Back to Journals » Drug Design, Development and Therapy » Volume 11
Experimental, molecular docking investigations and bioavailability study on the inclusion complexes of finasteride and cyclodextrins
Authors Mady FM ![]() , Farghaly Aly U
, Farghaly Aly U
Received 18 February 2017
Accepted for publication 20 April 2017
Published 7 June 2017 Volume 2017:11 Pages 1681—1692
DOI https://doi.org/10.2147/DDDT.S135084
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Fatma M Mady,1,2 Usama Farghaly Aly2
1Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Taibah University, Medina, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharmacy, Minia University, Minia, Egypt
Abstract: Finasteride (FIN) is a Class II candidate of the Biopharmaceutics Classification System (BCS). The lipophilic cavity of cyclodextrins (CyDs) enables it to construct a non-covalent inclusion complex with different insoluble drugs. Only β-cyclodextrin (β-CyD) and hydroxypropyl-β-CyD (HP-β-CyD) have been previously examined with FIN. This study aimed to investigate the consistence of FIN with different kinds of β-CyDs, including dimethyl-β-cyclodextrin (DM-β-CyD), carboxymethyl-β-cyclodextrin (CM-β-CyD), HP-β-CyD, sulfobutyl ether-β-cyclodextrin (SBE-β-CyD), and β-CyD, by the coprecipitation method. The resultant inclusion systems were characterized by differential scanning calorimetry, infrared spectroscopy, X-ray diffractometry, and dissolution studies. Moreover, molecular docking for the selected inclusion systems was carried out to explore the suitable arrangements of FIN in the cavity of β-CyD or its derivatives. The results suggested that the DM-β-CyD inclusion system gave the higher complexation efficiency for improvement in solubility of FIN and hence enhancement of its bioavailability. Pharmacokinetic parameters displayed a higher absorption rate and higher area under the curve of the FIN/DM-β-CyD inclusion complex when compared with the drug alone, which indicates an improvement in the absorption and bioavailability of FIN in the DM-β-CyD inclusion system.
Keywords: finasteride, cyclodextrins, molecular docking, pharmacokinetics, bioavailability
Introduction
Finasteride (FIN) is a 4-aza analog of testosterone, with a synonym of (5alpha,17beta)-(1,1-dimethylethyl)-3-oxo-4-azaandrost-1-ene-17-carboxamide (Figure 1). It is a competitive antagonist of both tissue and hepatic 5-α reductase, which blocks the conversion of testosterone to dihydrotestosterone, the hormone responsible for prostatic hypertrophy; hence, it is used for the treatment of benign prostatic hyperplasia (BPH).1 Testosterone-related processes, such as productivity, muscle power, potency, and libido, are minimally influenced by FIN.2,3 FIN is also utilized for the treatment of male pattern alopecia and other androgenic-related disorders such as prostate cancer and hirsutism.3,4 The aim of using FIN as a treatment is to decrease the prostate size, raise the outflow of urine and relief symptoms, and stop the advancement of BPH.5 FIN is a steroidal drug having a molecular weight of 373 Da.6,7 It has poor aqueous solubility and good permeability (Class II Biopharmaceutics Classification System [BCS]) with a good solubility in polar organic solvents, such as ethanol, methanol, and chloroform. It has a mean bioavailability of 63%.8
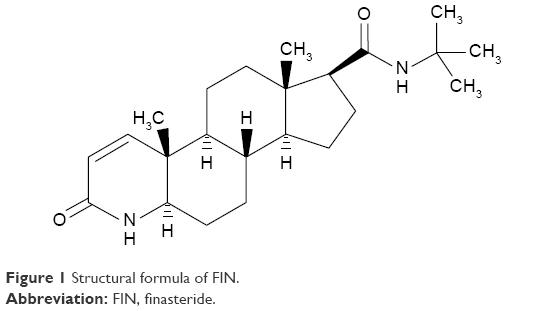 | Figure 1 Structural formula of FIN. |
Cyclodextrins (CyDs) are natural, cone-like shaped cyclic oligomers consisting of glucose components bonded by α-1,4-glycosidic links. All the CyDs contain a doughnut ring with hydroxyl functions on their external and a comparatively lipophilic cavity in their inside, which can engage a guest drug to construct an inclusion system.9 CyDs can improve many features, including aqueous solubility, stability, dissolution, and bioavailability of poorly water-soluble candidate drugs.9,10 The stability or binding constant, which is calculated from the phase solubility diagram, is very important to evaluate the strength of the formation of the inclusion complex.11 A drug of a suitable size can be caught within the CyD cavity, substituting the “high energy” aqueous molecules held in the bulk solution, and constructs a stable system. Thereby, the hydrophobic effect is considered as the essential activating power for the complex construction. Moreover, additional interactions, covalent bonds, and hydrogen bonds participate in the stability of the formed system.12 The drug can be present in various portions of the CyD according to the mode of inclusion, and thus, its spectral features may change.
In a previous literature, the researchers prepared FIN/hydroxypropyl-β-CyD (HP-β-CyD) binary and FIN/HP-β-CyD/chitosan and FIN/HP-β-CyD/polyvinylpyrrolidone (PVP) K30 ternary complexes. They concluded that the method of preparation and the nature of the added polymers that were utilized to prepare the ternary systems do not exert any considerable effect on the physicochemical properties of the complexes. Moreover, values of Kc and complexation efficiency (CE) decreased with chitosan and PVP K30 ternary systems, indicating that they do not have a good influence on the solubility of FIN.13 However, they did not try another derivative of CyDs and compare them with HP-β-CyD.
In our study, we investigated the inclusion complexes between FIN and different derivatives of β-cyclodextrin (β-CyD), such as HP-β-CyD, sulfobutyl ether-β-cyclodextrin (SBE-β-CyD), carboxymethyl-β-cyclodextrin (CM-β-CyD), and dimethyl-β-cyclodextrin (DM-β-CyD), prepared by the coprecipitation technique and characterized the prepared complexes using infrared (IR) spectrometry, X-ray diffractometry (XRD), and differential scanning calorimetry (DSC) analysis. Then, we studied the distribution of the supermolecular structures of the best formulated complex, which will have the higher Kc and CE, using a molecular docking simulation. In addition, pharmacokinetic studies were performed on the best formulated FIN inclusion complex to test the complexation influence on the bioavailability and pharmacokinetic parameters of FIN.
Materials and methods
Materials
FIN was donated from Adwia Pharmaceuticals Co. (Cairo, Egypt) as a gift. β-CyD, average molecular weight (Mw) =1,134.98 Da; 2-HP-β-CyD, average Mw =1,396 Da (average degree of substitution =4.7); SBE-β-CyD, average Mw =2,163 Da (average degree of substitution =6.6); CM-β-CyD sodium salt, average Mw=1,541.24 Da (average degree of substitution =~3); and DM-β-CyD, average Mw =1,331.36 Da (average degree of substitution =2) were purchased from Sigma-Aldrich (St Louis, MO, USA). All other materials were of analytical reagent grade.
