Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Exosomal miR-2110 from PM2.5-Exposed Lung Epithelial Cells Targets SRSF1 to Promote M1 Macrophage Polarization and Inflammatory Responses in COPD
Authors Miao Y, Gu J, Li J, Sheng D
Received 8 December 2025
Accepted for publication 11 March 2026
Published 24 March 2026 Volume 2026:21 584536
DOI https://doi.org/10.2147/COPD.S584536
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
Yehong Miao,1 Jianjun Gu,1 Juan Li,1 Danye Sheng2
1Department of Pulmonary and Critical Care Medicine, The Affiliated Hospital of Nantong University, Nantong, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Zhangjiagang First People’s Hospital, Zhangjiagang, People’s Republic of China
Correspondence: Danye Sheng, Email [email protected]
Purpose: Exposure to PM2.5 is a known contributor to the development of chronic obstructive pulmonary disease (COPD). As direct targets of PM2.5, bronchial epithelial cells participate in intercellular communication with macrophages and induce phenotypic changes in these immune cells. This study aimed to investigate the role and underly mechanism of epithelial cell-derived exosomes in regulating macrophage polarization.
Methods: Following PM2.5 exposure, exosomes were isolated from BEAS-2B cells and subsequently co-cultured with M0 macrophages. We measured the production of inflammatory cytokines and macrophage markers. The phenotypic effect of miR-2110 on macrophage polarization was examined in a COPD murine model, with subsequent exploration of relevant molecular pathways. The binding specificity of miR-2110 was assessed utilizing a luciferase reporter assay.
Results: In vitro analyses confirmed that P-Exo (PM2.5-exposed BEAS-2B cells exosomes) triggered M1 polarization, as evidenced by elevated expression of IL-6, TNF-α, and iNOS-2. Additionally, miR-2110 expression was upregulated in P-Exo. Inhibition of miR-2110 reduced M1 polarization and decreased inflammatory cytokine production both in vitro and in vivo. Luciferase assays confirmed that miR-2110 targeted SRSF1 expression. Overexpression of SRSF1 mitigated the regulatory role of miR-2110 in promoting M1 phenotype transition and pro-inflammatory cytokine production.
Conclusion: This work clarifies that exosomal miR-2110, which is produced from lung epithelial cells treated with PM2.5, exacerbates COPD and might be a viable target for COPD prevention and therapy.
Keywords: chronic obstructive pulmonary disease, PM2.5, exosomes, macrophage, miR-2110, SRSF1
Introduction
The pathophysiology of chronic obstructive pulmonary disease (COPD), a diverse set of lung disorders marked by a progressive deterioration of persistent respiratory symptoms, is directly linked to aberrant inflammatory reactions of the airways to dangerous particles and gases.1 Among the various hazardous pollutants in haze, PM2.5 is a major component. According to pertinent research, PM2.5 can considerably raise the risk of lung cancer death, impede lung function, and increase the prevalence of asthma and COPD, especially at concentration grades 3 and 4.2,3 As direct target cells of PM2.5, bronchial epithelial cells can be induced to initiate inflammatory responses and undergo cell death.4,5 A growing number of global populations‑based cohort studies have provided evidence that long‑term exposure to ambient PM2.5 is an independent risk factor for COPD incidence, acute exacerbations, hospitalization, and mortality.6,7 Furthermore, an increasing body of preclinical studies employs PM2.5 exposure in animals or cells to establish COPD‑like models.8,9 However, current treatment strategies for COPD remain largely palliative.10,11 In order to find innovative treatment strategies, future research should concentrate on clarifying the pathogenic processes of COPD.
Fundamental regulators of pulmonary homeostasis, bronchial epithelial cells and macrophages are central to mediating PM2.5-driven lung disease development including COPD.12,13 Exhibiting dual capacity in COPD, bronchial epithelial cells provide essential gatekeeper functions while coordinating inflammatory and immune regulation.14,15 As the primary immune effector cells in lung tissue and airways, macrophages exhibit high plasticity in response to different microenvironments. Demonstrating remarkable plasticity, macrophages differentiate into M1 or M2 polarized forms with distinct functional attributes.16 M1 macrophages are primarily pro-inflammatory and drive inflammatory processes, whereas M2 macrophages exert anti-inflammatory effects.17 For instance, through epithelial-macrophage crosstalk, cigarette smoke extract orchestrated M1-polarized activation that amplified lung tissue damage.18 These findings point to a dynamic exchange of signals between bronchial epithelial cells and macrophages, underscoring their reciprocal signaling. Nevertheless, the underlying mechanisms of epithelial-macrophage communication remain to be fully elucidated.
As nanoscale particles (40–160 nm), exosomes enable molecular transfer between cells by delivering functional components-proteins, various RNA species, and DNA-to recipient cells.19 For instance, in COPD, exosomes derived from airway epithelium delivered miRNA-125a-5p to macrophages, promoting their polarization toward the M1 phenotype and enhancing activation.20 Under cigarette smoke exposure, exosomes secreted by bronchial epithelial cells carrying miR-107 could induce macrophage polarization into a pro-inflammatory phenotype, thereby exacerbating acute lung injury.21 The study reveals exosomes facilitate bidirectional communication along the epithelial-macrophage regulatory axis through genetic content transfer. Based on this evidence, we hypothesize that exosomes may influence the progression of COPD through their packaged miRNAs.
In this study, we conducted in vitro and in vivo investigations to examine the role of exosomes derived from PM2.5-exposed lung epithelial cells in macrophage polarization and inflammatory responses in COPD. Furthermore, we demonstrated that these exosomes delivered miR-2110 into macrophages to alter their polarization state. Our findings provide deeper insights into the mechanisms of exosome-mediated communication between epithelial cells and macrophages and suggest a potential therapeutic target for COPD.
Materials and Methods
Cell Culture and Treatment
Three human cell lines-BEAS-2B (lung epithelial; CRL-3588), THP-1 (monocytic; TIB-202), and HEK293T were purchased from American Type Culture Collection (ATCC, Mansfield, Virginia, USA) and cultured under standard conditions (37°C, 5% CO2). BEAS-2B and HEK293T in complete DMEM (10% FBS, 1% penicillin-streptomycin; 21969035; Gibco, Grande Island, USA), and THP-1 in RPMI-1640 (11879020; Gibco) with 50 μM β-mercaptoethanol (21985023; Invitrogen, Waltham, MA, USA).
THP-1 cells were exposed to 100 ng/mL phorbol 12-myristate 13-acetate (PMA) for 48 hours in order to generate M0 macrophages.22 A stock solution of 5 μg/μL PM2.5 (SRM 1648a, NIST, Gaithersburg, Maryland, USA) was produced for PM2.5 exposure in accordance with earlier research.23 BEAS-2B cells were then exposed to a final concentration of 0, 100, 200 and 400 μg/mL PM2.5 for 48 hours.23 This exposure was repeated over five passages to simulate chronic PM2.5 treatment.
Isolation and Characterization of Exosomes
Using the ExoQuick-TC kit (EQPL10TC-1; System Biosciences, USA), exosomes from BEAS-2B cells that had either been left untreated or treated with PM2.5 were separated, and designated as control exosomes (C-Exo) and PM2.5- exosomes (P-Exo), respectively. Briefly, the supernatant from BEAS-2B cultures was centrifuged at 3000 × g for 30 minutes, followed by a second centrifugation at 10,000 × g for 20 minutes. To eliminate microparticles, the supernatant was then passed through a 100 kDa ultrafiltration apparatus (Amicon Ultra-15, Millipore, Billerica, Massachusetts, USA). Following overnight incubation with ExoQuick-TC, the mixture was sequentially centrifuged at 1500 × g (30 min) and 3000 × g (5 min) to isolate exosomal pellets after supernatant aspiration. Finally, the exosome pellet was resuspended in 200 μL of phosphate buffer saline (PBS) for subsequent analysis.
The morphological features of the isolated exosomes were examined using transmission electron microscopy (TEM) (HT7800, Hitachi, Tokyo, Japan). The size distribution of the exosomes was analyzed with a nanoparticle tracking analysis (ZetaView PMX 110, Particle Metrix, Amsterdam, Munich, Germany). Protein expression levels of exosomal surface markers (CD63 and CD9) and the negative marker Calnexin were detected by Western blotting.
