Back to Journals » Journal of Experimental Pharmacology » Volume 13
Experimental Disease-Modifying Agents for Frontotemporal Lobar Degeneration
Authors Giunta M, Solje E, Gardoni F, Borroni B, Benussi A
Received 22 January 2021
Accepted for publication 9 March 2021
Published 24 March 2021 Volume 2021:13 Pages 359—376
DOI https://doi.org/10.2147/JEP.S262352
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Marcello Giunta,1 Eino Solje,2 Fabrizio Gardoni,3 Barbara Borroni,1 Alberto Benussi1
1Neurology Unit, Department of Clinical and Experimental Sciences, University of Brescia, Brescia, Italy; 2Institute of Clinical Medicine - Neurology, University of Eastern Finland, Kuopio, Finland; 3Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
Correspondence: Alberto Benussi
Clinica Neurologica, Università degli Studi di Brescia, P.le Spedali Civili, 1, Brescia 25100, Italy
Tel +39 0303995632
Email [email protected]
Abstract: Frontotemporal dementia is a clinically, genetically and pathologically heterogeneous neurodegenerative disorder, enclosing a wide range of different pathological entities, associated with the accumulation of proteins such as tau and TPD-43. Characterized by a high hereditability, mutations in three main genes, MAPT, GRN and C9orf72, can drive the neurodegenerative process. The connection between different genes and proteinopathies through specific mechanisms has shed light on the pathophysiology of the disease, leading to the identification of potential pharmacological targets. New experimental strategies are emerging, in both preclinical and clinical settings, which focus on small molecules rather than gene therapy. In this review, we provide an insight into the aberrant mechanisms leading to FTLD-related proteinopathies and discuss recent therapies with the potential to ameliorate neurodegeneration and disease progression.
Keywords: frontotemporal dementia, frontotemporal lobar degeneration, therapy, TDP-43, tau, C9orf72, GRN, MAPT
Introduction
Frontotemporal Dementia (FTD), the second most common cause of early-onset dementia,1 is a neurodegenerative disorder enclosing a wide range of different neuropathological entities and clinical presentations, sharing a main impact on behavioral, linguistic and executive functions, due to the progressive atrophy of frontal and temporal lobes. According to the latest criteria, three core clinical variants have been recognized, namely the behavioral variant of FTD (bvFTD),2 the agrammatic/non-fluent variant of primary progressive aphasia (avPPA/nfvPPA) and the semantic variant of PPA (svPPA).3 The occurrence of associated motor symptoms, as in progressive supranuclear palsy (PSP), corticobasal syndrome (CBS) and motor neuron disease (FTD-MND), enriches the spectrum of FTD-related disorders.4
Common to all FTD clinical syndromes is the underlying frontotemporal lobar degeneration (FTLD) which can be classified according to the predominant constituent proteins of cellular inclusions. While until 2006 only FTLD-Tau was well characterized, the following discovery of TAR DNA binding protein 43 (TDP-43) and FET family proteins within tau-negative, ubiquitin-positive inclusions of a vast majority of FTLD cases, led to the introduction, respectively, of FTLD-TDP and FTLD-FET neuropathology.5,6
Along with heterogeneity in both clinical presentations and pathological hallmarks, an increasing literature depicts the complex figure of the genetic determinants of FTD, with up to 20–30% of the cases with mutations in progranulin (GRN), microtubule-associated protein tau (MAPT) and chromosome 9 open reading frame 72 (C9orf72) genes.7 This heterogeneity,8 as well the lack of a clear-cut relationship between clinical phenotypes,9 genetic traits and neuropathological features, represent the main obstacle hampering the development of a unifying pathogenetic model and, as consequence, of disease-modifying strategies of intervention.
In this paper, we review the most appealing candidates for pharmacological and non-pharmacological interventions and discuss the current achievements and challenges of novel therapeutic approaches in FTD.
FTLD: Crosstalk Between Genetics, Pathology and Clinical Features
FTLD is affected by a high heterogeneity, limiting the establishment of a one-to-one relationship between genetics, neuropathological and clinical correlates.10,11
FTLD-tau, defined by tau-immunopositive inclusions, accounts for about 40% of all FTLD cases and 20% of those with high heritability, related to mutations in the MAPT gene, with more than 60 pathogenic variations reported.7,12 According to the predominant species in the inclusions, which differ from each other by the number of 31–32 aminoacidic repeats in the microtubule-binding domain (3-repeat or 4-repeat tau),13 the actual molecular classification of FTLD-tau accomplishes four main histological subtypes, including Pick’s disease (PiD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and globular glial tauopathy (GGT). Among FTD syndromes, there is a strong association with FLTD-tau and clinical syndromes of bvFTD and nfvPPA, but especially common in CBS and PSP, while only a few cases of FTD-MND have been reported.14,15
Before the discovery of TDP-43 protein in a large group of ubiquitin-positive, tau-negative FTLD samples, in 2006,16 FTLD-tau was considered the main FTLD neuropathological type. To date, FTLD-TDP pathology has been recognized as the most common neurobiology, underlying about 50% of all FTLD cases including both sporadic and familial forms.6 Despite the common molecular substrate, FTLD-TDP covers a wide range of anatomopathological changes. As a consequence, and in consideration of the important clinicopathological and genetic associations, the reviewed criteria for FTLD distinguish four subtypes, with respect to morphology, abundance/presence of distinct inclusion types and their distribution across cortical laminar layers.17–20 Indeed, unlike FTLD-tau, genetic etiology of TDP-43 proteinopathies is complex, involving mutations in four main genes, demonstrating an autosomal dominant heritability.21
Mutations in the GRN gene, whose list is enriched with more than 110 pathogenic variants,7,12 account for 5–20% of familiar and 1–5% of sporadic FTD patients.22,23 In spite of the highly phenotypical heterogeneity, most GRN mutation carriers receive a clinical diagnosis of FTD, with bvFTD as a more frequent presentation than the language variant, that is nevertheless more prevalent than in sporadic forms and consistent with nfvPPA, as well as CBS.24–26
The hexanucleotide repeat expansion of GGGGCC in a non-coding region of the C9orf72 gene is the most common genetic cause of both sporadic and familial cases of FTD, MND and FTD-MND.27,28 The expansion carrier status influences the susceptibility of specific neuronal populations, producing a more severe loss of motor neurons.29 Clinical presentation matches with the distribution of TDP-43 pathology, explaining a more widespread involvement of frontal and temporal neocortices in cases with FTD or FTD-MND rather than MND only.30,31
Accounting for up to 4% of familiar FTD,32 mutations in the valosin-containing protein (VCP) gene, have been associated with the development of the autosomal dominant inherited inclusion body myopathy associated with Paget’s disease of bone and FTD (IBMPFD). Pathologically classified as FTLD-TDP type D,33 the clinical presentation is dominated by myopathy, found in about 90% of the patients,34 and FTD or ALS phenotype was observed in approximately 30% of the patients carrying VCP mutations.35
Finally, mutations in the gene encoding TANK-binding kinase 1 protein (TBK1) have been reported as probably the fourth most common genetic cause of FTD overall.36 Few pathological cases have been described that, although confirming a TDP-43 pathology recognized both a subtype A and B pattern.37,38 ALS is the predominant clinical syndrome with the majority of cases having either ALS or FTD-ALS syndrome, with a cognitive profile covering bvFTD and PPA (both nfvPPA and svPPA).39
Finally, FLDT-FET accounts for about 5–10% of the cases, and this histopathological subgroup was defined since the identification of the protein fused/translocated in sarcoma (FUS) within neural inclusions described in some FTLD patients.40 However, the possible coaggregation of proteins belonging to the FET family, including TATA-box binding protein-associated factor 15 (TAF15), Ewing’s sarcoma (EWS) and transportin 1, was reported only in FTLD-FET cases, not associated with FUS mutations.41 Indeed, while FUS-related ALS is mostly caused by FUS mutations, FTLD-FET tends to be sporadic.42 In turn, within FTLD-FET pathology, three subtypes are distinguished, named atypical FTLD-U, neuronal intermediate filament inclusion disease (NIFID) and basophilic inclusion body disease (BIBD). The corresponding clinical spectrum, for each subtype, ranges from early-onset bvFTD for atypical FTLD-U to early-onset FTD with MND and/or extrapyramidal motor symptoms for NIFID; it is more heterogeneous for BIBD, covering FTD/ALS, parkinsonism, chorea, dysarthria and gaze palsy.6
Tau Targeting Strategies
Tau protein, encoded by MAPT gene on chromosome 17, is a microtubule-associated protein, mainly enriched in the axonal compartment of neurons and primarily involved in the regulation of cytoskeletal turnover, by promoting microtubule polymerization and stabilization.43 The protein exists as six major alternative splicing variants, with the inclusion of exon 10 determining whether the protein contains either three or four microtubule-binding repeat regions. Variants are simply distinguished between three-repeat (3R) and four-repeat (4R) isoforms: their relative amount varies in the course of human brain development and is related to the pathological profile of the different tauopathies, with 3R in Pick’s disease, 4R in PSP and FTD while mixed forms in AD and chronic traumatic encephalopathy.44 Once synthesized, tau undergoes many post-translational modifications that, besides phosphorylation, include acetylation, glycation, nitration, O-GlcNAcylation, oxidation, polyamination, SUMOylation, and ubiquitination.45
With regard to the milestones of tau pathophysiology, the actual therapeutical approaches under investigation are articulated around five main strategies, including 1) modulation of MAPT expression, 2) post-translational modification, 3) modulation of protein aggregation and clearance, 4) immune neutralization (active and passive immunization), and 5) microtubule stabilization (see Table 1 and Figure 1).
