Back to Journals » Infection and Drug Resistance » Volume 14
Evolution of Acinetobacter baumannii in Clinical Bacteremia Patients
Authors Li H ![]() , Zhang J, Wang Z, Yin Y, Gao H, Wang R, Jin L, Wang Q, Zhao C, Wang Z, Wang H
, Zhang J, Wang Z, Yin Y, Gao H, Wang R, Jin L, Wang Q, Zhao C, Wang Z, Wang H
Received 18 May 2021
Accepted for publication 20 August 2021
Published 31 August 2021 Volume 2021:14 Pages 3553—3562
DOI https://doi.org/10.2147/IDR.S320645
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Henan Li, Jiangang Zhang, Zhiren Wang, Yuyao Yin, Hua Gao, Ruobing Wang, Longyang Jin, Qi Wang, Chunjiang Zhao, Zhanwei Wang, Hui Wang
Department of Clinical Laboratory, Peking University People’s Hospital, Beijing, People’s Republic of China
Correspondence: Hui Wang
Department of Clinical Laboratory, Peking University People’s Hospital, Beijing, 100044, People’s Republic of China
Tel +86-10-88326300
Email [email protected]
Introduction: Colonization of the respiratory tract by Acinetobacter baumannii has been established as an independent risk factor for bacteremia. However, within-host evolution of A. baumannii in bacteremia has not been extensively investigated.
Methods: We performed whole-genome sequencing to discover the evolutionary characteristics that accompany the transition from respiratory tract carriage to bloodstream infection in three patients with A. baumannii bacteremia.
Results: Within-host genetic diversity was identified. A total of 21 single nucleotide variants (SNVs) were detected. Genic and intergenic evolution occurred particularly in secretion system, DNA recombination, and cell motility genes. Intergenic SNVs occurred more frequently compared to synonymous and non-synonymous SNVs, which indicated potential transcription or translation regulation. Non-synonymous mutations mostly occurred during the transition from respiratory tract carriage to bloodstream infection. Isolates of clonal complex 208 (CC208) had lower substitution rate with approximately 10− 6 nucleotide substitutions per site year− 1, compared with non-CC208 isolates (approximately 10− 5). We found evidence for the occurrence of recombination in one patient. A total of 259 genes were found to be gained or lost during the within-host evolution, and 231 genes were only detected in one patient. Gene function annotation results suggested that most genes (71/259) were related to replication, recombination, and repair. Universal bloodstream specific genes were not found in all three patients, and only one putative membrane protein related gene was lost in two patients.
Conclusion: Our results indicated that within-host evolution of A. baumannii bacteremia was driven by mutations, gene content changes, and limited effect of recombination. Gene content diversity between different patients was identified, which suggested interplay of both host and pathogen factors in within-host genetic diversity. Secretion system-related genes showed higher frequency of genomic variations during the within-host evolution. Our findings enhanced our understanding of within-host evolution of A. baumannii bacteremia and provided a framework for discovering novel genomic changes and pathogenicity genes important for bacteremia, which will be validated in future studies.