Phase solubility studies
FIN, in large excess beyond its aqueous solubility, was hold into stoppered flasks containing 25 mL of distilled water to which different concentrations of the tested kinds of CyDs (0–15 mM) were added. The suspended mixtures were mechanically stirred at room temperature until equilibrium for 72 h. Then, they were undergone filtration (0.45 μm pore size) and assayed using UV spectrophotometer (Agilent 8453; Agilent Technologies, Santa Clara, CA, USA) at λmax 228.5 nm using first derivatives. The phase solubility diagrams were plotted. Then, the apparent stability constants (Kc) of each FIN/CyD complex and the CE were calculated using the slope and the aqueous solubility of the drug (S0) according to the following equations14:
|
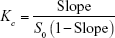
|
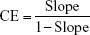
Preparation of the inclusion complex
The solid complexes of FIN (373 g/mol) and/or its derivatives HP-β-CyD, SBE-β-CyD, CM-β-CyD, and DM-β-CyD were prepared by the coprecipitation method. Molar ratio was maintained as 1:1 according to the data obtained from the phase solubility diagram. The tested type of CyDs was dissolved in methanol or water or a mixture of them, per their aqueous solubility, and then, 1:1 molar proportion of FIN was added. These solutions were mechanically stirred for 24 h until equilibrium. Then, they were allowed to evaporate using a rotary evaporator (Heidolph, Schwabach, Germany) at 45°C and 100 rpm and were removed and stored in vials in a desiccator until used. Physical mixtures were also prepared in a similar proportion by homogenous mixing of a sifted and accurately weighed FIN and CyDs.
Characterization of the complexes
Fourier transform infrared spectroscopy (FTIR)
The IR spectra were carried out using an FTIR-470 spectrophotometer (Shimadzu, Kyoto, Japan) in the range of 4,000–400 cm−1 by KBr disks at room temperature.
XRD analysis
The powder XRD patterns of FIN, CyDs, and their co-precipitated systems were carried out at room temperature by a Shimadzu XRD-6000 diffractometer. The employed scanning rate was 4°/min for an acquisition time of 1 h, with a diffraction angle of 2θ and a range of 4°–100°. The samples were set at 40 kV voltage with Cu·Kα radiation.
DSC analysis
The DSC thermograms of FIN, tested CyDs, and their coprecipitated complexes were achieved using DSC-50 (Shimadzu, Japan) by heating in the range of 30°C–400°C at a rate of 10°C/min under a N2 gas flow of 40 mL/min.
Molecular docking studies
To rationalize our experimental results, a molecular docking study for FIN with β-CyD, HP-β-CyD, SBE-β-CyD, CM-β-CyD, and DM-β-CyD was performed using Molecular Operating Environment 2015.10 (Chemical Computing Group, Montreal, QC, Canada). The 3D structure of β-CyD was extracted from the protein complex of alpha-amylase (Protein Data Bank [PDB] code: 1jl8),15 which was retrieved from the Brookhaven PDB and used as a template for building other CyDs. The structures of CM-β-CyD, HP-β-CyD, SBE-β-CyD, and DM-β-CyD were built by MOE Builder function via a substitution of primary hydroxyl groups with the appropriate radicals.16 MOE QuickPrep protocol was used to add hydrogen and minimize the structure. FIN was drawn by ChemBioDraw Ultra Ver.14 Suite (PerkinElmer, Waltham, MA, USA); its potential energy was diminished applying the proper force field AMBER 10 (University of California, San Francisco, CA, USA).17 The default docking protocol implemented in MOE was applied.18 Conformations of ligand were fitted in the position with the Triangle Matcher method and ordered with the London ΔG scoring function. The produced poses were ranked as per their docking scores. Finally, we choose the best energy pose.
Dissolution rate studies
The dissolution rates of FIN (5 mg) and their equivalent amount of coprecipitated CyD complexes were evaluated using apparatus 2, United States Pharmacopeia (USP) Dissolution Tester (Hanson, Chatsworth, CA, USA), in 900 mL distilled water maintained at 37°C±0.5°C with a stirring speed of 50 rpm.6 At predetermined periods, aliquots of 5 mL were withdrawn, microfiltered (0.45 μm pore size), and assayed using UV Spectrophotometer at λmax 228.5 nm. A sink condition was achieved by substitution of the withdrawn samples with an identical volume of the test medium. Results from FIN release were analyzed utilizing two dissolution parameters, the % cumulative released amount at time intervals and the similarity factor (f2) using the raw drug as a reference.19
Pharmacokinetic study
Chromatographic system
The chromatographic system composed of a Dionex UltiMate 3000 high performance liquid chromatography (HPLC) System (Thermo Fisher Scientific, Waltham, MA, USA), which contained a HPG-3400SD solvent delivery pump, WPS-3000(T)SL Analytical Autosampler, and Dionex UltiMate 3000 RS fluorescence detector. The manipulation of data was carried out by Thermo Fisher Scientific Dionex™ Chromeleon™ 7.2 Chromatography Data System (CDS) integration software.
The segregation was achieved using a Luna RP-C18 column (250×4.6 mm, 5 μm; Phenomenex, Torrance, CA, USA) conserved with a guard column (Phenomenex) equipped just before the inlet junction of the analytical column.
Standard solutions
A methanolic stock solution of FIN (100 μg/mL) was prepared. The secondary standard solutions of FIN were prepared by diluting the stock solutions with water. The working standard solutions of FIN were produced by spiking blank rabbit plasma with the secondary standard solutions. The six calibration standards of FIN in plasma final concentrations of 2, 5, 10, 25, 50, and 100 ng/mL were prepared independently. All solutions were stored at 4°C until assayed.
Study design
Pharmacokinetic studies in rabbits
The in vivo pharmacokinetic studies were performed using ten Albino rabbits divided into two groups. Animal utility was ratified by the local institutional review board of Preclinical & Clinical Research, Faculty of Medicine, Assiut University, Egypt, who warranted the protection and using of animals fitted to the European Union (EU) Directive 2010/63/EU on the care of animals applied for scientific objectives and Guiding Principles in the Care and Use of Animals (Department of Health, Education and Welfare Publication National Institutes of Health [NIH] 80-23) and keep on the “Principles of Laboratory Animal Care” (NIH publication 85-23, revised in 1985). These rabbits were supplied by the animal house of Faculty of Medicine, Assiut University, Egypt. They were housed individually under normal conditions. Each group was given 1 mg of FIN/kg as a raw material or its equivalent amount from the DM-β-CyD inclusion complex (chosen as the highest solubility results, eg, highest Kc and CE).
Collection and processing of blood samples
In all, 1 mL samples of blood were withdrawn from the marginal ear vein of the rabbits into heparinized tubes at predetermined time intervals, 0.5, 1, 1.5, 2, 2.5, 3, 4, 8, 12, 14, and 24 h, after drug administration. Samples were centrifuged at 1,500 rpm for 15 min for separation of plasma. The collected plasma was maintained at or below −20°C until used.
Procedure for plasma extraction
In all, 500 mL of rabbit plasma or the calibration standards, 100 μL of methanol, and 100 μL of perchloric acid (70%) were mixed for 30 s into a glass tube and centrifuged at 3,000 rpm for 10 min. The determination of FIN in plasma was performed by injecting 20 μL of the supernatant into the HPLC system.20
Analysis of plasma samples
Plasma samples were analyzed for the raw FIN using the following HPLC method. A mixture of acetonitrile, 30 mM phosphate buffer in a ratio of 65:35 v/v, was prepared as a mobile phase. Its pH was rendered 3 using orthophosphoric acid.
