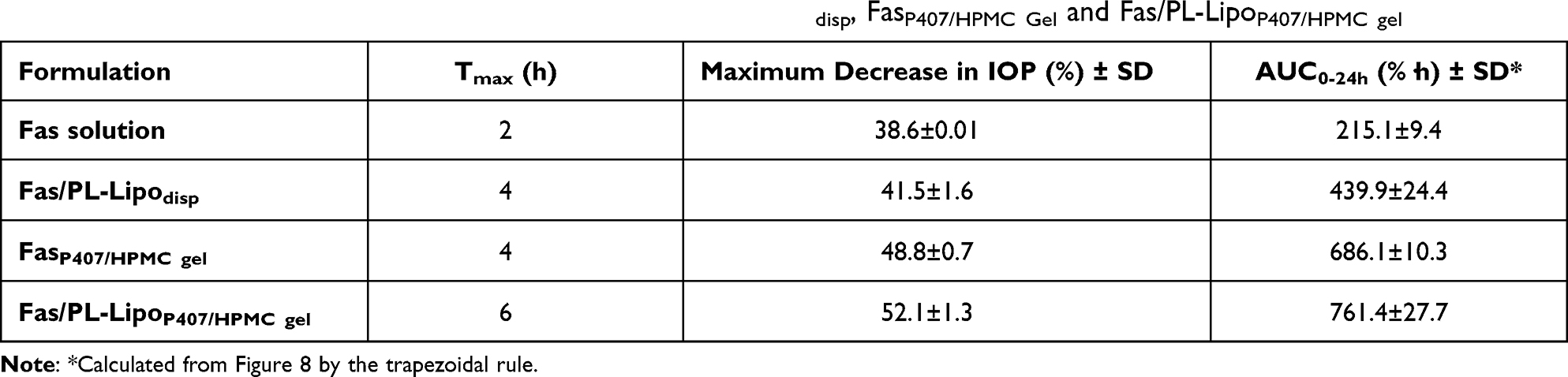 |
Table 6 Pharmacodynamic Parameters of Fas Solution, Fas/PL-Lipodisp, FasP407/HPMC Gel and Fas/PL-LipoP407/HPMC gel |
The question is often raised as to what extent ex vivo permeation data across a bio-membrane can provide a meaningful assessment of formulation performance. This question was tackled in the present study by investigating the correlation between the two sets of ex vivo and in vivo data per formulation. The ex vivo data used were the amounts of fasudil permeated to the receiver chamber at different time points. The in vivo data were the area under the pharmacodynamic curve, calculated by the trapezoidal rule, at the same time points.
The results (Figure 9) indicated strong correlation between the two sets of data, judging by the coefficient of determination (R2). The lowest R2 value was shown by Fas solution where the data points suggest low AUC values and flattening of the pharmacodynamic AUC values while the amounts permeated continued to increase. Such a pattern was not shared by the other three formulations whether liposomal dispersion or gel, whether Fas or Fas/PL complex; no flattening in AUC data was noted resulting in higher R2 values.
