Back to Journals » Drug Design, Development and Therapy » Volume 12
Experimental and molecular modeling approach to optimize suitable polymers for fabrication of stable fluticasone nanoparticles with enhanced dissolution and antimicrobial activity
Authors Ahmed S, Govender T, Khan I, ur Rehman N, Ali W, Shah SMH ![]() , Khan S
, Khan S ![]() , Hussain Z
, Hussain Z ![]() , Ullah R, Alsaid MS
, Ullah R, Alsaid MS
Received 11 August 2017
Accepted for publication 13 November 2017
Published 5 February 2018 Volume 2018:12 Pages 255—269
DOI https://doi.org/10.2147/DDDT.S148912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Shaimaa Ahmed,1 Thirumala Govender,1 Inamullah Khan,2 Nisar ur Rehman,2 Waqar Ali,2 Syed Muhammad Hassan Shah,3 Shahzeb Khan,4 Zahid Hussain,5 Riaz Ullah,6,7 Mansour S Alsaid6
1Discipline of Pharmaceutical Sciences, School of Health Sciences, University of KwaZulu-Natal, Durban, South Africa; 2Department of Pharmacy, COMSATS Institute of Information Technology (CIIT), Abbotabad, 3Department of Pharmacy, Sarhad University of Science and Technology, Peshawar, 4Department of Pharmacy, University of Malakand Dir (Lower), Chakdara, Khyber Pakhtunkhwa, Pakistan; 5Department of Pharmaceutics, Faculty of Pharmacy, Universiti Teknologi Mara, Puncak Alam, Selangor, Malaysia; 6Department of Pharmacognosy and Medicinal, Aromatic & Poisonous Plants Research Center (MAPPRC), College of Pharmacy, King Saud University, Riyadh, Saudi Arabia; 7Department of Chemistry, Government College Ara Khel FR, Kohat, Khyber Pakhtunkhwa, Pakistan
Background and aim: The challenges with current antimicrobial drug therapy and resistance remain a significant global health threat. Nanodrug delivery systems are playing a crucial role in overcoming these challenges and open new avenues for effective antimicrobial therapy. While fluticasone (FLU), a poorly water-soluble corticosteroid, has been reported to have potential antimicrobial activity, approaches to optimize its dissolution profile and antimicrobial activity are lacking in the literature. This study aimed to combine an experimental study with molecular modeling to design stable FLU nanopolymeric particles with enhanced dissolution rates and antimicrobial activity.
Methods: Six different polymers were used to prepare FLU nanopolymeric particles: hydroxyl propyl methylcellulose (HPMC), poly (vinylpyrrolidone) (PVP), poly (vinyl alcohol) (PVA), ethyl cellulose (EC), Eudragit (EUD), and Pluronics®. A low-energy method, nanoprecipitation, was used to prepare the polymeric nanoparticles.
Results and conclusion: The combination of HPMC-PVP and EUD-PVP was found most effective to produce stable FLU nanoparticles, with particle sizes of 250 nm ±2.0 and 280 nm ±4.2 and polydispersity indices of 0.15 nm ±0.01 and 0.25 nm ±0.03, respectively. The molecular modeling studies endorsed the same results, showing highest polymer drug binding free energies for HPMC-PVP-FLU (-35.22 kcal/mol ±0.79) and EUD-PVP-FLU (-25.17 kcal/mol ±1.12). In addition, it was observed that Ethocel® favored a wrapping mechanism around the drug molecules rather than a linear conformation that was witnessed for other individual polymers. The stability studies conducted for 90 days demonstrated that HPMC-PVP-FLU nanoparticles stored at 2°C–8°C and 25°C were more stable. Crystallinity of the processed FLU nanoparticles was confirmed using differential scanning calorimetry, powder X-ray diffraction analysis and TEM. The Fourier transform infrared spectroscopy (FTIR) studies showed that there was no chemical interaction between the drug and chosen polymer system. The HPMC-PVP-FLU nanoparticles also showed enhanced dissolution rate (P<0.05) compared to the unprocessed counterpart. The in vitro antibacterial studies showed that HPMC-PVP-FLU nanoparticles displayed superior effect against gram-positive bacteria compared to the unprocessed FLU and positive control.
Keywords: fluticasone, nanoparticles, drug delivery systems, antimicrobial, molecular modeling, molecular dynamics
Introduction
In drug discovery, the number of poorly soluble drugs has been amplified, with nearly 70% of new drug molecules discovered in recent years with poor water solubility.1 Due to such physicochemical properties, formulation of the drug into an effective medicine with acceptable bioavailability and therapeutic activity is one of the major problems encountered with formulation development of new chemical entities (NCEs) as well as for generic medicine development. More than 40% of NCEs developed in the pharmaceutical industry are practically insoluble in water.2 Oral bioavailability depends on various factors including aqueous solubility, drug permeability, dissolution rate, first-pass metabolism and presystemic metabolism.2 The most common causes of impaired oral bioavailability are poor solubility and low permeability.2 Therefore, in vitro dissolution has been recognized as a key element in drug development and thus enhancing the dissolution rate of poorly water soluble drugs may have a significant impact on their bioavailability and hence their therapeutic activity.3,4
Fluticasone (FLU), a potent corticosteroid with high topical activity, is a member of a wide class of synthetic glucocorticoids that have been extensively used to treat many diseases, such as autoimmune disorders, inflammation, asthma and allergies, adrenal insufficiency, cancer and many other health-related conditions.5–7 FLU propionate (FP; Heifi Evergreen Chemical Industry Co. Ltd, Hefei, China) is used to treat asthma, allergic rhinitis, nasal polyps, various skin disorders,8 Crohn’s disease,9 ulcerative colitis10 and eosinophilic esophagitis.11 FP has also been shown to have potential antimicrobial activities and found to be effective against a wide range of bacterial strains,12,13 and that its antimicrobial activity was more effective than other steroids.13 In addition to its use as a single entity for treating bacterial infections,12,13 it is also being included as an adjunct with other antibiotics for enhancing antimicrobial drug therapy.14 Approaches to enhance its dissolution profile and antimicrobial potential will contribute in enhancing its role in treating antibacterial infection.