A vacuum filtration unit (Phenomenex) with a 0.2 mm membrane filter was utilized to filter the mobile phase, and then, an ultrasonic cleaner (Cole-Parmer, Chicago, IL, USA) was used for degassing. A 20 μL sample was injected with an adjusted flow rate of 1 mL/min at room temperature. The detector wavelength was set at 215 nm.
Calibration curve
FIN standard solutions in plasma were extracted and analyzed according to the described procedures. Three injections of each standard solution were made. Peak areas of varying amounts of FIN versus the corresponding FIN concentrations were used to obtain the calibration curve.
Pharmacokinetic analysis (data analysis)
After measuring FIN concentration in plasma, FIN pharmacokinetics in plasma was assessed by fitting the plasma concentration–time data to the suitable model using WinNonlin™ standard version 1.5 software (Science Consulting Inc, Apex, NC, USA). Ordinary least-squares sum was used as a criterion function in the fitting process. Goodness of fit was estimated by visual inspection of the fitted curve and correlation coefficient, which exceeded 0.95.
Absorption rate constant (Ka), elimination rate constant (Kel), elimination half-life (t1/2), and area under the plasma FIN concentration versus time curve (area under the curve [AUC]) were calculated according to conventional algorithms. Maximum concentration (Cmax) and time to reach maximum concentration (Tmax) were reported as observed.
Based on the measured concentrations of FIN in rabbit plasma, individual concentration–time profiles following single oral administration of 1 mg FIN/kg as a raw material and its equivalent amount of the DM-β-CyD inclusion complex were constructed. Concentration time course in plasma is presented in Figure 2. Mean concentration time courses were fitted to a one-compartment model with first-order absorption and elimination. Mean (±SD) values for the pharmacokinetic parameters in plasma are listed in Table 1.
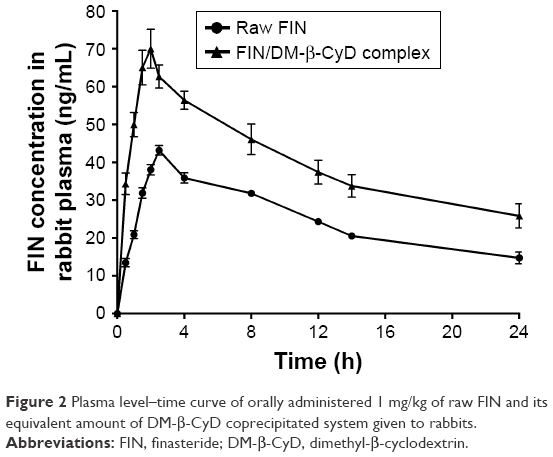 | Figure 2 Plasma level–time curve of orally administered 1 mg/kg of raw FIN and its equivalent amount of DM-β-CyD coprecipitated system given to rabbits. |
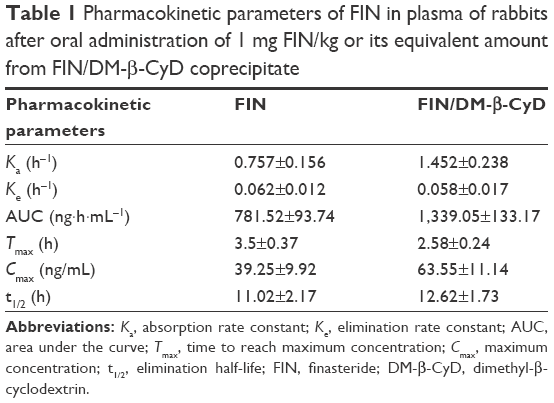 | Table 1 Pharmacokinetic parameters of FIN in plasma of rabbits after oral administration of 1 mg FIN/kg or its equivalent amount from FIN/DM-β-CyD coprecipitate |
Statistical analysis
Data were displayed as mean ± SD. Differentiation between two groups was calculated using Student’s t-test. Variations were considered as significant differences if P-value was <0.05. The statistical analyses were carried out by the GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA).
Results and discussion
Phase solubility studies
Figure 3 shows the effect of increasing concentrations of the tested types of CyDs (β-CyD, HP-β-CyD, SBE-β-CyD, CM-β-CyD, and DM-β-CyD) on the aqueous solubility of FIN. All the phase solubility diagrams were Higuchi AL type, which is depicted by an increase in the solubility of drug linearly relative to CyD concentration, suggesting the construction of a first-order complex relative to CyD. Concerning that all solubility diagrams gave a slope with a value of <1, a 1:1 stoichiometry can be supposed.21 The assessed values of S0, the slopes of the phase solubility diagrams, and the Kc are shown in Table 2.
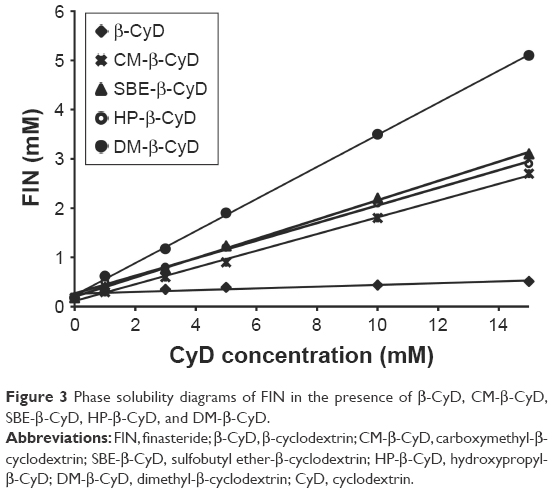 | Figure 3 Phase solubility diagrams of FIN in the presence of β-CyD, CM-β-CyD, SBE-β-CyD, HP-β-CyD, and DM-β-CyD. |
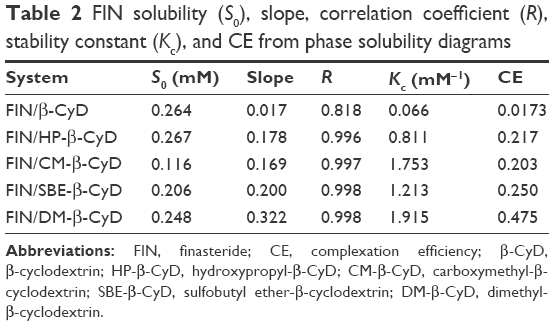 | Table 2 FIN solubility (S0), slope, correlation coefficient (R), stability constant (Kc), and CE from phase solubility diagrams |
According to literatures, only complexes with a Kc value between 0.1 and 2 mol·L−1 have industrial applications. Complexes with a lower Kc value than 0.1 mol·L−1 represent unstable drug–CyD systems, whereas complexes with a Kc value >2 mol·L−1 could inversely influence the absorption of drugs. If the complex was very weak, there will be a little enhancement in the drug solubility. Furthermore, if the complex was very strong, as denoted by a >10,000 M−1 value of stability constant, the complex may not be dissociated appreciably and consequently is not absorbed.22,23 The Kc values found for all the studied systems indicate that inclusion complexes with suitable stability were constructed except for β-CyD. Loftsson et al24 offered a different way to determine the solubilizing influences of CyDs, which depends on evaluating the CE. As observed in Table 2, the CE is higher in case of the DM-β-CyD complex and the lowest CE is conducted with β-CyD. All other derivatives of CyD are in-between DM-β-CyD and β-CyD.