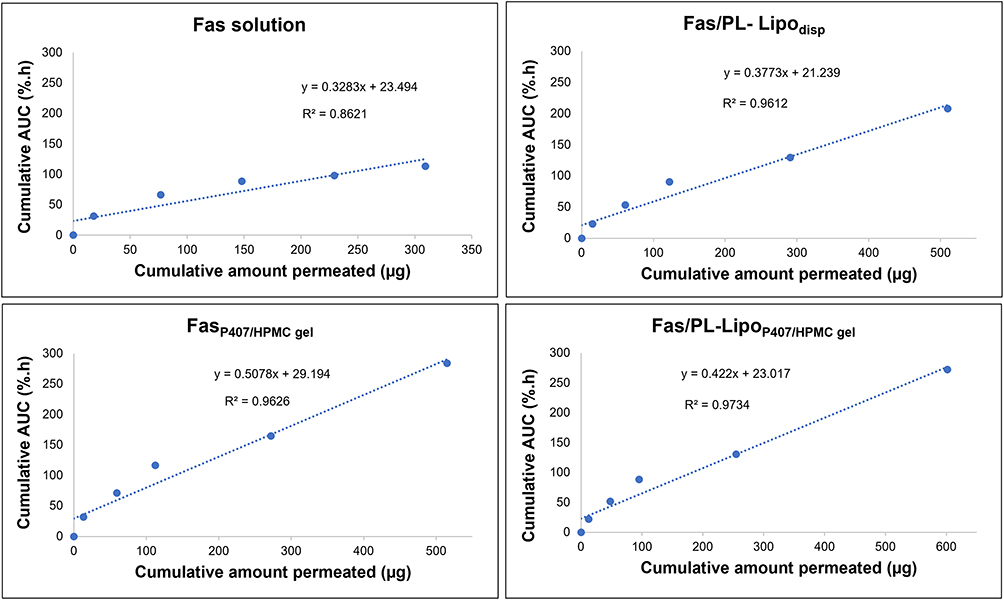 |
Figure 9 Correlation between ex vivo cumulative amount permeated (Figure 6A) and in vivo cumulative area under the pharmacodynamic curve (Figure 8) at corresponding times over 24 h. |
Conclusion
Fasudil, a novel ROCK inhibitor with proved efficacy in glaucoma management suffers a major drawback which is its highly hydrophilic nature leading to low ocular bioavailability. In the current work, Fas was formulated into liposomes to enhance its performance. In order to overcome the low entrapment efficiency and drug loading capability, Fas was complexed with phospholipid prior to liposome formation. This resulted in a pronounced increase in drug loading which is of great value to allow for an effective dose fulfillment. Since local administration of eye drops is usually accompanied by a rapid washout of the applied drug and hence low efficacy with the need for repeated instillation, the prepared liposomal formulation was further incorporated into a thermosensitive in situ gel. This allows for easy administration with prolonged residence on the ocular surface. In vitro mucoadhesion, ex vivo corneal permeation, ocular tolerance and in vivo efficacy of the optimized formulation all promoted Fas/PL-LipoP407/HPMC gel as a promising formulation with enhanced efficacy and tolerability for the management of glaucoma.
Acknowledgments
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Leite MT, Sakata LM, Medeiros FA. Managing glaucoma in developing countries. Arq Bras Oftalmol. 2011;74(2):83–84. doi:10.1590/s0004-27492011000200001
2. Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121(11):2081–2090. doi:10.1016/j.ophtha.2014.05.013
3. Braunger BM, Fuchshofer R, Tamm ER. The aqueous humor outflow pathways in glaucoma: a unifying concept of disease mechanisms and causative treatment. Eur J Pharm Biopharm. 2015;95(2):173–181. doi:10.1016/j.ejpb.2015.04.029
4. Tanna AP, Johnson M. Rho kinase inhibitors as a novel treatment for glaucoma and ocular hypertension. Ophthalmology. 2018;125(11):1741–1756. doi:10.1016/j.ophtha.2018.04.040
5. Pattabiraman PP, Toris CB. The exit strategy: pharmacological modulation of extracellular matrix production and deposition for better aqueous humor drainage. Eur J Pharmacol. 2016;787:32–42. doi:10.1016/j.ejphar.2016.04.048
6. Mietzner R, Breunig M. Causative glaucoma treatment: promising targets and delivery systems. Drug Discov Today. 2019;24(8):1606–1613. doi:10.1016/j.drudis.2019.03.017
7. Garnock-Jones KP. Ripasudil: first global approval. Drugs. 2014;74(18):2211–2215. doi:10.1007/s40265-014-0333-2
8. Hoy SM. Netarsudil ophthalmic solution 0.02%: first global approval. Drugs. 2018;78(3):389–396. doi:10.1007/s40265-018-0877-7
9. Honjo M, Tanihara H. Impact of the clinical use of ROCK inhibitor on the pathogenesis and treatment of glaucoma. Jpn J Ophthalmol. 2018;62(2):109–126. doi:10.1007/s10384-018-0566-9
10. Wang SK, Chang RT. An emerging treatment option for glaucoma: rho kinase inhibitors. Clin Ophthalmol. 2014;8:883–890. doi:10.2147/OPTH.S41000
11. Pakravan M, Naderi Beni A, Ghahari E, et al. The ocular hypotensive efficacy of topical fasudil, a Rho-associated protein kinase inhibitor, in patients with end-stage glaucoma. Am J Ther. 2016;24:1.
