Back to Archived Journals » Advances in Genomics and Genetics » Volume 4
Exome sequencing: what clinicians need to know
Authors Sastre L
Received 11 December 2013
Accepted for publication 23 January 2014
Published 31 March 2014 Volume 2014:4 Pages 15—27
DOI https://doi.org/10.2147/AGG.S39108
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Leandro Sastre
Instituto de Investigaciones Biomédicas, CSIC/UAM, C/Arturo Duperier 4, Madrid, Spain; Terapias Experimentales y Biomarcadores en Cáncer, IdiPaz, Madrid, Spain; CIBER de Enfermedades Raras, CIBERER, Valencia, Spain
Abstract: The recent development of high throughput methods of deoxyribonucleic acid (DNA) sequencing has made it possible to determine individual genome sequences and their specific variations. A region of particular interest is the protein-coding part of the genome, or exome, which is composed of gene exons. The principles of exome purification and sequencing will be described in this review, as well as analyses of the data generated. Results will be discussed in terms of their possible functional and clinical significance. The advantages and limitations of exome sequencing will be compared to those of other massive sequencing approaches such as whole-genome sequencing, ribonucleic acid sequencing or selected DNA sequencing. Exome sequencing has been used recently in the study of various diseases. Monogenic diseases with Mendelian inheritance are among these, but studies have also been carried out on genetic variations that represent risk factors for complex diseases. Cancer is another intensive area for exome sequencing studies. Several examples of the use of exome sequencing in the diagnosis, prognosis, and treatment of these diseases will be described. Finally, remaining challenges and some practical and ethical considerations for the clinical application of exome sequencing will be discussed.
Keywords: massively parallel sequencing, RNA sequencing, whole-genome sequencing, genetic variants, molecular diagnosis, pharmacogenomics, personalized medicine, NGS, SGS, SNP, SNV
Introduction
Numerous diseases have a genetic basis. Some are the consequence of an absence or dysfunction of a given protein due to mutations in the encoding gene. This is the case with diseases of Mendelian inheritance, such as Huntington’s disease, thalassemia, and approximately 1,000 other inherited rare diseases.1 Many diseases have a genetic basis, even if they are not exclusively due to the mutation of a single gene, and an increasing number of genetic variants and polymorphisms are being identified as risk factors for complex diseases.2 Cancer is a genetic disease caused by the mutation of one or more genes that either increase the risk of cancer (such as germ line mutations), or they promote cancer (oncogenes), or they impair the cellular mechanisms that control cell proliferation (suppressor genes), as occurs with somatic mutations.3
The identification of the genetic basis of these diseases has been a labor-intensive and challenging project, up until a few years ago. These projects often started with the identification of a genome region possibly involved in the transmission of the disease by genetic association studies.1 The analysis of large families with several affected members is usually required to define a genome region that is highly related to disease transmission. Generally, this region contains several genes that must be sequenced to identify a gene mutation present in all the affected individuals and not in their healthy relatives, in the case of dominant inheritance. In the case of recessive transmission, the mutation should be present in both alleles of the affected members and in one, or none, of the alleles of the unaffected relatives.
Diagnosis of genetic diseases was, and in most cases still is, equally laborious. In the best-case scenario, the disease can originate in a mutation in only one gene. Diagnosis would require determining the nucleotide sequence of that gene only. Typically, the gene is amplified as several fragments by polymerase chain reactions and the nucleotide sequence of each is determined. Often, the disease can be caused by mutations in any of several genes, and all of them must be amplified and sequenced in order to find the genetic origin of the disease in the affected patients. For example, in dyskeratosis congenita, mutations can be found in any of the genes dkc, tert, terc, NOP10, NH2, or TINF2, and the number of affected genes can be even larger since there is a fraction of patients in whom the causative mutation has not been identified.4,5 The nucleotide sequence of all these genes must be determined for the molecular diagnosis of each patient. We find several mutated genes in various types of cancers.3 Molecular diagnosis requires determining the nucleotide sequence of several of these genes. Currently, this is a laborious and expensive process that cannot be used for a large population of patients. In practice, only a few genes that are mutated in an important proportion of patients affected by some cancer types are sequenced for diagnosis and treatment.
Only in the last few years have techniques been developed for the simultaneous detection of multiple sequence variants in a given sample. Many of them are based on deoxyribonucleic acid (DNA) microarray technology. In genotyping arrays, oligonucleotides containing the actively identified mutations related to a given disease are spotted on a slide. A patient’s DNA sample is added on top of the slide and the hybridizing oligonucleotides are identified. Millions of known mutations can be tested in a single microarray hybridization.6 Copy number variations can also be analyzed using DNA microarrays designed to detect the presence of DNA regions that are duplicated or deleted in the DNA of the patient.7 These techniques are frequently used in medical research and for clinical diagnosis.8
However, a major step in molecular medicine has been the recent development of massive sequencing technologies that allow the nucleotide sequence of a patient’s DNA to be determined in a short period of time and at an affordable price.9,10 These methodologies have been in use since 2005 and are based on the simultaneous determination of the nucleotide sequence of millions of DNA fragments. They have been named as second generation sequencing, next generation sequencing, deep sequencing, or massively parallel sequencing. Thousands of millions of nucleotide sequences are determined in no more than 2 weeks using these machines. As an example of the capacity of these new sequencing systems, note that the milestone sequencing of the first human genome, published in 2001,11 required the coordinated work of 23 laboratories, which took 13 years, with a total cost of about US$3 billion. With the new methodologies, sequencing a human genome takes one laboratory and about 2 weeks, with an approximated cost of US$4,000.
The availability of modern sequencing methodologies is producing the exponential growth of our knowledge about the human genome, variability among individuals, and the identification of genetic variants in diseases. For example, these methodologies are the foundation of an ongoing 1000 Genomes Project,12 aimed at determining the complete nucleotide sequence of about 1,000 persons from different geographic and ethnical origins to determine the average sequence variation among individuals and to identify the most frequent polymorphisms.