According to the Biopharmaceutical Drug Classification System (BCS), Class II drugs, eg, FLU, prednisolone, etc, have poor aqueous solubility and good permeability. Approaches to enhance the solubility of poorly soluble drugs, such as micronization,15 nanonization,16,17 drug polymer complexation (eg, cyclodextrins),18 liposomal formulations19,20 and solid dispersions,21–23 have been applied to improve the dissolution profile and oral drug bioavailability. Interest has increased considerably over the past few decades in developing biodegradable nanoparticles as a drug delivery system. Nanodrug delivery system (NDDS) improves drug therapy outcomes by multiple mechanisms.24 They include: 1) targeted delivery to infected and inflamed sites, 2) inhibited and competitive binding to drug efflux pumps, such as P-gp,25 3) direct interaction with cell membranes leading to enhanced cellular drug entry,26,27 4) promoted drug uptake via endocytosis mechanisms,28 5) sustained drug release profiles to decrease frequency of administration, and 6) improved physicochemical properties.29,30
Research related to nanoparticle formulations of FLU appears to be a current gap in the literature. One study reported the use of nanoparticle agglomerates of FLU as a unique approach to obtain high performance aerosols for localized treatment of lung diseases and as an alternative to systemic drug therapy.31,32 Another study reported the design of nanosized FP nasal spray formulation to overcome the limited nasal permeability.33 For the topical delivery of FP, nanostructured lipid carriers (NLCs) were developed to further improve its safety profile and decrease the adverse side effects commonly reported in topical corticotherapy.34 Co-nanopolymeric/multipolymeric nanoparticles can enhance the performance of drug-loaded nanoparticles by improving the physicochemical features of the resulting drug–polymer complex, such as drug release, solubility and stability profiles.35,36 To date, no study has reported the design of co-nanopolymeric FLU formulations with enhanced dissolution rate for antimicrobial potential.
The aim of the current study was to: 1) identify optimal polymeric combinations to prepare FLU nanopolymeric particles with enhanced dissolution and release profiles for potential antimicrobial activity, and 2) to characterize the physicochemical properties of these polymeric particles in terms of their size, surface charge, thermal profile, drug release, morphology and antibacterial activity. Additionally, this study aimed to investigate the antibacterial potential of the FLU nanoparticles compared to their unprocessed form. Hybrid experimental and molecular modeling approaches were therefore applied in order to design stable FLU nanopolymeric particles with enhanced dissolution rate and antimicrobial activity.
In this study, a range of polymers were used and included hydroxyl propyl methylcellulose (HPMC; BASF, Ludwigshafen, Germany), Eudragit RS100® (EUD; Anhui Sunhere Pharmaceutical Excepients Co., Ltd, Huainan, China), ethyl cellulose (EC) and poly (vinyl alcohol) (PVA; Sigma-Aldrich, St Louis, MO, USA), with poly (vinylpyrrolidone) (PVP; BASF) and Pluronics® (Sigma-Aldrich) being used to investigate the impact of polymers on the FLU properties, and the nanoparticles being fabricated with individual polymers and combinations of polymers. The particle size and zeta potential of various nanoparticle formulations were determined, and the surface morphology of selected formulations was determined using a scanning electron microscope (SEM). Fourier transform infrared spectroscopy (FTIR) of selected formulations was done to check for any drug polymer interaction. The crystallinity of FLU in selected formulations was found by performing X-ray diffraction. The comparative in vitro dissolution and antibacterial studies were carried out to evaluate and compare the performance of unprocessed and processed nanoparticles. Molecular modeling studies were conducted to rationalize and investigate the structural and dynamic features of these nanoparticles, as well as the interactions between the drugs and polymers.
The comprehensive information gained from such extensive combined experimental and molecular modeling approaches will provide a deeper insight into the effect of nanopolymeric formulations on the antimicrobial activity of FLU.
Materials and methods
This section is divided into: 1) an experimental part, which includes details related to the preparation and analysis of polymeric nanoparticles, as well as the antibacterial activity, and 2) molecular modeling.
Experimental
FLU was gifted by Valor Pharmaceuticals, Islamabad, Pakistan. EUD, HPMC, Ethocel® (Dow Chemical Company, Midland, MI, USA), Pluronics, PVA and PVP were gifted by Focus and Rulz Pharmaceuticals (Pvt) Ltd., Islamabad, Pakistan. Other materials, such as mannitol (cryoprotectants), acetone, methanol, ethanol, n-hexane, ethyl acetate, dichloromethane, distilled water etc, were of analytical grade.
Preparation of FLU nanoparticles
Eudragit L100 drug-loaded nanoparticles
The Eudragit L100 drug-loaded nanoparticles of FLU were formulated by a solvent displacement or nanoprecipitation method.37–39 The drug and polymer were dissolved in a 20 mL mixture of acetone and methanol at different ratios (1:3). The polymer–drug solutions (internal phase) were added dropwise into 200 mL of the aqueous solution (external phase) and the mixtures were then stirred at 1,000 rpm for 6 hours. Through rotary vacuum evaporation under a water bath at 32°C, the organic phase was fully evaporated.
EC drug-loaded nanoparticles
The EC drug-loaded nanoparticles of FLU were prepared by a solvent displacement or nanoprecipitation method.40–42 The drug and polymer were dissolved in a 20 mL mixture of acetone and methanol at different ratios (1:3). The organic internal phase was added at a slow rate dropwise into 200 mL of the aqueous solution as an external phase containing 1% sodium dodecyl sulfate and 1.5% of poloxamer, and then stirred at 1,000 rpm for 6 hours.
HPMC-EUD drug-loaded nanoparticles
Nanosuspensions of FLU were synthesized by the following two-step procedure.41 First, the drug and polymer were dissolved in 20 mL of a mixture of acetone and methanol at a ratio of 1:3. Water was used as an antisolvent, with the ratio of acetone and water being 1:20. Second, the clear drug solution was injected slowly into 200 mL aqueous solution containing HPMC (1% w/w) under continuous magnetic stirring at a speed of 1,000 rpm for 3 hours. The organic phase was completely removed using a vacuum rotary evaporator at room temperature for 2 hours.
HPMC-PVP drug-loaded nanoparticles
Nanosuspensions of FLU were synthesized using a similar two-stage procedure.41 First, the FLU solution was prepared in 10 mL of acetone, with water being used as an antisolvent; the ratio of acetone and water being 1:20. Second, the clear drug solution was injected slowly into 200 mL aqueous solution containing HPMC and PVP K30 (1% w/w) under continuous magnetic stirring at a speed of 1,000 rpm for 3 hours. The organic phase was completely removed using a vacuum rotary evaporator at room temperature for 2 hours.
Eudragit (EUD)-PVA and EUD-PVP drug-loaded nanoparticles
Nanosuspensions of FLU were also synthesized using the following two-step procedure.41 The drug and polymer (EUD) were dissolved in 20 mL of a mixture of acetone and methanol (1:3), with water being used as an antisolvent; the ratio of acetone and water being 1:20. Second, the clear drug solution was injected slowly into 200 mL aqueous solution containing HPMC (1% w/w) and PVA under continuous magnetic stirring at a speed of 1,000 rpm for 3 hours. For EUD-PVP, the aqueous phase contained 1% PVP solution, and the organic phase was completely removed using a vacuum rotary evaporator at room temperature for 2 hours.