12. Tetko IV, Gasteiger J, Todeschini R, et al. Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des. 2005;19(6):453–463. doi:10.1007/s10822-005-8694-y
13. Qin X, Yang Y, Fan TT, Gong T, Zhang XN, Huang Y. Preparation, characterization and in vivo evaluation of bergenin-phospholipid complex. Acta Pharmacol Sin. 2010;31(1):127–136. doi:10.1038/aps.2009.171
14. Ding D, Sun B, Cui W, et al. Integration of phospholipid-drug complex into self-nanoemulsifying drug delivery system to facilitate oral delivery of paclitaxel. Asian J Pharmaceut Sci. 2019;14(5):552–558. doi:10.1016/j.ajps.2018.10.003
15. Ghate D, Edelhauser HF. Barriers to glaucoma drug delivery. J Glaucoma. 2008;17(2):147–156. doi:10.1097/IJG.0b013e31814b990d
16. Bachu RD, Chowdhury P, Al-Saedi ZHF, Karla PK, Boddu SHS. Ocular drug delivery barriers-role of nanocarriers in the treatment of anterior segment ocular diseases. Pharmaceutics. 2018;10(1):28. doi:10.3390/pharmaceutics10010028
17. Shafaa MW, Sabra NM, Fouad RA. The extended ocular hypotensive effect of positive liposomal cholesterol bound timolol maleate in glaucomatous rabbits. Biopharm Drug Dispos. 2011;32(9):507–517. doi:10.1002/bdd.778
18. Monem AS, Ali FM, Ismail MW. Prolonged effect of liposomes encapsulating pilocarpine HCl in normal and glaucomatous rabbits. Int J Pharm. 2000;198(1):29–38. doi:10.1016/s0378-5173(99)00348-8
19. Lombardo D, Calandra P, Barreca D, Magazù S, Kiselev MA. Soft interaction in liposome nanocarriers for therapeutic drug delivery. Nanomaterials. 2016;6(7):125. doi:10.3390/nano6070125
20. Fabiano A, Bizzarri R, Zambito Y. Thermosensitive hydrogel based on chitosan and its derivatives containing medicated nanoparticles for transcorneal administration of 5-fluorouracil. Int J Nanomedicine. 2017;12:633–643. doi:10.2147/ijn.s121642
21. Soliman KA, Ullah K, Shah A, Jones DS, Singh TRR. Poloxamer-based in situ gelling thermoresponsive systems for ocular drug delivery applications. Drug Discov Today. 2019;24(8):1575–1586. doi:10.1016/j.drudis.2019.05.036
22. Patel N, Thakkar V, Metalia V, Baldaniya L, Gandhi T, Gohel M. Formulation and development of ophthalmic in situ gel for the treatment ocular inflammation and infection using application of quality by design concept. Drug Dev Ind Pharm. 2016;42(9):1406–1423. doi:10.3109/03639045.2015.1137306
23. Cao F, Zhang X, Ping Q. New method for ophthalmic delivery of azithromycin by poloxamer/carbopol-based in situ gelling system. Drug Deliv. 2010;17(7):500–507. doi:10.3109/10717544.2010.483255
24. Kurniawansyah IS, Gozali D, Sopyan I, Iqbal M, Subarnas A. Physical study of chloramphenicol in situ gel with base hydroxypropyl methylcellulose and poloxamer 188. J Pharm Bioallied Sci. 2019;11(Suppl4):S547–S550. doi:10.4103/jpbs.JPBS_201_19
25. Hüsch J, Dutagaci B, Glaubitz C, et al. Structural properties of so-called NSAID-phospholipid-complexes. Eur J Pharm Sci. 2011;44(1–2):103–116. doi:10.1016/j.ejps.2011.06.010
26. Kuche K, Bhargavi N, Dora CP, Jain S. Drug-phospholipid complex-A go through strategy for enhanced oral bioavailability. AAPS PharmSciTech. 2019;20(2):43. doi:10.1208/s12249-018-1252-4