Massive sequencing technologies are presently evolving at a fast pace. Smaller, faster machines are being developed, and new sequencing methods are being introduced. An important aim, for example, is to sequence a single DNA molecule from an individual cell.13,14 Aside from the technical challenges, advances are steadily decreasing the price of DNA sequencing so that the goal of sequencing an individual human genome for US$1,000 seems to be within reach in a few years. Presently, sequencing a whole human genome and analyzing all the sequence data generated is complex, expensive, and time consuming, and many studies are being carried out in a smaller part of the genome. In particular, much attention is currently being paid to sequencing the protein-coding region of the genome, which is known as the exome. Exome sequencing is far more affordable than whole-genome sequencing, and the possibilities, advantages, and limitations of this technique will be discussed in this review.
What is an exome?
Almost all human protein-coding genes have a discontinuous structure. The protein-coding region is fragmented into several pieces, called exons. Exons are connected by nonprotein-coding DNA fragments, or introns, as schematically shown in Figure 1. Genes are transcribed from the promoter region under the control of several regulatory regions, which are present in different locations in relation to the gene, upstream, downstream, or even inside the gene. Transcription creates a primary transcript that contains exons and introns. Subsequent ribonucleic acid (RNA) splicing processes delete the introns and join the exons to generate the mature messenger RNA (mRNA) that contain only one continuous protein-coding region. Recent studies show that the primary transcripts of most genes can be spliced in several ways, giving rise to various mature mRNAs containing specific combinations of exons, known as alternative splicing variants (Figure 1). These mRNAs code for protein isoforms that have some common regions, but that also differ from other ones, depending on the exons incorporated.15
Analysis of the human genome has shown that protein-coding genes represent a small proportion of DNA, only about 3%.16 Exons represent an even smaller fraction at 1% of the genome.16 A summary of this data is shown in Table 1. The human genome is composed of 3.3 ×109 base pairs (bp) and contains 20,078 protein-coding genes.17 Each gene is divided in an average number of eight exons, each about 170 bp long. All the exons as a whole contain about 3 ×107 bp. However, sequencing all the exons provides the same information about the amino acid sequence of the encoded proteins as sequencing the whole genome, with the exception of mutations that alter mRNA splicing, as will be discussed in the Exome sequencing and data analysis section. This system of sequencing all the exons has been named exome sequencing and has become a valid method of detecting variations in the amino acid sequence of all human proteins.18 The very marked size difference makes exome sequencing much cheaper than genome sequencing, and this facilitates computational and functional analyses of the sequence data generated.
 | Figure 1 Schematic representation of gene structure and expression. |
 | Table 1 General characteristics of the human genome and exome |
Exome capture techniques
The first and most critical step in exome sequencing is the isolation or capture of the exons. The methods utilized are based on DNA hybridization. The analysis of the human genome has made the identification of all gene exons possible, and it facilitates the design of oligonucleotide probes specific to each of them. The probes are used for purification of the exons from the DNA.19 Fragmentation of the DNA into pieces no bigger than 500 bp is the first step. The DNA is then hybridized to the exon-specific oligonucleotide probes and the hybridized fragments are purified. Hybridization can be performed in the liquid phase. In this case, oligonucleotides are labeled so that DNA–oligonucletide complexes can be separated from the bulk of nonhybridized DNA. In a common example, oligonucleotides are covalently bound to biotin so that DNA–oligonucleotide hybrids can be isolated using the biotin-binding molecule, streptavidin, coupled to magnetic beads. DNA fragments that do not contain exons will not bind to the streptavidin beads and can be efficiently removed after several washing steps. The fragments containing exons, bound to the beads, can be recovered after dissociation of the DNA–oligonucleotide hybrids under low-ionic strength conditions.
Exons can also be isolated by hybridization to a solid support where the exon-specific oligonucleotides have been spotted, as with DNA microarrays. In this case, the fragmented DNA is spread over the oligonucleotides to allow hybridization. Later, nonhybridized DNA is washed away and the exon-enriched DNA is eluted under low-ionic conditions.
Various commercial suppliers offer kits for exome isolation using liquid phase hybridization protocols, including Agilent Technologies (Santa Clara, CA, USA), Roche NimbelGen, Inc. (Madison, WI, USA), Illumina, Inc. (San Diego, CA, USA), and Life Technologies (Carlsbad, CA, USA). These kits allow for the isolation of over 90% of the exons present in the genome, with over 90% specificity at an approximate price of US$150 per exome. Several authors have compared these exome capture platforms,20–22 and the data obtained by Clark et al22 comparing the SureSelect Human All Exon 50 Mb (Agilent Technologies), Roche NimbleGen, Inc.’s SeqCap EZ Exome Library v2.0, and Illumina, Inc.’s TruSeq Exome Enrichment kits are summarized in Table 2. Some of the kits cover the untranslated regions of mRNA, in addition to the protein-coding regions, and this allows for the analysis of regulatory regions such as microRNA (miRNA) binding sites. The inclusion of 5′ untranslated regions also allows for the analysis of the proximal promoter regions.22 In addition, most of the kits cover up to 80% of the miRNA coding regions.21 Recently, improved kits have been developed by these and other suppliers so that the data shown in Table 2 should be considered an indication only. It is important to note that exon purification is a critical step. Recovering 100% of the exons is difficult, and exons are frequently lost or underrepresented in the isolated exome. For example, if the exome of a patient is being analyzed and 10% of the exons are lost during purification, the probability of missing a relevant mutation will be about 10% due to this technical error. Therefore, the use of highly efficient exon-capture procedures is of critical importance in exome sequencing.
 | Table 2 Comparison of three major exome-capture platforms |
Exome sequencing and data analysis
Exon-containing fragments are sequenced using any of the presently available massive sequencing equipment systems or technologies. As mentioned in the introduction, these platforms determine the nucleotide sequence of millions of DNA fragments simultaneously. The determined length of the sequence of each fragment in exome sequencing is not long, typically between 35 bp and 100 bp. However, since the DNA was initially fragmented randomly, each individual nucleotide will be present in many overlapping fragments. Therefore, if a high enough number of sequences is obtained, even if short, every base will be independently sequenced in several DNA fragments. The number of times that each base is sequenced is called coverage or sequencing depth. The coverage is directly related to the quality and confidence of the nucleotide sequence generated. In general, coverage of 20×−30× is considered necessary to obtain reliable results in exome sequencing.59 This sequencing depth means that a possible sequence variation would have been sequenced independently in 20–30 different DNA fragments.