Western Blotting
Exosomes were manually homogenized using a glass homogenizer and lysed on ice with Radio Immunoprecipitation Assay (RIPA) buffer for 30 minutes to extract proteins. Following 10% SDS-PAGE separation (P0012A; Beyotime, Shanghai, China) and wet transfer to PVDF membranes (0.22 μm), blots were blocked with skim milk/TBS (1 hour) and incubated with primary antibodies (4 °C, overnight). After washing with TBST, the membrane was incubated with a horseradish peroxidase (HRP)-conjugated rabbit IgG secondary antibody (1:500; ab288151, Abcam, Cambridge, UK). Protein bands were visualized using an enhanced chemiluminescence (ECL) detection kit (P0018S; Beyotime). Antibodies used in this study included anti-CD63 (1:1000; ab134045, Abcam), anti-CD9 (1:1000; ab236630, Abcam), anti-Calnexin (1:1000; ab22595, Abcam), and GAPDH (1:1000; ab9485, Abcam).
Co-Culture System of Exosomes or BEAS-2B Cells with Macrophages
A Transwell™ system with 0.4 μm pore-sized membranes (Corning, Conyngham, New York, USA) was used to establish the co-culture of macrophages and BEAS-2B cells. THP-1 cells were seeded in the upper chamber and differentiated into M0 macrophages by treatment with PMA. In a 6-well transwell setup, BEAS-2B cells were cultured in the basal compartment with M0 macrophages in the upper chamber insert. After 48 hours of co-culture, macrophages were collected for further examination.
For the exosome-macrophage co-culture group, M0 macrophages were treated with 200 μL of C-Exo or P-Exo and incubated for 48 hours. Subsequently, macrophages were harvested for subsequent experiments.
Uptake Assay of Exosomes
According to established methodology, exosomes were labeled with the PKH26 Red Fluorescent Cell Linker Kit (MX4021; Sigma, USA).24 Following PBS washes, THP-1 cells were incubated with PKH26-labeled exosomes. To assess cellular uptake, nuclei were counterstained with DAPI and fluorescence imaging was performed using a Zeiss microscope (Zeiss, Oberkochen, Baden-Württemberg, Germany).
Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
Total RNA was extracted from cells or tissues using TransZol Up (ET111-01-V2; TransGen Biotech, Beijing, China). mRNA analysis involved cDNA synthesis with PrimeScript RT kit (RR014A; TaKaRa, Tokyo, Japan) followed by SYBR Green-based qPCR (4385617; Invitrogen). For miRNA quantification, we employed the TaqMan miRNA Reverse Transcription Kit and Expression Assay Kit (A28007; Invitrogen) for RT-qPCR detection. U6 and GAPDH were used as internal controls for miRNA and mRNA normalization, respectively. Primer sequences are listed in Supplementary Table 1.
Enzyme-Linked Immunosorbent Assay (ELISA)
Supernatants from macrophage cultures, blood samples, and bronchoalveolar lavage fluid were collected and centrifuged. The levels of IL-6 (ab178013), IL-10 (ab185986), IL-17 (ab216167), and IL-22 (ab216170) were measured using the following commercial ELISA kits (Abcam).
Analysis of GEO Databases
Data were sourced from the Gene Expression Omnibus database using the identifiers GSE156572 and GSE218571. GSE156572 contains miRNA expression profiles of extracellular vesicles (EVs) isolated from the conditioned medium of BEAS-2B cells. GSE218571 includes miRNA expression data of EVs obtained from bronchoalveolar lavage fluid of 24 subjects with stable mild or moderate COPD and 20 healthy controls. Venn analysis was performed to identify miRNAs commonly enriched in EVs from both datasets. The research protocol was approved by the Ethics Committee of The Affiliated Hospital of Nantong University (Approval No. 2025-L037). All experiments and procedures were performed according to the Declaration of Helsinki (as revised in 2013).
Cell Transfection
All oligonucleotides (miR-2110 mimic, miR-2110 inhibitor, corresponding negative controls) and plasmids (oe-SRSF1, oe-NC) were provided by GenePharma (Shanghai). For transfection, BEAS-2B or THP-1 cells were transfected at 80% confluency using Lipofectamine 3000 (L3000150; Invitrogen) per manufacturer’s protocol.
Construction and Grouping of Mouse COPD Model
All experiments used weight-matched female C57BL/6 mice (6–7 weeks) maintained in SPF facilities with ad libitum water and standard diet. The mice were randomly divided into six groups (n=6 each):
- Control group (Con): Air exposure and no exosome treatment was administered.
- PM2.5 exposure group (PM2.5)
- PM2.5 exposure + scrambled antagomir treatment (PM2.5 + Scr)
- PM2.5 exposure + miR-2110–specific antagomir treatment (PM2.5 + miR-2110 Ant)
- Air exposure + exosomes from NC inhibitor-transfected BEAS-2B cells after PM2.5 treatment (PM2.5 + NC inhibitor-Exo)
- Air exposure + exosomes from miR-2110 inhibitor -transfected BEAS-2B cells after PM2.5 treatment (PM2.5 + miR-2110 inhibitor-Exo)
Using an inhalation exposure system (Raymain, Shanghai, China), mice in the clean air and PM2.5 groups were housed in polycarbonate chambers and exposed to filtered air or 600 μg·m−3 PM2.5 respectively.25 The exposure was conducted for 4 hours per day, 5 days per week, over a period of 12 weeks. Starting from the second week, a subset of mice received weekly tail vein injections of 12 μL antagomir (1 × 109 TU/mL) for 12 weeks. The exosome-treated groups underwent a 12-week regimen of weekly tail vein injections at a dosage of 100 μg per mouse.25 The mice were euthanized by cervical dislocation after intraperitoneal injection of pentobarbital (150 mg/kg) and whole lung tissues were collected. All experimental procedures were approved by the Institutional Ethical Committee of the Nantong University (Approval ID: SYXK [SU] 2025–0037). The animal experiments were conducted in accordance with the Regulation of Animal Research Ethics in China (GB/T35892-2018).
Histology
Paraformaldehyde-fixed lung tissues were dehydrated, embedded, and sectioned at 5 μm for H&E and Masson trichrome staining (C0105S; Beyotime). Pathological assessment was performed with an Olympus IX73 microscope (IX73, Olympus, Japan).
Protein-Protein Interaction (PPI) Network and Hub Gene Screening
Using the STRING database with high-confidence threshold (score ≥ 0.700) and Homo sapiens restriction, PPI analysis was performed on putative miR-2110 target genes. Network visualization was achieved with Cytoscape 3.10.1. To identify hub genes among the potential targets, the top 10 central genes were screened using the BETWEENNESS, CLOSENESS, and DEGREE algorithms via the cytoHubba plugin in Cytoscape 3.10.1.
Luciferase Reporter Assay
SRSF1 3′-UTR fragments (wild-type/mutant) were ligated into pmirGLO luciferase vectors. Mutagenesis employed the Easy Mutagenesis System (Transgen). HEK293T transfections included miR-2110/NC mimics with corresponding reporters, with Dual-Luciferase® assays (E1910; Promega, USA) at 48h post-transfection and Renilla normalization.
Statistical Analysis
All in vitro experiments were repeated three times. For animals’ experiments, data are from six biological replicates. Data are presented as mean ± standard deviation (SD). Statistical significance was determined by one-way ANOVA followed by Dunnett’s multiple comparison test in GraphPad Prism 8.0, with P < 0.05 considered significant.
Results
Characterization of BEAS-2B-Derived Exosomes
Intercellular communication within the microenvironment coordinates the assembly and function of diverse cell types.26 During PM2.5 exposure, lung epithelial cells directly interact with harmful particles, leading to alterations in macrophage behavior within the microenvironment, which may contribute to airway remodeling and inflammation in COPD.27 A comparative analysis of exosomal biological properties was conducted using exosomes isolated from BEAS-2B cells under control (C-Exo) and PM2.5-exposed (P-Exo) conditions, to assess their role in epithelial-macrophage crosstalk. Both types of exosomes exhibited typical bilayer membrane vesicles (Figure 1A) and a size distribution within the range of 50–150 nm (Figure 1B). Western blot analysis confirmed the presence of characteristic exosome markers CD63 and CD9, while Calnexin was undetectable (Figure 1C). When THP-1 cells were incubated with PKH26-labeled C-Exo or P-Exo, fluorescence microscopy revealed co-localization of both exosome types with THP-1 cells, indicating that they were effectively taken up by THP-1 cells (Figure 1D). In summary, these results demonstrate the successful isolation of C-Exo and P-Exo and confirm their uptake by target cells.