The CE mean values for β-CyD and its derivatives were 0.0173, 0.217, 0.203, 0.250, and 0.475 (for β-CyD, HP-β-CyD, CM-β-CyD, SBE-β-CyD, and DM-β-CyD, respectively). This confirms that β-CyD derivatives are better solubilizers than native β-CyD. Moreover, DM-β-CyD seems to be the best solubilizer for FIN.
FTIR
FTIR spectrum for FIN, β-CyD, HP-β-CyD, CM-β-CyD, SBE-β-CyD, and DM-β-CyD with the physical mixtures and the coprecipitated inclusion complexes of these CyDs are shown in Figure 4. As it is seen, the FIN has characteristic peaks/bands (cm−1) attributed to vibration of N-H stretching (3,428), aromatic C-H stretching (3,116), C=O stretching (1,670), N-H bending (1,599), C=N stretching (1,363), C=C stretching (1,449 and 1,506), C-H bending (686), and C-N-H bending (2,840). In addition, the characteristic peaks/bands for DM-β-CyD are OH stretching (3,407), C-H stretching (2,933), C=O stretching (1,735), C=C stretching (1,640), C-O stretching (1,051–1,158), =C-H bending (702–967), and C-O-H bending (666). For physical mixtures, spectrum was approximately characterized by the superimposition of the individual absorption bands of FIN and the tested CyDs with no remarkable alterations.
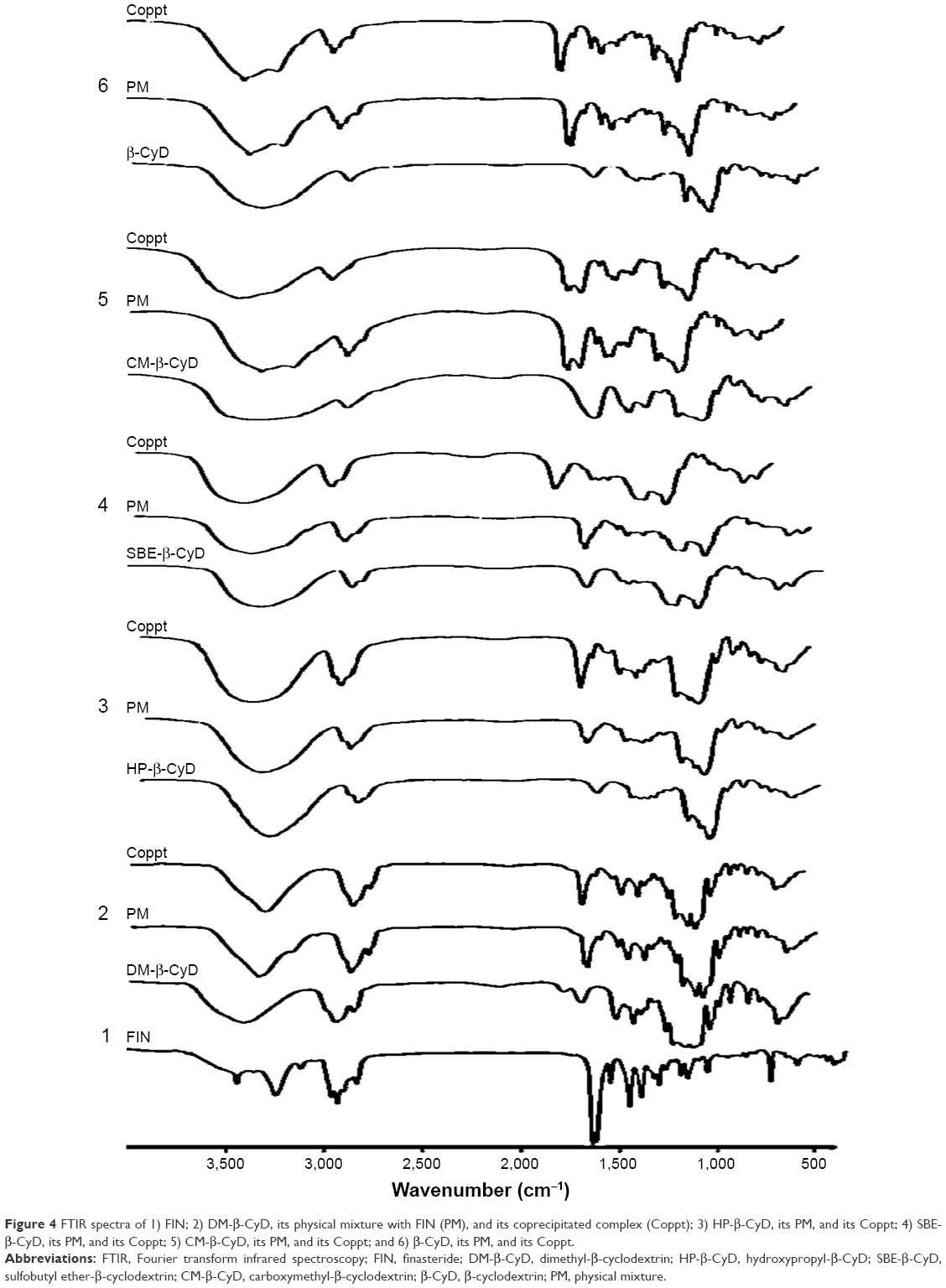 | Figure 4 FTIR spectra of 1) FIN; 2) DM-β-CyD, its physical mixture with FIN (PM), and its coprecipitated complex (Coppt); 3) HP-β-CyD, its PM, and its Coppt; 4) SBE-β-CyD, its PM, and its Coppt; 5) CM-β-CyD, its PM, and its Coppt; and 6) β-CyD, its PM, and its Coppt. |
Comparison of the IR spectra of all CyDs coprecipitated systems displayed a significant change (either disappearance, decrease in intensity, or shift) in case of DM-β-CyD and HP-CyD systems. The vanishing of both N-H stretching band and the C=O stretching of FIN was observed in the FIN/DM-β-CyD coprecipitated complex, which indicates the possible interaction between them. Moreover, the broad hydroxyl band of pure DM-β-CyD at 3,407.5 and 3,411.07 cm−1 for HP-β-CyD was found to be narrowed and slightly shifted to 3,416 and 3,386 cm−1, respectively, in the FTIR spectrum of their coprecipitate system, which is a good indication of the construction of the inclusion complex; this is a common phenomenon detected by many literatures in the formation of the inclusion complex between the host CyDs and guest compounds.25,26 On the other hand, no observable change in the IR spectra of SBE-β-CyD, CM-β-CyD, and β-CyD physical mixtures and their coprecipitated systems was detected.
