Back to Journals » The Application of Clinical Genetics » Volume 9
Examining genotypic variation in autism spectrum disorder and its relationship to parental age and phenotype
Authors Geier D, Kern J, Sykes L, Geier M
Received 13 May 2016
Accepted for publication 15 June 2016
Published 28 July 2016 Volume 2016:9 Pages 121—129
DOI https://doi.org/10.2147/TACG.S112712
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
David A Geier,1,2 Janet K Kern,1,3 Lisa K Sykes,2 Mark R Geier1,2
1Research Department, The Institute of Chronic Illnesses, Inc, 2Research Department, CoMeD, Inc, Silver Spring, MD, 3Research Department, CONEM US Autism Research Group, Allen, TX, USA
Background: Previous studies on genetic testing of chromosomal abnormalities in individuals diagnosed with autism spectrum disorder (ASD) found that ~80% have negative genetic test results (NGTRs) and ~20% have positive genetic test results (PGTRs), of which ~7% were probable de novo mutations (PDNMs). Research suggests that parental age is a risk factor for an ASD diagnosis. This study examined genotypic variation in ASD and its relationship to parental age and phenotype.
Methods: Phenotype was derived from detailed clinical information, and genotype was derived from high-resolution blood chromosome and blood whole-genome copy number variant genetic testing on a consecutive cohort (born: 1983–2009) of subjects diagnosed with ASD (N=218).
Results: Among the subjects examined, 80.3% had NGTRs and 19.7% had PGTRs, of which 6.9% had PDNMs. NGTR subjects were born more recently (the risk of PDNMs decreasing by 12% per more recent birth year) and tended to have an increased male–female ratio compared to PDNM subjects. PDNM subjects had significantly increased mean parental age and paternal age at subject’s birth (the risk of a PDNM increasing by 7%–8% per year of parental or paternal age) compared to NGTR subjects. PGTR and NGTR subjects showed significant improvements in speech/language/communication with increasing age. PGTR subjects showed significant improvements in sociability, a core feature of an ASD diagnosis, with increasing age, whereas NGTR subjects showed significant worsening in sociability with increasing age.
Conclusion: This study helps to elucidate different phenotypic ASD subtypes and may even indicate the need for differential diagnostic classifications.
Keywords: genotype, phenotype, autism spectrum disorder, microarray, advanced age, gene, pervasive developmental disorder, parental age
Introduction
Significant progress has been made in understanding the genetic pathogenesis of autism spectrum disorder (ASD) in recent years.1 Studies examining heritability have confirmed a role of genetic factors as risk for this disorder. A better understanding of genetic factors in ASD was facilitated by technical innovations allowing for genome-wide surveys of a range of possible sequence variations from common single-nucleotide polymorphisms to basically private chromosomal abnormalities. Copy number variant (CNV) studies have revealed important roles for recurrent and nonrecurrent large dosage imbalances. However, these studies have seldom delineated the individual genes responsible.1
As stated by Talkowski et al,1 rare point mutations and characterization of balanced chromosomal abnormalities have identified individual ASD-related genes of reasonably robust effect. These have included loci with substantive a priori biological significance, as well as those that were not necessarily suspected as biological targets of significant consequence. Many common variants, each adding a small individual contribution to ASD risk, have recently become apparent. Taken collectively, more recent genetic findings provide persuasive evidence that the genetic basis of ASD is very heterogeneous. There are apparently hundreds of genes involved, depending on their nature and their predisposing genetic difference, which can present variable amounts of risk.