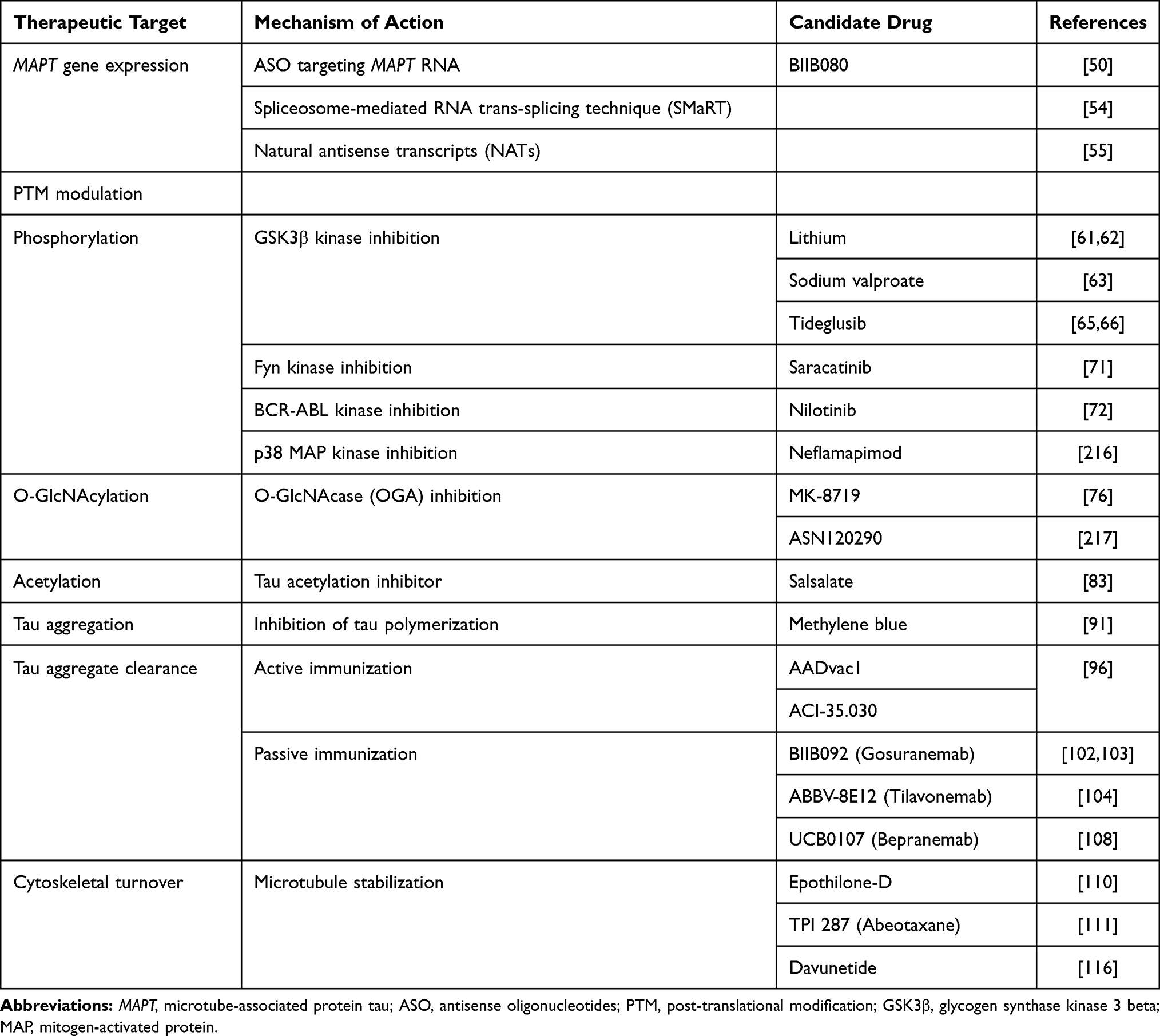 |
Table 1 Principal Mechanism of Action and Possible Candidate Drugs for the Treatment of FTLD-Tau |
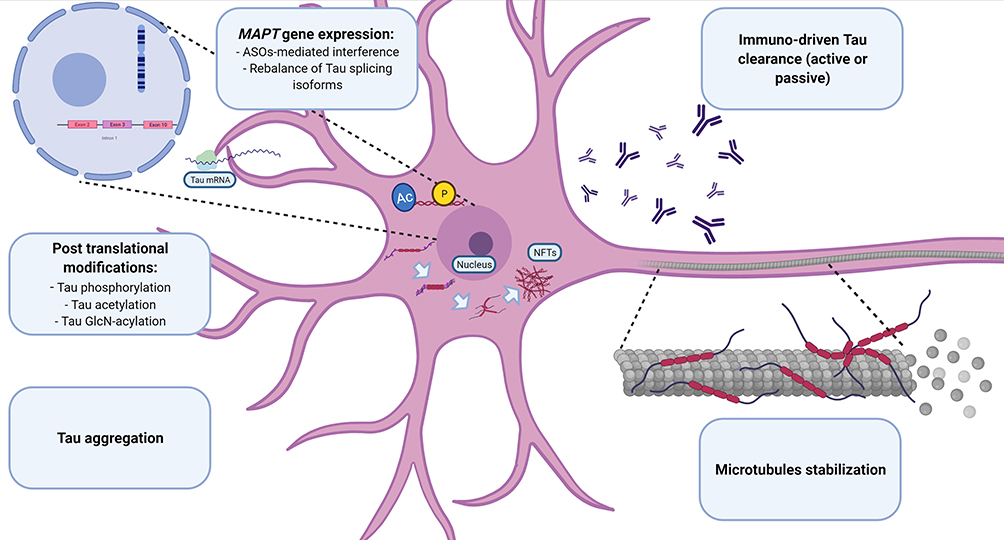 |
Figure 1 Principal pathogenic steps toward tau-related neurodegeneration. Pathophysiological transformations leading to tau dysfunction. The related potential therapeutic strategies are reported in the boxes. Images created with BioRender.com Abbreviations: ASOs, antisense oligonucleotides; AC, acetylation; MAPT, microtubule-associated protein tau; NFTs, neurofibrillary tangles; P, phosphorylation. |
Modulation of MAPT Expression
MAPT mutations, mainly concentrated in exons 9–12 and introns flanking exon 10,12,46–48 affect the ability of tau to interact with microtubules and alter the normal ratio among isoforms, enhancing its propensity to aggregate.49
Reduction of Tau in pathogenic mouse models has been shown to ameliorate seizure phenotypes and prevent neurodegeneration,50 identifying antisense oligonucleotides (ASOs) as potential therapeutics against Tau. Indeed, encouraged by the results achieved in multiple neurodegenerative diseases, such as spinal muscular atrophy and Duchenne muscular dystrophy,51 the use of small, single-stranded sequences of DNA in transgenic tauopathy mice have been demonstrated to significantly restore the balance between tau species and to revert the associated neuropathological and clinical phenotypes.52,53 Based on the preclinical data of the study by Devos et al, the drug BIIB080 is actually being administered in MCI due to AD patients in a Phase 1/2 randomized clinical trial (RCT) (NCT03186989). Another trial with ASOs (NIO752) is currently planned in PSP (NCT04539041).
Moreover, still aimed at modulating MAPT expression, novel experimental approaches have been proposed, relying on RNA‐guided mechanisms. Based on RNA reprogramming, spliceosome-mediated RNA trans-splicing technique (SMaRT) has provided an alternative method to correct the impaired alternative splicing caused by MAPT pathogenic mutations.54 Moreover, the use of natural antisense transcripts (NATs) is recently emerging as a potential, physiological tool to suppress tau protein levels.55,56
Post-Translational Modifications
Phosphorylation
Since the hyperphosphorylated tau state makes the protein susceptible to aggregation with the loss of cytoskeletal microtubule-stabilizing properties, leading to neural toxicity,57 research has strongly focused on compounds able to prevent this modification. Tau protein carries 85 mapped phosphorylation sites,58 which are tightly regulated by a plethora of protein kinases and phosphatases, which represent potential therapeutic targets.
Among this class of proteins, glycogen synthase kinase 3 beta (GSK3β) exerts a pleiotropic effect on neural homeostasis besides the direct action on tau, including axonal transport and synaptic function, adult neurogenesis, cell survival and neuro-inflammation.59 Moreover, neuropathological evidence has validated the pathogenetic role of GSK3β in tauopathies,60 fostering the discovery of several chemical classes of GSK3β inhibitors, some of them reaching human application. However, despite the results of preclinical studies showing the effect of lithium, a non-competitive, non-specific GSK3β inhibitor, on the state of tau phosphorylation and aggregation,61 a Phase 2 RCT demonstrated no improvement of clinical or biological outcomes in AD patients.62 Whilst no data are available, due to intolerability, in a phase 1/2 RCT in patients with PSP and CBD (NCT00703677), another RCT (NCT02862210) is currently ongoing to assess the efficacy of lithium for the treatment of neuropsychiatric symptoms in bvFTD. Another non-specific inhibitor of GSK3β, sodium valproate, demonstrated poor tolerability, with a possible clinical worsening when administered to PSP patients (NCT00385710),63 in conflict with preclinical findings.64 In order to ameliorate their safety profile, new specific GSK3β inhibitors have been discovered. Among these, tideglusib, a non-ATP-competitive GSK3β inhibitor, though failing in achieving primary clinical outcomes, has shown to reduce brain atrophy progression in PSP patients in the TAUROS study (NCT01049399).65,66
Besides GSK3β, a number of kinases have proved to be catalytically active on tau and to be associated with pathological epitopes of the phosphorylated protein, including CDK5, TAOKs, TTBK.67–69 In this view, expanding research in oncology, focused on inhibitors of the human kinome, has identified novel therapies that can be repurposed for the treatment of neurodegenerative disorders,70 making a wide spectrum of target kinase inhibitors available. Recently, three small molecules, saracatinib (NCT02167256), nilotinib (NCT02947893) and neflamapimod (NCT03402659), designed to inhibit the tau kinases Fyn, BCR-ABL and p38α, respectively, were assessed in AD patients in phase 2 trials. With the exception of the former, limited by a low availability of CSF samples, nilotinib and neflamapimod provided at least a biological proof of efficacy, with an improvement of neurodegeneration markers.71,72
Tolfenamic acid, which has been shown to lower tau mRNA and protein, as well as the levels of its phosphorylated form and CDK5,73,74 is currently being planned for testing in PSP patients (NCT04253132).
Others Post-Translational Modifications
In addition to the intervention of specific kinases, the state of phosphorylation of tau is also regulated by the eventual addition of O-linked N-acetylglucosamine (O-GlcNAc) moieties. Regulated by two enzymes, O-GlcNAc transferase (OGT) inserts O-GlcNAc, while O-GlcNAcase (OGA) removes the moieties on serine and threonine residues, thus creating a competition with the phosphorylation process.75 In line with the aim of upregulating the O-GlcNAcylation, two small OGA inhibitors, MK-8719 and ASN120290, shown to reduce tau-related degeneration hallmarks,76,77 have been announced in clinical trials for PSP patients.78
Acetylation, another post-translational modification (PTM), is also able to interfere with tau pathogenetic potential, acting on over than 30 lysine residues.79 The high acetylation rate found in post-mortem neuropathological samples of tauopathy patients80 could promote neurodegeneration by different mechanisms, such as fibrillar aggregation, microtubule detachment and transneuronal spreading.81 In this regard, salsalate, an anti-inflammatory salicylate, protects against neurodegeneration in FTLD-tau mice by the inhibition of p300, an acetyltransferase that targets tau.82 An open-label phase 1 study in PSP patients has recently been concluded, reporting non-significant effects on disease progression, that could be due to the low rate of CNS penetrance of salsalate.83
Modulation of Tau Aggregation and Clearance
As described above, PTMs can drive tau aggregation by oligomerization of non‐bound hyperphosphorylated tau into pre-tangles, followed by the subsequent formation of filaments such as paired helical filaments or straight filaments, until the assembly of neurofibrillary tangles (NFTs). In line with the development of a disease-modifying strategy, a direct approach to tau aggregation might be more successful, considering the difficulties in avoiding the disruption of kinases’ physiological signaling pathways, together with the heterogeneity of phosphorylation sites on tau, with some promoting and others inhibiting tau aggregation.84
Methylene blue (MB) has gained attention for its anti-aggregating properties, achieved by modification of cysteine residues critical for aggregation,85 although recent studies have broadened its pharmacodynamic spectrum to proteosome function,86 autophagy and oxidative stress.87,88 Encouraged by the results of a phase 2 trial showing an improvement in mild to moderate AD,89 the same results were not replicated in two subsequent Phase 3 trials in AD and bvFTD patients.90,91 The failure of these trials, as recently reported,92 could be due to the ability of MB to hinder the synthesis of fibrillar species only, increasing instead neurotoxic tau granule formation.