Data analysis is the last step in exome-sequencing projects (Figure 2). As mentioned above, data from millions of sequences are generated, and their analyses require specific and complex computer programs and expertise.19,23 A preliminary step is an analysis of the quality of the sequence generated. The accuracy of sequence reading at various sequence lengths, the average length of the reads, as well as other parameters, are tested. If the quality is good enough, each sequence is compared to a reference sequence, which is usually the last available version of the human genome sequence. Typically, over 80% of the sequences generated can be aligned with the reference genome.22 This step allows a small degree of nucleotide variation with respect to the reference genome. The next step in the analysis is to identify sequence variations between the reference sequence and the exome sequence obtained in our study. The subsequent analyses of these variants could provide the desired information on the medical problem under study.
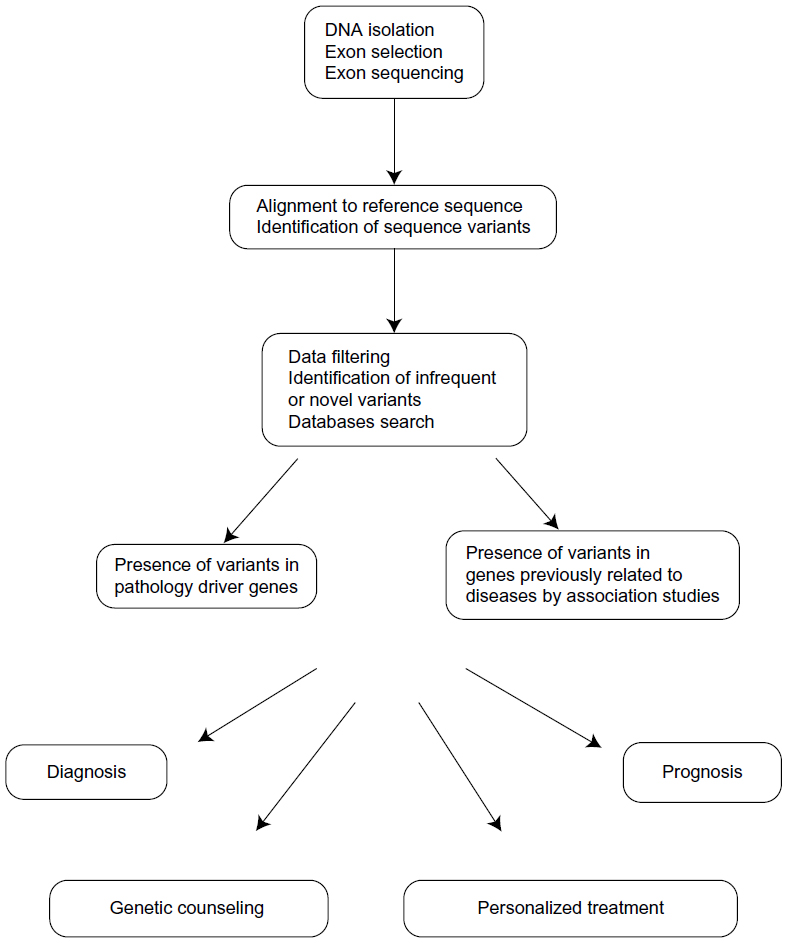 | Figure 2 Exome sequencing data analysis. |
Exome sequencing can detect several types of genetic variations. One of the most frequently found differences is a change of one nucleotide into another, for example, A for G (ATA codon to ATG). These variations are called single nucleotide variants (SNVs), although they are considered single nucleotide polymorphisms (SNPs) when their frequency in the population is larger than 1%–5% and there is no strong effect on the risk of any disease. Most SNVs are silent, or also known as synonymous, because both sequence variants code for the same amino acid (for example, a variation of GCA to GCC, since both are alanine codons). Most of these polymorphisms do not represent any difference for the encoded protein, are not under evolutionary selection, and represent the most frequently found variations in the human exome. The exception are some silent mutations that affect splicing-regulatory signals, or even transcription regulatory sites, altering mRNA splicing or expression even if they do not change the encoded amino acids. In other cases, the nucleotide variation has a consequence in the encoded protein and these are nonsilent or nonsynonymous variants. These changes can result in variations in the encoded amino acid (for example, GAT to GAG changes aspartic acid to glutamic acid), and are called missense mutations. More drastic alterations are produced when the nucleotide variation creates a translation stop codon (for example, TGC to TGA changes a cysteine codon to a stop codon), which is called a nonsense mutation. There is also a type of SNV that can be detected by exome sequencing even if it does not affect the protein codons. Since exons are selected after random fragmentation of the DNA, they can also contain contiguous DNA regions, including neighboring intron sequences and even gene promoters if untranslated regions were captured.24 Intron regions contain the regulatory signals required for mRNA splicing. SNVs in these regions can alter splicing in various ways.15 For example, the affected intron might be retained in the mature mRNA, or the contiguous exon might be spliced out (exon skipping). These alterations change the nucleotide sequence of the mature mRNA and, therefore, the encoded protein downstream from the SNV.25 Exome sequencing can also detect sequence variations due to small insertions or deletions (indels).22 These variations can result in a frame shift, except when they affect three or a multiple of three nucleotides. In that case, small deletions or insertions of amino acids would be produced.