27. Domazou AS, Luigi Luisi P. Size distribution of spontaneously formed liposomes by the alcohol injection method. J Liposome Res. 2002;12(3):205–220. doi:10.1081/LPR-120014758
28. Gao D, Tang S, Tong Q. Oleanolic acid liposomes with polyethylene glycol modification: promising antitumor drug delivery. Int J Nanomedicine. 2012;7:3517–3526. doi:10.2147/ijn.s31725
29. Eldesouky LM, El-Moslemany RM, Ramadan AA, Morsi MH, Khalafallah NM. Cyclosporine lipid nanocapsules as thermoresponsive gel for dry eye management: promising corneal mucoadhesion, biodistribution and preclinical efficacy in rabbits. Pharmaceutics. 2021;13(3):360. doi:10.3390/pharmaceutics13030360
30. Morsi N, Ghorab D, Refai H, Teba H. Ketoroloac tromethamine loaded nanodispersion incorporated into thermosensitive in situ gel for prolonged ocular delivery. Int J Pharm. 2016;506(1–2):57–67. doi:10.1016/j.ijpharm.2016.04.021
31. Fathalla MA, Vangala A, Longman M, et al. Poloxamer-based thermoresponsive ketorolac tromethamine in situ gel preparations: design, characterisation, toxicity and transcorneal permeation studies. Eur J Pharmaceut Biopharmaceut. 2017;114:119–134. doi:10.1016/j.ejpb.2017.01.008
32. Alomrani A, Badran M, Harisa GI, et al. The use of chitosan-coated flexible liposomes as a remarkable carrier to enhance the antitumor efficacy of 5-fluorouracil against colorectal cancer. Saudi Pharmaceut J. 2019;27(5):603–611. doi:10.1016/j.jsps.2019.02.008
33. Gupta N, Al-Saikhan FI, Patel B, Rashid J, Ahsan F. Fasudil and SOD packaged in peptide-studded-liposomes: properties, pharmacokinetics and ex-vivo targeting to isolated perfused rat lungs. Int J Pharm. 2015;488(1):33–43. doi:10.1016/j.ijpharm.2015.04.031
34. Tan G, Yu S, Pan H, et al. Bioadhesive chitosan-loaded liposomes: a more efficient and higher permeable ocular delivery platform for timolol maleate. Int J Biol Macromol. 2017;94:355–363. doi:10.1016/j.ijbiomac.2016.10.035
35. Fahmy HM, Saad EAE-MS, Sabra NM, El-Gohary AA, Mohamed FF, Gaber MH. Treatment merits of latanoprost/thymoquinone – encapsulated liposome for glaucomatus rabbits. Int J Pharm. 2018;548(1):597–608. doi:10.1016/j.ijpharm.2018.07.012
36. Tan G, Yu S, Li J, Pan W. Development and characterization of nanostructured lipid carriers based chitosan thermosensitive hydrogel for delivery of dexamethasone. Int J Biol Macromol. 2017;103:941–947. doi:10.1016/j.ijbiomac.2017.05.132
37. Huang W, Zhang N, Hua H, et al. Preparation, pharmacokinetics and pharmacodynamics of ophthalmic thermosensitive in situ hydrogel of betaxolol hydrochloride. Biomed Pharmacother. 2016;83:107–113. doi:10.1016/j.biopha.2016.06.024
38. Lee C-A, Kim B-S, Cho C-W. Quantitative evaluation of mucoadhesive polymers to compare the mucoadhesion. J Pharmaceut Investig. 2016;46(2):189–194. doi:10.1007/s40005-016-0233-4
39. Pecora TMG, Ragazzo B, Bertin W, et al. Rheological behavior of a new mucoadhesive oral formulation based on Sodium Chondroitin Sulfate, Xyloglucan and Glycerol. J Funct Biomater. 2021;12(2):28. doi:10.3390/jfb12020028
40. Aggarwal D, Garg A, Kaur IP. Development of a topical niosomal preparation of Acetazolamide: preparation and evaluation. J Pharm Pharmacol. 2004;56(12):1509–1517. doi:10.1211/0022357044896