Pluronic, PVA, PVP and PVP-PVA-loaded nanoparticles
Likewise, the nanosuspensions of FLU were synthesized by the following two-step procedure.41 Initially, the FLU solution was prepared in 10 mL of acetone, with water being used as an antisolvent; the ratio of acetone and water being 1:20. Second, the clear drug solution was injected slowly into each 200 mL aqueous solution containing PVA, PVP K30 and Pluronics under continuous magnetic stirring at a speed of 1,000 rpm for 3 hours. For the PVP-PVA-FLU nanoparticles, the antisolvent phase was composed of a PVA and PVP solution (1% w/v). The organic phase was completely removed using a vacuum rotary evaporator at room temperature for 2 hours.
Collection of nanoparticles
For each formulation, the prepared milky suspension was centrifuged at 10,000 rpm for 15 minutes using a Sigma 1-14K, UK microcentrifuge, with the supernatant being decanted. The sediments were redispersed in 0.5% mannitol solution, centrifuged at 10,000 rpm for 10 minutes, and the supernatant fluid discarded. Mannitol solution was used as a cryoprotectant, with the final nanoparticles being frozen at −20°C, then lyophilized at −50°C for 5 hours to obtain a dry powder, and stored at 40°C.
Particle size and zeta potential measurements
Dynamic light scattering (DLS) (Zeta Sizer Nano-ZS90; Malvern Instruments, Malvern, UK) was used to measure the average particle size and zeta potential of the FLU nanoparticles at room temperature. The nanoparticles were dispersed in deionized water and stirred with a magnetic stirrer at 2,000 rpm to decrease the aggregates, with all the samples being analyzed in triplicate.
Determination of drug incorporation efficiency
A total of 10 mg of lyophilized nanoparticles was dissolved in 20 mL dimethyl sulfoxide (a universal solvent), with the FLU in the solution being measured by ultraviolet (UV) spectroscopy at 235 nm (3000 UV/Visible Spectrophotometer; ORI, Hille Deutschland, Germany). The drug content (% w/w) and drug encapsulation efficiency (%) were calculated from equations (1) and (2), respectively, with the individual values for the two replicate determinations and their mean values being reported.
|
|
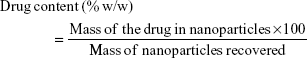
|
|
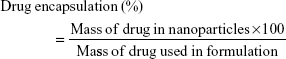
Scanning electron microscopy
The surface morphology of particles was imaged using an SEM (Nova NanoSEM; FEI Company, Hillsboro, OR, USA), after which the nanoparticles were placed on a cover glass slide that was coated with gold, and dried in a desiccator. The samples were examined at 10–15 kV to determine the morphology and shape of nanoparticles of FLU.
Fourier transform infrared spectrometry (FTIS)
FTIS spectra were obtained on an IRAffinity-1S FTIR spectrometer (Shimadzu, Japan), and the samples for the FTIR analysis were prepared in potassium bromide (KBr) disks (having 2 mg sample in 200 mg KBr). The sample measurement was done in the range from 500 to 4,000 cm−1 using a resolution of 1 cm−1. The FTIR data were analyzed using Origin 6.1 Software (version 6.1; OriginLab Corporation, Northampton, MA, USA) to identify the characteristic peaks.
Differential scanning calorimetry (DSC)
The heat of fusion and melting point determinations of the unprocessed FLU and optimized nanoparticles of the FLU were performed using a Differential Scanning Calorimeter (Mettler Toledo®; Columbus, OH, USA). The sample was placed in an empty aluminum pan and sealed, and then heated at a heating rate of 10°C min−1 from 25°C to 350°C at a dry nitrogen flow rate of 50 mL/minute, with all samples being analyzed in triplicate.
Powder X-ray diffraction analysis (PXRD)
The crystalline and amorphous nature of the unprocessed and optimized nanoparticles of the FLU was identified by PXRD studies, with the diffractometer (D8 ADVANCE; Bruker Corporation, Billerica, MA, USA) being used to perform this characterization. The unprocessed and processed samples were analyzed by plastic and silicon well sample holders, respectively. The sample and the unprocessed drug were scanned in the range of 0° ≤2θ ≤50° in triplicate by using copper Kα as a radiation source with 1 mm slit at 1.542 Å wavelength. The step size was 0.05°, and the time lapse between the steps was 2 seconds.
In vitro dissolution studies
In vitro drug release studies were performed using a USP Apparatus 2 paddle method (Curio DL 2020), and the lyophilized FLU nanoparticles (HPMC-PVP-FLU and EUD-PVP-FLU) were added to a microdialysis tube having (2–4 mg) FLU. Each sample was put into a vessel containing 500 mL dissolution medium with a temperature of 37°C±0.5°C (pH 6.8 phosphate buffer) at a paddle speed of 100 rpm, as previously reported.43 Each sample (5 mL) was processed at 5, 10, 15, 30, 45 and 60 minutes, and an equal volume of fresh medium was added to the vessel. The samples were filtered and quantified using UV spectroscopy at a wavelength of 235 nm (3000 UV/Visible Spectrophotometer; ORI), with all samples being analyzed in triplicate.
Antibacterial testing
Preparation of bacterial and fungal cultures
Strains of gram-positive (Staphylococcus aureus and Bacillus subtilis) and gram-negative bacteria (Escherichia coli, Salmonella sp.) were cultured, respectively, on nutrient agar, with Sheep Mueller–Hinton Agar media being used for Streptococcus pneumoniae. The produced cultures were then tested for antimicrobial effects under the standard environmental conditions.
Antimicrobial disk diffusion assay
The culture media of nutrient agar were prepared and microbes cultivated by a spreading technique using the method reported by Appalasamy et al.44 For the negative and positive controls, filter paper disks, impregnated with 1 μL of HPMC-PVP solution in water and streptomycin, were used as a negative and positive control, respectively. HPMC-PVP-FLU nanoparticles and unprocessed FLU were impregnated on the filter paper disks, and the produced cultures were then incubated under the standard environmental conditions. During the incubation period of 48 hours, the diameters of the zones of inhibition were measured every 6 hours, with all tests being performed in triplicate.
Minimum inhibition concentration (MIC) measurement
The MIC was determined for each microbe, based on the smallest concentrations of FLU nanosuspensions (processed) and its unprocessed form. Dilutions of FLU nanosuspensions, and its unprocessed form, were prepared using the sample at a concentration of 0.1–3 mg/mL. On each plate of test microbes, six disks of all the six concentrations were impregnated, with all tests being done in triplicate. The minimum bactericidal concentration (MBC) value was determined by subculturing the test dilution (which showed no visible turbidity) onto freshly prepared nutrient agar media, after which the plates were incubated for a further 18–42 hours at 37°C. The highest dilution that yielded no single bacterial colony on the nutrient agar plates was taken as the MBC.