Identification of causative mutations
The functional relevance of the sequence variants detected must be determined in the next data analysis step. Even if all humans are almost identical from a genetic point of view, the number of nucleotide sequence differences among individuals is considerable.26 This heterogeneity complicates the interpretation of the data obtained in individual sequencing projects. Some general data on individual sequence variations are shown in Table 3. When considering the whole genome, the number of sequence differences among individuals has been estimated at 4 × 106, according to the data obtained in the 1000 Genomes Project and smaller whole-genome sequencing projects.27 Exomes show a smaller, but still considerable, number of sequence variations, numbering about 20,000–25,000 between any two unrelated individuals.27,28 Most of these genetic variations are silent, as discussed earlier. The number of nonsilent sequence differences among individuals has been estimated at 10,000. Most of these variants exist in the general population and are transmitted for generations. It has been estimated that less than one nonsilent SNV appears de novo in each individual.29
The data obtained in exome sequencing projects are frequently filtered to identify all the SNPs that are present in other individuals and that are not, therefore, related to the disease being studied.2,19,23 This process can be done by comparison with public databases where the SNPs that are found in the sequencing projects are compiled. A caveat to be taken into account is that all large databases contain a number of proven mutations causing relatively frequent diseases. About 400–700 novel, and possibly relevant, SNVs remain after this filtering step (Table 3).28 The next challenge is to determine which of the SNVs that are not present in the global population, if any, are at the origin of the disease under study. Many of the differences observed will not be associated with any incidence of the disease, and these are known as passenger changes.23 In contrast, one or a few changes might have a causative role and they are called driver changes. The approach used to identify these driver changes will depend on the particular circumstances of the study. In diseases with a Mendelian pattern of inheritance, it is usually necessary to analyze a number of affected and nonaffected individuals to find gene variations that perfectly segregate with the disease. This comparison is more informative in large families with well characterized pedigrees. In the absence of sufficiently large affected families, the comparison of a number of unrelated patients and controls also allows for the identification of driver genes. Additional criteria are used to select possible SNVs related to the disease, including in silico algorithms, which predict the possible importance of the mutated amino acid based on evolutionary conservation, and on the predicted impact on the structure and function of the protein. The predicted function of the mutated protein and its tissue-specific expression pattern are also criteria used in selecting putative causative mutations.
 | Table 3 Summary of the sequence variation among individuals |
Some examples of these types of studies will be provided in a later section. However, as more studies are being carried out, more gene variations are being identified as causative of inherited diseases, which makes it probable that some of the genes mutated in the patient would have already been described. These mutated genes can be found in the literature and in specialized databases such as the Online Mendelian Inheritance in Man database (http://www.omim.org). The possible relevance of the mutations found in various genes can also be searched in the Genome Ensemble page (http://www.ensembl.org/) if they have been previously described.
Cancer is probably the most prevalent group of diseases with a genetic basis. Many studies have been directed to determine the driver genes for various types of cancer.30 The emergent group of cancer driver genes can be consulted in databases such as the Catalogue of Somatic Mutations In Cancer (COSMIC; http://cancer.sanger.ac.uk) or The Cancer Genome Atlas (http://cancergenome.nih.gov/). Several more detailed examples will be shown in the Examples of the clinical use of exome sequencing section.
Comparison of exome sequencing to other massive sequencing approaches
Genome sequencing
As mentioned in the Introduction, sequencing the whole human genome is becoming more and more affordable. Compared to exome sequencing, whole-genome sequencing is a much more complex alternative. The number of sequencing reactions that must be carried out is much higher, as are the numbers of nucleotide sequence data generated. The computational analysis is greatly increased. In addition, many more genetic variants are found, as shown in Table 3, which makes the identification of driver genes more difficult. However, genome sequencing provides a complete view of the genetic alterations present in the patient, including large genome reorganizations. However, short-read sequencing of a genome at moderate depth will miss structural variations, especially in low-complexity regions. This information is summarized in Table 4, which compares exome sequencing to other sequencing approaches.
As mentioned earlier, protein-coding genes only represent 3% of the genome.16 Until recently, the rest of the genome was considered to be “bulk DNA” without much information value. However, recent studies have completely changed this point of view. A large genome-wide project is studying the function of all the regions of the genome, the Encyclopedia of DNA Elements (ENCODE) project.31 The presently available results show that over 70% of the genome is transcribed. Many of the transcripts generated do not code for proteins, but they seem to have a regulatory role in gene expression. Among them are the already known miRNAs, which regulate mRNA stability and translation (Figure 1), but also over 20,000 long noncoding RNAs that regulate transcription. In addition, many DNA regions that regulate gene expression have been identified, including many previously unknown promoter and transcription regulatory regions (Figure 1). This information is of clinical relevance because mutations in regulatory regions can affect the expression of specific genes and can have pathological results. In fact, a large proportion of genome-wide association studies have related DNA regions, where no protein-coding mutations have been found, with pathological conditions.32 The data generated in the ENCODE project have allowed for the revision of some cases, which found that mutations in gene-expression regulatory regions are responsible for the disease.31,32 Also, in a recent example, Weedon et al33 reported that mutations in a transcription regulatory region of the PTF1A gene cause isolated pancreas agenesis. Mutations in regulatory regions cannot be detected by exome sequencing since they do not affect the encoded protein, but instead its expression. Therefore, whole-genome sequencing provides more information than exome sequencing at the expense of increased complexity and economic cost.