Immune Neutralization
Parallel to strategies aimed to prevent tau aggregate formation and deposition, the enhancement of clearance systems represents a promising field of research. In this regard, anti-tau immunotherapy approaches turned out as feasible options for clearing toxic species and specific protein epitopes in tauopathies: they can be distinguished in active and passive strategies.93
Active Immunization
Given the devastating in vivo results of a first immunization approach against full-length recombinant human tau, the ongoing vaccine development has focused on tau fragments and phosphorylated tau peptides.94 Indeed, the first vaccine reaching human experimentation (AADvac1), which consisted of an antigenic peptide hypothesized to trigger misfolding and aggregation,95 reduced tau-related pathological alterations with a corresponding behavioral improvement in transgenic rats.96 Still confirming its neurobiological efficacy on neurodegeneration markers in the following ADAMANT phase 2 trial (NCT02579252), AADvac1 exhibited only a trend to slow the decline in mild AD patients.97–100 The same compound is actually being administered to nfvPPA patients in a phase 1 trial (NCT03174886).
Following this pivotal vaccine, AC Immune developed ACI-35, an alternative liposome-delivered agent, different from the former, instead directed against tau fragments enriched with phosphorylates residues. ACI-35.030, a second-generation compound redesigned to increase the immune response, is being tested in a phase 1/2 clinical trial in AD patients (NCT04445831), now in recruitment stage.
Passive Immunization
The consolidated experience in the management of monoclonal antibody (Ab)-based therapies, along with the expanding libraries of targetable altered forms of tau protein has made passive immune clearance one of the main areas of intervention in disease-modifying drug research. Although Ab can recognize either the N-terminal, the proline-rich or C-terminal regions, published results of clinical trials came only from Ab against the N-terminal region.101 Indeed, BIIB092 (gosuranemab), a humanized monoclonal antibody engaging the extracellular N-terminal tau sequence, reduced CSF-free N-terminal tau in PSP patients in a phase 1 trial.102 Unfortunately, two following phase 2 trials, PASSPORT (NCT03068468) and TauBasket (NCT03658135), aimed at testing BIIB092 in PSP and in in four primary tauopathies (CBS, FTLD-tau, nfvPPA and traumatic encephalopathy syndrome), respectively, were prematurely interrupted due to lack of efficacy in the interim analysis.103
Similar characteristics were observed for C2N-8E12 (ABBV-8E12, tilavonemab), an IgG4 antibody recognizing an epitope mapped on an extracellular N-terminal region,104 which was proven safe in a phase 1 trial (NCT03413319), but was then discontinued for PSP after interim results of a phase 2 study (NCT02985879) showing no beneficial therapeutic effects.105 The failure of these trials, along with the evidence of a lower extracellular/intracellular tau ratio in non-AD tauopathies, suggests that targeting extracellular N-terminal fragments alone could be futile in this population.106
Moreover, based on spectroscopy studies showing the prevalence of terminals-lacking tau fragments in CSF,107,108 an extracellularly acting Ab directed against a mid-domain region could achieve better results. In this direction, UCB0107 (bepranemab), a monoclonal Ab binding to the mid-region of tau, interferes with transneuronal propagation and tau seeding activity in vivo and in vitro108,109 A phase 1 trial with an open-label extension (NCT04185415, NCT04658199) to evaluate the safety profile on PSP patients is now ongoing and completion is expected in November 2021.
Microtubule Stabilization
In addition to the neurotoxic gain-of-function, also tau loss-of-function may lead to neurodegeneration, primarily through the detachment from microtubule and consequent microtubule disassembly.84 In order to overcome this process, three microtubule-stabilizing agents have been developed.
Epothilone-D (EpoD), a taxol-derived small molecule, reduces axonal dystrophy, increased microtubule density and, more globally, ameliorated tau-pathology in PS19 tau transgenic mice.110 However, although translated to a human phase 1/2 clinical trial (NCT01492374) for AD patients completed in October 2013, results were not published.
More recently, TPI 287 (abeotaxane), another taxol-derived compound with a similar mechanism of action to EpoD, was tested in two phase 1 RCTs in PSP, CBS and AD patients. While being better tolerated in 4R-tauopathies, than in mixed 3R/4R pathology of AD, a motor and a dose-related worsening in exploratory cognitive outcomes was reported.111 Finally, davunetide (NAP), a derivative from the endogenous activity-dependent neuroprotective protein (ADNP), whose genetic deficiency is associated with tau pathology,112 was found to improve cognitive function in tau transgenic mice by enhancing tau–microtubule interaction.113,114 Firstly approved for a phase 2 trial (NCT00422981) in MCI patients, it produced an improvement in memory and attention-based tasks.115 Unfortunately, a following phase 2/3 RCT (NCT01110720) on PSP patients did not confirm these results,116 possibly explained by a recent in vitro study demonstrating the preferential interaction of davunetide with 3R-tau in comparison with 4R-tau.117
TDP-43 Targeting Strategies
Together with FUS, TDP-43 is a ubiquitous protein belonging to the class of nuclear ribonucleoproteins (hnRNPs), with whom it shares two N-terminal located RNA recognition motifs (RRM1 and RRM2),118 that enable the binding and the regulation of several RNA processing pathways. The C-terminal sequence is a determinant of solubility state, cellular localization and interprotein interactions,119 supported by the clustering of many pathogenic FTD and ALS mutations in the corresponding region of the TARDBP gene.120,121 Predominantly located within the nucleus, when TDP-43 is shuttled to the cytoplasm the protein carries out various biological functions, including RNA translation, synaptic plasticity, autophagy and mitochondrial homeostasis.122 TDP-43 can drive the formation of stress granules in response to environmental stressors like other hnRNPs undergoing a liquid–liquid phase separation.123 Both the presence of mutations and aberrant post-translational modifications lead to the clearance of nuclear TDP-43, its mislocalization and, finally, to the classical TDP-43 cytoplasmic inclusions.124 In a similar way to tau, it relies on a simultaneous loss- and gain-of-function, secondary to the sequestration of TDP-43 and to the toxicity of the aggregates themselves.125 Paradoxically, if in vitro models show that a comparable degree of neural degeneration can be achieved in both inclusion-bearing and non-bearing neurons,126 it is also possible that aggregates might be protective at the early stages of the disease.127 However, two main factors have hindered the development of direct TDP-43-targeted strategies: 1) the ubiquitous expression together with the impairment of different cell type-specific pathways discourages the target of the protein in a generalized manner; and 2) the complexity of its biological properties, mostly unexplored, requires the implementation of a function-based intervention.128 As a direct consequence, monogenic forms of FTLD-TDP have received more attention for research purposes (see Table 2 and Figure 2).
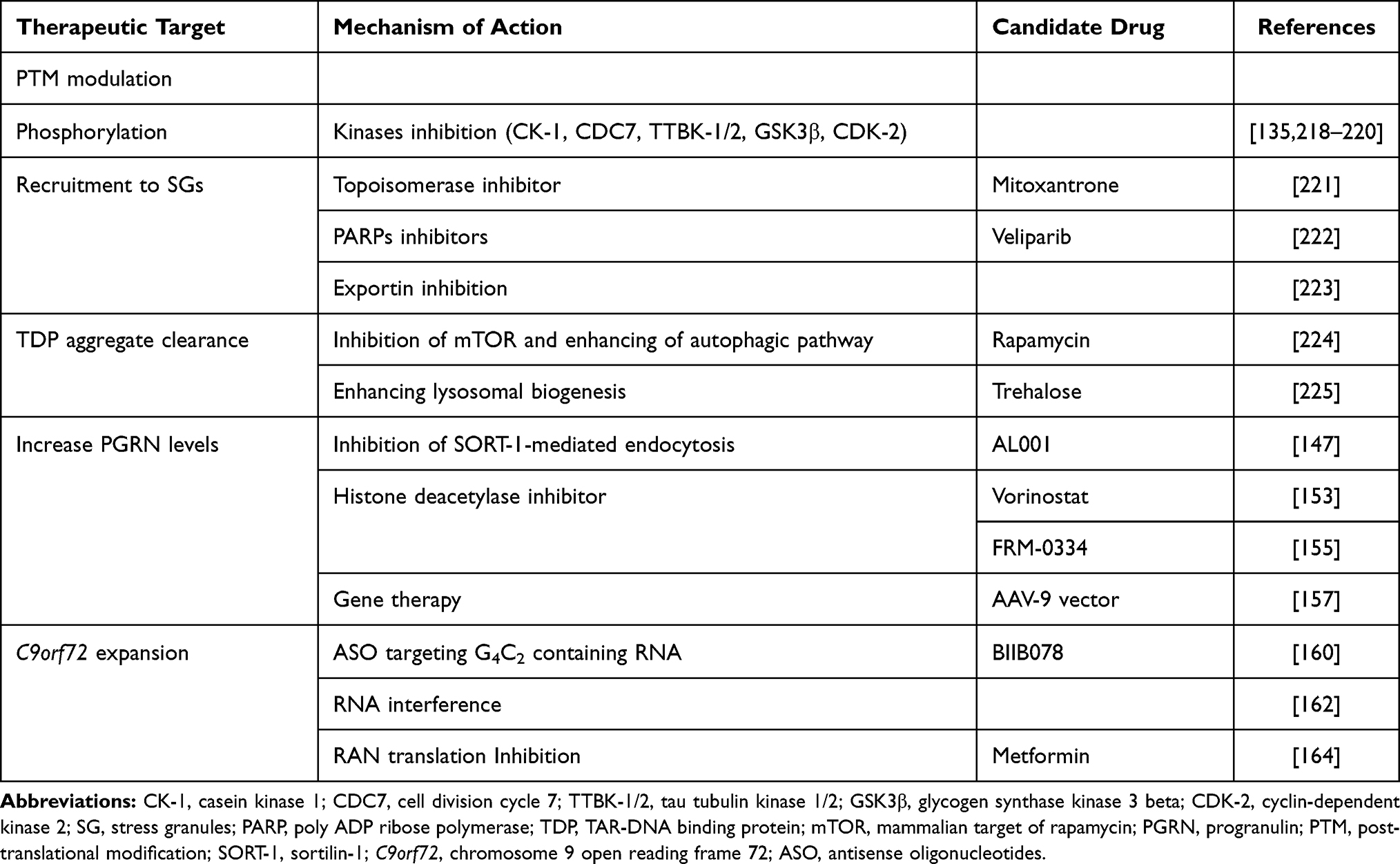 |
Table 2 Principal Mechanism of Action and Possible Candidate Drugs for the Treatment of FTLD-TDP |
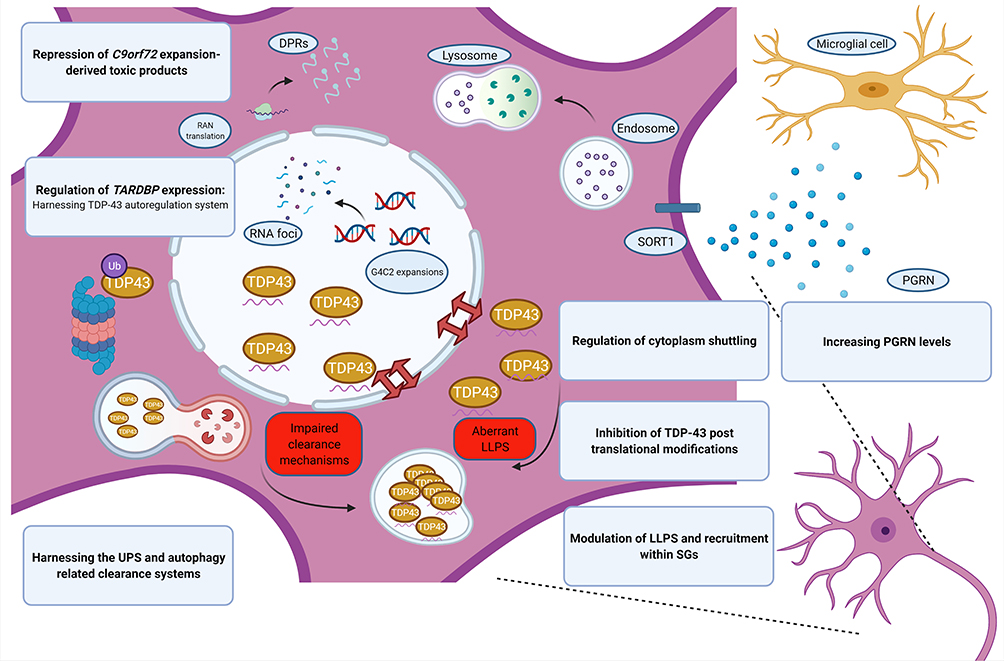 |
Figure 2 Aberrant TDP-43 biology in FTLD. Pathological model of TDP-43-associated neurodegeneration and the corresponding potential therapeutic approach (in blue boxes). Note the pathways involving C9Orf72 expansions and progranulin. Images created with BioRender.com. Abbreviations: C9orf72, chromosome 9 open reading frame 72; DPRs, dipeptide repeat proteins; LLPS, liquid–liquid phase separation; PGRN, progranulin; RAN translation, repeat-associated non-AUG translation. |
Targeting TDP-43 Pathophysiology
Still debating whether FTLD pathological mechanisms act primarily through gain- or loss-of-function effects, in vivo models show that both upregulation and downregulation of protein levels result in the triggering of a neurodegenerative cascade.129 Not surprisingly, TDP-43 levels are under tight control, including self-regulation based on a nonsense-mediated mRNA decay (NMD) mechanism.130 Given the role of its imbalance and the importance in maintaining protein levels, harnessing the NMD system might be a conserved, promising mechanism for TARDP-43 regulation, as recently reported.131
A hallmark of TDP-43 proteinopathies is the presence of aggregates predominantly composed of a protein extensively modified by post-translational mechanisms, not observed in healthy neurons and including ubiquitination, acetylation, SUMOylation, and phosphorylation and cleaved to generate C-terminal fragments (CTFs).124 As the most consistent feature of aggregates, also linked to disease-associated mutations,132 TDP phosphorylation represents a major area of research in FTLD therapy. Whilst this post-translational modification can occur in over 50 potential sites, some of them (especially at the C-terminus, with regard to serine residues at position 409/410)133 are believed to contribute to the aberrant behavior of TDP-43. The first TDP-directed kinase family described,134 casein kinase 1 (CK-1), has been recently identified as a potential therapeutic target to be an interesting molecular target. Its inhibition was shown to reduce TDP-43 phosphorylation and to restore its cellular nuclear localization in human-based cell models from FTLD-TDP patients.135