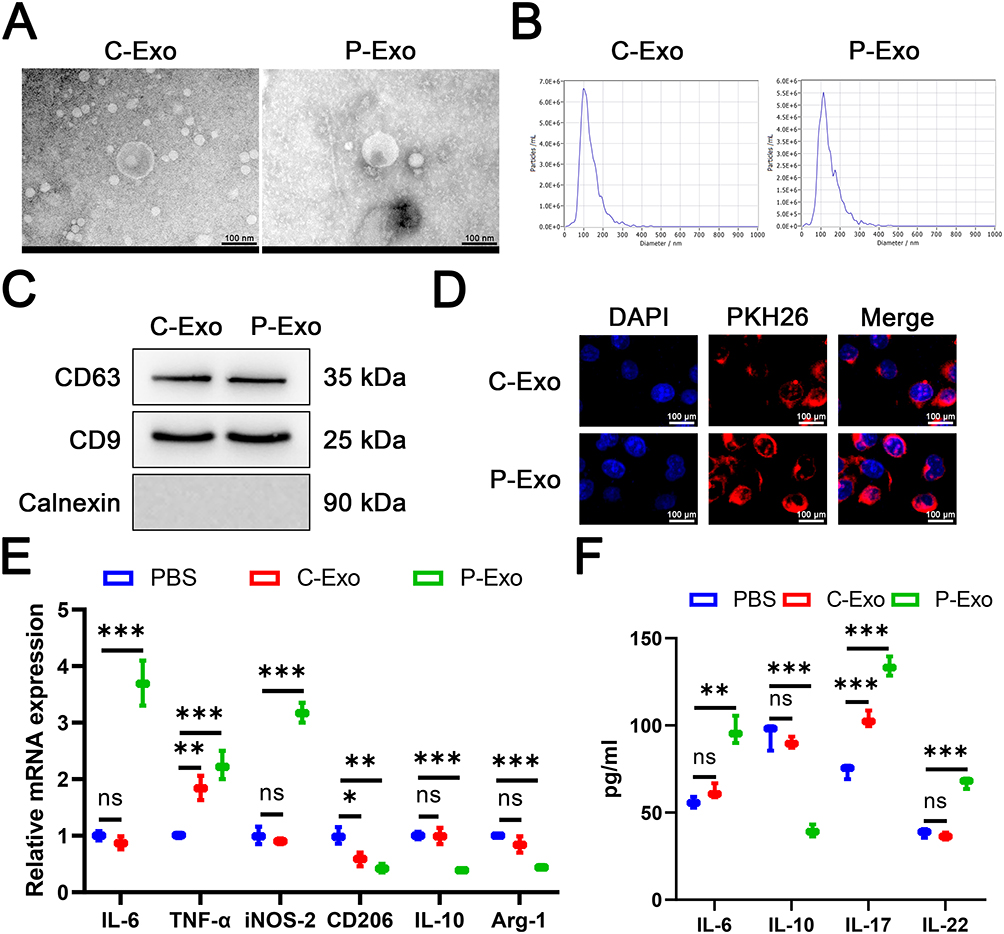 |
Figure 1 P-Exo promoted M1 macrophage polarization and inflammatory response. We isolated exosomes from BEAS-2B (C-Exo) or PM2.5-exposed BEAS-2B cells (P-Exo). (A) Transmission electron microscopy was employed to characterize the morphology of exosomes. (B) Nanoparticle tracking analysis was employed to characterize the particle size distribution of exosomes. (C) Western blot was performed to detect CD63, CD9, and Calnexin protein expression. (D) PHK26 labeling of exosomes was employed to detect exosomes uptake. (E) The mRNA expression of IL-6, TNF-α, iNOS-2, CD206, IL-10, and Arg-1was detected by RT-qPCR. (F) The level of IL-6, IL-17, IL-22, and IL-10 was detected by ELISA. Data are presented as mean ± SD. “ns” represents no statistical significance. *P<0.05, **P<0.01, ***P<0.001. |
P-Exo Promoted M1 Macrophage Polarization and Inflammatory Response
To further investigate the effect of P-Exo on macrophage polarization, M0 macrophages were generated from THP-1 cells through PMA stimulation. Co-culture with P-Exo induced a macrophage polarization shift, evidenced by concurrent elevation of M1 transcripts (IL-6, TNF-α, iNOS-2) and reduction of M2 markers (CD206, IL-10, Arg-1) relative to PBS-treated cells (Figure 1E). Exposure to C-Exo exerted a limited effect, inducing partial alteration of polarization markers with mild upregulation of select M1 markers and downregulation of some M2 markers (Figure 1E). ELISA analysis validated that P-Exo co-culture significantly enhanced secretory levels of the M1 marker IL-6 and pro-inflammatory cytokines IL-17/IL-22, while concurrently suppressing the M2 marker IL-10 (Figure 1F). Collectively, these findings indicate that P-Exo promotes macrophage polarization toward the M1 phenotype and enhances inflammatory responses upon co-culture with macrophages.
P-Exo Carried miR-2110 into Macrophages
In this study, we aimed to identify specific miRNAs carried by lung epithelial cell-derived exosomes into macrophages. Based on analysis of GEO datasets (GSE156572 and GSE218571), five miRNAs were found to be significantly enriched in EVs from both COPD patients and BEAS-2B cells (Figure 2A). Subsequent validation in BEAS-2B cells showed that, compared to normal cells, PM2.5-exposed BEAS-2B exhibited significantly elevated expression of miR-2110 and miR-345-5p, while the other three miRNAs (miR-182-5p, miR-200b-5p, and miR-191-5p) showed no notable change (Figure 2B). Furthermore, RT-qPCR analysis revealed that, among these two miRNAs, only miR-2110 was significantly upregulated in P-Exo compared to C-Exo, with no differential expression observed for miR-345-5p (Figure 2C). These findings suggest miR-2110 may be a key miRNA cargo in P-Exo. As shown in Figure 2D, in BEAS-2B cells exposed to varying doses, the expression of miR-2110 increased with higher PM2.5 concentrations, and the expression level of miR-2110 was highest at 400 μg/mL This strongly supports that miR-2110 is a key sensitive molecule in PM2.5-induced cellular stress responses. Furthermore, co-culture with P-Exo resulted in significantly elevated miR-2110 expression in M0 macrophages relative to both PBS controls and C-Exo treatment conditions (Figure 2E). Together, these results indicate that miR-2110 serves as an important mediator in exosome-mediated communication between PM2.5-exposed lung epithelial cell-derived exosomes and macrophages.