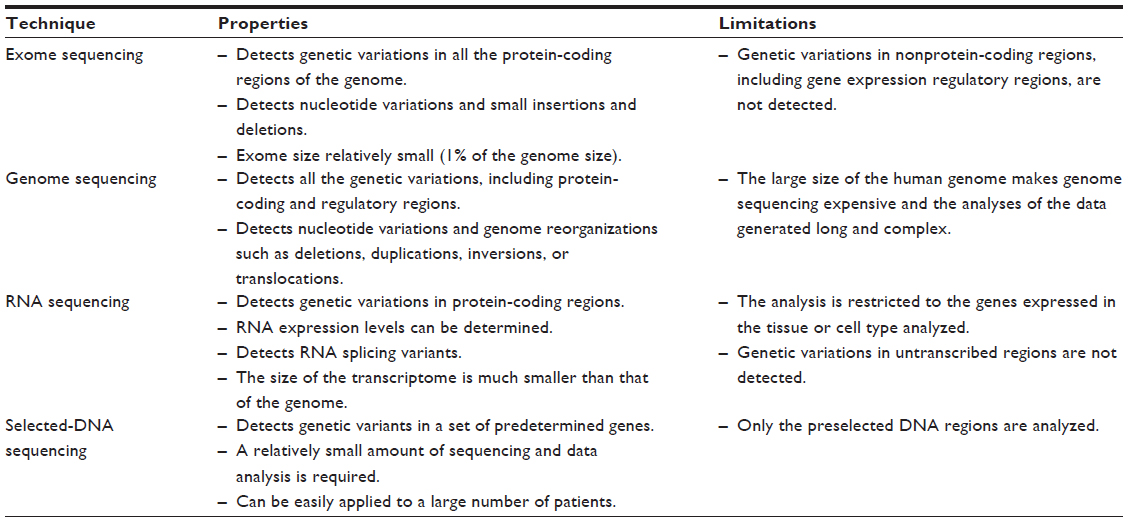 | Table 4 Comparison of massive sequencing techniques |
RNA sequencing
RNA sequencing techniques consist of the conversion of RNA populations to complementary DNA (cDNA) by reverse transcription and their subsequent sequencing.34,35 In the case of mRNA sequencing, the complete population of mRNAs expressed in a cell line or tissue sample (known as transcriptome) are converted to cDNA and sequenced. The process of mRNA sequencing provides information about the nucleotide sequence of the genes that are being transcribed in the sample analyzed and, therefore, on the amino acid sequence of the corresponding proteins. In addition, the number of sequences generated for each mRNA can be estimated and is proportional to its abundance. Therefore, gene expression levels can be determined and compared to those of other samples, including possible control samples (Table 4). Another specific advantage of mRNA sequencing is that it allows the study of alternative splicing events.36,37 As mentioned earlier, primary transcripts are often processed in multiple ways to give rise to mRNA that contain different exons (Figure 1). These mRNAs can be identified by mRNA sequencing and not by exome or genome sequencing, which determines the sequencing of the DNA being transcribed and not that of the mature transcript. Otherwise, mRNA and exome sequencing provide similar information about the protein-coding region of the genome. The difference is that exome sequencing includes all the genes and mRNA sequencing is restricted to the genes expressed in the sample analyzed. For example, a recent mRNA sequencing study of lymphoblastoid cell lines from 462 individuals determined the coding sequence of about 13,000 genes out of the 20,078 human genes.38 In this example, about 7,000 genes were not studied because they were not expressed in lymphoblastoid cell lines. However, in those cases where the cell type or tissue affected by a given disease is well known, mRNA sequencing would be equivalent to exome sequencing for the study of driver mutations. Another characteristic of mRNA sequencing is that it allows for the detection of sequence variations produced by RNA editing.39 A number of mRNAs are processed so that some nucleotides are changed, and adenosine to inosine changes are the most frequently produced. These alterations are detected by mRNA sequencing, but whether they are produced by RNA editing or as a consequence of genomic variations cannot be determined unless both the mRNA and genomic sequences are compared.
Determining mRNA expression levels can be very convenient in certain cases, since some diseases can be caused by the deregulated expression of one or more genes. Changes in expression levels can be very informative about the genetic origin of the disease. For example, alterations in the expression of one or more genes in a patient could indicate a dysfunction in the mechanisms that regulate their expression. This dysfunction could be due to mutations in the gene-transcription regulatory regions, as discussed in the genome sequencing section. It could also be due to alterations in the expression or structure of transcription regulatory factors.40 Changes in gene expression are frequently due to alterations in epigenetic mechanisms of gene expression regulation, such as DNA methylation, which cannot be detected by genome or exome sequencing.41 Methods for the study of whole-genome methylation that allow for the detailed study of this epigenetic information have been recently developed.42 Cancer is one of the diseases on which more studies in gene expression levels have been carried out. In an increasing number of cases, alterations in the expression of genes or a group of genes are related to a cancer diagnosis, prognosis, or a prediction of the response to anticancer drugs.43 These changes in gene expression are being used as biomarkers. Many of these studies are available through the Cancer Genome Anatomy Project (http://cgap.nci.nih.gov) database.
A specific type of RNA sequencing project is aimed at determining the nucleotide sequence and expression levels of small regulatory RNAs (miRNAs). Small RNAs regulate the expression of other genes by determining the stability and/or translation of their mRNAs (Figure 1). Changes in patterns of miRNA expression can, therefore, have a marked impact on the protein expression profile of cells and tissues. Protocols have been developed for the purification and sequencing of the complete population of miRNAs of a given sample and to determine their expression levels.44 Most exon-capture platforms also include up to 80% of the known miRNA-coding regions.21
Sequencing selected sets of genes
Some diseases have already been studied in such detail that most of the genes involved are known. This can be the case with diseases with a Mendelian pattern of inheritance, in which all the cases studied are due to mutations in any of a number of known genes. Other examples are some cancer types that are predominantly due to mutations in a reduced number of genes. In such cases, the more direct approach to characterize a patient’s sample would be to determine the sequence of the genes previously identified as causative of the disease. The classical approach would be to amplify all the exons of these genes and determine the nucleotide sequence of each. The alternative massive sequencing approach would be to purify all the putative genomic regions involved, and to simultaneously determine their nucleotide sequence in a single run.45–47 Two methods are generally used for the purification of the candidate DNA regions. The first is their amplification by polymerase chain reactions using a set of specific oligonucleotides as primers. The second method consists of fragmentation of the sample’s DNA and the purification of the relevant fragments by hybridization to specific oligonucleotides, either in solution or fixed to a solid support, as previously described for exons’ purification.48 The selected regions can contain protein-coding exons and also other DNA regions, such as transcription regulatory regions. These regions usually correspond to a few hundred genes and, therefore, the analysis of the sequence data generated is much easier than in other massive sequencing approaches. The main limitation is that this is a hypothesis-driven approach that does not allow for the detection of mutations in genes not previously related to the studied disease (Table 4).