In addition, many of the genes considered to be risk factors for ASD also appear to be risk factors for other neurodevelopmental disorders and for a range of neuropsychiatric disorders. It is clear that some genes associated with an ASD diagnosis, such as synaptic proteins (eg, SHANKs, neuroligins, and neurexins), as well as Fragile X mental retardation-associated proteins, have more obvious functional importance. Other genes associated with an ASD diagnosis do not offer a plausible mechanism for specific neuropsychiatric dysfunction (eg, regulators of chromatin modification and global gene expression). Overall, genetic studies to date have yielded some notable results. However, a consensus in the understanding of ASD pathogenesis or its heterogeneous clinical expression has not been achieved.1
Recently, some have postulated that advancing parental age, both of the father and mother, increases the risk of ASD in their children.2 Whole-genome sequencing testing has shown an association between older fathers with elevated rates of de novo mutations and increased risk of ASD. However, according to Lee and McGrath,2 the mechanisms related to increasing maternal age and a higher risk of an ASD diagnosis may be different from those operating with increasing paternal age. In each case, multiple risk factors may be associated with multiple interacting factors. Even though the pathogenesis of ASD-associated mutations is not well understood, research on paternal age may offer useful information as to the possible environmental and/or genetic risks involved in ASD.
The purpose of this study was to build upon previous genetic studies by examining the genotypic variation in ASD and its relationship to parental age and phenotype. In particular, this study examined the hypothesis that genetic mutations in individuals diagnosed with ASD are important to phenotypic variation.
Methods
The institutional review board (IRB) of the Liberty IRB, Inc (Deland, FL, USA; #12.08.0023) approved this study to retrospectively examine existing clinical data. Liberty IRB, Inc is fully accredited with the Association for the Accreditation of Human Research Protection Programs (AAHRPP). The Liberty IRB determined that this retrospective examination of de-identified data was not human subject research and, as a result, did not require informed consent from the subjects.
Study subjects
This study examined de-identified data derived from a cohort of subjects diagnosed with ASD (N=218). This study examined consecutive qualifying subjects who were recruited from retrospective examination of medical charts. The subjects in this study had presented at a medical clinic for outpatient genetic consultations. All of the study subjects were previously diagnosed by a trained professional using the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) criteria for autism or ASD.
Clinical evaluation
The examined subjects had extensive medical histories taken by trained health care professionals at the time of their initial clinical visit. Subject’s age at initial clinical visit, date of birth, sex, ASD classification (any type of developmental regression after birth or no developmental regression after birth), maternal, and paternal age at subject’s birth were obtained. In addition, a parent/guardian-completed Autism Treatment Evaluation Checklist (ATEC) was collected for each subject.
The ATEC was used to measure the phenotypic severity (ie, ASD symptom severity) of subjects in this study diagnosed with ASD by measuring ASD severity (ie, the worse the ASD phenotypic severity, the higher the ATEC severity score). Total severity, speech/language/communication, sociability, sensory/cognitive awareness, and health/physical/behavior scores were determined quantitatively from the ATEC evaluation of each subject.
Genetic testing
All subjects subsequently underwent a blood draw for genetic testing by the Laboratory Corporation of America (LabCorp). The specimens were collected at LabCorp specimen collection locations following their standard collection procedures. The genetic testing for each subject consisted of a high-resolution blood chromosome and blood whole-genome single-nucleotide polymorphism (SNP) CNV microarray analysis.3 The blood whole-genome CNV microarray analysis was conducted by LabCorp either using 1.8 million or 2.695 million genotyping targets.
The 1.8 million genotyping target analysis was performed by LabCorp using the Affymetrix 6.0 platform, which uses >900,000 SNP probes and 900,000 NPCN probes with a median spacing of 0.7 kbp. Five hundred nanogram of total genomic DNA extracted from lymphocytes was digested with NspL and Styl and then ligated to Nspl or Styl adaptors, respectively, and amplified using the GeneAmp Polymerase Chain Reaction (PCR) System 9700. PCR products were purified using AMPure beads and quantified using NanoDrop 8000. Purified DNA was fragmented and biotin labeled and hybridized to the Affymetrix 6.0 GeneChip. Data were analyzed using the Affymetrix Genotyping Console Browser (Version 3.01). Positive evaluation criteria included 1) DNA copy gain/loss within known clinically significant gene region of 50 kbp; 2) DNA copy number loss outside the known clinically significant regions of >200 kbp with <100.0% CNV, a stringency of >50 SNPs/CN probes/200 kbp segment, at least one Online Mendelian Inheritance in Man (OMIM®; Johns Hopkins University School of Medicine, Baltimore, MD, US) annotated gene; or 3) a single chromosome with long contiguous segment of homozygosity >15 Mbp) – extended allele homozygosity >10 Mbp per chromosome on multiple chromosomes suggests homozygosity due to common descent.