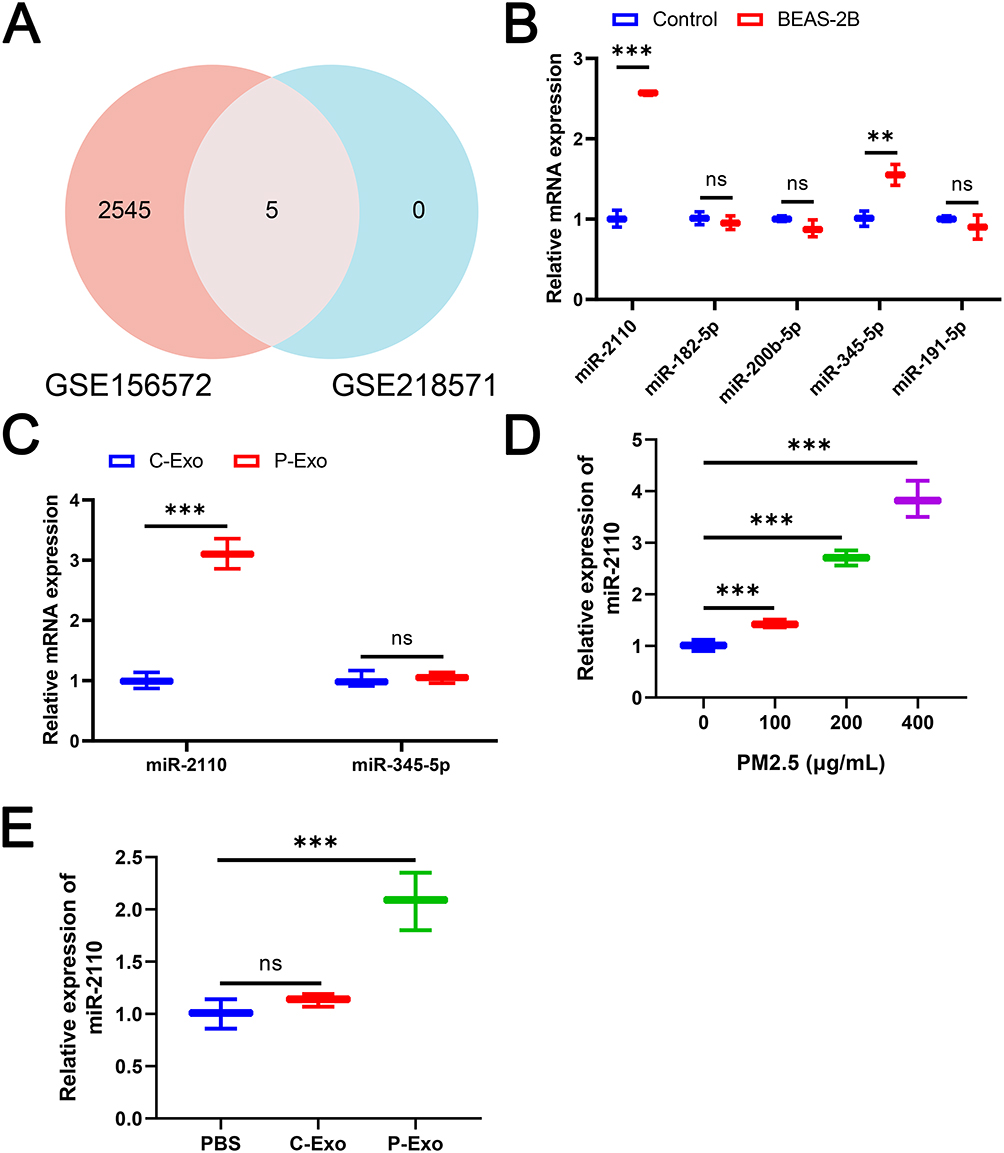 |
Figure 2 P-Exo promoted M1 macrophage polarization and inflammatory response. (A) Venn Diagram of intersecting genes from GSE156572 database and GSE218571 database. (B) The expression of miR-2110, miR-345-5p, miR-182-5p, miR-200b-5p, and miR-191-5p in PM2.5-treated or untreated BEAS-2B cells was detected by RT-qPCR. (C) The expression of hsa-miR-2110 and miR-345-5p in C-Exo and P-Exo was detected by RT-qPCR. (D) The expression of hsa-miR-2110 in 0, 100, 200 and 400 μg/mL PM2.5-treated BEAS-2B was detected by RT-qPCR. (E) The expression of miR-2110 in M0 macrophages treated with C-Exo or P-Exo was detected by RT-qPCR. Data are presented as mean ± SD. “ns” represents no statistical significance. **P<0.01, ***P<0.001. |
Exosomal miR-2110 Derived from PM2.5-Stimulated Lung Epithelial Cells Induced M1 Macrophage Polarization and Inflammatory Responses
Given the significant enrichment of miR-2110 in exosomes from PM2.5-exposed lung epithelial cells, we further investigated its role in regulating macrophage polarization and inflammatory responses. miR-2110 inhibitor was transfected into BEAS-2B cells to suppress miR-2110 expression, which significantly reduced miR-2110 level as confirmed by RT-qPCR (Figure 3A). Subsequently, BEAS-2B cells were treated with PM2.5 to establish an in vitro COPD model, and co-cultured with M0 macrophages using a Transwell system to evaluate macrophage polarization. BEAS-2B cells under PM2.5 exposure promoted M1 marker expression and inhibited M2 markers in macrophages (Figure 3B). Transfection with miR-2110 inhibitor reversed these polarization changes (Figure 3B). ELISA results further demonstrated that inhibition of miR-2110 in BEAS-2B cells significantly attenuated the PM2.5-induced secretion of IL-6, IL-17, and IL-22, while enhancing the production of IL-10 (Figure 3C). In summary, therapeutic targeting of miR-2110 in pulmonary epithelial cells effectively alleviates PM2.5-triggered M1 polarization and subsequent inflammatory cascades.
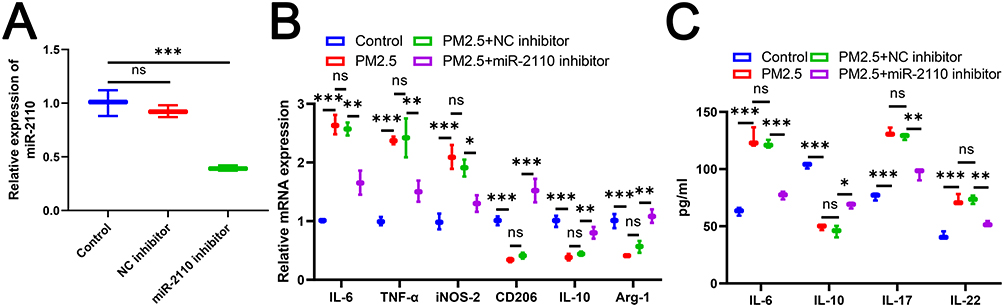 |
Figure 3 Exosomal miR-2110 Derived from PM2.5-Stimulated Lung Epithelial Cells Induced M1 Macrophage Polarization and Inflammatory Responses. BEAS-2B cells were co-cultured with M0 macrophages after transfection using PM2.5, miR-2110 inhibitor or NC inhibitor. (A) The expression of miR-2110 in BEAS-2B cells was detected by RT-qPCR. (B) The mRNA expression of IL-6, TNF-α, iNOS-2, CD206, IL-10, and Arg-1 in M0 macrophages was detected by RT-qPCR. (C) The level of IL-6, IL-17, IL-22, and IL-10 in M0 macrophages was detected by ELISA. Data are presented as mean ± SD. “ns” represents no statistical significance. *P<0.05, **P<0.01, ***P<0.001. |
Exosomal miR-2110 Promoted Airway Inflammation by Inducing M1 Macrophage Polarization
To investigate the role of miR-2110 in vivo, we established an in vivo COPD model. Mice were administered via tail vein injection either a miR-2110-specific antagomir (miR-2110 Ant) or exosomes from miR-2110 inhibitor or NC inhibitor -transfected BEAS-2B cells after PM2.5 treatment (PM2.5+miR-2110 inhibitor-Exo or PM2.5+NC inhibitor-Exo). RT-qPCR results showed that PM2.5 exposure significantly increased miR-2110 expression, which was suppressed by miR-2110 Ant (Figure 4A). Furthermore, PM2.5+NC inhibitor-Exo, but not those transfected with miR-2110 inhibitor, elevated miR-2110 levels in mouse lung tissue (Figure 4A). H&E and Masson staining revealed that inhibition of miR-2110 expression attenuated PM2.5-induced lung tissue damage, inflammation, and airway obstruction (Figure 4B and C). In contrast, the PM2.5+NC inhibitor-Exo group exhibited enhanced inflammatory cell infiltration and airway obstruction, similar to the PM2.5-only group (Figure 4B and C). These pathological changes were reversed by treatment with PM2.5+miR-2110 inhibitor-Exo (Figure 4B and C). Similarly, levels of IL-6, IL-17, and IL-22 in plasma and bronchoalveolar lavage fluid were significantly upregulated in the PM2.5 and PM2.5+NC inhibitor-Exo groups compared to controls, while IL-10 was downregulated (Figure 4D and E). These alterations were reversed both by miR-2110 Ant and PM2.5+miR-2110 inhibitor-Exo treatment (Figure 4D and E). Moreover, miR-2110 Ant and PM2.5+miR-2110 inhibitor-Exo downregulated the expression of M1 macrophage markers and upregulated M2 markers in lung tissues compared to the PM2.5 and PM2.5+NC inhibitor-Exo groups (Figure 4F). In conclusion, these results demonstrate that exosomal miR-2110 drives M1 polarization to promote airway inflammation.