Statistical analysis
All required tests were performed in triplicate, and the results were expressed as mean ± standard mean error. Mean values were compared using analysis of variance (ANOVA), and the difference was considered statistically significant at P<0.05, using Statistics 8.1 software (Version 8.1; SAS Institute Inc., Cary, NC, USA).
Molecular modeling
Polymeric structures modeling and docking calculations
Twelve structures of FLU polymers were built up and the energy minimized using Schrodinger’s Maestro Molecular modeling suite.45 First, pKa calculations were performed at the experimental pH to assign the correct protonation states for the studied molecules. Two different tools were used to compute the pKa at the experimental pH (neutral): MarvinSketch46 and Maestro,45 to ensure the validity and reliability of the results, with both tools predicting the same protonation states. AutoDock Vina was used for docking calculation,47 and Geister partial chargers were allocated during docking. Chimera modeling suite48 was used for visualizing and analyzing the docking calculations. The grid box was set to cover the entire polymer to ensure that all possible interactions with the drug were searched. The Lamarckian Genetic Algorithm in AutoDock Vina was used for the docking calculations.
Molecular dynamics (MD) and postdynamic analysis
Unrestrained all-atom MD simulations were performed using the GPU version of the PMEMD engine in Amber14 software package.49 The restrained electrostatic potential (RESP) procedure50 was used to calculate the partial atomic charges for the ligands at the HF/6-31G* level using the Gaussian 09 package.51 The systems were solvated in a cubic box of TIP3P52 water, such that all atoms were within 10 Å of a box edge. Long-range electrostatic interactions were treated by the Ewald method53 and a van der Waals cutoff of 12 Å. Each of the systems was minimized for 5,000 steps (2,500 steepest descent followed by 2,500 steps of the conjugate gradient). The Langevin thermostat, with a collision frequency of 1.0 ps−1 with harmonic restrained of 5 kcal/mol Å on the solutes, was applied during the gradual heating up of the systems to a temperature of 300.00 K in the canonical ensemble for 50 ps. This was followed by 50 ps of density equilibration in NPT ensemble and a final 500 ps equilibration at 300.00 K, 1 bar pressure and a coupling constant of 2 ps, and by a MD production run of 20 ns. All the bond lengths involving hydrogen atoms were constrained using SHAKE algorithm.54 Chimera,55 VMD,56 Ligplus57 and Pymol58 packages were used for visualization and graphical representations, while the polymers were set as a receptor (host) and the FLU was set as a ligand drug (guest).
Thermodynamic calculations
The molecular mechanics/generalized born surface area (MM/GBSA) method was used to calculate the binding affinities for ligand–receptor complexes.59–62 MM/GBSA has been proven to be more computationally efficient, hence widely used in drug design.63 The following set of equations describes the calculation of the binding free energy (ΔG):
|
|
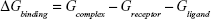
|
|
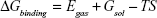
|
|
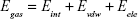
|
|
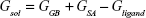
|
|
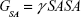
where Egas signifies gas-phase energy; Eint signifies internal energy and Eele and Evdw represent the electrostatic and van der Waals contributions, respectively. The Egas was directly evaluated from the FF12SB force field terms. The solvation energy (Gsol) is the summation of contributions from the polar states, GGB, and nonpolar states, GSA. The GGB is derived from solving the GB equation, whereas GSA contribution is estimated from the solvent accessible surface area (SASA) determined using a water probe radius of 1.4 Å. T and S represented the temperature and total solute entropy, respectively.
Results and discussion
Experimental
Preparation and characterization of various polymeric FLU nanoparticles
Different polymeric nanoparticles of FLU were fabricated by the antisolvent precipitation method. The polymers were either solubilized in aqueous medium or in organic phase, followed by infusion into a respective antisolvent phase. The particle sizes of the FLU nanoparticles produced by single and different combinations of polymers were determined. The combinations of HPMC-PVP and EUD-PVP were found to be the most suitable combinations to produce the FLU nanoparticles, with smallest particle sizes of 250.0 nm ±2.0 and 280±4.0, respectively (Table 1 and Figure 1).
 | Figure 1 Particle size measurements of HPMC-PVP-FLU nanoparticles (A) and EUD-PVP-FLU nanoparticles (B). |
 | Table 1 Particle size measurements of different polymer-FLU nanoparticles |
This demonstrated that the combinations of HPMC-PVP and EUD-PVP were able to be sufficiently adsorbed onto the surface of FLU nanoparticles, which would lead to stabilization of the nanoparticles by strong repulsion and consequently prevent their aggregation.64 Additionally, the said combinations of polymers resulted in a narrow size distribution of the produced FLU nanoparticles with polydispersity index (PDI) values, of 0.15±0.01 and 0.25±0.03, respectively. The range of PDI value for a stable dispersion has been reported to be <0.5.65 The zeta potential values for the HPMC-PVP-FLU and EUD-PVP-FLU nanoparticles were found to be −45.0 and −40.0 mV, respectively. These values also support the stability of the produced polymeric nanoparticles, as for stable nanosuspensions, the zeta potential value for electrostatically stabilized nanosuspensions should be over ±30.0 mV, which prevents agglomeration of the approaching particles.65 In contrast to other polymeric nanoparticles, HPMC-PVP-FLU and EUD-PVP-FLU showed maximum encapsulation efficiency (>90.0%) with maximum drug loading. Thus, initial screening identified HPMC-PVP-FLU and EUD-PVP-FLU as optimized formulations for further characterization.
Morphology of HPMC-PVP-FLU and EUD-PVP-FLU nanoparticles
Figure 2 shows the SEM micrographs of HPMC-PVP-FLU and EUD-PVP-FLU nanoparticles.
 | Figure 2 SEM micrographs of HPMC-PVP-FLU (A) and EUD-PVP-FLU (B) nanoparticles. |
It is evident that EUD-PVP-FLU nanoparticles were more crystalline compared to the HPMC-PVP-FLU nanoparticles. The EUD-PVP-FLU nanoparticles were cubic, rod, and rectangular in shape, while the HPMC-PVP-FLU nanoparticles were spherical and prism shaped. This indicates that the HPMC-PVP-FLU was less crystalline than the EUD-PVP-FLU, which may be due to the solvent, FLU and polymer interactions that can potentially change the morphology of the produced nanoparticles.66,67 Furthermore, all the particles were found to be homogeneously distributed, and no deposition of the produced nanoparticles was observed, which is in strong agreement with the Zetasizer data.