The 2.695 million genotyping target analysis was performed by LabCorp using the Affymetix Cytoscan HD platform, which uses >743,000 SNP probes and 1,953,000 NPCN probes with a median spacing of 0.88 kbp. Two hundred fifty nanogram of total genomic DNA extracted from lymphocytes was digested with Nspl and then ligated to Nspl adaptors, respectively, and amplified using Titanium Taq with the GeneAmp PCR System 9700. PCR products were purified using AMPure beads and quantified using NanoDrop 8000. Purified DNA was fragmented and biotin labeled and hybridized to the Affymetrix Cytoscan HD GeneChip. Data were analyzed using the Chromosome Analysis Suite. The analysis is based on the GRCh37/hg19 assembly. Positive evaluation criteria included: 1) DNA copy gain/loss within a known clinically significant gene region of ≥50 kbp; 2) DNA copy number loss of >200 kbp or gain >500 kbp outside known clinically significant regions with at least one OMIM annotated gene or within a region of clear clinical significance; 3) a long contiguous region of homozygosity in a single chromosome >20 Mbp interstitially or >10 Mbp telomerically (15 Mbp and 8 Mbp, respectively, for imprinted chromosomes); 4) contiguous homozygosity >8 Mbp within multiple chromosomes suggesting common descent – these are regions of potential recessive allele risk; or 5) a high level of allele homozygosity due to numerous contiguous short runs associated with geographically or socially limited gene pool at the 99th percentile is reported.
The laboratory technician was blinded to the subject’s clinical information. The results of LabCorp genetic testing were classified as negative genetic test results (NGTRs; these included subjects who were classified by LabCorp as not having any changes from either high-resolution blood chromosome or blood whole-genome SNP CNV microarray analysis), overall positive genetic test results (PGTRs; these included subjects who were classified by LabCorp as having identified genetic changes from either the high-resolution blood chromosome or blood whole-genome SNP CNV microarray analysis), or positive for possible de novo variant genetic test results (these included subjects who were classified by LabCorp as having identified genetic changes from either the high-resolution blood chromosome or blood whole-genome SNP CNV microarray analysis that were confirmed to not be present in the subject’s parents, not classified by LabCorp as possible familial variants, or not classified by LabCorp as a normal population variant).
Statistical evaluation
The statistical package contained in StatsDirect (Version 2.8.0) was utilized for all statistical tests, and in all conducted statistical tests, a two-sided P-value <0.05 was considered statistically significant.
In order to examine the subcohorts (NGTRs, overall PGTRs, and positive for possible de novo variant genetic test results), the Fisher’s exact statistical test (for the categorical variables of sex and classification) or the nonparametric Mann–Whitney U-test statistic (for continuous variables of date of birth, parental age at subject’s birth, and ATEC scores) were utilized. For those continuous variables that were significantly different between the different subcohorts examined, the logistic regression statistic was then used. The null hypothesis means there would be no differences between the various subcohorts examined for any of the continuous or categorical variables. In addition, the potential relationship between the subject’s age and ATEC scores was examined in the NGTRs and overall PGTR subcohorts using the Spearman’s rank coefficient (rho) statistic. The null hypothesis means there would be no relationship between the subject’s age and ATEC scores in either the negative genetic testing or the overall positive genetic testing subcohorts.