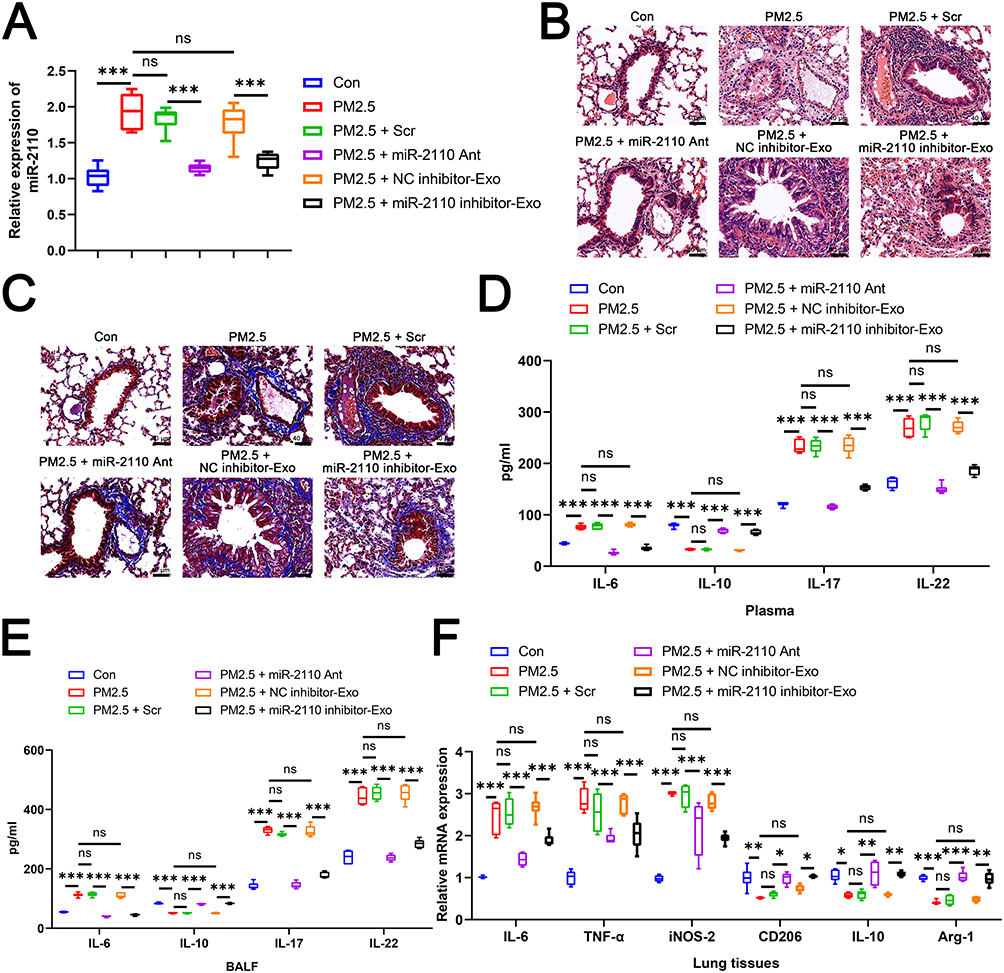 |
Figure 4 Exosomal miR-2110 Promoted Airway Inflammation by Inducing M1 Macrophage Polarization. The mice were then randomly divided into six weight-matched groups (n = 6 per group) as follows: (1) Control group (Con); (2) PM2.5 exposure group (PM2.5); (3) PM2.5 exposure + scrambled antagomir treatment (PM2.5 + Scr); (4) PM2.5 exposure + miR-2110–specific antagomir treatment (PM2.5 + miR-2110 Ant); (5) Air exposure + exosomes derived from PM2.5-exposed BEAS-2B cells transfected with NC inhibitor (PM2.5 + NC inhibitor-Exo); (6) Air exposure + exosomes derived from PM2.5-exposed BEAS-2B cells transfected with miR-2110 inhibitor (PM2.5 + miR-2110 inhibitor-Exo). (A) The RNA expression of hsa-miR-2110 in lung tissues was detected by RT-qPCR. (B) H&E staining was used to detect the lung tissue morphology of each group. (C) Masson staining was used to detect the lung tissue morphology of each group. (D) The level of IL-6, IL-17, IL-22, and IL-10 in plasma was detected by ELISA. (E) The level of IL-6, IL-17, IL-22, and IL-10 in alveolar lavage fluid was detected by ELISA. (F) The mRNA expression of IL-6, TNF-α, iNOS-2, CD206, IL-10, and Arg-1in lung tissues was detected by RT-qPCR. Data are presented as mean ± SD. “ns” represents no statistical significance. *P<0.05, **P<0.01, ***P<0.001. |
MiR-2110 Targeted and Suppressed SRSF1 Expression
We used the miRDB, mirDIP 3, and TargetScan databases to predict potential downstream targets of miR-2110. By intersecting the predicted targets with genes known to be downregulated in COPD, we identified 42 candidate target genes associated with COPD pathogenesis (Figure 5A) and PPI network was constructed (Figure 5B), and the top 16 hub genes were selected using the cytoHubba plugin in Cytoscape based on BETWEENNESS, CLOSENESS, and DEGREE centrality measures (Figure 5C–E). The intersection of these hub genes yielded six common core nodes: PRKACA, SRSF1, MAPK1, SFPQ, AR, and QKI (Figure 5F). Based on literature review, SRSF1,28,29 MAPK1,30,31 SFPQ,32 and QKI33,34 were selected for further investigation due to their reported roles in COPD and macrophage function. Subsequently, we transfected THP-1 cells with miR-2110 mimic to validate the expression of the candidate genes. Transfection with miR-2110 mimic significantly increased the expression of miR-2110 in THP-1 cells (Figure 5G). Moreover, overexpression of miR-2110 markedly suppressed the expression of SRSF1 and MAPK1, while it had no significant effect on the expression of QKI or SFPQ (Figure 5H). Among the affected genes, the inhibition of SRSF1 was more pronounced. SRSF1 was therefore investigated as a miR-2110 target. The predicted binding site (Figure 5I) and direct 3’UTR binding were confirmed by luciferase reporter assays (Figure 5J). In summary, miR-2110 targets SRSF1 and inhibits its expression.