41. Cañadas C, Alvarado H, Calpena AC, et al. In vitro, ex vivo and in vivo characterization of PLGA nanoparticles loading pranoprofen for ocular administration. Int J Pharm. 2016;511(2):719–727. doi:10.1016/j.ijpharm.2016.07.055
42. Kaskoos RA. Investigation of moxifloxacin loaded chitosan-dextran nanoparticles for topical instillation into eye: in-vitro and ex-vivo evaluation. Int J Pharm Investig. 2014;4(4):164–173. doi:10.4103/2230-973X.143114
43. Gupta H, Aqil M, Khar RK, Ali A, Bhatnagar A, Mittal G. Sparfloxacin-loaded PLGA nanoparticles for sustained ocular drug delivery. Nanomedicine. 2010;6(2):324–333. doi:10.1016/j.nano.2009.10.004
44. McKenzie B, Kay G, Matthews KH, Knott RM, Cairns D. The hen’s egg chorioallantoic membrane (HET-CAM) test to predict the ophthalmic irritation potential of a cysteamine-containing gel: quantification using Photoshop® and ImageJ. Int J Pharm. 2015;490(1–2):1–8. doi:10.1016/j.ijpharm.2015.05.023
45. Shokry M, Hathout RM, Mansour S. Exploring gelatin nanoparticles as novel nanocarriers for Timolol Maleate: augmented in-vivo efficacy and safe histological profile. Int J Pharm. 2018;545(1):229–239. doi:10.1016/j.ijpharm.2018.04.059
46. Morsi N, Ibrahim M, Refai H, El Sorogy H. Nanoemulsion-based electrolyte triggered in situ gel for ocular delivery of Acetazolamide. Eur J Pharmaceut Sci. 2017;104:302–314. doi:10.1016/j.ejps.2017.04.013
47. Ebada HMK, Nasra MMA, Elnaggar YSR, Abdallah OY. Novel rhein–phospholipid complex targeting skin diseases: development, in vitro, ex vivo, and in vivo studies. Drug Deliv Transl Res. 2021;11(3):1107–1118. doi:10.1007/s13346-020-00833-1
48. Pathak S, Mishra R, Kumar S, Prakash GS, Parthasarthy R. Effect of cholesterol concentration on size of liposome. IOSR J Pharm Biol Sci. 2012;1(1):50–53. doi:10.9790/3008-0115053
49. Jaafar-Maalej C, Diab R, Andrieu V, Elaissari A, Fessi H. Ethanol injection method for hydrophilic and lipophilic drug-loaded liposome preparation. J Liposome Res. 2010;20(3):228–243. doi:10.3109/08982100903347923
50. Vázquez-González ML, Bernad R, Calpena AC, Domènech O, Montero MT, Hernández-Borrell J. Improving ex vivo skin permeation of non-steroidal anti-inflammatory drugs: enhancing extemporaneous transformation of liposomes into planar lipid bilayers. Int J Pharm. 2014;461(1):427–436. doi:10.1016/j.ijpharm.2013.12.009
51. Yu S, Wang Q-M, Wang X, et al. Liposome incorporated ion sensitive in situ gels for ophthalmic delivery of timolol maleate. Int J Pharm. 2015;480(1–2):128–136. doi:10.1016/j.ijpharm.2015.01.032
52. Mura P, Mennini N, Nativi C, Richichi B. In situ mucoadhesive-thermosensitive liposomal gel as a novel vehicle for nasal extended delivery of opiorphin. Eur J Pharm Biopharm. 2018;122:54–61. doi:10.1016/j.ejpb.2017.10.008
53. Da Silva JB, Cook MT, Bruschi ML. Thermoresponsive systems composed of poloxamer 407 and HPMC or NaCMC: mechanical, rheological and sol-gel transition analysis. Carbohydr Polym. 2020;240:116268. doi:10.1016/j.carbpol.2020.116268
54. Venkatesh DMP, Kamlesh L, Kumar P. Development and evaluation of chitosan based thermosensitive in situ gels of pilocarpine. Int J Pharm Pharmaceut Sci. 2013;5:164–169.