The impairment of cellular clearance mechanisms represents a core pathogenetic element in FTLD, as supported by the possible occurrence of mutations in genes encoding critical proteins in the ubiquitin–proteasome system (UPS) and autophagy–lysosome pathway (ALP) mediated degradation, such as VCP, CHMP2B, TBK1, OPTN, p62/SQSTM1 and UBQLN2.125 Both these processes regulate the clearance of TDP-43, although soluble and aggregated TDP are degraded primarily by UPS and ALP, respectively.136 Among modules within the autophagy system, the dysfunction of the endosomal sorting complexes required for transport (ESCRT) machinery results in TDP-43 accumulation and mutations of CHMP2B, a key component of this system, have been identified as causative of familiar FTD.137,138 While the disruption of CHMP2B perturbs endo-lysosomal trafficking, vesicle fusion and autophagic degradation, it also promotes TDP-43 hyperphosphorylation and insolubility by the control of the UPS-dependent turnover of CK-1.139,140 Given that therapeutics have been identified for CHMP2B this could represent a promising therapeutic axis.139
On the contrary, TDP-43 itself plays an active role in the regulation of these mechanisms, modulating the expression of the phagosome machinery.141 Indeed, TDP-43 depletion disrupts the fine control on the mTOR-complex overwhelming the last stages of autophagic discharge and the accumulation of immature autophagic vesicles, ultimately inducing neurotoxicity as pointed out in animal models.142
Targeting Progranulin Protein Levels
Progranulin is synthesized in the CNS by different cells, including neurons, astrocytes, microglia, endothelial cells.143 It is a secreted protein and biologically active on its own or as a cleavage product (granulins), possibly having opposite effects, with anti-inflammatory properties for the former and pro-inflammatory for the latter.144,145 Therefore, as well as acting as microglial modulator, progranulin is also involved in neuronal survival, by trophic and neuroprotective functions, synaptogenesis and lysosomal dynamics.146
Besides the already known high prevalence of GRN mutations in the FTD population, since haploinsufficiency of PGRN is the predominant mechanism leading to FTD, the rescue of its deficiency is an attractive objective for drug development. Both in vitro and in vivo models, targeting sortilin-1 (SORT-1) have identified SORT-1 inhibition as a positive regulator of progranulin protein levels. SORT-1, a Vps10 family member, is a PGRN binding protein involved in the extracellular uptake and delivery to the endolysosomal compartment.
Targeting cellular pathways addressing progranulin to lysosomal degradation by inhibition of SORT-1 has demonstrated, in vitro and in vivo models, to influence progranulin brain and serum plasma levels.147,148 According to the results in preclinical settings, healthy subjects and GRN mutation carriers were enrolled in a phase 1 trial (NCT03636204) testing anti-human SORT-1 monoclonal IgG1 antibodies (AL001). The study showed a dose-dependent effect of AL001 in increasing progranulin levels to normal ranges.149 While the study of AL001 in FTD patients carrying GRN mutation is ongoing in a phase 2 trial (INFRONT-2, NCT03987295), in July 2020 a phase 3 trial (INFRONT-3, NCT04374136) started enrolling both at risk and symptomatic FTD patients.
A different protein involved in lysosome trafficking, prosaposin, has been identified as a regulator of progranulin levels.150 AZP2006, a compound thought to prevent tau-phosphorylation and to stabilize prosaposin-progranulin complexes, was approved for a phase 2 trial (NCT04008355) in PSP patients, which is currently ongoing.151
Also aimed at enhancing progranulin levels, the epigenetically active class of histone deacetylase inhibitors has emerged as a possible disease-modifying agent in FTD.152 The first of this class identified as a possible modulator of progranulin levels, suberanilohydroxamic acid (vorinostat), was shown to normalize both the mRNA and protein levels in cellular models of GRN haploinsufficiency.153,154
Still belonging to the same pharmacological class, but characterized by a better distribution within the CNS,155 FRM-0334 was assessed in a phase 2 trial (NCT02149160) in 28 individuals with GRN mutations. The treatment did not produce significant changes in progranulin concentrations, although this failure might be explained by an insufficient FRM‐0334 exposure.156
Finally, genetic cases of FTD related to GRN haploinsufficiency represent an ideal field of application for vector-based gene therapies. Recently, the results of an intraventricular adeno-associated viral (AAV) vector administration in nonhuman primates paved the way for the development of AAV-based gene therapy in genetic FTD.157 A phase 1/2, multicenter study (PROCLAIM, NCT04408625) is currently ongoing, evaluating the safety and efficacy of intra-cisternal PR006 administration, an AAV-9 vector designed to deliver the GRN gene.158
Targeting the C9orf72 Expansion
The exact way by which the pathological length C9orf72 repeat expansions drives neurodegeneration is still debated, but three pathomechanisms are being suggested, including loss of physiological functions and gain of toxicity by RNA foci and DPRs, both derived by repeat-containing RNAs.159
Directly affecting the production of ribonucleoprotein toxic species, the first in vivo study employing ASOs directed against repeat-enriched RNAs, showed their ability to mitigate RNA foci and DPR burden, with a corresponding improvement of phenotypical signatures.160 From these encouraging results, a phase 1 trial (NCT03626012, NCT04288856) started enrolling C9orf72 repeat expansion carrying ALS patients, to assess BIIB078, an ASO specifically designed to reduce only the repeat-containing C9orf72 transcripts. Other efforts, focused on contrasting repetitions-containing RNAs, engage small molecules that stabilize G4C2 RNA repeats reducing RNA foci and RAN translation.161 Furthermore, some strategies are meant to target aberrantly upregulated components of the transcription machinery, while others work by harnessing RNA interference.162,163 Interestingly, the diabetes drug metformin has recently expanded its pharmacodynamic profile as a potential treatment in FTD-ALS. In C9orf72 transgenic mice, metformin was shown to inhibit the RNA-dependent protein kinase (PKR) phosphorylation, decreasing RAN translation and downregulating the effects of eukaryotic initiation factor 2α (eIF2α) phosphorylation, which is known to impair protein synthesis.164 Since January 2020 a phase 2 trial (NCT04220021) is ongoing to evaluate metformin safety and efficacy in C9orf72 ALS-FTD patients.
Therapeutic Approaches Based on Non-Invasive Brain Stimulation
Considering the high clinical and pathological heterogeneity of FTD, exacerbated by the lack of biomarkers accurately predicting in vivo tau or TDP pathology, an alternative approach to pharmacological treatments may be found in non-invasive brain stimulation.165 Several techniques have been recently developed to enhance cortical plasticity, including repetitive transcranial magnetic stimulation (rTMS), transcranial direct current stimulation (tDCS), or to entrain and modulate cerebral rhythms with transcranial alternate current stimulation (tACS).
Repetitive Transcranial Magnetic Stimulation
rTMS is a method in which externally produced repetitive magnetic pulses lead to depolarization of cortical neurons. rTMS can be applied at various stimulation frequencies or as a patterned train of pulses, and has a modulatory effect on cortical excitability and may induce long-term potentiation (LTP)-like cortical plasticity.166
There are only few reports describing the efficacy of this technique as a treatment in FTD.
In an open-label study, Antczak et al delivered 10 daily sessions of 10 Hz rTMS over the dorsolateral prefrontal cortex bilaterally in 11 patients with FTD (9 bvFTD, 2 nfvPPA), and observed an improvement in cognitive functions and frontal behavioral inventory scores.167
In another study, Cotelli et al applied a 20 Hz stimulation to both dorsolateral prefrontal cortices (DLPFC) in 10 patients with nfvPPA, in a sham-controlled design. They observed an improvement in action-naming possibly due to the modulation of DLPFC pathways and a facilitation effect on lexical retrieval processes.168
Other small case studies have shown similar positive effects of rTMS or deep rTMS in PPA patients.169–171
Several trials with rTMS are currently underway and awaiting completion in PPAs (NCT04188067, NCT03580954, NCT03153540, NCT04431401, NCT03406429).
Transcranial Direct and Alternate Current Stimulation
tDCS is a method based on the modulation of cortical excitability by a weak electrical current, which is delivered through scalp electrodes by a portable battery-powered stimulator. Repeated stimulation sessions are thought to generate long-lasting effects on cortical structures, induced by cortical plasticity.172
tDCS has shown initial positive effects in a sham-controlled trial in 13 patients with bvFTD. By stimulating frontotemporal cortices bilaterally for 5 days, researchers showed an improvement in neuropsychiatric symptoms and visual reaction times up to 1 month after stimulation.173
A recent much larger study, performed in 70 participants, has shown that a 2-week course with left prefrontal cortex stimulation may improve several clinical scores, particularly attention and executive functions, compared to sham stimulation, for up to 6 months.174 Moreover, tDCS restored intracortical connectivity measures, evaluated with TMS, which have been shown to be impaired early in the disease course.175–191 Interestingly researchers found that presymptomatic mutation carriers improved in cognitive tests after tDCS.175
tDCS has been found to enhance theory of mind, the ability to understand and predict other people’s behavior by attributing independent mental states to them, in patients with bvFTD.192
Several smaller studies have also shown an improvement in language in patients with PPA and PSP, also combined with individualized speech therapy, compared to sham stimulation.193–208
In a recent meta-analysis, tDCS was shown to be more effective than rTMS in the treatment of PPA.209 Overall, the meta-analysis suggested significant benefits of both methods in PPA patients, with the optimal treatment protocol remaining unknown.