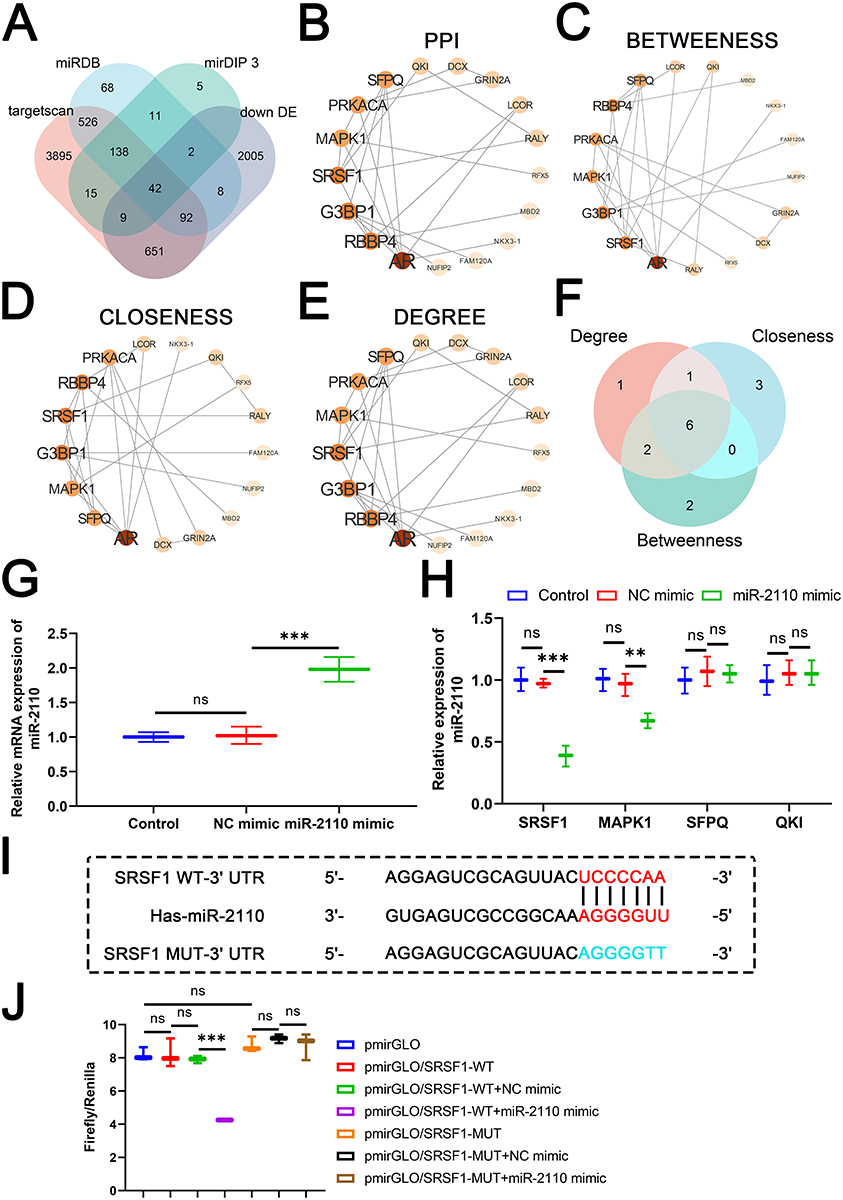 |
Figure 5 MiR-2110 Targeted and Suppressed SRSF1 Expression. (A) Venn Diagram of target gene intersection across four databases. (B) PPI network relationship diagram of candidate genes. (C) Network relationship diagram of the top 10 hub genes obtained by filtering using the BETWEENNESS centrality measure. (D) Network relationship diagram of the top 10 hub genes obtained by filtering using the CLOSENESS centrality measure. (E) Network relationship diagram of the top 10 hub genes obtained by filtering using the DEGREE centrality measure. (F) Venn Diagram of the intersection of top 10 hub genes identified based on BETWEENNESS, CLOSENESS, and DEGREE centrality measures. (G) The RNA expression of hsa-miR-2110 in THP-1 cells was detected by RT-qPCR. (H) The mRNA expression of SRSF1, MAPK1, SFPQ, and QKI in THP-1 cells was detected by RT-qPCR. (I) Prediction of binding targets of miR-2110 to SRSF1. (J) In 293T cells, dual luciferase assay was used to validate the targeting relationship between miR-2110 and SRSF1. Data are presented as mean ± SD. “ns” represents no statistical significance. *P<0.01, ***P<0.001. |
MiR-2110 Promoted M1 Macrophage Polarization and Inflammatory Responses by Targeting SRSF1
To investigate whether miR-2110 regulates macrophage polarization through SRSF1, we first transfected THP-1 cells with oe-SRSF1 to achieve stable overexpression of SRSF1. The results confirmed a significant increase in SRSF1 expression following transfection (Figure 6A). The THP-1 cells were then differentiated into M0 macrophages using PMA. Compared to the NC mimic group, transfection with miR-2110 mimic markedly upregulated the expression of M1 macrophage markers (IL-6, TNF-α, and iNOS-2) and downregulated M2 markers (CD206, IL-10, and Arg-1) (Figure 6B). However, overexpression of SRSF1 counteracted the polarization effects induced by miR-2110 mimic (Figure 6B). ELISA results further showed that miR-2110 mimic significantly enhanced the secretion of Inflammatory factors, while reducing the level of IL-10, which were also reversed by oe-SRSF1 (Figure 6C). In conclusion, these findings demonstrate that miR-2110 promotes M1 macrophage polarization and inflammatory responses by negatively regulating SRSF1 expression.
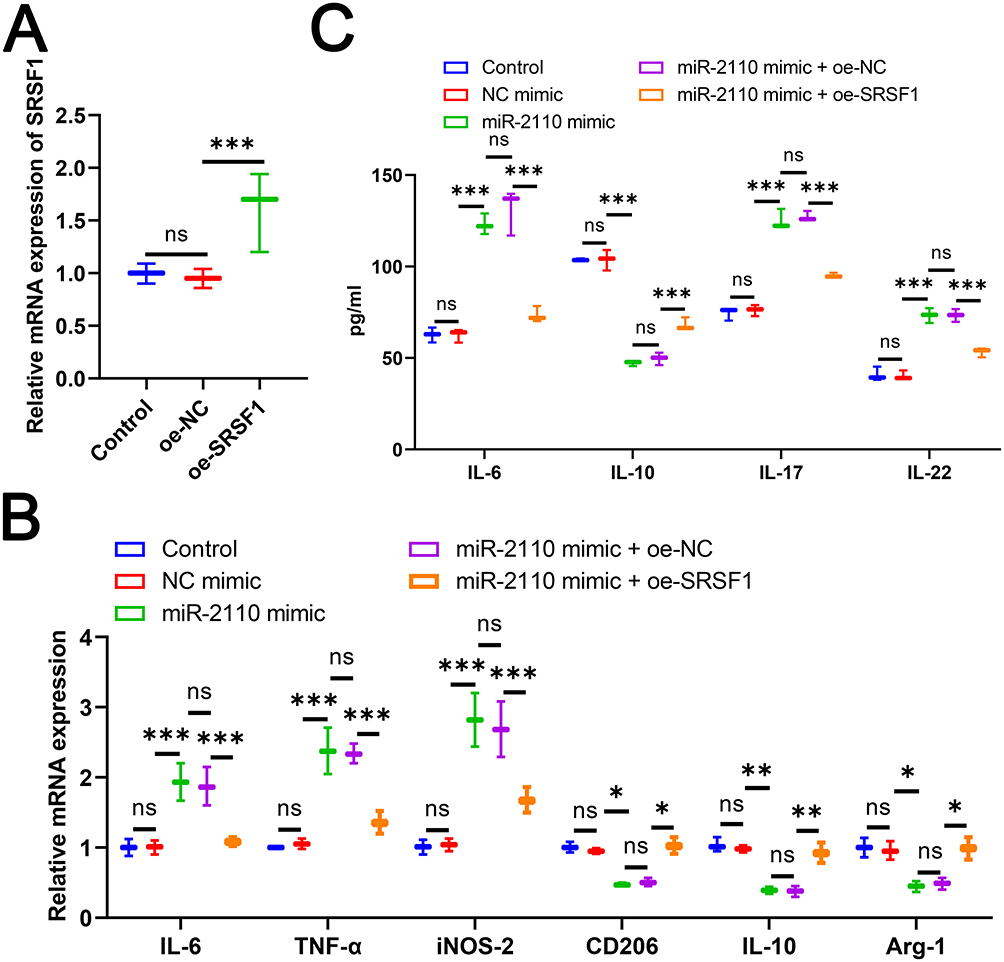 |
Figure 6 MiR-2110 Promoted M1 Macrophage Polarization and Inflammatory Responses by Targeting SRSF1. M0 macrophage differentiation was induced by PMA after transfection of oe-SRSF1 or miR-2110 mimic in THP-1 cells. (A) The mRNA expression of SRSF1 in THP-1 cells was detected by RT-qPCR. (B) The mRNA expression of IL-6, TNF-α, iNOS-2, CD206, IL-10, and Arg-1in M0 macrophages was detected by RT-qPCR. (C) The level of IL-6, IL-17, IL-22, and IL-10 in M0 macrophages was detected by ELISA. Data are presented as mean ± SD. “ns” represents no statistical significance. *P<0.05, **P<0.01, ***P<0.001. |
Discussion
With ongoing social development, PM2.5 pollution poses an increasing threat to human health. Chronic exposure to PM2.5 causes inflammation in the lungs and airways, which aids in the development of COPD.35 As a direct target of PM2.5, bronchial epithelial BEAS-2B cells exposed to PM2.5 exhibit increased secretion of inflammatory cytokines.27,36 Furthermore, PM2.5-induced oxidative stress, airway inflammation, and macrophage polarization are implicated in the pathogenesis of COPD and can lead to emphysema. Previous studies have shown that PM2.5-exposed BEAS-2B cells triggered macrophage polarization toward a pro-inflammatory phenotype, representing a key factor in COPD development.23 The molecular basis of epithelial-macrophage communication remains incompletely characterized. To address this knowledge gap, we utilized complementary in vitro and in vivo approaches to delineate the mechanisms of pulmonary intercellular crosstalk.