Results
Table 1 summarizes the general composition of the overall cohort of subjects diagnosed with ASD examined (N=218). Among the subjects examined, it was observed that the average age at initial clinical evaluation was 10.23±5.14 years old, and they were generally born in the late 1990s (range: 1983–2009). Overall, there were far more males relative to females (male:female ratio =4.89). A majority (57%) of the subjects examined experienced some degree of regression in skills following birth. The parental age at the time of birth of the subjects examined was generally in their early 30s (range: 17–59 years old). The ATEC scores showed that the overall severity of the subjects examined was moderate (50th–59th percentile of severity) with study subjects being least impacted in the area of speech/language/communication skills (30th–39th percentile of severity) and most impacted in the area of sensory/cognitive awareness skills (80th–89th percentile of severity).
 | Table 1 An overview of the entire study cohort (N=218) examined Notes: aSubjects who experienced any type of regression in skills following birth. bIncreasing scores and increasing percentiles of severity are associated with increasing severity of symptoms. Abbreviations: ATEC, Autism Treatment Evaluation Checklist; DOB, date of birth; SD, standard deviation. |
Table 2 reveals the genetic test results observed for those in the subcohort with PGTRs (n=43). Overall, blood chromosome microarray and high-resolution blood chromosome genetic testing identified a frequency of 19.72% for the subjects examined having some type of PGTRs. Among those with PGTRs, the most common findings were the detection of deletions (48.8%) or duplications (39.5%) using blood chromosome microarray testing and inversions (14%) using high-resolution blood chromosome testing. A majority of the PGTRs were possible familial variants/normal population variants (65.1%) in comparison to possible de novo variants (34.9%), and the overall frequency of positive genetic testing for possible de novo variants in the cohort of subjects examined was 6.9%.
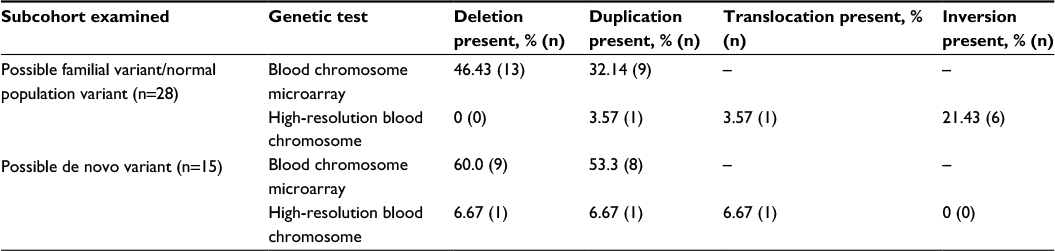 | Table 2 An overview of the overall positive genetic testing subcohort (n=43) examineda Notes: aSubjects may have findings present on more genetic test results, and they may have more than one type of genetic changes present on the same genetic test results. |
Table 3 provides an overview of the categorical and continuous variables evaluated among the various subcohorts examined. The mean subject’s date of birth in years at initial clinical evaluation was significantly further in the past in the positive for possible de novo variant genetic test results subcohort (1995.93±6.1) than that in the NGTRs subcohort (1999.78±4.92). Follow-up logistic regression analysis revealed an odds ratio =0.878 (95% confidence interval [CI] =0.798–0.965, P<0.01) for decreasing risk of testing positive for possible de novo variant genetic test results in comparison to testing negative for genetic test results for each more recent year of birth. Also evident was the fact that the positive for possible de novo variant genetic test results subcohort had significantly increased mean parental age and paternal age at subject’s birth when compared to the NGTRs subcohort. Follow-up logistic regression analysis also revealed an increased odds ratio =1.078 (95% CI=1.0038–1.158, P<0.05) for each increasing year of parental age and an increased odds ratio =1.078 (95% CI=1.0169–1.1353, P<0.05) for each increasing year of paternal age at subject’s birth, with regard to the risk of subject being positive for possible de novo variant genetic test results in comparison to the subcohort with NGTRs. Finally, the positive for possible de novo variant genetic test results subcohort had significantly increased parental age at subject’s birth when compared to the overall PGTRs. Follow-up logistic regression analysis revealed an odds ratio =1.069 (95% CI=1–1.14, P=0.05) for mean parental age at subject’s birth among subjects in the subcohort with positive for possible de novo variant genetic test results in comparison to the subcohort with overall PGTRs. No other significant differences were observed among the various subcohorts examined for sex, regression classification, parental age at subject’s birth, or ATEC scores.