55. Ricci EJ, Lunardi LO, Nanclares DM, Marchetti JM. Sustained release of lidocaine from Poloxamer 407 gels. Int J Pharm. 2005;288(2):235–244. doi:10.1016/j.ijpharm.2004.09.028
56. Baranowski P, Karolewicz B, Gajda M, Pluta J. Ophthalmic drug dosage forms: characterisation and research methods. Sci World J. 2014;2014:861904. doi:10.1155/2014/861904
57. Zhang B, Chen J, Lu Y, Qi J, Wu W. Liposomes interiorly thickened with thermosensitive nanogels as novel drug delivery systems. Int J Pharm. 2013;455(1–2):276–284. doi:10.1016/j.ijpharm.2013.07.020
58. El-Kamel AH. In vitro and in vivo evaluation of Pluronic F127-based ocular delivery system for timolol maleate. Int J Pharm. 2002;241(1):47–55. doi:10.1016/s0378-5173(02)00234-x
59. Zhang ZJ, Osmałek T. Deformable liposomal hydrogel for dermal and transdermal delivery of meloxicam. Int J Nanomedicine. 2020;15:9319–9335. doi:10.2147/ijn.s274954
60. Leriche G, Cifelli J, Sibucao K, et al. Characterization of drug encapsulation and retention in archaea-inspired tetraether liposomes. Org Biomol Chem. 2017;15(10):2157–2162. doi:10.1039/C6OB02832B
61. Mehanna MM, El-Kader NA, Samaha MW. Liposomes as potential carriers for ketorolac ophthalmic delivery: formulation and stability issues. Braz J Pharmaceut Sci. 2017;53(2). doi:10.1590/s2175-97902017000216127
62. Paarakh MP, Jose PA, Setty C, Christoper GVP. Release kinetics – concepts and applications. Int J Pharm Res Tech. 2018;8:12–20.
63. Nie S, Hsiao WL, Pan W, Yang Z. Thermoreversible Pluronic F127-based hydrogel containing liposomes for the controlled delivery of paclitaxel: in vitro drug release, cell cytotoxicity, and uptake studies. Int J Nanomedicine. 2011;6:151–166. doi:10.2147/ijn.s15057
64. Silva MM, Calado R, Marto J, Bettencourt A, Almeida AJ, Gonçalves LMD. Chitosan nanoparticles as a mucoadhesive drug delivery system for ocular administration. Mar Drugs. 2017;15(12). doi:10.3390/md15120370
65. Li H, Liu Y, Zhang Y, et al. Liposomes as a novel ocular delivery system for brinzolamide: in vitro and in vivo studies. AAPS PharmSciTech. 2016;17(3):710–717. doi:10.1208/s12249-015-0382-1
66. Spielmann H, Castle J, Gomez M. In Vitro Methods in Pharmaceutical Research. San Diego, CA: Academic Press; 1997.
67. Worakul N, Robinson JR. Ocular pharmacokinetics/pharmacodynamics. Eur J Pharmaceut Biopharmaceut. 1997;44(1):71–83. doi:10.1016/S0939-6411(97)00064-7
68. Mohsen AM, Salama A, Kassem AA. Development of Acetazolamide loaded bilosomes for improved ocular delivery: preparation, characterization and in vivo evaluation. J Drug Deliv Sci Technol. 2020;59:101910. doi:10.1016/j.jddst.2020.101910
69. He W, Guo X, Feng M, Mao N. In vitro and in vivo studies on ocular vitamin A palmitate cationic liposomal in situ gels. Int J Pharm. 2013;458(2):305–314. doi:10.1016/j.ijpharm.2013.10.033
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.