Exosomes play a crucial role in intercellular communication by transferring functional genetic material to recipient cells.19 Recent studies have highlighted their involvement in the progression of lung diseases. For example, Previous work by Guo et al established the contribution of exosomal crosstalk in bronchial epithelial-fibroblast communication to PM2.5-exacerbated airway remodeling in COPD via promoted myofibroblast activation.25 In this study, we found that P-Exo were taken up by recipient macrophages, promoting their polarization toward the M1 phenotype and enhancing inflammatory responses. Through dynamic adaptation to microenvironmental signals, macrophages polarize into M1 subtypes that initiate inflammatory cascades via inflammatory factors secretion. This response correlates with lung parenchymal damage and COPD pathogenesis exacerbation.37 Therefore, inhibiting M1 macrophage infiltration represents a critical strategy for mitigating airway and pulmonary inflammation in COPD.38 Our findings were consistent with previous reports showing that CSE-treated BEAS-2B-derived EVs significantly enhanced the expression of CD86 and CD80 and promoted the secretion of TNF-α, IL-6, IL-1β, iNOS, and IL-12, thereby inducing M1 macrophage polarization.39 These results suggest that BEAS-2B-derived exosomes/EVs facilitate M1 macrophage polarization, underscoring their role as key mediators of intercellular communication and important contributors to the pathogenesis of COPD.
MiRNAs are small single-stranded non-coding RNA molecules involved in diverse cellular processes.40 Studies have shown that exosomes or EVs derived from lung epithelial cells are enriched with numerous miRNAs.41 These vesicles stably transport encapsulated miRNAs to recipient cells, where they exert biological effects. For instance, in COPD, epithelial-derived exosomes delivered miRNA-125a-5p to macrophages, promoting M1 macrophage polarization.20 Expression of miR-2110 was significantly elevated in EVs isolated from bronchoalveolar lavage fluid of COPD patients, and it demonstrated excellent predictive performance in distinguishing between healthy individuals and those with mild COPD.42 In this study, we identified miR-2110 as highly enriched in P-Exo. P-Exo delivered miR-2110 into macrophages, leading to increased intracellular expression of miR-2110. Previous studies reported that miR-2110 was significantly enriched in EVs from COPD patients and was closely associated with disease pathogenesis.42 However, the role of miR-2110 in macrophage polarization had not been previously investigated. Our results found that inhibition of miR-2110 expression reduced M1 macrophage polarization and alleviated PM2.5-induced pulmonary inflammatory infiltration and tissue damage. Furthermore, exosomes derived from miR-2110-inhibited BEAS-2B cells reversed the P-Exo-driven M1 polarization and inflammatory response. These findings indicate that P-Exo promotes pro-inflammatory responses in macrophages via the delivery of miR-2110, and that miR-2110 acts as a genetic mediator in epithelial-macrophage crosstalk that exacerbates COPD progression.
The canonical function of miRNAs involves 3’UTR binding to exert regulatory effects at post-transcriptional or translational levels.43 To identify the target genes regulated by miR-2110, we employed bioinformatic tools and identified SRSF1 as a promising candidate. Current evidence confirmed that miR-2110 bound to the 3’UTR of SRSF1 and suppressed its expression. Serine/arginine-rich splicing factors (SRSFs) constitute a large family of RNA-binding proteins with diverse physiological functions.44 Studies have shown that SRSFs serve as potential biomarkers in chronic pulmonary inflammatory diseases and are involved in inflammatory responses.28 For example, SRSF1 attenuated inflammatory progression by restricting the production of interferon-gamma (IFN-γ).45 Furthermore, SRSF1 is closely associated with the regulation of macrophage polarization. Ting Gan et al demonstrated that SRSF1 protein expression suppressed pro-inflammatory activity by inhibiting M1 macrophage polarization.29 These findings collectively highlight the anti-inflammatory potential of SRSF1. In this study, we found that overexpression of SRSF1 reversed the pro-inflammatory effects of miR-2110, notably by inhibiting both M1 macrophage polarization and inflammatory activity in macrophages. These results further support the mechanism whereby miR-2110, upon entering macrophages, promotes inflammatory progression by suppressing the expression of the anti-inflammatory protein SRSF1.
Although this research clarifies key aspects of epithelial-macrophage communication in COPD, the complexity of the airway microenvironment necessitates broader investigation. Future studies should examine epithelial interactions with diverse immune populations (including T and NK cells) and determine the full spectrum of mechanisms (eg., protein/ncRNA transfer) by which PM2.5-induced exosomes regulate macrophage polarization. Furthermore, whether exosomes derived from PM2.5-exposed lung epithelial cells alter macrophage polarization through additional mechanisms-such as carrying proteins or other non-coding RNAs-requires further in-depth investigation.
Conclusion
In summary, our study identified that P-EXO delivers miR-2110 to promote M1 macrophage polarization, potentially through the miR-2110/SRSF1 axis. These results establish exosomes as critical mediators of epithelial-macrophage communication in COPD pathogenesis, highlighting the miR-2110/SRSF1 pathway as a potential therapeutic strategy.
ARRIVE Guidelines Statement
This work adhered to the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments).
Data Sharing Statement
All the results are presented in the article. Further inquiries can be directed to the corresponding authors.
Ethics Statement
All the experimental procedures involving animals were conducted in accordance with ARRIVE guidelines. The welfare of the laboratory animals was conducted in accordance with the Regulation of Animal Research Ethics in China (GB/T35892-2018). The animal experiments were approved by the Institutional Ethical Committee of the Nantong University (Approval ID: SYXK [SU] 2025-0037). The research protocol was approved by the Ethics Committee of The Affiliated Hospital of Nantong University (Approval No. 2025-L037). All experiments and procedures were performed according to the Declaration of Helsinki (as revised in 2013).
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Qian Y, Cai C, Sun M, Lv D, Zhao Y. Analyses of factors associated with acute exacerbations of chronic obstructive pulmonary disease: a review. Int J Chron Obstruct Pulmon Dis. 2023;18:2707–15. doi:10.2147/copd.S433183
2. Qibin L, Yacan L, Minli J, et al. The impact of PM2.5 on lung function in adults with asthma. Int J Tuberc Lung Dis. 2020;24(6):570–576. doi:10.5588/ijtld.19.0394
3. Li R, Zhou R, Zhang J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol Lett. 2018;15(5):7506–7514. doi:10.3892/ol.2018.8355
4. Wang Y, Liao S, Pan Z, et al. Hydrogen sulfide alleviates particulate matter-induced emphysema and airway inflammation by suppressing ferroptosis. Free Radic Biol Med. 2022;186:1–16. doi:10.1016/j.freeradbiomed.2022.04.014
5. Shan H, Li X, Ouyang C, et al. Salidroside prevents PM2.5-induced BEAS-2B cell apoptosis via SIRT1-dependent regulation of ROS and mitochondrial function. Ecotoxicol Environ Saf. 2022;231:113170. doi:10.1016/j.ecoenv.2022.113170
6. Gou A, Tan G, Ding X, et al. Urban-rural difference in the lagged effects of PM2.5 and PM10 on COPD mortality in Chongqing, China. BMC Public Health. 2023;23(1):1270. doi:10.1186/s12889-023-16113-9
7. Yang X, Zhang T, Zhang Y, Chen H, Sang S. Global burden of COPD attributable to ambient PM2.5 in 204 countries and territories, 1990 to 2019: a systematic analysis for the Global Burden of Disease Study 2019. Sci Total Environ. 2021;796:148819. doi:10.1016/j.scitotenv.2021.148819
8. Niu Y, Zhang L, Guo S, Wu S. In vivo and in vitro models of PM2.5 induced COPD: focus on the role of RTA-408. Int J Chron Obstruct Pulmon Dis. 2024;19:2239–2257. doi:10.2147/copd.S475281
9. Fan X, Dong T, Yan K, Ci X, Peng L. PM2.5 increases susceptibility to acute exacerbation of COPD via NOX4/Nrf2 redox imbalance-mediated mitophagy. Redox Biol. 2023;59:102587. doi:10.1016/j.redox.2022.102587
10. Iyer AS, Sullivan DR, Lindell KO, Reinke LF. The role of palliative care in COPD. Chest. 2022;161(5):1250–1262. doi:10.1016/j.chest.2021.10.032
11. Celli BR, Anderson JA, Cowans NJ, et al. Pharmacotherapy and lung function decline in patients with chronic obstructive pulmonary disease. A systematic review. Am J Respir Crit Care Med. 2021;203(6):689–698.
12. Tao F, Kobzik L. Lung macrophage–epithelial cell interactions amplify particle-mediated cytokine release. Am J Respir Cell Mol Biol. 2002;26(4):499–505.