PXRD and DSC studies
The comparative DSC and PXRD studies showed that the HPMC-PVP-FLU nanoparticles were crystalline in nature and maintained their physical form. A sharp melting peak for unprocessed FLU (287°C) was observed; however, the melting point for HPMC-PVP-FLU (278°C) nanoparticles was slightly reduced, with a broader peak (Figure 3). The broadening of the melting point peaks and slight reduction for the nanoparticles could be due to the small particle size and lower packing density.64,68 In addition, the adsorbed polymer can also cause a broadening and a reduction of the melting point for the nanoparticles.69 Moreover, lower chemical potential of the drug in polymer blend leads to a lower melting point compared to the pure drug.70 The melting point of the processed drug in solid dispersion can also be reduced due to dissolution of the drug in polymer matrix.71
 | Figure 3 DSC studies of unprocessed FLU and processed (HPMC-PVP-FLU) nanoparticles. |
The PXRD patterns of the unprocessed and processed FLU nanoparticles are shown in Figure 4, which clearly indicates the crystalline nature of the produced nanoparticles. Nevertheless, for the processed samples, the peaks were less intense and some of the peaks disappeared. The reduction of peak intensity and their disappearance have been previously reported and are due to the small size and polymer traces.67,68,72
 | Figure 4 XRD studies of unprocessed FLU and processed FLU (HPMC-PVP-FLU). |
FTIR
The comparative FTIR studies showed the stable nature of the FLU, as no chemical interaction of the FLU and the chosen polymeric system was observed (Figure 5).
 | Figure 5 FTIR studies of unprocessed FLU and polymeric FLU nanoparticles. |
The FTIR spectra of both unprocessed and processed samples were compared and analyzed, with Figure 5 showing the FTIR spectrum of FLU and the processed FLU. The stretching of the −OH peak at 3,318.65 cm−1 suggested an external hydrogen bonding in the crystalline FLU. The peak at 1,740.33 cm−1 is attributed to the carbonyl functional group (C=O) attached to the aliphatic ring, and the carbonyl group attached to sulfur (S−C=O) showed a peak at 1,698.29 cm−1. The peak at 1,658.25 cm−1 showed the stretching of the carbonyl group (C=O), while the vibrational stretching of the quinonoid aromatic ring was recorded at 1,608.65 cm−1. The F−C−S stretching vibrations peak was denoted at 1,028.96 cm−1, while the C−F stretching vibrations peak was shown at 1,271.8 cm−1. The aromatic deformation (OOC/CCH) could be denoted at 882.14 cm−1.73 Both spectra for the processed and unprocessed FLU are similar, indicating no change in the structure. However, a little broadening of the peak and shift toward lower frequency (<3,318.65 cm−1), which exhibits hydrogen bond formation, have been observed. For hydrogen bond formation, the broadening of peak and shift toward lower frequency have also been previously reported in polymeric nanoparticles.74
Physical stability studies
Nanoparticles, due to their small size and large surface area, have high free energies, and hence controlling their size during nucleation and post nucleation is very challenging.75,76 Owing to their high free energies, the nanoparticles tend to aggregate and agglomerate, with subsequent fast particle growth due to Ostwald ripening.77 It is still an important issue to control the size of nanoparticles produced by the bottom-up method, where nucleation is the key stage to manipulate the particle sizes.78 Suitable polymeric medium can be optimized to control the particle size during the nucleation process,79 with Table 2 showing the particle sizes of different polymeric nanoparticles produced by a range of polymers over a period of 90 days.
 | Table 2 Monitoring of particle sizes of different FLU nanoparticles for 90 days at room temperature (25°C) |
The combination of HPMC-PVP was found to be the most suitable to effectively control the particle growth of FLU nanoparticles during the antisolvent precipitation process. The HPMC-PVP-FLU nanoparticles remained relatively stable for 90 days when stored at room temperature (25°C) (Table 2).
This study demonstrated that sufficient adsorption of the polymers occurred on the surfaces of the produced nanoparticles, which resulted in strong repulsion of the particles approaching each other and subsequent stabilization of the growing particles. The molecular modeling studies also suggested that HPMC-PVP-FLU had higher binding free energy (−35.22 kcal/mol) compared to other complexes, which can potentially produce the stable nanoparticles. In addition, EUD-PVP-FLU nanoparticles were found to be stable for 90 days compared to other nanoparticles, but comparatively less stable than HPMC-PVP-FLU nanoparticles. Owing to the little difference in binding free energies of the EUD-PVP-FLU (−25.17 kcal/mol), the particle size was large and unstable compared to the HPMC-PVP-FLU nanoparticles (−35.22 kcal/mol). It has been reported that a higher temperature can cause instability of the nanosuspensions, because the Brownian motion and kinetic energies of the suspended particles increase at an elevated temperature, which results in the aggregation and agglomeration of the particles.64 Furthermore, at higher temperature, viscosity is decreased80 and solubility of the suspended particles increases, which potentially leads to increase in interparticulate interaction, when particle growth can occur. For maximum stability of the nanosuspensions, Freitas and Müller81 recommended storing the nanosuspensions at a temperature range of 2°C–8°C.
Dissolution studies
The comparative dissolution studies of the unprocessed, processed and micronized FLU are presented in Figure 6.
 | Figure 6 Comparative dissolution studies of different forms of FLU particles. |
A substantial increase in the dissolution rate was observed in the HPMC-PVP-FLU nanoparticles when compared to the unprocessed FLU. Moreover, due to the small particle size, the dissolution rate of the processed FLU was also higher than that of the FLU microsuspension (8.0±2.5 μm) (Figure 6). Additionally, it was evident from the dissolution studies that more than approximately 70% and 68% of the FLU dissolved from the HPMC-PVP-FLU and EUD-PVP-FLU nanoparticles, respectively, in the first 2 minutes. The dissolution rate of the FLU from HPMC-PVP-FLU nanoparticles was slightly higher than that of the EUD-PVP-FLU nanoparticles, which is attributed to the greater value of binding free energy of the HPMC-PVP-FLU (−32.22 kcal/mol) compared to the EUD-PVP-FLU (−25.17 kcal/mol) (Table 3). In a similar study, we previously reported the relationship between the binding energy and the drug release,82 while this study showed that the polymeric nanoparticles of the FLU led to a faster release of the drug.
 | Table 3 MM/GBSA binding free energy calculations for different FLU polymeric complexes |
Antimicrobial assay
The zone of inhibition shows that the HPMC-PVP-FLU nanoparticles were more effective against gram-positive bacteria than the unprocessed FLU and positive control, whereas no significant difference was observed in the effects of the unprocessed FLU and standard drug (Table 4). However, both the unprocessed FLU and nanoformulation were found to be inactive against the gram-negative bacteria but not against the positive control. Both the unprocessed FLU and nanoformulation showed no activity against Escherichia coli and Shigella, while the positive control was found active against these microorganisms. These results clearly indicated that the processed nanoformulation was found highly effective, and a significant difference was observed among the values of the unprocessed FLU and its nanoformulation.