Examples of the clinical use of exome sequencing
The most common use of exome sequencing is probably for the diagnosis of monogenic diseases. Over 3,000 monogenic disorders have been described, although the molecular genetic causes of most of them are still unknown.1 Exome sequencing can be used to identify these mutations, as discussed by Kuhlenbäumer et al1 in a recent review. In some of the first studies, exome sequencing was used to identify genetic mutations responsible for familiar diseases such as the Kabuki’s,49 Schinzel–Giedion,50 Joubert,51 and hyperphosphatasia mental retardation syndromes,52 severe brain malformations,53 or familiar amyotrophic lateral sclerosis.54 Exome sequencing has also been used to discover novel mutations present in a sporadic case of mental retardation.29 In addition, this technique has been used for the diagnosis, for example, of congenital chloride diarrhea,55 inflammatory bowel disease,56 Charcot–Marie–Tooth disease,57 neonatal diabetes mellitus,58 or the Brown–Vialetto–van Laere syndrome.59 The study reported by Worthey et al56 represents a relevant example of the clinical application of exome sequencing. A male child presented with a Crohn’s disease-like illness without a definitive diagnosis, despite a comprehensive clinical evaluation. The authors decided to use an exome sequencing approach to identify the causative mutation(s). Analysis of the sequence data detected 16,124 variants in the patient. Filtering the data while considering novel variants present in homozygosity, hemizygosity, or compound heterozygosity, and while affecting highly conserved amino acid residues predicted to be damaging to protein function, allowed the authors to select a mutation in the X-linked inhibitor of apoptosis gene (XIAP). Functional studies demonstrated the relevance of this mutation in the proinflammatory response observed in the patient. Based on the identification of this mutation, an allogenic hematopoietic progenitor transplant of cells was performed. Therefore, exome sequencing allowed for the identification of an uncharacterized mutation to make a molecular diagnosis for an individual patient, in the setting of a novel disease, which resulted in a management plan. The use of exome sequencing in the discovery of new causative mutations and in diagnosis has been recently reviewed.60,61
The study of common and complex diseases has also been approached through exome sequencing. Genome-wide association studies have shown that some genetic variants confer risk for a number of diseases. Well characterized examples are apolipoprotein E in Alzheimer’s disease, complement factor H in macular degeneration, or glucocerebrosidase/leucine rich repeat kinase 2 in Parkinson’s disease.62–64 The possible use of exome sequencing for the study of complex diseases has been discussed.2,28 One limitation of the use of exome sequencing in these studies is that most of the phenotype-associated variants lie distal to protein-coding regions, which would make whole-genome sequencing a better approach.32 Some of these genetic variants can affect the functionality of transcription regulatory regions that control gene expression. The ENCODE project31,65 has performed a genome-wide analyses of these regulatory regions, and it was found that several genetic variants in specific regions of chromosome 5 (for example) are binding sites for the transcription factor, GATA2, which are strongly associated with Crohn’s disease and other inflammatory diseases.
Cancers are diseases caused by the accumulation of genomic changes that result in the alteration of multiple biological processes.19 In contrast to the monogenic genetic alterations discussed earlier, most cancer driver mutations are not present in the normal tissue of the patient; a large proportion of these mutations resides in protein-coding regions and can be detected by exome sequencing.19 However, another important group of genetic alterations are large genomic reorganizations such as deletions, inversions, or translocations that cannot be detected by exome sequencing.66 Despite this limitation, exome sequencing has been applied to the discovery of cancer driver genes using two general strategies: the comparison of the exome of the tumors to that of healthy tissues from the same patient; or the comparison of a number of unrelated patients’ exomes to that of a similar number of healthy controls.67–70 Currently, extensive studies are being carried out that involve the exome or genome sequencing of a large cohort of cancer patients and controls to identify all the cancer driver genes.19,71,72 The 5,000 cancer genomes project is one example,73 as it aims to sequence the genome of 50 of the most common cancer types. The available data have already provided a general genomic landscape of the more common cancers, as reviewed by Vogelstein et al.3 About 140 genes have been identified that promote tumorigenesis when altered, and this can be found in the previously mentioned COSMIC database.3 Detecting the mutation of one of these genes in the exome of a cancer sample can be an important step towards the proper diagnosis and treatment of the patient. Present data also give an idea of the complexity of the cancer genome.3 Common solid tumors present an average number of 33 to 66 nonsilent somatic mutations.3 This number increases to over 200 in tumors induced by mutagenic agents, such as lung cancer and melanoma, and even to more than 1,000 in tumors deficient in DNA repair mechanisms or in the DNA polymerase E.3 In contrast, liquid and pediatric tumors present with less than ten somatic mutations.3 An important characteristic of tumors is that they evolve quickly and become heterogeneous, so that different mutations can be found in samples from the same patient collected in different regions or in different periods of time along the treatment, as recently shown by exome sequencing.74,75 Despite this complexity, some unifying concepts are emerging, and most of the known cancer driver genes participate in one or more of the 12 pathways that regulate cell survival, cell fate, and genome maintenance.3,19 In this scenario, exome sequencing is beginning to be used for cancer diagnosis through the identification of driver mutations, for example, in prostate cancer.76
Exome sequencing can also be useful for cancer treatment. The presence of some gene mutations can confer sensitivity or resistance to a given drug, which has been named pharmacogenomics. For example, the use of protein tyrosine kinase inhibitors in cancers that overexpress the Abelson murine leukemia viral oncogene homolog 1 (ABL) or epidermal growth factor receptor (EGFR) proteins has been known for several years. However, exome and genome sequencing approaches are revealing many more mutation responses to treatment associations (as highlighted in one review77). An informative example is the recent publication of the exome of the NCI-60 panel of cells.78 This panel contains 60 well-characterized cell lines from nine cancer types and has been used in a broad range of biological and pharmacological studies.79 The nucleotide sequence of the exome of these cells was determined to establish the cancer driver genes mutated in each of them. In addition to providing a list of putative novel cancer driver genes, the authors studied the possible correlation between the genotype of each cell line and the previously determined response to a large number of anticancer agents. A correlation was found between specific gene mutations and the response to several drugs, revealing the possible importance of exome sequencing in the selection of a personalized treatment. Exome sequencing can also be used to predict cancer predisposition. Some examples can be found in a recent review centered on colorectal cancer and using whole-genome sequencing.72
Medical challenges of exome sequencing
Exome sequencing promises significant improvements in patients’ diagnoses, prognoses, and personalized treatments. However, the extensive application of this technology still requires a number of improvements, as well as the definition of important ethical and medical considerations, as has been discussed in recent reviews.23,27,60,61,71,77 Technical challenges include the development of more efficient techniques of exon capture, sequencing, and alignment to obtain a complete and even representation of all the exons in the sequence. Improvements in the data analysis software tools for quick and accurate detection of pathological variants are also needed. Extensive exome sequencing will require the implementation of specialized equipment and hiring teams of specialists with adequate expertise to generate the sequences and to analyze and interpret the data obtained.