Keywords: Acinetobacter baumannii, within-host evolution, mutation, secretion system, bacteremia
Introduction
Acinetobacter baumannii is an important opportunistic pathogen that causes severe nosocomial infections worldwide.1 In China, A. baumannii is one of the predominant pathogens of bacteremia.2 Bacteremia is a prevalent cause of patient mortality, and the mortality rate in patients with A. baumannii bacteremia is 32.5% to 63.5%.3,4 An increasing trend in the mortality of bacteremia patients infected with A. baumannii has been observed in China.2 Furthermore, due to increased exposure to antibiotics, incidence of multidrug resistant (MDR) and carbapenem-resistant A. baumannii have been predictably increasing in recent years. MDR A. baumannii is one of the most frequently detected MDR pathogens in intensive care unit-acquired bloodstream infections.5 The incidence of imipenem-resistant A. baumannii increased from 31.0% in 2005 to 70.7% in 2017 in China.6 With limited treatment options available, infections caused by MDR and carbapenem-resistant A. baumannii may result in higher mortality. A meta-analysis showed that patients with carbapenem-resistant A. baumannii infection had a significantly higher risk of mortality compared to patients with carbapenem-susceptible A. baumannii infection.7
A. baumannii is widely distributed in natural and hospital environments and can colonize the human skin, mouth, and nasopharynx.8 Colonization on human mucosal surfaces and medical devices can lead to the formation of biofilms, increasing the risk of developing A. baumannii respiratory infections.9,10 The progression of bacteria from colonization to infection is the result of within-host evolution.11 A surveillance study found that MDR A. baumannii may be carried for long durations, up to 42 months.8 Meanwhile, a case-control study showed that previous respiratory tract colonization with species including A. baumannii was an independent risk factor for bacteremia.12 A 7-year observational study in a general hospital found that 47 of 441 (10.7%) A. baumannii colonized patients developed a bloodstream infection.13
Several studies have demonstrated the evolution of A. baumannii in vivo. A study of four patients with long-term colonization of A. baumannii found that multilocus sequence typing remained unchanged during colonization in three patients. The study found that A. baumannii undergoes parallel evolution during colonization. One efflux pump gene and two phage-related genes showed genetic variation in all patients. Studies have shown that antibiotic use and host environment exert selective pressure on colonized A. baumannii, prompting rapid molecular evolution in vivo.14 A total of 24 A. baumannii strains isolated successively from the blood of a single patient were subjected to whole-genome sequencing in another study. The strains were differentiated into three groups based on single nucleotide variants (SNVs), indicating that the patient suffered from re-infections or co-infections by similar but different strains. The results also showed that A. baumannii strains in each group were rather stable at the genomic level.15 Another genomic analysis of long-term infection with A. baumannii showed that mutations acquired during infection were over-represented in transcriptional regulators, notably pmrAB and adeRS, which could mediate resistance to the last-line therapies colistin and tigecycline, respectively.16 A study have found that half of the A. baumannii isolates from the same patients’ sputum harboured different sequence types (STs). Within some patients, the isolates evolved a more mucoid phenotype via genetic changes in the putative protein tyrosine kinase gene ptk.17 Mutations of pmrB and the pmrC homologue eptA were identified among pairs of colistin-susceptible and colistin-resistant A. baumannii, which were sequentially isolated from the same patients before and after colistin treatment.18,19
However, the molecular evolution of A. baumannii bacteremia in vivo remains unclear. Dynamic genomic variations during respiratory tract carriage and transition to bloodstream infection have not yet been demonstrated. In our study, we analysed genomic variation in different isolates isolated from the same patient in order to elucidate the molecular mechanism of within-host evolution of A. baumannii bacteremia.
Materials and Methods
Bacterial Strain Isolation and Phenotype Tests
A. baumannii isolates were obtained from three A. baumannii bacteremia patients sequentially during hospitalization at Peking University People’s Hospital (Table S1). All three patients were admitted to the intensive care unit with tracheal intubation. The respiratory tract samples were collected via sputum suction with bronchoscope when the patients had symptoms of respiratory tract infection. The blood samples were collected using blood culture bottle when the patients experienced abnormal body temperature. A. baumannii was identified using the VITEK2 system (bioMérieux, Marcy l’ toile, France), and confirmed by the presence of blaOXA51-like gene through polymerase chain reaction and sequencing. Minimal inhibitory concentrations were determined using the broth microdilution method. The antimicrobial breakpoints were interpreted according to the Clinical and Laboratory Standards Institute criteria M100.20 The cefoperazone-sulbactam susceptibility results were interpreted following the cefoperazone breakpoint for Enterobacteriaceae. Escherichia coli ATCC 25922 and Pseudomonas aeruginosa ATCC 27853 were used as quality-control strains.