XRD analysis
Powder XRD is a helpful technique to detect CyD complexation. The diffractogram of the complex should be distinctly clear from those procured by the superposition of each component, if an inclusion complex constructs.27,28 The XRD pattern of FIN manifested a sharp style reflecting the diffraction peaks at 12.8°, 14.11°, 15.6°, 9.5°, and 26.9° (2θ), which indicated a crystalline feature. The diffractograms of the physical mixtures displayed a crystalline pattern of FIN with all the tested types of CyDs and the appearance of the sharp peaks of the drug, which indicates the absence of interaction with physical mixing, whereas a hollow style and disappearance of the characteristic sharp peaks of FIN were registered for coprecipitated systems of both DM-β-CyD and HP-β-CyD (Figure 5). On the other hand, the characteristic peaks of FIN did not vanish in case of the coprecipitated systems of CM-β-CyD, SBE-β-CyD, or β-CyD, which detected a crystalline profile of FIN, indicating an absence of interaction with these types of CyDs.
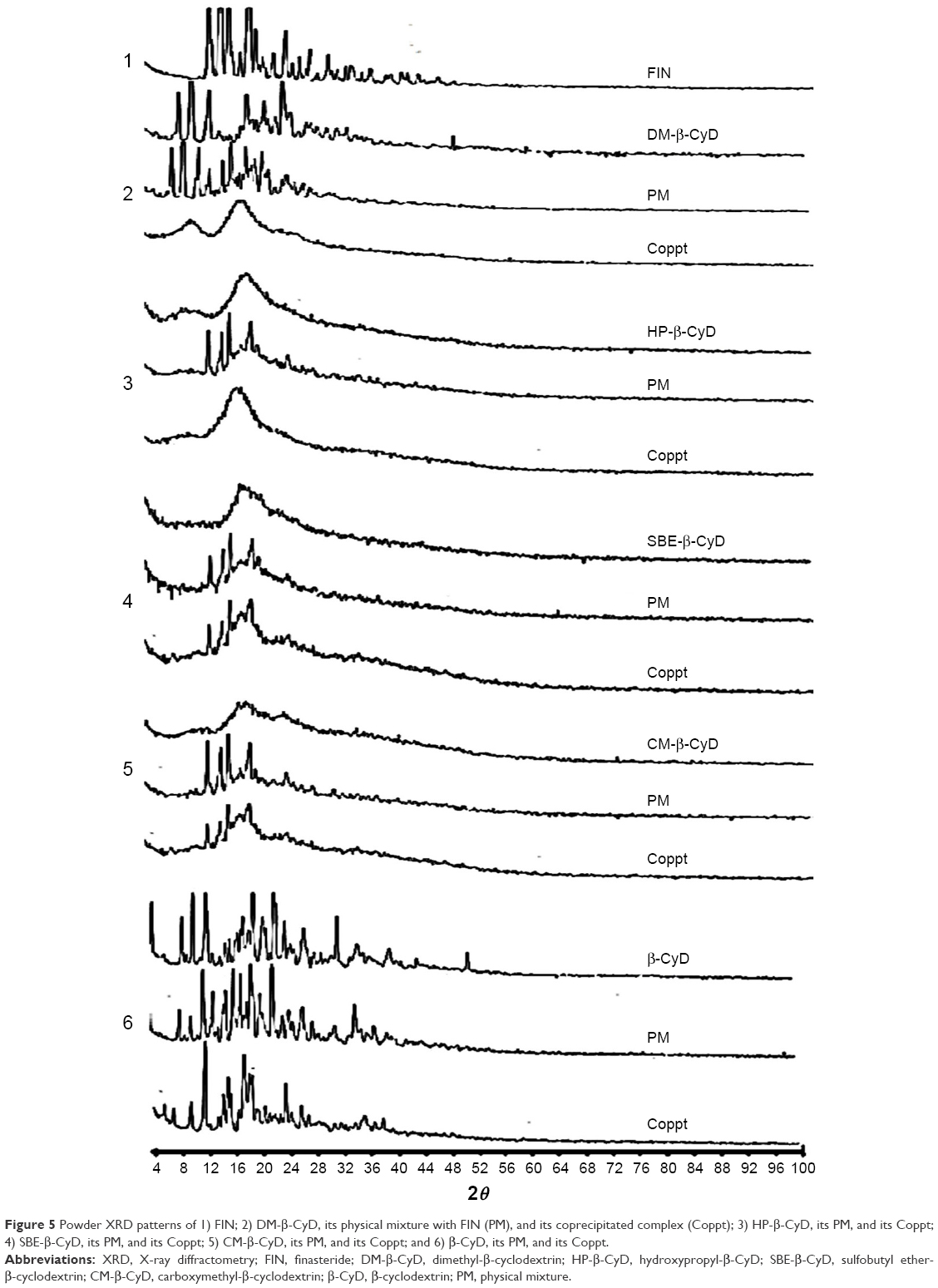 | Figure 5 Powder XRD patterns of 1) FIN; 2) DM-β-CyD, its physical mixture with FIN (PM), and its coprecipitated complex (Coppt); 3) HP-β-CyD, its PM, and its Coppt; 4) SBE-β-CyD, its PM, and its Coppt; 5) CM-β-CyD, its PM, and its Coppt; and 6) β-CyD, its PM, and its Coppt. |
DSC analysis
DSC was utilized to recognize FIN complexes in the solid condition and to attain additional conforming proof of construction of complexes. Forasmuch, the thermal profile of raw FIN was typically crystalline anhydrous, with a sharp endothermic peak at 260.35°C, indicating the melting point of FIN, and this agrees with the previous report.29 Broad endothermal peaks provided with water loss were registered for the tested types of CyDs.30 Comparison of DSC thermograms from the coprecipitated CyD systems indicated that the endothermic incident related to the fusion of FIN was shifted to 290.14°C, broadened and decreased in intensity in case of the coprecipitate of DM-β-CyD, elucidating the construction of amorphous entities and/or inclusion complexes. Moreover, the endothermic peak of FIN was shifted with a lower intensity in the HP-β-CyD coprecipitated system, which also manifests the formation of amorphous entities and/or inclusion complexes. However, the DSC curves of CM-β-CyD, SBE-β-CyD, and β-CyD coprecipitated systems showed clearly the characteristic fusion peak of FIN, which indicates the presence of FIN in a crystalline form or absence of interaction (Figure 6). On the other hand, comparison of DSC curves from physical mixtures of FIN and the tested CyDs indicated that the characteristic peak of FIN (corresponding to melting of the drug) was distinctly recognizable in all physical mixtures.