 | Table 4 Comparative antimicrobial activity of HPMC-PVP-FLU nanoparticles (mg/mL) and unprocessed drug using streptomycin (mg/mL) as positive control and HPMC-PVP in water as negative control by disk diffusion assay |
As shown in Table 5, the MIC values of the unprocessed FLU were found to be low at 0.62 mg/mL in the case of gram-positive bacteria, whereas against the gram-negative strains, both unprocessed and processed FLUs were found to be ineffective. The produced HPMC-PVP-FLU nanoparticles were found to be highly effective against the gram-positive bacterial strains, including Shigella, Streptococci, Staphylococci and Pneumocystis pneumonia. In the case of the MBC, in general, significantly higher values were recorded compared to the MIC (Table 5). It was noted that the HPMC-PVP-FLU nanoparticles were highly effective compared to the unprocessed FLU in terms of the MBC. Different strains of bacteria have different cell permeability, which plays a key role in the rate of large molecules penetration.83 In addition, nanoparticles have unique physicochemical properties, including large surface to mass ratio, ultra small size, high reactivity and unique interactions with biological systems.
 | Table 5 MIC and MBCs of HPMC-PVP-FLU nanoparticles and unprocessed drug |
Molecular modeling studies
MD simulations and system stability
MD simulations provide a useful tool to study biological systems in terms of their structural and dynamic features. In order to understand the binding mode between the drug and polymers used in this study and to explore the interaction mechanism and complex stability, MD simulations were performed for the docked complexes. The stability of the simulated systems was assessed by measuring the differences in the root mean square deviation (RMSD) in relation to the minimized structures, as well as the potential energy fluctuations of the complexes over the entire simulation time. To ensure a thorough investigation of the stability of the simulated structures, RMSD, kinetic energy and potential energy were monitored along the MD trajectories. All simulated systems under investigation were well equilibrated, and the conformational stability was observed throughout the simulations.
Binding free energy calculations and conformational analysis
The relative binding free energies between the FLU and different polymeric molecules were computed using MM/GBSA, and are shown in Table 3. Conformational analysis was performed to identify the lowest energy conformations.
The binding affinity calculation between a polymer (host) and a drug molecule (guest) will estimate the strength of the interactions between them, and subsequently the drug release rate. Generally speaking, tighter interactions between the drug molecules and polymer might lead to a stable drug–polymer complex and may result in a more sustained drug release profile than when compared to looser interaction/binding.84
As indicated in Table 3, a general observation was that the co-polymeric forms exhibited enhanced binding affinity to the drug when compared to monopolymeric structures (synergistic effect). It was also evident that the inclusion of PVP in the co-polymeric forms tends to significantly improve the binding affinity of the drug compared to the corresponding monopolymeric form. For instance, −35.22 versus −4.34 kcal/mol for HPMC-PVP and HPMC alone, respectively, and −25.17 versus −8.04 kcal/mol for EUD-PVP and EUD alone, respectively. Such significant improvement in binding affinity may be primarily due to the hydrophobic interactions of PVP (Figures 7 and 8). As the antibacterial activity was assessed for the HPMC-PVP-FLU, the 3D representation of this complex is provided in Figure 7.
 | Figure 7 The lowest energy conformation of HPMC-PVP-FLU complex from MD simulations: (A) showing how the FLU is encapsulated/wrapped within the HPMC/PVP cage and (B) showing the hydrogen bonding interactions between FLU and PVP/HPMC. |
 | Figure 8 The lowest energy conformation of EUD-PVP-FLU complex from MD simulations: (A) showing how the FLU is encapsulated/wrapped within the EUD/PVP cage and (B) showing the hydrogen bonding interaction between FLU and PVP/EUD. |
Among the monopolymeric structures, Ethocel exhibited the highest binding affinity (−20.89 kcal/mol), which may be due to the fact that the long chain of Ethocel allowed the molecule to wrap around the drug, achieving the highest possible binding interactions (Figure 9). On the extreme side, the PVA showed the least binding affinity, as the molecule was found to preserve linear conformation, which does not allow favorable interactions with the drug.
 | Figure 9 Ethocel®-FLU complex showing Ethocel chain wrapped around the FLU with a hydrogen bonding interaction between the drug (ball and sticks) and polymer (sticks). |
Nevertheless, co-polymeric nanoparticles showed enhanced physicochemical properties when compared to monopolymeric forms, placing the focus of the antimicrobial studies on the co-polymeric particles.
It is interesting to notice that the computational results are in reasonable agreement with the experimental data. For instance, HPMC-PVP and EUD-PVA were found to have a relatively higher binding affinity to the drug and more able to be sufficiently adsorbed onto the surface of the FLU, producing more stable nanoparticles, as is evident from the zeta potential values. Moreover, when compared to other nanopolymeric formulations, HPMC-PVP showed maximum encapsulation efficiency (>90%) with maximum drug loading.
Conclusion
This study concluded that the selection of suitable polymers to produce stable polymeric nanoparticles is the key step, which consequently led to polymeric nanoparticles with high dissolution and enhanced antibacterial activity. The combinations of HPMC-PVP and EUD-PVP were found to be the most suitable in controlling the particle size and stability of the produced nanoparticles. The molecular modeling studies supported our experimental results and found high binding free energies for the said complexes of the polymers. This study also identified the strong antibacterial potential of FLU, which needs to be further explored.
Molecular modeling studies confirmed that the co-polymeric structures show better binding with the drug compared to the individual respective polymers. Compared to PVA, which favors a linear conformation, Ethocel was found to wrap its chain around the drug, allowing favorable interactions with it. The molecular level insight from the molecular modeling studies provided a useful platform to understand the interaction mechanisms between the drug and the polymers, which would be useful for designing and optimizing drug delivery systems.
Optimization of the produced FLU nanoparticles shows potential for development into an oral solid dosage form with subsequent improved therapeutic outcomes.
Acknowledgments
S Khan acknowledges the continuous efforts of the Research Centre, College of Pharmacy, and Deanship of Scientific Research at King Saud University, Riyadh, Saudi Arabia. S Khan also thanks the Lahore University of Management Sciences, Pakistan for providing the characterization facilities for the produced nanoparticles.
Disclosure
The authors report no conflicts of interest in this work.