13. Dong Y, Dong Y, Zhu C, et al. Targeting CCL2-CCR2 signaling pathway alleviates macrophage dysfunction in COPD via PI3K-AKT axis. Cell Commun Signal. 2024;22(1):364. doi:10.1186/s12964-024-01746-z
14. Bissonnette EY, Lauzon-Joset J-F, Debley JS, Ziegler SF. Cross-talk between alveolar macrophages and lung epithelial cells is essential to maintain lung homeostasis. Front Immunol. 2020;11:583042.
15. Kotlyarov S. Involvement of the innate immune system in the pathogenesis of chronic obstructive pulmonary disease. Int J Mol Sci. 2022;23(2):985.
16. Yamasaki K, Eeden SFV. Lung macrophage phenotypes and functional responses: role in the pathogenesis of COPD. Int J Mol Sci. 2018;19(2). doi:10.3390/ijms19020582
17. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090. doi:10.1016/j.ejphar.2020.173090
18. He S, Chen D, Hu M, et al. Bronchial epithelial cell extracellular vesicles ameliorate epithelial-mesenchymal transition in COPD pathogenesis by alleviating M2 macrophage polarization. Nanomedicine. 2019;18:259–271. doi:10.1016/j.nano.2019.03.010
19. Krylova SV, Feng D. The machinery of exosomes: biogenesis, release, and uptake. Int J Mol Sci. 2023;24(2). doi:10.3390/ijms24021337
20. Wang R, Zhu Z, Peng S, et al. Exosome microRNA-125a-5p derived from epithelium promotes M1 macrophage polarization by targeting IL1RN in chronic obstructive pulmonary disease. Int Immunopharmacol. 2024;137:112466. doi:10.1016/j.intimp.2024.112466
21. Ni X, Lv Y, Han L, et al. Exosomal miR-107 derived from cigarette smoking-exposed bronchial epithelial cells aggravates acute lung injury by polarizing macrophage to proinflammatory phenotype. J Biochem Mol Toxicol. 2025;39(2):e70139. doi:10.1002/jbt.70139
22. Yang L, Han P, Cui T, et al. M2 macrophage inhibits the antitumor effects of Lenvatinib on intrahepatic cholangiocarcinoma. Front Immunol. 2023;14:1251648. doi:10.3389/fimmu.2023.1251648
23. Xu H, Li X, Liu K, Huang P, Liu XJ. PM2.5 promotes macrophage-mediated inflammatory response through airway epithelial cell-derived exosomal miR-155-5p. J Inflamm Res. 2024;17:8555–8567. doi:10.2147/jir.S482509
24. Ning T, Li J, He Y, et al. Exosomal miR-208b related with oxaliplatin resistance promotes Treg expansion in colorectal cancer. Mol Ther. 2021;29(9):2723–2736.
25. Guo H, Fei L, Yu H, et al. Exosome-encapsulated lncRNA HOTAIRM1 contributes to PM(2.5)-aggravated COPD airway remodeling by enhancing myofibroblast differentiation. Sci China Life Sci. 2024;67(5):970–985. doi:10.1007/s11427-022-2392-8
26. Fujita Y, Araya J, Ito S, et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles. 2015;4:28388. doi:10.3402/jev.v4.28388
27. Liu G, Li Y, Zhou J, Xu J, Yang B. PM2.5 deregulated microRNA and inflammatory microenvironment in lung injury. Environ Toxicol Pharmacol. 2022;91:103832. doi:10.1016/j.etap.2022.103832
28. Maghsoudloo M, Azimzadeh Jamalkandi S, Najafi A, Masoudi-Nejad A. An efficient hybrid feature selection method to identify potential biomarkers in common chronic lung inflammatory diseases. Genomics. 2020;112(5):3284–3293. doi:10.1016/j.ygeno.2020.06.010
29. Gan T, Liu W, Wang Y, et al. LncRNA MAAMT facilitates macrophage recruitment and proinflammatory activation and exacerbates autoimmune myocarditis through the SRSF1/NF-κB axis. Int J Biol Macromol. 2024;278(Pt 1):134193. doi:10.1016/j.ijbiomac.2024.134193
30. Ye H, He B, Zhang Y, et al. Herb-symptom analysis of Erchen decoction combined with Xiebai powder formula and its mechanism in the treatment of chronic obstructive pulmonary disease. Front Pharmacol. 2023;14:1117238. doi:10.3389/fphar.2023.1117238
31. Benatzy Y, Palmer MA, Lütjohann D, et al. ALOX15B controls macrophage cholesterol homeostasis via lipid peroxidation, ERK1/2 and SREBP2. Redox Biol. 2024;72:103149. doi:10.1016/j.redox.2024.103149
32. Traxler P, Reichl S, Folkman L, et al. Integrated time-series analysis and high-content CRISPR screening delineate the dynamics of macrophage immune regulation. Cell Syst. 2025;16(8):101346. doi:10.1016/j.cels.2025.101346
33. Li X, Noell G, Tabib T, et al. Single cell RNA sequencing identifies IGFBP5 and QKI as ciliated epithelial cell genes associated with severe COPD. Respir Res. 2021;22(1):100. doi:10.1186/s12931-021-01675-2
34. Zhai D, Wang W, Ye Z, et al. QKI degradation in macrophage by RNF6 protects mice from MRSA infection via enhancing PI3K p110β dependent autophagy. Cell Biosci. 2022;12(1):154. doi:10.1186/s13578-022-00865-9
35. Wang Q, Liu S. The effects and pathogenesis of PM2.5 and its components on chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2023;18:493–506. doi:10.2147/copd.S402122
36. Fan X, Gao Y, Hua C, Peng L, Ci X. Daphnetin ameliorates PM2.5-induced airway inflammation by inhibiting NLRP3 inflammasome-mediated pyroptosis in CS-exposed mice. Biomed Pharmacother. 2023;165:115047. doi:10.1016/j.biopha.2023.115047
37. Xue L, Xu J, Gong H, et al. Overexpression of BPIFB4 alleviates COPD inflammatory damage by inhibiting M1 macrophage activation via the PI3K/AKT pathway. Lung. 2025;203(1):69. doi:10.1007/s00408-025-00824-4
38. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
39. Chen Z, Wu H, Fan W, et al. Naringenin suppresses BEAS-2B-derived extracellular vesicular cargoes disorder caused by cigarette smoke extract thereby inhibiting M1 macrophage polarization. Front Immunol. 2022;13:930476. doi:10.3389/fimmu.2022.930476
40. Lu Q, Wu R, Zhao M, Garcia-Gomez A, Ballestar E. miRNAs as therapeutic targets in inflammatory disease. Trends Pharmacol Sci. 2019;40(11):853–865. doi:10.1016/j.tips.2019.09.007
41. Zhang J, Li S, Li L, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinf. 2015;13(1):17–24. doi:10.1016/j.gpb.2015.02.001
42. Burke H, Cellura D, Freeman A, et al. Pulmonary EV miRNA profiles identify disease and distinct inflammatory endotypes in COPD. Front Med. 2022;9:1039702. doi:10.3389/fmed.2022.1039702
43. Li J, Zeng X, Wang W. miR-122-5p downregulation attenuates lipopolysaccharide-induced acute lung injury by targeting IL1RN. Exp Ther Med. 2021;22(5):1278.
44. Sertznig H, Roesmann F, Wilhelm A, et al. SRSF1 acts as an IFN-I-regulated cellular dependency factor decisively affecting HIV-1 post-integration steps. Front Immunol. 2022;13:935800. doi:10.3389/fimmu.2022.935800
45. Katsuyama T, Li H, Krishfield SM, Kyttaris VC, Moulton VR. Splicing factor SRSF1 limits IFN-γ production via RhoH and ameliorates experimental nephritis. Rheumatology. 2021;60(1):420–429. doi:10.1093/rheumatology/keaa300
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Bone Marrow Mesenchymal Stem Cells-Derived Exosomes Inhibit Apoptosis of Pulmonary Microvascular Endothelial Cells in COPD Mice Through miR-30b/Wnt5a Pathway

Song Q, Zhou A, Cheng W, Zhao Y, Liu C, Zeng Y, Lin L, Zhou Z, Peng Y, Chen P
International Journal of Nanomedicine 2025, 20:1191-1211
Published Date: 30 January 2025