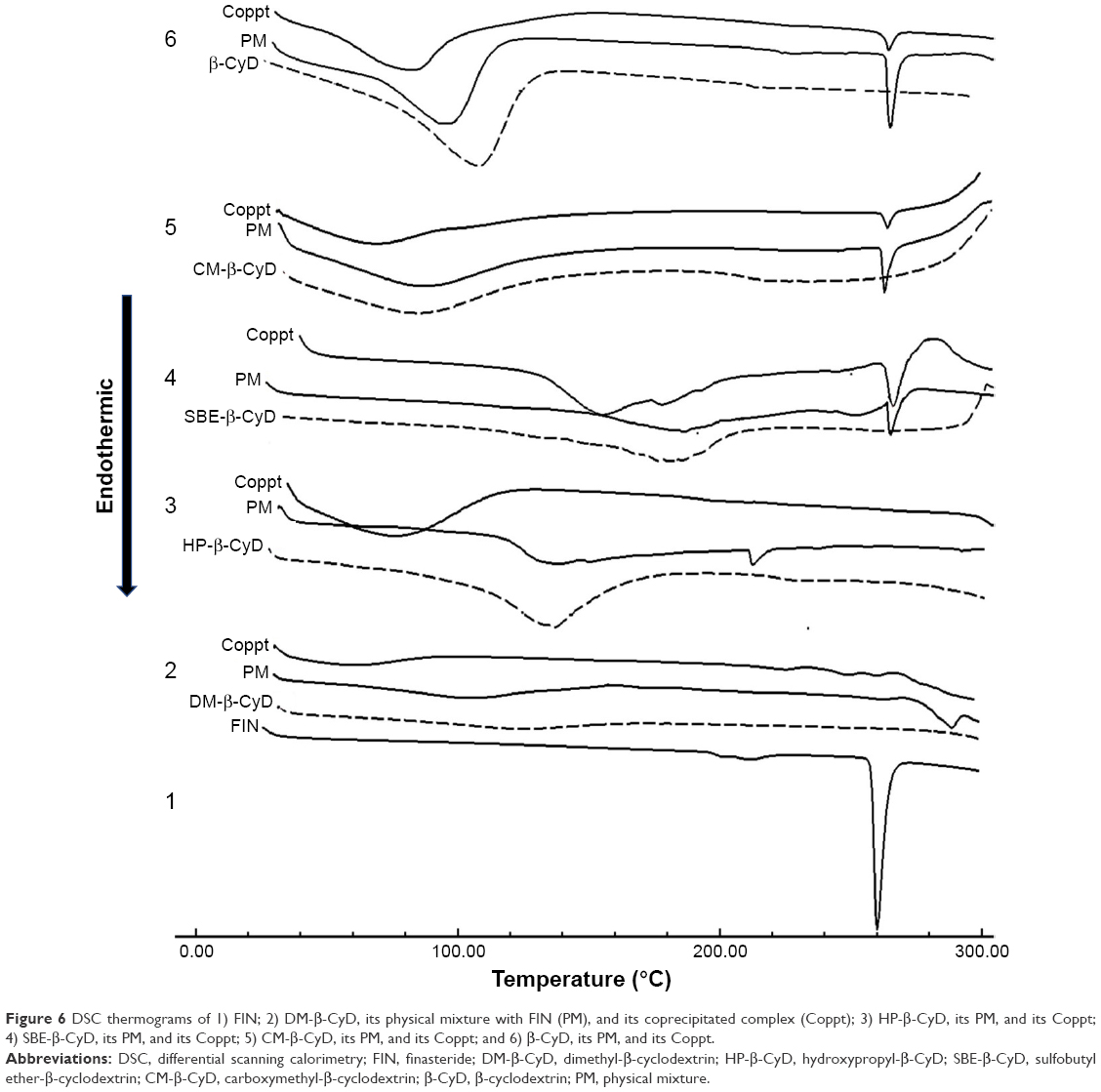 | Figure 6 DSC thermograms of 1) FIN; 2) DM-β-CyD, its physical mixture with FIN (PM), and its coprecipitated complex (Coppt); 3) HP-β-CyD, its PM, and its Coppt; 4) SBE-β-CyD, its PM, and its Coppt; 5) CM-β-CyD, its PM, and its Coppt; and 6) β-CyD, its PM, and its Coppt. |
Molecular docking studies
The focal profit of implementing molecular modeling computations is to provide some perspicacity on the orientation and interaction of drug/guest molecule with CyD cavity, which may assist the experimental feedbacks.29 The longer the hydrophobic chain linked to the β-CyD, the better the assembly of the drug within the complex.31 Moreover, it is well known that complexation with CyDs is affected by guest hydrophobicity.32,33
In this study, we have performed docking studies to propose the possible interaction of FIN with CyD cavity. Our results indicate that FIN interacts with DM-β-CyD in 1:1 ratio (Table 2) with the best stability constant (Kc) and the best CE. It was evident that the drug has been included in the CyD cavity in a 1:1 M ratio, which is in agreement with the conclusions of the DSC, XRD, and FTIR studies. In case of docking with DM-β-CyD (Figure 7A), the binding energies were −6.3, −5.5, −4.1, −3, and −3.1 kcal/mol with DM-β-CyD, HP-β-CyD, SBE-β-CyD, CM-β-CyD, and β-CyD, respectively. Figure 7 demonstrates that the binding is stronger in case of DM-β-CyD and is more favorable due to the hydrophobic properties of DM-β-CyD pocket that has additional methyl groups compared to the other β-CyDs. This hydrophobic pocket gives a more stable complex when it sandwiches the hydrophobic and water insoluble molecule FIN.34 Therefore, it was not surprising to get a high solubilizing effect for FIN by complexation with DM-β-CyD rather than the other tested CyD derivatives, and our computational calculations were in accordance with the experimental analysis data.
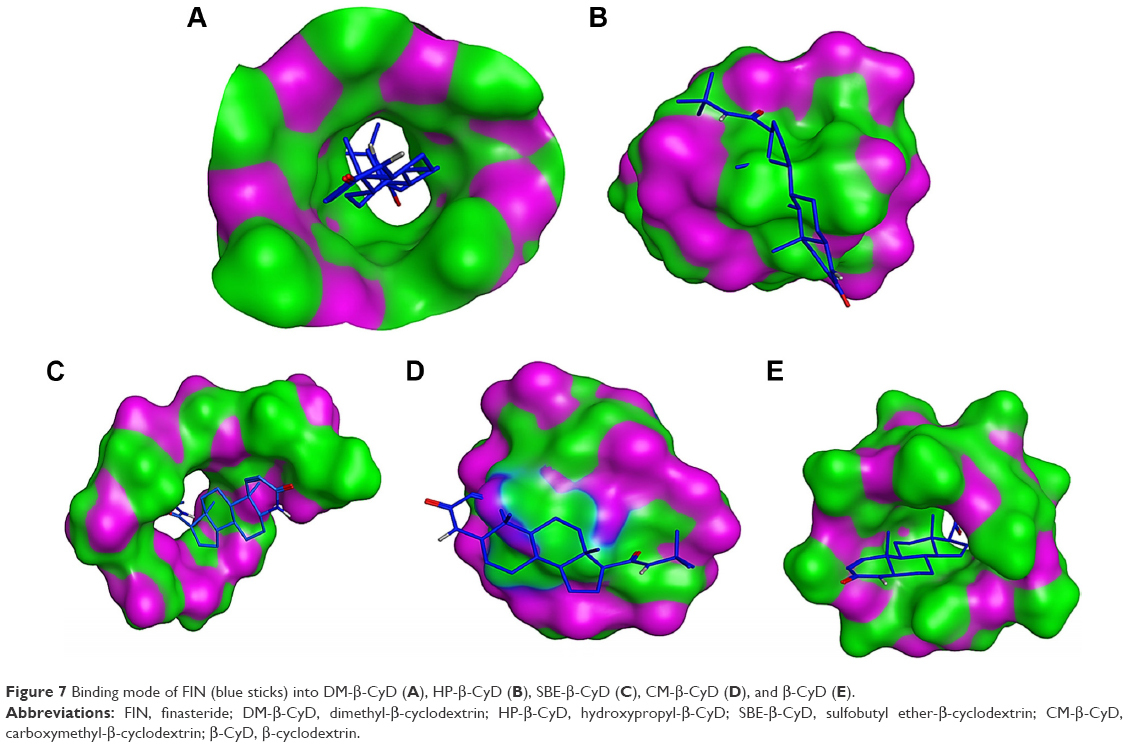 | Figure 7 Binding mode of FIN (blue sticks) into DM-β-CyD (A), HP-β-CyD (B), SBE-β-CyD (C), CM-β-CyD (D), and β-CyD (E). |
Dissolution rate studies
The hydrophobicity of the drug prohibited its contact with the dissolution milieu, causing it to float on the surface and thus disturbing its dissolution.