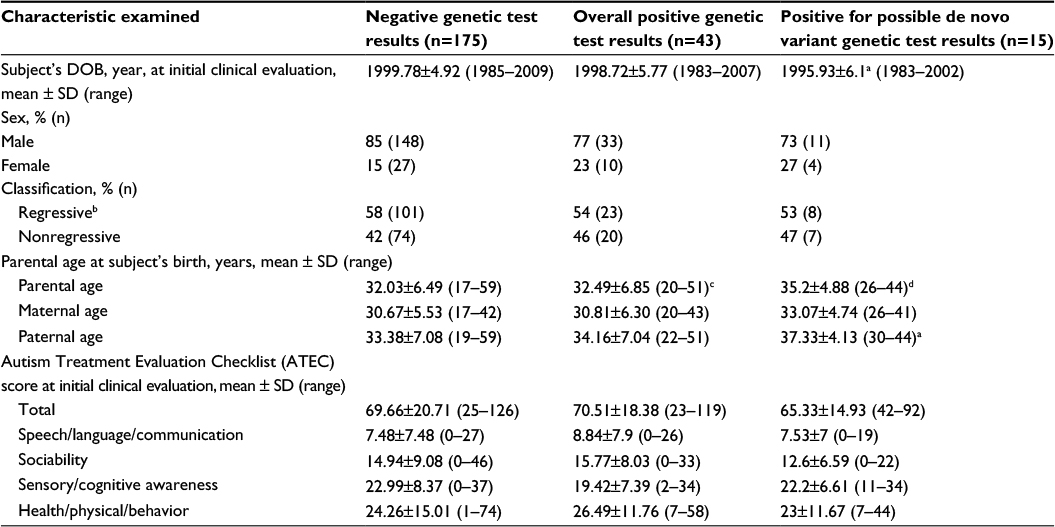 | Table 3 An overview of the study subcohorts examined Notes: aThe positive probable de novo variant genetic test results subcohort (n=15) was significantly (P<0.05) different than the negative genetic test results negative subcohort (n=175) using the nonparametric Mann–Whitney U-test statistic. bSubjects who experienced any type of regression in skills following birth. cThe positive probable de novo variant genetic test results subcohort (n=15) was significantly (P<0.05) different than the overall positive for genetic test results subcohort (n=43) using the nonparametric Mann–Whitney U-test statistic. dThe positive probable de novo variant genetic test results subcohort (n=15) was significantly (P<0.005) different than the negative genetic test results (n=175) using the nonparametric Mann–Whitney U-test statistic. Abbreviations: ATEC, Autism Treatment Evaluation Checklist; DOB, date of birth; SD, standard deviation. |
Table 4 reveals a summary of the Spearman’s rank correlation coefficient between a subject’s age and ATEC scores in the NGTRs and overall PGTRs subcohorts examined. In both subcohorts examined, ATEC scores in the areas of speech/language/communication skill scores improved as a function of increasing age of the subject. It was also observed that sociability skill scores significantly worsened with increasing age of the subject in the NGTRs subcohort, whereas sociability skill scores significantly improved with increasing subject’s age in the overall PGTRs subcohort.
 | Table 4 A summary of the relationship between the subject’s age and ATEC scores based upon genetic testing status Notes: Values shown in bold are statistically significant. Abbreviation: ATEC, Autism Treatment Evaluation Checklist. |
Discussion
The present hypothesis testing study evaluated the relationship between increasing parental age and its association with the presence of genetic mutations in subjects diagnosed with ASD. The results of this study reveal direct clinical evidence to support the hypothesis that subjects diagnosed with ASD and positive for possible de novo variant genetic test results had significantly increased mean parental ages and paternal ages in comparison to those with NGTRs. Logistic regression analysis revealed that the risk of a subject diagnosed with ASD and positive for possible de novo variant genetic test results in comparison to a subject diagnosed with ASD and NGTRs by ∼7%–8% per year of increased parental or paternal age. By contrast, subjects diagnosed with ASD and positive for possible de novo variant genetic tests results did not have significantly increased mean maternal ages in comparison to those with NGTRs.