References
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | ||
Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012;2012:195727. | ||
Costa P, Sousa Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. | ||
Hu J, Johnston KP, Williams RO. Nanoparticle engineering processes for enhancing the dissolution rates of poorly water soluble drugs. Drug Dev Ind Pharm. 2004;30(3):233–245. | ||
McKay LI, Cidlowski JA [webpage on the Internet]. Corticosteroids in the Treatment of Neoplasms. 2003. Available from: https://www.ncbi.nlm.nih.gov/books/NBK13383/. Accessed June 29, 2017. | ||
Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335(1):2–13. | ||
Wood AJJ, Barnes PJ. Inhaled glucocorticoids for asthma. N Engl J Med. 1995;332(13):868–875. | ||
Roeder A, Schaller M, Schäfer-Korting M, Korting HC. Safety and efficacy of fluticasone propionate in the topical treatment of skin diseases. Skin Pharmacol Physiol. 2004;18(1):3–11. | ||
de Kaski MC, Peters AM, Lavender JP, Hodgson HJ. Fluticasone propionate in Crohn’s disease. Gut. 1991;32(6):657–661. | ||
Angus P, Snook JA, Reid M, Jewell DP. Oral fluticasone propionate in active distal ulcerative colitis. Gut. 1992;33(5):711–714. | ||
Alexander JA, Katzka DA. Therapeutic options for eosinophilic esophagitis. Gastroenterol Hepatol (N Y). 2011;7(1):59–61. | ||
Philip L, Agbay R, Samantha C. In vitro antibacterial activity of mometasone furoate, fluticasone propionate and fluticasone furoate nasal preparations against Streptococcus pneumoniae, Haemophilus influenzae, Streptococcus viridians, Staphylococcus aureus, Pseudomonas aeruginosa, and E. Philipp J Otolaryngol Head Neck Surg. 2010;25:13–16. | ||
Chernuskiy VG, Govalenkova OL, Letyago GV. A concomitant antimicrobial activity of methylated and halogenated glucocorticosteroids against microorganisms isolated from the sputum of children with bronchial asthma. J V N Karazin Kharkiv Natl Univ Ser Med. 2015;29:22–26. | ||
Shahani SR, Jerajani HR, Sharma K, Cooverji ND. Comparative biorelease study of fluticasone in combination with antibacterial (Neomycin) and or antifungal (coltrimazol, miconazole) agents by histamine percutaneous reaction method in healthy volunteers. Indian J Dermatol Venereol Leprol. 2017;63(3):173–177. | ||
Feng S-S, Chien S. Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. | ||
Feng S-S. New-concept chemotherapy by nanoparticles of biodegradable polymers: where are we now? Nanomedicine. 2006;1(3):297–309. | ||
Feng SS. Nonoparticles of biodegradable polymers for new-concept chemotherapy. Expert Rev Med Devices. 2004;1(1):115–125. | ||
Ammar HO, Salama HA, Ghorab M, Mahmoud AA. Implication of inclusion complexation of glimepiride in cyclodextrin-polymer systems on its dissolution, stability and therapeutic efficacy. Int J Pharm. 2006;320(1–2):53–57. | ||
Kim H, Kim Y, Lee J. Liposomal formulations for enhanced lymphatic delivery. Asian J Pharm Sci. 2013;8(2):100–109. | ||
Guéritte-Voegelein F, Guénard D, Lavelle F, Le Goff MT, Mangatal L, Potier P. Relationships between the structure of taxol analogues and their antimitotic activity. J Med Chem. 1991;34(3):992–998. | ||
Bikiaris D, Papageorgiou GZ, Stergiou A, et al. Physicochemical studies on solid dispersions of poorly water-soluble drugs: evaluation of capabilities and limitations of thermal analysis techniques. Thermochim Acta. 2005;439(1–2):58–67. | ||
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. | ||
Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12(23–24):1068–1075. | ||
Martinez LR, Han G, Chacko M, et al. Antimicrobial and healing efficacy of sustained release nitric oxide nanoparticles against Staphylococcus aureus skin infection. J Invest Dermatol. 2009;129(10):2463–2469. | ||
Rahman A, Husain SR, Siddiqui J, et al. Liposome-mediated modulation of multidrug resistance in human HL-60 leukemia cells. J Natl Cancer Inst. 1992;84(24):1909–1915. | ||
Riganti C, Voena C, Kopecka J, et al. Liposome-encapsulated doxorubicin reverses drug resistance by inhibiting P-glycoprotein in human cancer cells. Mol Pharm. 2011;8(3):683–700. | ||
Kilchrist KV, Evans BC, Brophy CM, Duvall CL. Mechanism of enhanced cellular uptake and cytosolic retention of MK2 inhibitory peptide nano-polyplexes. Cell Mol Bioeng. 2016;9(3):368–381. | ||
Zheng C, Xu J, Yao X, Xu J, Qiu L. Polyphosphazene nanoparticles for cytoplasmic release of doxorubicin with improved cytotoxicity against Dox-resistant tumor cells. J Colloid Interface Sci. 2011;355(2):374–382. | ||
Duchene D, Cavalli R, Gref R. Cyclodextrin-based polymeric nanoparticles as efficient carriers for anticancer drugs. Curr Pharm Biotechnol. 2016;17(3):248–255. | ||
Feng X, Vo A, Patil H, et al. The effects of polymer carrier, hot melt extrusion process and downstream processing parameters on the moisture sorption properties of amorphous solid dispersions. J Pharm Pharmacol. 2016;68(5):692–704. | ||
El-Gendy N, Pornputtapitak W, Berkland C. Nanoparticle agglomerates of fluticasone propionate in combination with albuterol sulfate as dry powder aerosols. Eur J Pharm Sci. 2011;44(4):522–533. | ||
Ali R, Mittal G, Ali R, et al. Development, characterisation and pharmacoscintigraphic evaluation of nano-fluticasone propionate dry powder inhalation as potential antidote against inhaled toxic gases. J Microencapsul. 2013;30(6):546–558. | ||
Dai J, Ruan B, Zhu Y, Liang X, Su F, Su W. Preparation of nanosized Fluticasone Propionate nasal spray with improved stability and uniformity. Chem Ind Chem Eng Q. 2015;21(3):457–464. | ||
Doktorovová S, Araújo J, Garcia ML, Rakovský E, Souto EB. Formulating fluticasone propionate in novel PEG-containing nanostructured lipid carriers (PEG-NLC). Colloids Surf B Biointerfaces. 2010;75(2):538–542. | ||
Kadajji VG, Betageri GV. Water soluble polymers for pharmaceutical applications. Polymers (Basel). 2011;3(4):1972–2009. | ||
Ahmed S, Vepuri SB, Jadhav M, Kalhapure RS, Govender T. Identifying the interaction of vancomycin with novel pH-responsive lipids as antibacterial biomaterials via accelerated molecular dynamics and binding free energy calculations. Cell Biochem Biophys. Epub 2017 Mar 9. | ||
Ubrich N, Schmidt C, Bodmeier R, Hoffman M, Maincent P. Oral evaluation in rabbits of cyclosporin-loaded Eudragit RS or RL nanoparticles. Int J Pharm. 2005;288(1):169–175. | ||