Figure 8 shows the magnitude of FIN that dissolved as a raw material in CyDs inclusion complexes. All the inclusion formulations displayed a more rapid dissolution rate and extent of FIN than the free drug, with a significant improvement in FIN dissolution from both HP-β-CyD and DM-β-CyD coprecipitated complexes. Moreover, the increase in the dissolution rate of FIN in the inclusion complexes prepared by the coprecipitation method could be imputed to the lack of crystallinity, improved wettability and dispersibility, and particle size reduction. Moreover, the hydrophilic influence of CyDs will enhance the solubility of the drug in the solid–liquid boundary and can decrease the interfacial tension between FIN and the dissolution medium, consequently producing a faster dissolution rate. The rank order of solubilization improvement was DM-β-CyD>HP-β-CyD>CM-β-CyD>SBE-β-CyD>β-CyD. Moreover, to evaluate the extent of enhancement in the rate of dissolution of FIN from its complexes, the similarity factor (f2) was calculated for all the tested CyDs in the coprecipitated complex taking the pure FIN as a reference. The calculated f2 values indicate a significant difference between all tested types of CyD coprecipitated systems and the pure drug with a different extent. The release profiles of FIN/DM-β-CyD and FIN/HP-β-CyD are significantly different from pure FIN (f2 values are 4 and 11, respectively), which explains that complexes with these two types of CyDs give better dissolution results than CM-β-CyD, SBE-β-CyD, and β-CyD (with f2 values of 34, 41, and 49.2, respectively). From the dissolution curves displayed in Figure 8 and considering the diversity of conformations that FIN can be assumed to form complexes with CyDs of varying stabilities, as suggested by phase solubility diagrams, IR, XRD, DSC, and docking simulations, it can be assumed that there are complex mechanisms involved in FIN solubilization.35
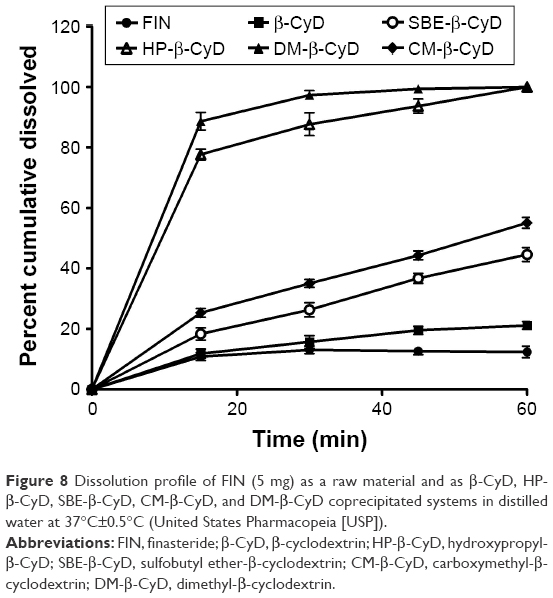 | Figure 8 Dissolution profile of FIN (5 mg) as a raw material and as β-CyD, HP-β-CyD, SBE-β-CyD, CM-β-CyD, and DM-β-CyD coprecipitated systems in distilled water at 37°C±0.5°C (United States Pharmacopeia [USP]). |
Pharmacokinetic study
Peak areas of varying amounts of FIN versus the corresponding FIN concentrations were used to obtain the calibration curve (data not shown). The limit of detection (LOD) and limit of quantification (LOQ) were 3.14 and 7.49 ng/mL, respectively.
It has been declared that improving the dissolution and absorption of poorly soluble drugs will enhance their pharmacokinetic parameters and consequently the drug bioavailability.36–38 Plasma levels following the administration of FIN (1 mg/kg) as a raw material or its equivalent of FIN-DM-β-CyD inclusion complex prepared by the coprecipitation method utilized in the pharmacokinetic study are shown in Figure 2. The pharmacokinetic study revealed that the FIN/DM-β-CyD inclusion complex displays higher Cmax (63.55 ng·mL−1) and slightly shorter Tmax (2.58 h) accompanied by a higher Ka (1.452 h−1) when it was compared with Cmax, Tmax, and Ka of raw FIN suspension (39.25 ng·mL−1, 3.5 h, and 0.757 h−1, respectively; Table 1). Moreover, the DM inclusion complex had a higher AUC (1,339.05 ng·h·mL−1) compared with that of the oral raw drug (781.52 ng·h·mL−1). The higher Ka and higher AUC revealed a better absorption rate and extent of the drug as a result of the enhancement of solubility made by inclusion complex formation. These results confirm that the FIN-DM-β-CyD inclusion complex leads to a significant improvement in the relative bioavailability of FIN compared with the raw drug.
Conclusion
The results of this study showed that the FIN/DM-β-CyD coprecipitated complex gave the highest aqueous solubility displayed by higher Kc and CE when compared with other β-CyD systems (HP-β-CyD, CM-β-CyD, SBE-β-CyD, and β-CyD). There was a great increase in the solubility and dissolution of FIN/DM-β-CyD confirmed by higher binding energy between FIN and DM-β-CyD in the molecular docking simulation, which led to an improvement in the rate and extent of FIN absorption and hence an increase in its oral bioavailability.
Acknowledgments
The authors express their heartfelt gratitude to Professor Dr Masami Otsuka, Graduate School of Pharmaceutical Sciences, Kumamoto University, Japan, and to Dr Mohammed Osman for their support in performing molecular modeling in this research.
Disclosure
The authors report no conflicts of interest in this work.