The findings made in this study provide direct clinical support to a recent hypothesis that postulated among subjects diagnosed with ASD, increases in mutation rates and de novo mutations occurring at an increased frequency among older fathers, might make paternal age a factor in ASD etiology.4 In addition, children of older mothers were reported to have increased maternal recombinations as compared to younger mothers.5 However, transmission of mutations to offspring occurs among men (regardless of the sex of the offspring) more than women.5 Therefore, it was concluded, since increased parental age and an increased genetic frequency of mutations are both reported to be associated with a higher risk of developing ASD, an association between increasing genetic mutation frequency and increasing parental age in ASD pathogenesis exists.5
This study is based on our own previous study of subjects diagnosed with ASD, where we tested the theory that if increased genetic frequency of mutations from increasing paternal age is important to ASD pathogenesis, then there should be a significant relationship between increasing parental age and increasing ASD phenotypic.6 The results of our previous study revealed that increasing ASD phenotypic severity did not correlate with increasing parental age. We concluded our previous study by suggesting that, despite the lack of findings between increasing ASD phenotypic severity and increasing parental age, future research should investigate the relationship between genetic mutations and increasing parental age in ASD. The results of this study confirm the importance of examining individuals diagnosed with ASD and positive for possible de novo variant genetic tests, as a means to evaluate the importance of parental age on the presence of genetic mutations.
This study also tested the hypothesis that subjects diagnosed with ASD and genetic mutations would have a distinct clinical phenotype in comparison to subjects diagnosed with ASD and without genetic mutations. Results demonstrated that the date of birth for subjects positive for possible de novo variant genetic test results were further in the past (1995.93) in comparison to subjects with NGTRs (1999.78). Logistic regression analysis revealed that the risk of testing positive for possible de novo variant genetic test results decreased by ∼12% for each more recent year of birth. In addition, a nonsignificant, ∼twofold, decrease in male–female ratio from the subcohort with NGTRs =5.48 in comparison to the subcohort which was positive for possible de novo variant genetic test results =2.75. These findings suggest the existence of an increasing environmental component contributing to ASD etiology during the 1990s; this component more preferentially impacted males in comparison to females, in the absence of genetic mutations. This phenomenon is supported by previous research observing evidence for increasing environmental triggers for ASD in the 1990s.7 Interestingly, it was also observed that there were no differences in the NGTRs subcohort in comparison to the positive for possible de novo variant genetic test results subcohort for the frequency of any type of regression after birth or overall ASD severity/clinical features as measured by ATEC scores.
In order to further evaluate the hypothesis that subjects diagnosed with ASD and genetic mutations would have a distinct clinical phenotype, in comparison to subjects diagnosed with ASD and without genetic mutations, an assessment was undertaken to assess how ASD severity/clinical features, as measured by ATEC scores, varied in relation to the subject’s age. It was observed that the subcohort with NGTRs and the overall PGTRs subcohort showed significant improvements in ATEC speech/language/communication scores with increasing age of the subject. This type of observation was made in previous research of subjects diagnosed with ASD.6
However, it was observed in this study that the subcohort with NGTRs showed significant worsening of ATEC sociability scores with increasing age of the subject, whereas the overall PGTRs subcohort showed significant improvements in ATEC sociability scores with increasing age of the subject. A similar phenomenon was observed in previous genetic studies.8 This result suggests that sociability (a key hallmark diagnostic feature of ASD) actually significantly worsened in those with NGTRs, which, again, points toward an ongoing environmental component continuing to contribute to their increasingly worsening sociability ASD clinical phenotype as the subjects grow older. In contrast, this result also suggests that improvements in sociability among those with overall PGTRs indicates that whatever the role the positive genetic tests results played in the ASD clinical phenotype, it is contributing significantly less to their sociability ASD clinical phenotype as the subjects grow older.