Genome Sequencing and Bioinformatic Analysis
Bacterial DNA was extracted using a DNA purification kit (Qiagen, Hilden, Germany). Illumina sequencing libraries were prepared using Nextera kits with indexed encoded adapters from Illumina, according to the manufacturer’s instructions. Libraries were pooled for sequencing on a MiSeq sequencer. The resulting FASTQ files were quality-trimmed and assembled de novo using Velvet (Ridom GmbH, Münster, Germany) (https://github.com/dzerbino/velvet).21 The assembled contigs were annotated with Prokka (https://github.com/tseemann/prokka).22 Multiple sequence alignments were generated using Roary (http://sanger-pathogens.github.io/Roary/).23 After recombination region removal, RAxML was used to build a phylogenetic tree (https://github.com/stamatak/standard-RAxML).24 Whole-genome sequencing data were also used to determine the STs using the Center for Genomic Epidemiology Platform (https://cge.cbs.dtu.dk/services/MLST/).25 Multiple sequence alignment diagrams were generated using progressiveMAUVE (http://www.darlinglab.org/mauve/user-guide/progressivemauve.html).26 Homologous recombination was assessed using Gubbins (https://github.com/sanger-pathogens/gubbins).27 The clusters of orthologous genes functional categories were identified using eggNOG-Mapper (http://eggnog-mapper.embl.de/).28 Detailed comparative analysis of SNVs was performed using SnpEff (http://pcingola.github.io/SnpEff/).29 SNVs of the genomes were identified by mapping sequence reads for each isolate against the first isolates in each patient (A1 for patient A, C2 for patient C, and L6 for patient L). All annotations were visualized using iTOL (https://itol.embl.de/).30
Nucleotide Sequence Accession Numbers
This whole-genome shotgun project has been deposited at GenBank under the accession numbers CP060732 (A1), JACKXF000000000 (A2), JACKXG000000000 (A3), JACKXH000000000 (A14), JACKXI000000000 (C2), JACKXJ000000000 (C4), JACKXK000000000 (C6), JACATT000000000 (L6), JACATS000000000 (L7), JACATR000000000 (L8), JACATQ000000000 (L9), JACATP000000000 (L10).
Ethics Approval
The study was reviewed and approved by the Ethical Review Committee of Peking University People’s Hospital (No. 2018PHB187). Individuals younger than the age of 16 were not involved. Informed consent was waived by the Ethical Review Committee of Peking University People’s Hospital. The retrospective study was considered exempt from the ethical review because only bacterial isolates were taken from patients and the patient confidentiality was fully guaranteed. This study was conducted in accordance with the Declaration of Helsinki.
Results
Description of Isolates
A. baumannii isolates were obtained from three A. baumannii bacteremia patients. The number of isolates per patient was in the range of 3–5, and there were over 8–30 days between the first respiratory tract isolate and last bloodstream isolate (Figure 1). All isolates belonged to the predominant clonal complex 2 (CC2). Different Oxford STs were identified in three patients, including ST2211 in Patient A, ST469 in Patient C, and ST195 in Patient L. The pattern of antimicrobial susceptibility indicated that all isolates were carbapenem-resistant A. baumannii, and were only susceptible to colistin (Table S1).
 |
Figure 1 Phylogenetic analysis of A. baumannii collected from respiratory tract carriage to bloodstream infection in three patients. The phylogenetic tree was constructed based on core single nucleotide variants. The scale bar represented the number of nucleotide substitutions per sites. The colour of the outer of the trees denoted different patient. The red circle denoted isolates from bloodstream and the white circle denoted isolates from respiratory tract. The sampling time interval was listed on the right, and the first isolate in each patient was set day 1. |
Within-Host Genetic Diversity from Respiratory Tract Carriage to Bloodstream Infection
To facilitate detailed analysis of strain relationships, we developed a robust phylogeny based on SNVs present in core regions of the genome to represent ancestral relationships among isolates (Figure 1). The phylogenetic tree analysis indicated that isolates collected from different patients were grouped into distinct clades. Isolates in each patient showed different genomic characteristics. Bloodstream isolates A14, C4, C6, L9, and L10 were clustered with isolates from the respiratory tract of each patient.