References
Parsons JK, Mougey J, Lambert L, et al. Lower urinary tract symptoms increase the risk of falls in older men. BJU Int. 2009;104(1):63–68. | ||
Stoner E. The clinical development of a 5-alpha-reductase inhibitor, finasteride. J Steroid Biochem Mol Biol. 1990;37:375–378. | ||
Snyder PJ. Androgens. In: Bruton LL, Lazo JS, Parker KL, editors. Goodman and Gilman’s the Pharmacological Basis of Therapeutics. 11th ed. New York, NY: McGraw-Hill; 2006:1573–1585. | ||
Rathnayake D, Sinclair R. Male androgenetic alopecia. Expert Opin Pharmacother. 2010;11(8):1295–1304. | ||
Parsons JK, Schenk JM, Arnold KB, et al. Finasteride reduces the risk of incident clinical benign prostatic hyperplasia. Eur Urol. 2012;62(2):234–241. | ||
The United States Pharmacopoeia/The National Formulary. 30th ed. Rockville: US Pharmacopeial Convention; 2007:2133–2135. | ||
European Pharmacopoeia. 4th ed. Paris: Council of Europe; 2002:1188–1189. | ||
Maynard RL. The Index Merck (Monograph Number: 4113). 13th ed. New York, NY: Merck; 2001:721–722. | ||
Loftsson T, Duchêne D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329(1–2):1–11. | ||
Mady FM, Abo-Taleb A, Khalid KA, et al. Evaluation of carboxymethyl-beta-cyclodextrin with acid function: improvement of chemical stability, oral bioavailability and bitter taste of famotidine. Int J Pharm. 2010;397(1–2):1–8. | ||
Jambhekar SS, Breen P. Cyclodextrins in pharmaceutical formulations II: solubilization, binding constant, and complexation efficiency. Drug Discov Today. 2016;21(2):363–368. | ||
Valle EMMD. Cyclodextrins and their uses: a review. Process Biochem. 2004;39:1033–1046. | ||
Asbahr ACC, Franco L, Barison A, Silva CWP, Ferraz HG, Rodrigues LNC. Binary and ternary inclusion complexes of finasteride in HP-β-CD and polymers: preparation and characterization. Bioorg Med Chem. 2009;17(7):2718–2723. | ||
Higuchi T, Connors KA. Advances in Analytical Chemistry and Instrumentation. Vol. IV. New York, NY: Wiley Interscience; 1965. | ||
Yokota T, Tonozuka T, Shimura Y, Ichikawa K, Kamitori S, Sakano Y. Structures of Thermoactinomyces vulgaris R-47 alpha-amylase II complexed with substrate analogues. Biosci Biotechnol Biochem. 2001;65(3):619–626. | ||
Shityakov S, Salmas RE, Durdagi S, et al. Characterization, in vivo evaluation, and molecular modeling of different propofol–cyclodextrin complexes to assess their drug delivery potential at the blood–brain barrier level. J Chem Inf Model. 2016;56(10):1914–1922. | ||
Case DA, Cheatham TE 3rd, Darden Ti S, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | ||
Radwan MO, Sonoda S, Ejima T, et al. Zinc-mediated binding of a low-molecular-weight stabilizer of the host anti-viral factor apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G. Bioorg Med Chem. 2016;24(18):4398–4405. | ||
Kane RN, Kuchekar BS. Preparation, physicochemical characterization, dissolution and formulation studies of telmisartan cyclodextrin inclusion complexes. Asian J Pharm. 2010;4(1):52–59. | ||
Mousavi SHH, Kobarfard F, Husain SW, et al. A rapid, simple, liquid chromatographic-electrospray ionization, ion trap mass spectrometry method for the determination of finasteride in human plasma and its application to pharmacokinetic study. Iran J Pharm Res. 2012;11(1):59–67. | ||
Brewster ME, Loftsson T. Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 2007;59(7):645–666. | ||
Carrier RL, Miller LA, Ahmed I. The utility of cyclodextrins for enhancing oral bioavailability. J Control Release. 2007;123(2):78–99. | ||
Rama ACR, Veiga F, Figueiredo IV, Sousa A, Caramona M. Aspectos biofarmacêuticos da formulação de medicamentos para neonatos. Fundamentos da complexação de indometacina com hidroxipropil-β-ciclodextrina para tratamento oral do fechamento do canal arterial [Biopharmaceutical aspects of formulation of medicines for neonates. Basics of Indomethacin complexation with hydroxypropyl-β-cyclodextrin for oral treatment of ductus arteriosus closure]. Braz J Pharm Sci. 2005;41:281. Portuguese. | ||
Loftsson T, Hreinsdóttir D, Másson M. Evaluation of cyclodextrin solubilization of drugs. Int J Pharm. 2005;302(1–2):18–28. | ||
Li N, Liu J, Zhao X, et al. Complex formation of ionic liquid surfactant and β-cyclodextrin. Colloids Surf A. 2007;292:196–201. | ||
Li W, Lu B, Sheng A, Yang F, Wang Z. Spectroscopic and theoretical study on inclusion complexation of beta-cyclodextrin with permethrin. J Mol Struct. 2010;981:194–203. | ||
Ribeiro L, Loftsson T, Ferreira D, Veiga F. Investigation and physicochemical characterization of vinpocetine-sulfobutyl ether beta-cyclodextrin binary and ternary complexes. Chem Pharm Bull. 2003;51(8):914. | ||
Zingone G, Rubessa F. Preformulation study of the inclusion complex warfarin-beta-cyclodextrin. Int J Pharm. 2004;291(1–2):3. | ||
Ahmadi P, Ghasemi JB. 3D-QSAR and docking studies of the stability constants of different guest molecules with beta-cyclodextrin. J Incl Phenom Macrocycl Chem. 2013;79:401–413. | ||
Aly UF. Enhancement of solubility and dissolution of atorvastatin calcium: in-vitro and in-vivo evaluation. Alex J Pharm Sci. 2011;25(2):83–90. | ||
De-Chasteign S, Fessi H, Davissaguet JP, Puisieusx F. Comparative study of the association of Itraconazole with colloidal drug carrier. Drug Dev Res. 1996;38(2):125–133. | ||
Astray G, Mejuto JC, Morales J, Rial-Otero R, Simal-Gándara J. Factors controlling flavors binding constants to cyclodextrins and their applications in foods. Food Res Int. 2010;43:1212–1218. | ||
Kfoury M, Auezova L, Fourmentin S, Greige-Gerges HJ. Investigation of monoterpenes complexation with hydroxypropyl-β-cyclodextrin. J Incl Phenom Macrocycl Chem. 2014;80:51–60. | ||
Mathiron D, Marçon F, Dubaele JM, Cailleu D, Pilard S, Djedaïni-Pilard F. Benefits of methylated cyclodextrins in the development of midazolam pharmaceutical formulations. J Pharm Sci. 2013;102(7):2102–2111. | ||
Corrigan OI, Stanley CT. Mechanism of drug dissolution rate enhancement from beta-cyclodextrin-drug systems. J Pharm Pharmacol. 1982;34(10):621–626. | ||
Ahmed TA, El-Say KM, Mahmoud MF, Samy AM, Badawi AA. Miconazole nitrate oral disintegrating tablets: in vivo performance and stability study. AAPS PharmSciTech. 2012;13(3):760–771. | ||
Nair AB, Attimarad M, Al-Dhubiab BE, Wadhwa J, Harsha S, Ahmed M. Enhanced oral bioavailability of acyclovir by inclusion complex using hydroxypropyl-β-cyclodextrin. Drug Deliv. 2014;21(7):540–547. | ||
Balakrishnan P, Song CK, Cho H-J, et al. Inclusion complex effect on the bioavailability of clotrimazole from poloxamer-based solid suppository. Arch Pharm Res. 2012;35(7):1169–1175. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.