The use of exome sequencing for diagnosis will also require the implementation of technical guidelines and regulations. Parameters such as the sequencing depth, exon coverage, quality metrics for nucleotide sequence data, or alignment calling will have to be normalized. Data storage should also be regulated.
There are also a number of complex ethical issues. An important issue is related to the information that should be provided to the patient. Exome sequencing might detect genetic variations that are not related to the disease under diagnosis. The patient might present with genetic variants that represent risk factors or might be causative of other diseases. What information should be returned to the patient? What would be the evidence required to consider a genetic variant linked to a disease? The ownership, access, and storage of the data are other relevant issues. Should the data generated be kept for possible future use during the patient’s lifetime? These and other ethical considerations will probably raise considerable controversy80 and will require extensive discussion to reach an agreement on the criteria to be used in clinical practice.
Conclusion
Exome sequencing is already a powerful tool used to determine the molecular basis of genetic diseases. The depth of the genetic analysis is less than that of whole-genome sequencing since genetic variations in nonprotein-coding regions are not detected. However, the reduced number of sequences and sequence-analyses required for exome sequencing makes it a more affordable approach in clinical practice. Therefore, exome sequencing will probably be the technique of choice for the initial analysis of patients, at least until the price of whole-genome sequencing decreases and the considerable data analysis procedure is improved. An important limitation of the application of exome sequencing in clinical practice is that the functional significance of most of the expected genetic variants is still unknown. This situation is quickly changing, as an increasing number of disease-associated genetic variants are determined and made available in public databases. It is plausible that in a few years, most of the genetic variations related to the risk of acquiring a disease, with precise molecular diagnostics, a prediction of the disease evolution, and a pharmacological response, will be known. The precise knowledge of the patient’s exome, or genome, will then be a determining factor in medical practice.
Acknowledgments
I gratefully thank Rosario Perona and Juliette Siegfried (ServingEdit.com) for critical review of the manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Kuhlenbäumer G, Hullmann J, Appenzeller S. Novel genomic techniques open new avenues in the analysis of monogenic disorders. Hum Mutat. 2011;32(2):144–151. | |
Kiezun A, Garimella K, Do R, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44(6):623–630. | |
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. | |
Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008;73(2):103–112. | |
Walne AJ, Dokal I. Advances in the understanding of dyskeratosis congenita. Br J Haematol. 2009;145(2):164–172. | |
Brady PD, Vermeesch JR. Genomic microarrays: a technology overview. Prenat Diagn. 2012;32(4):336–343. | |
Hehir-Kwa JY, Pfundt R, Veltman JA, de Leeuw N. Pathogenic or not? Assessing the clinical relevance of copy number variants. Clin Genet. 2013;84(5):415–421. | |
Simons A, Sikkema-Raddatz B, de Leeuw N, Konrad NC, Hastings RJ, Schoumans J. Genome-wide arrays in routine diagnostics of hematological malignancies. Hum Mutat. 2012;33(6):941–948. | |
Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11(1):31–46. | |
Sastre L. New DNA sequencing technologies open a promising era for cancer research and treatment. Clin Transl Oncol. 2011;13(5):301–306. | |
Lander ES, Linton LM, Birren B, et al. International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. | |
Abecasis GR, Auton A, Brooks LD, et al. 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. | |
Yang Y, Liu R, Xie H, et al. Advances in nanopore sequencing technology. J Nanosci Nanotechnol. 2013;13(7):4521–4538. | |
Chen YS, Lee CH, Hung MY, Pan HA, Chiou JC, Huang GS. DNA sequencing using electrical conductance measurements of a DNA polymerase. Nat Nanotechnol. 2013;8(6):452–458. | |
Lu ZX, Jiang P, Xing Y. Genetic variation of pre-mRNA alternative splicing in human populations. Wiley Interdiscip Rev RNA. 2012;3(4):581–592. | |
Pruitt KD, Harrow J, Harte RA, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19(7):1316–1323. | |
Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22(9):1760–1774. | |
Teer JK, Mullikin JC. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19(R2):R145–R151. | |
Liu X, Wang J, Chen L. Whole-exome sequencing reveals recurrent somatic mutation networks in cancer. Cancer Lett. 2013;340(2):270–276. | |
Parla JS, Iossifov I, Grabill I, Spector MS, Kramer M, McCombie WR. A comparative analysis of exome capture. Genome Biol. 2011; 12(9):R97. | |
Sulonen AM, Ellonen P, Almusa H, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12(9):R94. | |
Clark MJ, Chen R, Lam HY, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29(10):908–914. | |
Gullapalli RR, Desai KV, Santana-Santos L, Kant JA, Becich MJ. Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics. J Pathol Inform. 2012;3:40. | |
Samuels DC, Han L, Li J, et al. Finding the lost treasures in exome sequencing data. Trends Genet. 2013;29(10):593–599. | |
Taneri B, Asilmaz E, Gaasterland T. Biomedical impact of splicing mutations revealed through exome sequencing. Mol Med. 2012;18:314–319. | |
Fu W, O’Connor TD, Jun G, et al; NHLBI Exome Sequencing Project. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216–220. | |
Marian AJ. Challenges in medical applications of whole exome/genome sequencing discoveries. Trends Cardiovasc Med. 2012;22(8):219–223. | |
Singleton AB. Exome sequencing: a transformative technology. Lancet Neurol. 2011;10(10):942–946. | |
Vissers LE, de Ligt J, Gilissen C, et al. A de novo paradigm for mental retardation. Nat Genet. 2010;42(12):1109–1112. | |
Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, et al. IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods. 2013;10(11):1081–1082. | |
Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M; ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. | |
Hardison RC. Genome-wide epigenetic data facilitate understanding of disease susceptibility association studies. J Biol Chem. 2012;287(37):30932–30940. | |
Weedon MN, Cebola I, Patch AM, et al. International Pancreatic Agenesis Consortium. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nat Genet. 2014;46(1):61–64. | |
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. | |
Mutz KO, Heilkenbrinker A, Lönne M, Walter JG, Stahl F. Transcriptome analysis using next-generation sequencing. Curr Opin Biotechnol. 2013;24(1):22–30. | |
Hitzemann R, Bottomly D, Darakjian P, et al. Genes, behavior and next-generation RNA sequencing. Genes Brain Behav. 2013;12(1):1–12. | |
Costa V, Aprile M, Esposito R, Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur J Hum Genet. 2013;21(2):134–142. | |
Lappalainen T, Sammeth M, Friedländer MR, et al; Geuvadis Consortium; Geuvadis Consortium. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013; 501(7468):506–511. | |
Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013;5:105. | |
Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–1251. | |
Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339(6127):1567–1570. | |
Li P, Demirci F, Mahalingam G, Demirci C, Nakano M, Meyers BC. An integrated workflow for DNA methylation analysis. J Genet Genomics. 2013;40(5):249–260. | |
Chibon F. Cancer gene expression signatures - the rise and fall? Eur J Cancer. 2013;49:2000–2009. | |
Dedeoğlu BG. High-throughput approaches for microRNA expression analysis. Methods Mol Biol. 2014;1107:91–103. | |
Ni T, Wu H, Song S, Jelley M, Zhu J. Selective gene amplification for high-throughput sequencing. Recent Pat DNA Gene Seq. 2009; 3(1):29–38. | |
Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. | |
Garnett MJ, Edelman EJ, Heidorn SJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–575. | |
Hoischen A, Gilissen C, Arts P, et al. Massively parallel sequencing of ataxia genes after array-based enrichment. Hum Mutat. 2010;31(4):494–499. | |
Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–793. | |
Hoischen A, van Bon BW, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42(6):483–485. | |
Edvardson S, Shaag A, Zenvirt S, et al. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86(1):93–97. | |
Krawitz PM, Schweiger MR, Rödelsperger C, et al. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42(10):827–829. | |
Bilgüvar K, Oztürk AK, Louvi A, et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature. 2010;467(7312):207–210. | |
Johnson JO, Mandrioli J, Benatar M, et al. ITALSGEN Consortium. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864. | |
Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–19101. | |
Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13(3):255–262. | |
Montenegro G, Powell E, Huang J, et al. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann Neurol. 2011;69(3):464–470. | |
Bonnefond A, Durand E, Sand O, et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One. 2010;5(10):e13630. | |
Johnson JO, Gibbs JR, Van Maldergem L, Houlden H, Singleton AB. Exome sequencing in Brown-Vialetto-van Laere syndrome. Am J Hum Genet. 2010;87(4):567–9; author reply 569. | |
Bras JM, Singleton AB. Exome sequencing in Parkinson’s disease. Clin Genet. 2011;80(2):104–109. | |
Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80(2):117–126. | |
Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. | |
Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. | |
Tan EK. Identification of a common genetic risk variant (LRRK2 Gly2385Arg) in Parkinson’s disease. Ann Acad Med Singapore. 2006;35(11):840–842. | |
Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3(4):e58. | |
Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. | |
Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. | |
Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. | |
Timmermann B, Kerick M, Roehr C, et al. Somatic mutation profiles of MSI and MSS colorectal cancer identified by whole exome next generation sequencing and bioinformatics analysis. PLoS One. 2010;5(12):e15661. | |
Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469(7331):539–542. | |
Ku CS, Cooper DN, Roukos DH. Clinical relevance of cancer genome sequencing. World J Gastroenterol. 2013;19(13):2011–2018. | |
Kilpivaara O, Aaltonen LA. Diagnostic cancer genome sequencing and the contribution of germline variants. Science. 2013;339(6127):1559–1562. | |
Hudson TJ, Anderson W, Artez A, et al; International Cancer Genome Consortium. International network of cancer genome projects. Nature. 2010;464(7291):993–998. | |
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. | |
Ren SC, Qu M, Sun YH. Investigating intratumour heterogeneity by single-cell sequencing. Asian J Androl. 2013;15(6):729–734. | |
Hieronymus H, Sawyers CL. Traversing the genomic landscape of prostate cancer from diagnosis to death. Nat Genet. 2012;44(6):613–614. | |
McLeod HL. Cancer pharmacogenomics: early promise, but concerted effort needed. Science. 2013;339(6127):1563–1566. | |
Abaan OD, Polley EC, Davis SR, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73(14):4372–4382. | |
Weinstein JN. Drug discovery: Cell lines battle cancer. Nature. 2012;483:544–545. | |
Shahmirzadi L, Chao EC, Palmaer E, Parra MC, Tang S, Gonzalez KD. Patient decisions for disclosure of secondary findings among the first 200 individuals undergoing clinical diagnostic exome sequencing. Genet Med. Epub October 10, 2013. | |
Flicek P, Amode MR, Barrell D, et al. Ensembl 2011. Nucleic Acids Res. 2011;39:D800–D806. | |
Pruitt KD, Tatusova T, Klimke W, Maglott DR. NCBI Reference Sequences: current status, policy and new initiatives. Nucleic Acids Res. 2009;37:D32–D36. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.