We defined the amount of genetic diversity as the number of SNVs between 12 isolates within the three patients. SNVs of genomes were identified by mapping sequence reads for each isolate against the first isolates in each patient (A1 for patient A, C2 for patient C, and L6 for patient L). A total of twenty-one SNVs were identified (Table S2). A complete list of all the genic mutations differing between the isolates was given in Table 1. Except for a hypothetical protein, the SNVs were found in srpA, gspJ, srmB, fimV, sca1, transposase, pilR, pcaJ, tniA genes, which encode for organic peroxide-dependent peroxidase, general secretion pathway protein, DEAD box helicase family, Tfp pilus assembly protein FimV, phage-related minor tail protein, transposase IS66 family, sigma-54 interaction domain protein, 3-oxoadipate CoA-transferase, and mu transposase, respectively. Two genes encode the same protein, Tfp pilus assembly protein FimV. The genes encoding for 3-oxoadipate CoA-transferase PcaJ and Tfp pilus assembly protein FimV had the highest density of SNVs. Two missense variants (Ile91Leu, Phe90Leu) and one synonymous variant (Arg89Arg) were detected in PcaJ. One missense variant (Thr4Lys) and two synonymous variants (Asp5Asp and Asp18Asp) were detected in FimV.
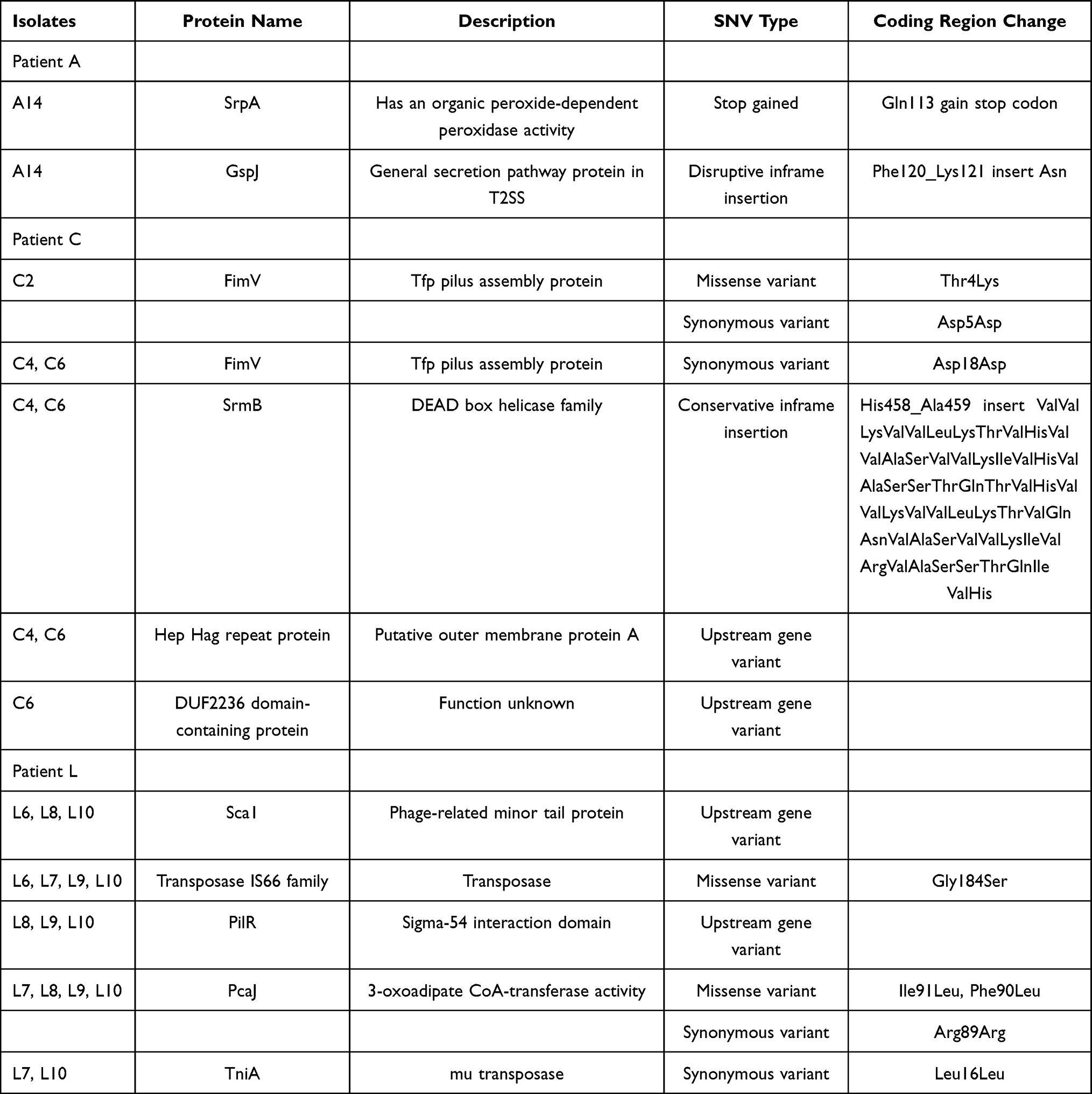 |