Several trials with tDCS are currently underway and awaiting completion in PPAs (NCT04046991, NCT02606422, NCT03728582, NCT04486586, NCT03887481, NCT04566731, NCT03805659).
Another approach is being used in the GIFTeD trial, which is currently evaluating the effects of alternate current stimulation at gamma frequencies (40 Hz) with tACS in patients with FTD (NCT04425148). So far, reports of tACS as the clinical disease modifier in FTD have not been published.
Conclusions
The last few years have seen an improvement in the histological comprehension of FTLD, allowing a more detailed classification among the main underlying proteinopathies. Moreover, the strong genetic footprint has enriched the understanding of the disease mechanisms. Together with the results of genome-wide association studies (GWAS),210,211 new potential pathogenetic mechanisms have emerged, enhancing the arsenal of possible therapeutic strategies, including new insights from the role of autoimmunity.191,212–214
However, despite this progress, a precise definition of the specific pathogenetic path is still far from reach. Indeed, as depicted in this review, although the biological cascades leading to the specific protein (ie, Tau, TDP-43, FET)-driven neurodegeneration are still to be fully elucidated, they occur along common pathophysiological pathways. These premises have prompted the development of strategies aimed at the recovery of the disrupted proteostatic microenvironment (ie, kinome, UPS, ALP), an alternative approach to the removal of pathological species through selective antibody-based therapies. Indeed, the multitargeted properties of small molecules are emerging as promising in reversing or correcting several pathological pathways involved in neurodegeneration, highlighting the role of a “one drug, many targets” approach.215 The persistent failure of therapies based on clearance of deposited pathological species, adopted from the experience in different diseases, such as AD, highlights how future therapies should not only target the epiphenomenon of the pathological cascade but also understand and prevent the lack of physiological functions too. In support of this concept, the best results have been achieved by strategies targeting well-defined pathogenetic pathways (ie, GRN regulation), even if based on a more traditional “one drug, one target” paradigm.
In this perspective, given the possible convergence in the same clinical phenotypes, another challenging point in clinical trial settings is the availability of biomarkers able to distinguish between different underlying neuropathologies.
While in these landscape new treatments (ie, TMS and tDCS) are also emerging to reverse the secondary disruption of neuronal functioning, basic mechanistic and diagnostic research still holds the key to success in understanding and effectively treating this disease.
Acknowledgments
This study is supported by the Airalzh-AGYR2020 grant (AB), JPND GENFI-prox (BB), Italian Ministry of Health (RF-2018-12366665) (BB), Sigrid Jusélius Foundation (ES), Finnish Brain Foundation (ES), Instrumentarium Science Foundation (ES) and Orion Research Foundation (ES).
Disclosure
Dr Eino Solje reports grants from Orion Research Foundation, outside the submitted work. The authors report no other potential conflicts of interest for this work.
References
1. Vieira RT, Caixeta L, Machado S, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Pract Epidemiol Ment Heal. 2013;9:88–95. doi:10.2174/1745017901309010088
2. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt9):2456–2477. doi:10.1093/Brain/Awr179
3. Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. doi:10.1212/WNL.0b013e31821103e6
4. Borroni B, Benussi A. Recent advances in understanding frontotemporal degeneration. F1000Research. 2019;8:2098. doi:10.12688/f1000research.20330.1
5. Mackenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. doi:10.1007/s00401-009-0612-2
6. Neumann M, Mackenzie IRA. Review: neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2019;45(1):19–40. doi:10.1111/nan.12526
7. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075–2086. doi:10.1007/s00415-019-09363-4
8. Cosseddu M, Benussi A, Gazzina S, et al. Progression of behavioural disturbances in frontotemporal dementia: a longitudinal observational study. Eur J Neurol. 2020;27(2):265–272. doi:10.1111/ene.14071
9. Benussi A, Premi E, Gazzina S, et al. Progression of behavioral disturbances and neuropsychiatric symptoms in patients with genetic frontotemporal dementia. JAMA Netw Open. 2021:1–17. doi:10.1001/jamanetworkopen.2020.30194.
10. Borroni B, Padovani A. Dementia: a new algorithm for molecular diagnostics in FTLD. Nat Rev Neurol. 2013;9(5):241–242. doi:10.1038/nrneurol.2013.72
11. Benussi A, Padovani A, Borroni B. Phenotypic heterogeneity of monogenic frontotemporal dementia. Front Aging Neurosci. 2015;7(SEP). doi:10.3389/fnagi.2015.00171
12. Moore KM, Nicholas J, Grossman M, et al. Age at symptom onset and death and disease duration in genetic frontotemporal dementia: an international retrospective cohort study. Lancet Neurol. 2020;19(2):145–156. doi:10.1016/S1474-4422(19)30394-1
13. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12(6):609–622. doi:10.1016/S1474-4422(13)70090-5
14. Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122(2):137–153. doi:10.1007/s00401-011-0839-6
15. Lashley T, Rohrer JD, Mead S, Revesz T. Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol. 2015;n/a-n/a.
16. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi:10.1126/science.1134108
17. Mackenzie IR, Neumann M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017;134(1):79–96. doi:10.1007/s00401-017-1716-8
18. Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169(4):1343–1352. doi:10.2353/ajpath.2006.060438
19. Mackenzie IRA, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112(5):539–549. doi:10.1007/s00401-006-0138-9
20. Cairns NJ, Bigio EH, Mackenzie IRA, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5–22. doi:10.1007/s00401-007-0237-2
21. Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73(18):1451–1456. doi:10.1212/WNL.0b013e3181bf997a
22. Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916–919. doi:10.1038/nature05016
23. Cruts M, Gijselinck I, van der Zee J, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442(7105):920–924. doi:10.1038/nature05017
24. Masellis M, Momeni P, Meschino W. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129(11):3115–3123. doi:10.1093/brain/awl276
25. Le ber I, Camuzat A, Hannequin D, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008;131(3):732–746. doi:10.1093/brain/awn012
26. Chen-Plotkin AS, Martinez-Lage M, Sleiman PMA, et al. Genetic and clinical features of progranulin-associated frontotemporal lobar degeneration. Arch Neurol. 2011;68(4):488–497. doi:10.1001/archneurol.2011.53
27. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;1–12.
28. Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. 2011;1–12.
29. Bigio EH, Weintraub S, Rademakers R, et al. Frontotemporal lobar degeneration with TDP-43 proteinopathy and chromosome 9p repeat expansion in C9ORF72: clinicopathologic correlation. Neuropathology. 2013;33(2):122–133. doi:10.1111/j.1440-1789.2012.01332.x
30. Hsiung G-YR, Dejesus-Hernandez M, Feldman HH, et al. Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012;135(3):709–722. doi:10.1093/brain/awr354
31. Murray ME, DeJesus-Hernandez M, Rutherford NJ, et al. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011;122(6):673–690. doi:10.1007/s00401-011-0907-y
32. Wong TH, Pottier C, Hondius DC, et al. Three VCP mutations in patients with frontotemporal dementia. J Alzheimer’s Dis. 2018;65(4):1139–1146. doi:10.3233/JAD-180301
33. Mackenzie IRA, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122(1):111–113. doi:10.1007/s00401-011-0845-8
34. Mehta SG, Khare M, Ramani R, et al. Genotype-phenotype studies of VCP-associated inclusion body myopathy with Paget disease of bone and/or frontotemporal dementia. Clin Genet. 2012;83(5):422–431. doi:10.1111/cge.12000
35. Spina S, Van Laar AD, Murrell JR, et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur J Neurol. 2013;20(2):251–258. doi:10.1111/j.1468-1331.2012.03831.x
36. de Majo M, Topp SD, Smith BN, et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol Aging. 2018;71:
37. Koriath CA, Bocchetta M, Brotherhood E, et al. The clinical, neuroanatomical, and neuropathologic phenotype of TBK1-associated frontotemporal dementia: a longitudinal case report. Alzheimer’s Dement Diagnosis, Assess Dis Monit. 2017.
38. Hirsch-Reinshagen V, Alfaify OA, Hsiung GYR, et al. Clinicopathologic correlations in a family with a TBK1 mutation presenting as primary progressive aphasia and primary lateral sclerosis. Amyotroph Lateral Scler Front Degener. 2019;20(7–8):568–575. doi:10.1080/21678421.2019.1632347
39. Swift IJ, Bocchetta M, Benotmane H, Woollacott IO, Shafei R, Rohrer JD. Variable clinical phenotype in TBK1 mutations: case report of a novel mutation causing primary progressive aphasia and review of the literature. Neurobiol Aging. 2020. doi:10.1016/j.neurobiolaging.2020.08.014
40. Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, MacKenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(11):2922–2931. doi:10.1093/brain/awp214
41. Neumann M, Bentmann E, Dormann D, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134(9):2595–2609. doi:10.1093/brain/awr201
42. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8(8):423–434. doi:10.1038/nrneurol.2012.117
43. Goedert M, Crowther RA, Garner CC. Molecular characterization of microtubule-associated proteins tau and map2. Trends Neurosci. 1991;14(5):193–199. doi:10.1016/0166-2236(91)90105-4
44. Goedert M, Eisenberg DS, Crowther RA. Propagation of Tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40(1):189–210. doi:10.1146/annurev-neuro-072116-031153
45. Morris M, Knudsen GM, Maeda S, et al. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci. 2015;18(8):1183–1189. doi:10.1038/nn.4067
46. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–7741. doi:10.1073/pnas.95.13.7737
47. Forrest SL, Kril JJ, Stevens CH, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2018;141(2):521–534. doi:10.1093/brain/awx328
48. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41(1):24–46. doi:10.1111/nan.12213
49. Rossi G, Tagliavini F. Frontotemporal lobar degeneration: old knowledge and new insight into the pathogenetic mechanisms of tau mutations. Front Aging Neurosci. 2015;7:34873. doi:10.3389/fnagi.2015.00192
50. DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33(31):12887–12897. doi:10.1523/JNEUROSCI.2107-13.2013
51. Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides: a primer. Neurol Genet. 2019.
52. Schoch KMM, DeVos S, Miller RLL, et al. Increased 4R-Tau induces pathological changes in a human-tau mouse model. Neuron. 2016;90(5):941–947. doi:10.1016/j.neuron.2016.04.042
53. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481. doi:10.1126/scitranslmed.aag0481
54. Rodriguez-Martin T, Anthony K, Garcia-Blanco MA, Mansfield SG, Anderton BH, Gallo J-M. Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum Mol Genet. 2009;18(17):3266–3273. doi:10.1093/hmg/ddp264
55. Simone R. antisense long non-coding RNA represses MAPT translation through an embedded mir repeat. Alzheimer’s Dement. 2017;13(7):P918. doi:10.1016/j.jalz.2017.07.347
56. Wanowska E, Kubiak MR, Rosikiewicz W, Makałowska I, Szcześniak MW. Natural antisense transcripts in diseases: from modes of action to targeted therapies. Wiley Interdiscip Rev RNA. 2018;9(2):e1461. doi:10.1002/wrna.1461
57. Lauretti E, Praticò D. Alzheimer’s disease: phenotypic approaches using disease models and the targeting of tau protein. Expert Opin Ther Targets. 2020;24(4):319–330. doi:10.1080/14728222.2020.1737012
58. Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112–119. doi:10.1016/j.molmed.2009.01.003
59. Llorens-Martín M, Jurado J, Hernández F, Ávila J. GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci. 2014;7. doi:10.3389/fnmol.2014.00046.