Finally, the results of this study provide important insights into the frequency of genetic variation in subjects diagnosed with ASD. Among the cohort of subjects diagnosed with ASD examined, the overall PGTRs were observed in 19.7%. Among those in the subcohort with PGTRs, many different genetic mutations were identified such as deletions, duplications, translocations, and inversions, and these genetic mutations were distributed across many different loci on many different chromosomes. Furthermore, analysis of the overall PGTRs subcohort revealed a further subcohort of subjects with positive possible de novo variants, but this subcohort represented only 6.9% of all the subjects examined in this study. As a consequence, the vast majority (80.3%) of the subjects diagnosed with ASD did not have any identifiable genetic findings from high-resolution blood chromosome or blood chromosome microarray testing.
Our results are entirely consistent with another previous large cohort of 933 subjects diagnosed with ASD who underwent high-resolution blood chromosome or blood chromosome microarray testing.9 These investigators described ∼19.4% of the subjects tested were positive for any type of blood chromosome or blood chromosome microarray testing (19.7% in this study), and this number was reduced to 7.0% when considering subjects with positive possible de novo variant genetic test results (6.9% in this study). These investigators observed, just as we did in this study, that among those with PGTRs, it was found that many different genetic mutations were identified such as deletions, duplications, translocations, and inversions, and these genetic mutations were distributed across many different loci on many different chromosomes.
Strengths/limitations
A central strength of this study was the study design. The study individuals in this study were recruited from retrospective examination of medical charts of subjects who came for genetic consultations on an outpatient basis. All of the individuals examined in this study had been previously diagnosed with ASD by a health care professional and then underwent an extensive initial clinical evaluation and subsequently underwent routinely clinically available chromosome microarray and high-resolution blood chromosome genetic testing from LabCorp. The clinical evaluations and laboratory testing were conducted blinded to each other’s findings, which minimized potential observer or study participation biases.
The use of DSM-IV criteria by health care professionals to diagnosis the cohort of subjects examined in this study was another strength of this study. Using the same diagnostic criteria for every subject in the study allowed for diagnostic consistency. It ensured that the individuals in the study had been diagnosed with ASD under uniform diagnostic criteria and that the diagnosis was made prior to the collection of data for this study, so that the influence of diagnostic status of the subjects would not have influenced the collection of the data examined.
The use of parental-completed ATECs to determine the phenotypic severity of the individuals examined in this study that were diagnosed with ASD was also a strength of this study. The ATEC is commonly used in research to measure ASD phenotypic severity. Quantitative total scores and subscale scores for specific domains in ASD symptoms are provided by the ATEC and, in recent studies, was found to significantly correlate with other well-established measures of ASD severity.10
The lack of neurotypical controls is a limitation of the study because the increased ASD risk associated with advanced paternal age must be validated with a population study analyzing the ASD incidence in subgroups with different parental ages. A neurotypical group would allow for the calculation of the rate of autism by age group. Future studies should explore this.
While it may be worthwhile to study parental ages among subjects diagnosed with ASD in comparison to neurotypical controls, this study went further than this simple comparison between these two populations for genomic or phenotypic variations. This study was able to directly demonstrate that increasing parental age in individuals diagnosed with ASD was statistically significantly associated with an increased frequency of positive for possible de novo variant genetic test results in comparison with those with NGTRs. Furthermore, the results of this study provide important statistically significant differences in phenotypic variations among individuals with ASD who had genetic mutations in comparison with those without genetic mutations.
Another possible limitation of this study was that only ATEC scoring was used to determine ASD phenotypic severity. Despite this limitation, the ATEC has been used and validated in previous research.10 In addition, this study evaluated individuals diagnosed with ASD under the DSM-IV criteria, which has been replaced with DSM-V criteria, since this study took place. Future studies should utilize other measures of ASD to observe their findings with our results.
An additional potential limitation of this study was the cohort of individuals derived only from one set of clinics. It is possible that the ASD population examined may be phenotypically or genetically different from other ASD populations. Despite this potential limitation, many of the phenotypic and genetic results observed among the ASD subjects examined are virtually identical to those observed in an even larger ASD cohort study examining phenotypic and genomic variations.7 We recommend in future studies that other cohorts of subjects diagnosed with ASD be examined to observe their consistency with the observations made in this study.