Yen FL, Wu TH, Lin LT, Cham TM, Lin CC. Naringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl4-induced acute liver failure. Pharm Res. 2009;26(4):893–902. | ||
Zili Z, Sfar S, Fessi H. Preparation and characterization of poly-epsilon-caprolactone nanoparticles containing griseofulvin. Int J Pharm. 2005;294(1–2):261–267. | ||
Ravikumara NR, Madhusudhan B, Nagaraj TS, Hiremat SR, Raina G. Preparation and evaluation of nimesulide-loaded ethylcellulose and methylcellulose nanoparticles and microparticles for oral delivery. J Biomater Appl. 2009;24(1):47–64. | ||
Suthar AK, Solanki SS, Dhanwani RK. Enhancement of dissolution of poorly water soluble raloxifene hydrochloride by preparing nanoparticles. J Adv Pharm Educ Res. 2011;2:189–194. | ||
Bilati U, Allémann E, Doelker E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur J Pharm Sci. 2005;24(1):67–75. | ||
Shah SMH, Ullah F, Khan S, et al. Smart nanocrystals of artemether: fabrication, characterization, and comparative in vitro and in vivo antimalarial evaluation. Drug Des Devel Ther. 2016;10:3837–3850. | ||
Appalasamy S, Lo KY, Ch’Ng SJ, Nornadia K, Othman AS, Chan LK. Antimicrobial activity of artemisinin and precursor derived from in vitro plantlets of Artemisia annua L. Biomed Res Int. 2014;2014:215872. | ||
DeLano WL [homepage on the Internet]. The PyMOL Molecular Graphics System. Schrödinger LLC; 2002. Version 1. Available from: http://www.pymol.org. Accessed November 25, 2017. | ||
Olsen L, Samavati FF, Sousa MC, Jorge JA. Sketch-based modeling: a survey. Comput Graph. 2009;33(1):85–103. | ||
Trott O, Olson AJ. AutoDock Vina. J Comput Chem. 2010;31:445–461. | ||
Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157(1):281–287. | ||
Case DA, Cheatham TE, Darden T, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. | ||
Fox T, Kollman PA. Application of the RESP methodology in the parametrization of organic solvents. J Phys Chem B. 1998;102(41):8070–8079. | ||
Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09. Wallingford, CT: Gaussian, Inc.; 2009:2–3. | ||
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926. | ||
Harvey MJ, De Fabritiis G. An implementation of the smooth particle mesh Ewald method on GPU hardware. J Chem Theory Comput. 2009;5(9):2371–2377. | ||
Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23(3):327–341. | ||
Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. | ||
Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):27–38. | ||
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. | ||
Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8. 2015. Available from: https://pymol.org/2/support.html. Accessed January 3, 2017. | ||
Homeyer N, Gohlke H. FEW: a workflow tool for free energy calculations of ligand binding. J Comput Chem. 2013;34(11):965–973. | ||
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate – DNA helices. J Am Chem Soc. 1998;120(37):9401–9409. | ||
Godschalk F, Genheden S, Söderhjelm P, Ryde U. Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Phys Chem Chem Phys. 2013;15(20):7731–7739. | ||
Foloppe N, Hubbard R. Towards predictive ligand design with free-energy based computational methods? Curr Med Chem. 2006;13(29):3583–3608. | ||
Hou T, Wang J, Li Y, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods: I. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Comput Sci. 2011;51(1):69–82. | ||
Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. | ||
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
ter Horst JH, Geertman RM, van Rosmalen GM. The effect of solvent on crystal morphology. J Cryst Growth. 2001;230(1–2):277–284. | ||
Khan S, de Matas M, Zhang J, Anwar J. Nanocrystal preparation: low-energy precipitation method revisited. Cryst Growth Des. 2013;13(7):2766–2777. | ||
Bunjes H, Koch MHJ, Westesen K. Effect of Particle Size on Colloidal Solid Triglycerides. Hamburg, Germany: Langmuir; 2000. | ||
Valleri M, Mura P, Maestrelli F, Cirri M, Ballerini R. Development and evaluation of glyburide fast dissolving tablets using solid dispersion technique. Drug Dev Ind Pharm. 2004;30(5):525–534. | ||
Lin D, Huang Y. A thermal analysis method to predict the complete phase diagram of drug-polymer solid dispersions. Int J Pharm. 2010;399(1–2):109–115. | ||
Liu X, Lu M, Guo Z, Huang L, Feng X, Wu C. Improving the chemical stability of amorphous solid dispersion with cocrystal technique by hot melt extrusion. Pharm Res. 2012;29(3):806–817. | ||
O’Mahony M, Leung AK, Ferguson S, Trout BL, Myerson AS. A process for the formation of nanocrystals of active pharmaceutical ingredients with poor aqueous solubility in a nanoporous substrate. Org Process Res Dev. 2015;19(9):1109–1118. | ||
Ali HR, Edwards HG, Kendrick J, Scowen IJ. Vibrational spectroscopic study of fluticasone propionate. Spectrochim Acta A Mol Biomol Spectrosc. 2009;72(2):244–247. | ||
Sun TM, Du JZ, Yan LF, Mao HQ, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29(32):4348–4355. | ||
Verma S, Kumar S, Gokhale R, Burgess DJ. Physical stability of nanosuspensions: Investigation of the role of stabilizers on Ostwald ripening. Int J Pharm. 2011;406(1–2):145–152. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. | ||
Voorhees PW. The theory of Ostwald ripening. J Stat Phys. 1985;38(1–2):231–252. | ||
Dalvi SV, Dave RN. Analysis of nucleation kinetics of poorly water-soluble drugs in presence of ultrasound and hydroxypropyl methyl cellulose during antisolvent precipitation. Int J Pharm. 2010;387(1–2):172–179. | ||
Anwar J, Khan S, Lindfors L. Secondary crystal nucleation: nuclei breeding factory uncovered. Angew Chem Int Ed Engl. 2015;54(49):14681–14684. | ||
Afifi SA, Hassan MA, Abdelhameed AS, Elkhodairy KA. Nanosuspension: an emerging trend for bioavailability enhancement of etodolac. Int J Polym Sci. 2015;2015:1–16. | ||
Freitas C, Müller RH. Effect of light and temperature on zeta potential and physical stability in solid lipid nanoparticle (SLN™) dispersions. Int J Pharm. 1998;168(2):221–229. | ||
Seedat N, Kalhapure RS, Mocktar C, et al. Co-encapsulation of multi-lipids and polymers enhances the performance of vancomycin in lipid–polymer hybrid nanoparticles: in vitro and in silico studies. Mater Sci Eng C. 2016;61:616–630. | ||
Lambert PA. Cellular impermeability and uptake of biocides and antibiotics in Gram-positive bacteria and mycobacteria. J Appl Microbiol. 2002;92(s1):46S–54S. | ||
Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.