60. Ferrer I, Barrachina M, Puig B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002;104(6):583–591. doi:10.1007/s00401-002-0587-8
61. Noble W, Planel E, Zehr C, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102(19):6990–6995. doi:10.1073/pnas.0500466102
62. Hampel H, Ewers M, Bürger K, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70(6):922–931. doi:10.4088/JCP.08m04606
63. Leclair-Visonneau L, Rouaud T, Debilly B, et al. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg. 2016;146:35–39. doi:10.1016/j.clineuro.2016.04.021
64. Tatebayashi Y, Haque N, Tung Y-C, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3β in the regulation of organelle transport. J Cell Sci. 2004;117(9):1653–1663. doi:10.1242/jcs.01018
65. Tolosa E, Litvan I, Höglinger GU, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29(4):470–478. doi:10.1002/mds.25824
66. Höglinger GU, Huppertz H-J, Wagenpfeil S, et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord. 2014;29(4):479–487. doi:10.1002/mds.25815
67. Kimura T, Ishiguro K, Hisanaga SI. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7. doi:10.3389/fnmol.2014.00065.
68. Giacomini C, Koo C-Y, Yankova N, et al. A new TAO kinase inhibitor reduces tau phosphorylation at sites associated with neurodegeneration in human tauopathies. Acta Neuropathol Commun. 2018;6(1). doi:10.1186/s40478-018-0539-8
69. Taylor LM, McMillan PJ, Kraemer BC, Liachko NF. Tau tubulin kinases in proteinopathy. FEBS J. 2019;286(13):2434–2446. doi:10.1111/febs.14866
70. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018. doi:10.1038/nrd.2018.21
71. van Dyck CH, Nygaard HB, Chen K, et al. Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease. JAMA Neurol. 2019;06510(10):1219–1229. doi:10.1001/jamaneurol.2019.2050
72. Turner RS, Hebron ML, Lawler A, et al. Nilotinib effects on safety, tolerability, and biomarkers in Alzheimer’s disease. Ann Neurol. 2020;88(1):183–194. doi:10.1002/ana.25775
73. Adwan L, Subaiea GM, Basha R, Zawia NH. Tolfenamic acid reduces tau and CDK5 levels: implications for dementia and tauopathies. J Neurochem. 2015;133(2):266–272. doi:10.1111/jnc.12960
74. Chang JK, Leso A, Subaiea GM, et al. Tolfenamic Acid: a Modifier of the Tau Protein and its Role in Cognition and Tauopathy. Curr Alzheimer Res. 2018;15(7):655–663. doi:10.2174/1567205015666180119104036
75. Yu Y, Zhang L, Li X, et al. Differential effects of an o-GlcNAcase inhibitor on tau phosphorylation. PLoS One. 2012. doi:10.1371/journal.pone.0035277
76. Wang X, Li W, Marcus J, et al. MK-8719, a novel and selective O-GlcNAcase inhibitor that reduces the formation of pathological tau and ameliorates neurodegeneration in a mouse model of tauopathy. J Pharmacol Exp Ther. 2020;374(2):252–263. doi:10.1124/jpet.120.266122
77. Treating Tau: Finally, Clinical Candidates Are Stepping into the Ring | ALZFORUM.
78. VandeVrede L, Boxer AL, Polydoro M. Targeting tau: clinical trials and novel therapeutic approaches. Neurosci Lett. 2020;731:134919. doi:10.1016/j.neulet.2020.134919
79. Kontaxi C, Piccardo P, Gill AC. Lysine-directed post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Front Mol Biosci. 2017;4. doi:10.3389/fmolb.2017.00056.
80. Irwin DJ, Cohen TJ, Grossman M, et al. Acetylated Tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol. 2013;183(2):344–351. doi:10.1016/j.ajpath.2013.04.025
81. Tracy T, Claiborn KC, Gan L. Regulation of Tau Homeostasis and Toxicity by Acetylation. Advan Exp Med Biol. 2019. doi:10.1007/978-981-32-9358-8_4
82. Min S-W, Chen X, Tracy TE, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21(10):1154–1162. doi:10.1038/nm.3951
83. VandeVrede L, Dale ML, Fields S, et al. Open-Label Phase 1 futility studies of salsalate and young plasma in progressive supranuclear palsy. Mov Disord Clin Pract. 2020;7(4):440–447. doi:10.1002/mdc3.12940
84. Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2015;17(1):22–35.
85. Akoury E, Pickhardt M, Gajda M, Biernat J, Mandelkow E, Zweckstetter M. Mechanistic basis of phenothiazine-driven inhibition of Tau aggregation. Angew Chemie - Int Ed. 2013. doi:10.1002/anie.201208290
86. Medina DX, Caccamo A, Oddo S. Methylene blue reduces Aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. doi:10.1111/j.1750-3639.2010.00430.x
87. Congdon EE, Wu JW, Myeku N, et al. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8(4):609–622. doi:10.4161/auto.19048
88. Vaccaro A, Patten SA, Ciura S, et al. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS One. 2012;7(7):e42117. doi:10.1371/journal.pone.0042117
89. Wischik CM, Staff RT, Wischik DJ, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–720. doi:10.3233/JAD-142874
90. Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–2884. doi:10.1016/S0140-6736(16)31275-2
91. Wilcock GK, Gauthier S, Frisoni GB, et al. Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: cohort analysis as modified primary outcome in a Phase III clinical trial. J Alzheimer’s Dis. 2018. doi:10.3233/JAD-170560
92. Soeda Y, Saito M, Maeda S, et al. Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: a plausible key to understanding failure of a clinical trial for Alzheimer’s disease. J Alzheimer’s Dis. 2019;68(4):1677–1686. doi:10.3233/JAD-181001
93. Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer’s disease. Nat Rev Immunol. 2006;6(5):404–416. doi:10.1038/nri1843
94. Rosenmann H, Grigoriadis N, Karussis D, et al. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006;63(10):1459–1467. doi:10.1001/archneur.63.10.1459
95. Kontsekova E, Zilka N, Kovacech B, Skrabana R, Novak M. Identification of structural determinants on tau protein essential for its pathological function: novel therapeutic target for tau immunotherapy in Alzheimer’s disease. Alzheimers Res Ther. 2014;6(4):45. doi:10.1186/alzrt277
96. Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau–tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther. 2014;6(4):44. doi:10.1186/alzrt278
97. AADvac1 | ALZFORUM.
98. Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16(2):123–134. doi:10.1016/S1474-4422(16)30331-3
99. Novak P, Schmidt R, Kontsekova E, et al. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimer’s Res Ther. 2018;10(1):1–16. doi:10.1186/s13195-018-0436-1
100. Novak P, Zilka N, Zilkova M, et al. AADvac1, an active immunotherapy for Alzheimer’s Disease and Non Alzheimer tauopathies: an overview of preclinical and clinical development. J Prev Alzheimer’s Dis. 2019;6(1):63–69. doi:10.14283/jpad.2018.45
101. Jadhav S, Avila J, Schöll M, et al. A walk through tau therapeutic strategies. Acta Neuropathol Commun. 2019;7(1):22. doi:10.1186/s40478-019-0664-z
102. Boxer AL, Qureshi I, Ahlijanian M, et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1B trial. Lancet Neurol. 2019;18(6):549–558. doi:10.1016/S1474-4422(19)30139-5
103. Sandusky-Beltran LA, Sigurdsson EM. Tau immunotherapies: lessons learned, current status and future considerations. Neuropharmacology. 2020;175:108104. doi:10.1016/j.neuropharm.2020.108104
104. Yanamandra K, Kfoury N, Jiang H, et al. Erratum to Anti-Tau antibodies that block tau aggregate seeding invitro markedly decrease pathology and improve cognition in vivo [Neuron, 74, (2012), 227-245]. Neuron. 2013;80(2):402–414. doi:10.1016/j.neuron.2013.12.007
105. AbbVie’s Tau Antibody Flops in Progressive Supranuclear Palsy | ALZFORUM.
106. Colin M, Dujardin S, Schraen-Maschke S, et al. From the prion-like propagation hypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol. 2020;139(1):3–25. doi:10.1007/s00401-019-02087-9
107. Barthélemy NR, Gabelle A, Hirtz C, et al. Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer’s disease, progressive supranuclear palsy, and dementia with lewy bodies. J Alzheimer’s Dis. 2016;51(4):1033–1043. doi:10.3233/JAD-150962
108. Albert M, Mairet-Coello G, Danis C, et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain. 2019;142(6):1736–1750. doi:10.1093/brain/awz100
109. Courade J-P, Angers R, Mairet-Coello G, et al. Epitope determines efficacy of therapeutic anti-Tau antibodies in a functional assay with human Alzheimer Tau. Acta Neuropathol. 2018;136(5):729–745. doi:10.1007/s00401-018-1911-2
110. Zhang B, Carroll J, Trojanowski JQ, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012. doi:10.1523/JNEUROSCI.4922-11.2012
111. Tsai RM, Miller Z, Koestler M, et al. Reactions to multiple ascending doses of the microtubule stabilizer TPI-287 in patients with alzheimer disease, progressive supranuclear Palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol. 2019.