A potential limitation of this study was that the genetic findings of subjects with PGTRs were not explored in detail. Among the 43 subjects with PGTRs, their genetic abnormalities were highly complex and as such the underlying genetic pathology is beyond the scope of this study. Future studies should further explore this phenomenon. In addition, while the sex and race breakdowns were similar in this study among the different groups examined, future studies should examine other potential confounders, such as socioeconomic status and their impact on the results observed.
Conclusion
The genetic etiology of ASD has been elucidated over a number of years by unprecedented technological innovations in genetics. Based on previous genetic studies, the results of this study demonstrate that increasing parental age, and in particular, increasing paternal age, in individuals diagnosed with ASD was significantly related to an increased frequency of positive for possible de novo variant genetic test results. Furthermore, this study also revealed that genetic mutations in subjects diagnosed with ASD were important to ASD phenotypic variation. Finally, this study showed that while overall PGTRs were observed in 19.7% of the subjects examined (80.3% of the subjects examined did not have any identifiable genetic findings from high-resolution blood chromosome or blood chromosome microarray testing), the subcohort of subjects with positive possible de novo variants represented only 6.9% of all the individuals examined in this study. In addition, among those in the subcohort with PGTRs, it was found that many different genetic mutations were identified such as deletions, duplications, translocations, and inversions, and these genetic mutations were distributed across many different loci on many different chromosomes.
Therefore, we recommend that future studies be conducted on large cohort populations to determine the exact frequency of an ASD diagnosis among specific types of genetic mutations in comparison to the general population, so as to determine in such populations the risk of an ASD diagnosis and to further evaluate potential differences in ASD phenotypic variation. We also suggest that our results emphasize the need for genetic testing among individuals with an ASD diagnosis and that future studies examine the issues of genetic testing and phenotypic presentation to help elucidate subtypes and clarify the possible need for diagnostic classification differences.
Acknowledgments
This study was supported by the nonprofit Institute of Chronic Illnesses, Inc, the nonprofit CoMeD, Inc, the Seltz Foundation, and the Dwoskin Family Foundation. Doctor Mark Geier and Mr David Geier are father and son, respectively.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Talkowski ME, Minikel EV, Gusella JF. Autism spectrum disorder genetics: diverse genes with diverse clinical outcomes. Harv Rev Psychiatry. 2014;22(2):65–75. | ||
Lee BK, McGrath JJ. Advancing parental age and autism: multifactorial pathways. Trends Mol Med. 2015;21(2):118–125. | ||
Shaikh TH. Oligonucleotide arrays for high-resolution analysis of copy number alteration in mental retardation/multiple congenital anomalies. Genet Med. 2007;9(9):617–625. | ||
Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488(7412):471–475. | ||
Buizer-Voskamp JE, Blauw HM, Boks MP, et al. Increased paternal age and the influence on burden of genomic copy number variation in the general population. Hum Genet. 2013;132(4):443–450. | ||
Geier DA, Hooker BS, Kern JK, Sykes LK, Geier MR. An evaluation of the effect of increasing parental age on the phenotypic severity of autism spectrum disorder. J Child Neurol. Epub 2014 Aug 27. | ||
McDonald ME, Paul JF. Timing of increased autistic disorder cumulative incidence. Environ Sci Technol. 2010;44(6):2112–2118. | ||
DiStefano C, Gulsrud A, Huberty S, et al. Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J Neurodev Disord. 2016;8:19. | ||
Shen Y, Dies KA, Holm IA, et al; Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125(4):e727–e735. | ||
Geier DA, Kern JK, Geier MR. A comparison of the Autism Treatment Evaluation Checklist (ATEC) and Childhood Autism Rating Scale (CARS) for the quantitative evaluation of autism. J Ment Health Res Intellect Disabil. 2013;6(4):255–267. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.