112. Vulih-Shultzman I, Pinhasov A, Mandel S, et al. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J Pharmacol Exp Ther. 2007;323(2):438–449. doi:10.1124/jpet.107.129551
113. Matsuoka Y, Jouroukhin Y, Gray AJ, et al. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer’s disease. J Pharmacol Exp Ther. 2008;325(1):146–153. doi:10.1124/jpet.107.130526
114. Oz S, Ivashko-Pachima Y, Gozes I. The ADNP derived peptide, NAP modulates the tubulin pool: implication for neurotrophic and neuroprotective activities. PLoS One. 2012;7(12):e51458. doi:10.1371/journal.pone.0051458
115. Morimoto BH, Schmechel D, Hirman J, et al. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement Geriatr Cogn Disord. 2013;35(5–6):325–339. doi:10.1159/000348347
116. Boxer AL, Lang AE, Grossman M, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014;13(7):676–685. doi:10.1016/S1474-4422(14)70088-2
117. Ivashko-Pachima Y, Maor-Nof M, Gozes I. NAP (davunetide) preferential interaction with dynamic 3-repeat Tau explains differential protection in selected tauopathies. PLoS One. 2019;14(3):e0213666. doi:10.1371/journal.pone.0213666
118. Buratti E, Baralle FE. Characterization and Functional Implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR Exon 9. J Biol Chem. 2001;276(39):36337–36343. doi:10.1074/jbc.M104236200
119. Ayala YM, Zago P, D’Ambrogio A, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(22):3778–3785. doi:10.1242/jcs.038950
120. Pesiridis GS, Lee VM-Y, Trojanowski JQ. Mutations in TDP-43 link glycine-rich domain functions to amyotrophic lateral sclerosis. Hum Mol Genet. 2009;18(R2):R156–R162. doi:10.1093/hmg/ddp303
121. Floris G, Borghero G, Cannas A, et al. Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J Neurol. 2015;262(2):375–384. doi:10.1007/s00415-014-7575-5
122. Birsa N, Bentham MP, Fratta P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin Cell Dev Biol. 2020;99:193–201. doi:10.1016/j.semcdb.2019.05.023
123. Molliex A, Temirov J, Lee J, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163(1):123–133. doi:10.1016/j.cell.2015.09.015
124. Buratti E. TDP-43 post-translational modifications in health and disease. Expert Opin Ther Targets. 2018;22(3):279–293. doi:10.1080/14728222.2018.1439923
125. Borroni B, Alberici A, Buratti E. Review: molecular pathology of frontotemporal lobar degenerations. Neuropathol Appl Neurobiol. 2019;45(1):41–57. doi:10.1111/nan.12534
126. Nana AL, Sidhu M, Gaus SE, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137(1):27–46. doi:10.1007/s00401-018-1942-8
127. Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4(1). doi:10.1038/ncomms3753
128. Budini M, Baralle FE, Buratti E. Targeting TDP-43 in neurodegenerative diseases. Expert Opin Ther Targets. 2014;18(6):617–632. doi:10.1517/14728222.2014.896905
129. Tsao W, Jeong YH, Lin S, et al. Rodent models of TDP-43: recent advances. Brain Res. 2012;1462:26–39. doi:10.1016/j.brainres.2012.04.031
130. Kishor A, Fritz SE, Hogg JR. Nonsense-mediated mRNA decay: the challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip Rev RNA. 2019;10(6). doi:10.1002/wrna.1548
131. Barmada SJ, Ju S, Arjun A, et al. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proc Natl Acad Sci U S A. 2015;112(25):7821–7826. doi:10.1073/pnas.1509744112
132. Buratti E. Functional significance of TDP-43 mutations in disease. Adv Genet. 2015. doi:10.1016/bs.adgen.2015.07.001
133. Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117(2):137–149. doi:10.1007/s00401-008-0477-9
134. Kametani F, Nonaka T, Suzuki T, et al. Identification of casein kinase-1 phosphorylation sites on TDP-43. Biochem Biophys Res Commun. 2009;382(2):405–409. doi:10.1016/j.bbrc.2009.03.038
135. Alquezar C, Salado IG, De La Encarnación A, et al. Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: a novel strategy for the treatment of frontotemporal dementia. Mol Neurodegener. 2016;11(1). doi:10.1186/s13024-016-0102-7
136. Scotter EL, Vance C, Nishimura AL, et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127(6):1263–1278. doi:10.1242/jcs.140087
137. Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol. 2007;179(3):485–500. doi:10.1083/jcb.200702115
138. Isaacs AM, Johannsen P, Holm I, Nielsen JE. Frontotemporal dementia caused by CHMP2B mutations. Curr Alzheimer Res. 2011;999(999):1–6. doi:10.2174/1567211212225992050
139. Ugbode C, West RJH. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. 2021;147:105144. doi:10.1016/j.nbd.2020.105144
140. Sun X, Deng X, Hu R, et al. CHMP2B regulates TDP-43 phosphorylation and proteotoxicity via modulating CK1 turnover independent of the autophagy-lysosomal pathway. bioRxiv. 2020. doi:10.1101/2020.06.04.133546
141. Bose JK, Huang CC, Shen CKJ. Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem. 2011;286(52):44441–44448. doi:10.1074/jbc.M111.237115
142. Xia Q, Wang H, Hao Z, et al. TDP -43 loss of function increases TFEB activity and blocks autophagosome–lysosome fusion. EMBO J. 2016;35(2):121–142. doi:10.15252/embj.201591998
143. Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48(7):999–1009. doi:10.1177/002215540004800713
144. Kessenbrock K, Fröhlich L, Sixt M, et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008. doi:10.1172/jci34694
145. Salazar DA, Butler VJ, Argouarch AR, et al. The progranulin cleavage products, granulins, exacerbate TDP-43 toxicity and increase TDP-43 levels. J Neurosci. 2015;35(25):9315–9328. doi:10.1523/JNEUROSCI.4808-14.2015
146. Townley RA, Boeve BF, Benarroch EE. Progranulin: functions and neurologic correlations. Neurology. 2018;90(3):118–125. doi:10.1212/WNL.0000000000004840
147. Hu F, Padukkavidana T, Vægter CB, et al. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68(4):654–667. doi:10.1016/j.neuron.2010.09.034
148. Lee WC, Almeida S, Prudencio M, et al. Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet. 2014;23(6):1467–1478. doi:10.1093/hmg/ddt534
149. Alector Announces Data from On-going Phase 1b Trial Demonstrating that AL001 Reverses Progranulin Deficiency in Frontotemporal Dementia Patients | Alector.
150. Nicholson AM, Finch NA, Almeida M, et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat Commun. 2016;7(1):11992. doi:10.1038/ncomms11992
151. AZP2006 | ALZFORUM.
152. She A, Kurtser I, Reis SA, et al. Selectivity and kinetic requirements of HDAC inhibitors as progranulin enhancers for treating frontotemporal dementia. Cell Chem Biol. 2017;24(7):892–906.e5. doi:10.1016/J.CHEMBIOL.2017.06.010
153. Cenik B, Sephton CF, Dewey CM, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. 2011;286(18):16101–16108. doi:10.1074/jbc.M110.193433
154. Almeida S, Gao F, Coppola G, Gao FB. Suberoylanilide hydroxamic acid increases progranulin production in iPSC-derived cortical neurons of frontotemporal dementia patients. Neurobiol Aging. 2016;42:35–40. doi:10.1016/j.neurobiolaging.2016.03.001
155. Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem. 2016;138:
156. Boxer AL, Moebius H, Harris B, et al. O4-02-01: PHASE 2A Randomized, Double-Blind, Placebo-Controlled Trial Of The Histone Deacetylase Inhibitor (HDACI), FRM-0334, in asymptomatic carriers of, or patients with frontotemporal lobar degeneration (FTLD) due to, progranulin gene mutations. Alzheimer’s Dement. 2019;15:P1231–P1232. doi:10.1016/j.jalz.2019.06.4746
157. Hinderer C, Miller R, Dyer C, et al. Adeno-associated virus serotype 1-based gene therapy for FTD caused by GRN mutations. Ann Clin Transl Neurol. 2020;7(10):1843–1853. doi:10.1002/acn3.51165
158. Heckman LD, Burstein SR, Brandes A, et al. Preclinical development of PR006, a gene therapy for the treatment of frontotemporal dementia with progranulin mutations. Alzheimer’s Dement. 2020;16(S2). doi:10.1002/alz.043632
159. Gagliardi D, Costamagna G, Taiana M, et al. Insights into disease mechanisms and potential therapeutics for C9orf72-related amyotrophic lateral sclerosis/frontotemporal dementia. Ageing Res Rev. 2020. doi:10.1016/j.arr.2020.101172
160. Jiang J, Zhu Q, Gendron TF, et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron. 2016;90(3):535–550. doi:10.1016/j.neuron.2016.04.006
161. Simone R, Balendra R, Moens TG, et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD / ALS pathology in vitro and in vivo. EMBO Mol Med. 2018;10(1):22–31. doi:10.15252/emmm.201707850
162. Martier R, Liefhebber JM, García-Osta A, et al. Targeting RNA-mediated toxicity in C9orf72 ALS and/or FTD by RNAi-based gene therapy. Mol Ther - Nucleic Acids. 2019;16:26–37. doi:10.1016/j.omtn.2019.02.001
163. Goodman LD, Prudencio M, Kramer NJ, et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat Neurosci. 2019;22(6):863–874. doi:10.1038/s41593-019-0396-1
164. Zu T, Guo S, Bardhi O, et al. Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proc Natl Acad Sci U S A. 2020;117(31):18591–18599. doi:10.1073/pnas.2005748117
165. Borroni B, Benussi A, Premi E, et al. Biological, neuroimaging, and neurophysiological markers in frontotemporal dementia: three faces of the same coin. J Alzheimers Dis. 2018;62(3):1113–1123. doi:10.3233/JAD-170584
166. Benussi A, Padovani A, Borroni B. Transcranial magnetic stimulation in Alzheimer’s disease and cortical dementias. J Alzheimer’s Dis Park. 2015;05:03. doi:10.4172/2161-0460.1000197
167. Antczak J, Kowalska K, Klimkowicz-Mrowiec A, et al. Repetitive transcranial magnetic stimulation for the treatment of cognitive impairment in frontotemporal dementia: an open-label pilot study. Neuropsychiatr Dis Treat. 2018;14:749–755. doi:10.2147/NDT.S153213
168. Cotelli M, Manenti R, Alberici A, et al. Prefrontal cortex rTMS enhances action naming in progressive non-fluent aphasia. Eur J Neurol. 2012;19(11):1404–1412. doi:10.1111/j.1468-1331.2012.03699.x
169. Bereau M, Magnin E, Nicolier M, et al. Left prefrontal repetitive transcranial magnetic stimulation in a logopenic variant of primary progressive aphasia: a case report. Eur Neurol. 2016;76(1–2):12–18. doi:10.1159/000447399
170. Finocchiaro C, Maimone M, Brighina F, Piccoli T, Giglia G, Fierro B. A case study of primary progressive aphasia: improvement on verbs after rTMS treatment. Neurocase. 2006;12(6):317–321. doi:10.1080/13554790601126203
171. Trebbastoni A, Raccah R, de Lena C, Zangen A, Inghilleri M. Repetitive deep transcranial magnetic stimulation improves verbal fluency and written language in a patient with primary progressive aphasia-logopenic Variant (LPPA). Brain Stimul. 2013;6(4):545–553. doi:10.1016/j.brs.2012.09.014
172. Lefaucheur JP, Antal A, Ayache SS, et al. Evidence-based guidelines on the therapeutic use of transcranial direct current stimulation (tDCS). Clin Neurophysiol. 2017;128(1):56–92. doi:10.1016/j.clinph.2016.10.087
173. Ferrucci R, Mrakic-Sposta S, Gardini S, et al. Behavioral and neurophysiological effects of transcranial direct current stimulation (tDCS) in fronto-temporal dementia. Front Behav Neurosci. 2018;12:
174. Benussi A, Dell’Era V, Cosseddu M, et al. Transcranial stimulation in frontotemporal dementia: a randomized, double-blind, sham-controlled trial. Alzheimer’s Dement Transl Res Clin Interv. 2020;6(1). doi:10.1002/trc2.12033
175. Benussi A, Dell’Era V, Cantoni V, et al. TMS for staging and predicting functional decline in frontotemporal dementia. Brain Stimul. 2020;13(2):386–392. doi:10.1016/j.brs.2019.11.009
176. Benussi A, Cosseddu M, Filareto I, et al. Impaired long-term potentiation-like cortical plasticity in presymptomatic genetic frontotemporal dementia. Ann Neurol. 2016;80(3):472–476. doi:10.1002/ana.24731
177. Padovani A, Benussi A, Cotelli MS, et al. Transcranial magnetic stimulation and amyloid markers in mild cognitive impairment: impact on diagnostic confidence and diagnostic accuracy. Alzheimers Res Ther. 2019;11(1):95. doi:10.1186/s13195-019-0555-3
178. Benussi A, Dell’Era V, Cantoni V, et al. Neurophysiological correlates of positive and negative symptoms in frontotemporal dementia. Arighi A, ed. J Alzheimers Dis. 2020;73(3):1133–1142. doi:10.3233/JAD-190986
179. Benussi A, Grassi M, Palluzzi F, et al. Classification accuracy of transcranial magnetic stimulation for the diagnosis of neurodegenerative dementias. Ann Neurol. 2020;87(3):394–404. doi:10.1002/ana.25677
180. Benussi A, Premi E, Gazzina S, et al. Neurotransmitter imbalance dysregulates brain dynamic fluidity in frontotemporal degeneration. Neurobiol Aging. 2020;94:176–184. doi:10.1016/j.neurobiolaging.2020.05.017
181. Benussi A, Karikari T, Ashton N, et al. Diagnostic and prognostic value of serum NfL and p-Tau 181 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2020;91(9):960–967. doi:10.1136/jnnp-2020-323487
182. Benussi A, Ashton NJ, Karikari TK, et al. Serum Glial Fibrillary Acidic Protein (GFAP) is a marker of disease severity in frontotemporal lobar degeneration. Arighi A, ed. J Alzheimer’s Dis. 2020;29:1–13. doi:10.3233/JAD-200608
183. Benussi A, Grassi M, Palluzzi F, et al. Classification accuracy of TMS for the diagnosis of mild cognitive impairment. Brain Stimul. 2021;14(2):241–249. doi:10.1016/j.brs.2021.01.004
184. Benussi A, Di Lorenzo F, Dell’Era V, et al. Transcranial magnetic stimulation distinguishes Alzheimer disease from frontotemporal dementia. Neurology. 2017;89(7):665–672. doi:10.1212/WNL.0000000000004232
185. Benussi A, Dell’Era V, Cantoni V, et al. Discrimination of atypical parkinsonisms with transcranial magnetic stimulation. Brain Stimul. 2018;11(2):366–373. doi:10.1016/j.brs.2017.11.013
186. Gazzina S, Benussi A, Premi E, et al. Neuroanatomical correlates of transcranial magnetic stimulation in presymptomatic granulin mutation carriers. Brain Topogr. 2018;31(3):488–497. doi:10.1007/s10548-017-0612-9
187. Padovani A, Benussi A, Cantoni V, et al. Diagnosis of mild cognitive impairment due to Alzheimer’s disease with transcranial magnetic stimulation. J Alzheimers Dis. 2018;65(1):221–230. doi:10.3233/JAD-180293
188. Benussi A, Alberici A, Ferrari C, et al. The impact of transcranial magnetic stimulation on diagnostic confidence in patients with Alzheimer disease. Alzheimers Res Ther. 2018;10(1):94. doi:10.1186/s13195-018-0423-6
189. Benussi A, Gazzina S, Premi E, et al. Clinical and biomarker changes in presymptomatic genetic frontotemporal dementia. Neurobiol Aging. 2019;76:133–140. doi:10.1016/j.neurobiolaging.2018.12.018
190. Benussi A, Dell’Era V, Cantoni V, et al. Stimulation over the cerebellum with a regular figure-of-eight coil induces reduced motor cortex inhibition in patients with progressive supranuclear palsy. Brain Stimul. 2019;12(5):1290–1297. doi:10.1016/j.brs.2019.05.017
191. Palese F, Bonomi E, Nuzzo T, et al. Anti-GluA3 antibodies in frontotemporal dementia: effects on glutamatergic neurotransmission and synaptic failure. Neurobiol Aging. 2020;86:143–155. doi:10.1016/j.neurobiolaging.2019.10.015
192. Cotelli M, Adenzato M, Cantoni V, et al. Enhancing theory of mind in behavioural variant frontotemporal dementia with transcranial direct current stimulation. Cogn Affect Behav Neurosci. 2018;18(6):1065–1075. doi:10.3758/s13415-018-0622-4
193. Cotelli M, Manenti R, Paternicò D, et al. Grey matter density predicts the improvement of naming abilities after tDCS intervention in agrammatic variant of primary progressive aphasia. Brain Topogr. 2016;29(5):738–751. doi:10.1007/s10548-016-0494-2
194. Gervits F, Ash S, Coslett HB, Rascovsky K, Grossman M, Hamilton R. Transcranial direct current stimulation for the treatment of primary progressive aphasia: an open-label pilot study. Brain Lang. 2016;162:35–41. doi:10.1016/j.bandl.2016.05.007
195. de Aguiar V, Zhao Y, Faria A, et al. Brain volumes as predictors of tDCS effects in primary progressive aphasia. Brain Lang. 2020;200(December):2018. doi:10.1016/j.bandl.2019.104707
196. Valero-Cabré A, Sanches C, Godard J, et al. Language boosting by transcranial stimulation in progressive supranuclear palsy. Neurology. 2019;93(6):E537–E547. doi:10.1212/WNL.0000000000007893
197. Harris AD, Wang Z, Ficek B, Webster K, Edden RA, Tsapkini K. Reductions in GABA following a tDCS-language intervention for primary progressive aphasia. Neurobiol Aging. 2019;79:75–82. doi:10.1016/j.neurobiolaging.2019.03.011
198. Roncero C, Kniefel H, Service E, Thiel A, Probst S, Chertkow H. Inferior parietal transcranial direct current stimulation with training improves cognition in anomic Alzheimer’s disease and frontotemporal dementia. Alzheimer’s Dement Transl Res Clin Interv. 2017;3(2):247–253. doi:10.1016/j.trci.2017.03.003
199. McConathey EM, White NC, Gervits F, et al. Baseline Performance Predicts tDCS-mediated improvements in language symptoms in primary progressive aphasia. Front Hum Neurosci. 2017;11:84. doi:10.3389/fnhum.2017.00347
200. Hung J, Bauer A, Grossman M, Hamilton RH, Coslett HB, Reilly J. Semantic feature training in combination with Transcranial Direct Current Stimulation (tDCS) for progressive anomia. Front Hum Neurosci. 2017;11:390. doi:10.3389/fnhum.2017.00253
201. Teichmann M, Lesoil C, Godard J, et al. Direct current stimulation over the anterior temporal areas boosts semantic processing in primary progressive aphasia. Ann Neurol. 2016;80(5):693–707. doi:10.1002/ana.24766
202. Ficek BN, Wang Z, Zhao Y, et al. The effect of tDCS on functional connectivity in primary progressive aphasia. NeuroImage Clin. 2018;19:703–715. doi:10.1016/j.nicl.2018.05.023
203. Fenner AS, Webster KT, Ficek BN, Frangakis CE, Tsapkini K. Written verb naming improves after tDCS over the left IFG in primary progressive aphasia. Front Psychol. 2019;10(JUN). doi:10.3389/fpsyg.2019.01396
204. Tsapkini K, Webster KT, Ficek BN, et al. Electrical brain stimulation in different variants of primary progressive aphasia: a randomized clinical trial. Alzheimer’s Dement Transl Res Clin Interv. 2018;4:461–472. doi:10.1016/j.trci.2018.08.002
205. Wang J, Wu D, Chen Y, Yuan Y, Zhang M. Effects of transcranial direct current stimulation on language improvement and cortical activation in nonfluent variant primary progressive aphasia. Neurosci Lett. 2013;549:29–33. doi:10.1016/j.neulet.2013.06.019
206. Cotelli M, Manenti R, Petesi M, et al. Treatment of primary progressive aphasias by transcranial direct current stimulation combined with language training. J Alzheimers Dis. 2014;39(4):799–808. doi:10.3233/JAD-131427
207. Zhao Y, Ficek B, Webster K, et al. White matter Integrity Predicts Electrical Stimulation (tDCS) and language therapy effects in primary progressive aphasia. Neurorehabil Neural Repair. 2021;35(1):44–57. doi:10.1177/1545968320971741
208. de Aguiar V, Zhao Y, Ficek BN, et al. Cognitive and language performance predicts effects of spelling intervention and tDCS in primary progressive aphasia. Cortex. 2020;124:66–84. doi:10.1016/j.cortex.2019.11.001
209. Nissim NR, Moberg PJ, Hamilton RH. Efficacy of noninvasive brain stimulation (tDCS or TMS) paired with language therapy in the treatment of primary progressive aphasia: an exploratory meta-analysis. Brain Sci. 2020;10(9):1–17. doi:10.3390/brainsci10090597
210. Pottier C, Ren Y, Perkerson RB, et al. Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD. Acta Neuropathol. 2019. doi:10.1007/s00401-019-01962-9
211. Pottier C, Bieniek KF, Finch N, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77–92. doi:10.1007/s00401-015-1436-x
212. Gardoni F, Stanic J, Scheggia D, Benussi A, Borroni B, Di LM. NMDA and AMPA receptor autoantibodies in brain disorders: from molecular mechanisms to clinical features. Cells. 2021;10(7):77. doi:doi.org/10.3390/cells10010077
213. Alberici A, Cristillo V, Gazzina S, Benussi A, Padovani A, Borroni B. Autoimmunity and frontotemporal dementia. Curr Alzheimer Res. 2018;15(7):602–609. doi:10.2174/1567205015666180119104825
214. Benussi A, Alberici A, Buratti E, et al. Toward a glutamate hypothesis of frontotemporal dementia. Front Neurosci. 2019;13:
215. Fowler AJ, Hebron M, Missner AA, et al. Multikinase Abl/DDR/Src inhibition produces optimal effects for tyrosine kinase inhibition in neurodegeneration. Drugs R D. 2019;19(2):149–166. doi:10.1007/s40268-019-0266-z
216. Scheltens P, Prins N, Lammertsma A, et al. An exploratory clinical study of p38 α kinase inhibition in Alzheimer’s disease. Ann Clin Transl Neurol. 2018;5(4):464–473. doi:10.1002/acn3.549
217. Ryan JM, Quattropani A, Abd-Elaziz K, et al. O1-12-05: phase 1 study in healthy volunteers of the O-glcnacase inhibitor ASN120290 as a novel therapy for progressive supranuclear palsy and related tauopathies. Alzheimer’s Dement. 2018;14(7S_Part_4):P251–P251. doi:10.1016/j.jalz.2018.06.2400
218. Rojas-Prats E, Martinez-Gonzalez L, Gonzalo-Consuegra C, et al. Targeting nuclear protein TDP-43 by cell division cycle kinase 7 inhibitors: a new therapeutic approach for amyotrophic lateral sclerosis. Eur J Med Chem. 2020;210. doi:10.1016/j.ejmech.2020.112968
219. Moujalled D, James JL, Parker SJ, et al. Kinase Inhibitor screening identifies cyclin-dependent kinases and glycogen synthase kinase 3 as potential modulators of TDP-43 cytosolic accumulation during cell stress. PLoS One. 2013;8(6):e67433. doi:10.1371/journal.pone.0067433
220. Taylor LM, McMillan PJ, Liachko NF, et al. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol Neurodegener. 2018;13(1). doi:10.1186/s13024-018-0237-9
221. Fang MY, Markmiller S, Vu AQ, et al. Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron. 2019;103(5):802–819.e11. doi:10.1016/j.neuron.2019.05.048
222. McGurk L, Gomes E, Guo L, et al. Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell. 2018;71(5):703–717.e9. doi:10.1016/j.molcel.2018.07.002
223. Archbold HC, Jackson KL, Arora A, et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8(1). doi:10.1038/s41598-018-22858-w
224. Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A. 2012;109(37):15024–15029. doi:10.1073/pnas.1206362109
225. Holler CJ, Taylor G, McEachin ZT, et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 2016;11(1):538. doi:10.1186/s13024-016-0114-3
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.