Table 1 The Genic Single Nucleotide Variants (SNVs) Found from Respiratory Tract Carriage to Bloodstream Infection in Three A. baumannii Bacteremia Patients. SNVs of the Genomes Were Identified by Mapping Sequence Reads for Each Isolate Against the First Isolates in Each Patient |
The mean number of SNVs between consecutively sampled isolates ranged from 0.67 to 8 for different patients (Figure 2A). To assess whether the accumulation of substitutions was time-dependent, we fitted a linear regression model of the number of accrued substitutions against the corresponding time (Figure 2B). In general, A. baumannii isolates accumulated mutations over time at different rates in three patients. With the exception of Patient C, who’s within-host substitution rate was 4.35114×10−5 SNVs site−1 year−1, the other two patients showed lower within-host substitution rate ranging from 6.46753×10−6 to 7.52486×10−6 SNVs site−1 year−1 (Table 2). Oxford STs of Patient A (ST2211) and L (ST195) belonged to clonal complex 208 (CC208), which is the predominant CC in China. Isolates of CC208 had similar substitution rate, and such within-host substitution rate resulted in introducing approximately 29 substitutions per year. Non-CC208 isolates showed faster substitution rate (182.5 substitutions per year).
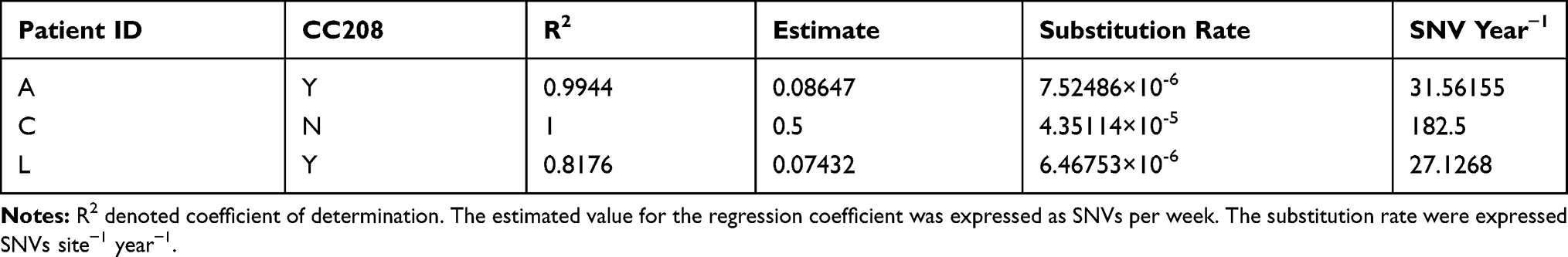 |
Table 2 Within-Host Nucleotide Substitution Rates from Respiratory Tract Carriage to Bacteremia |
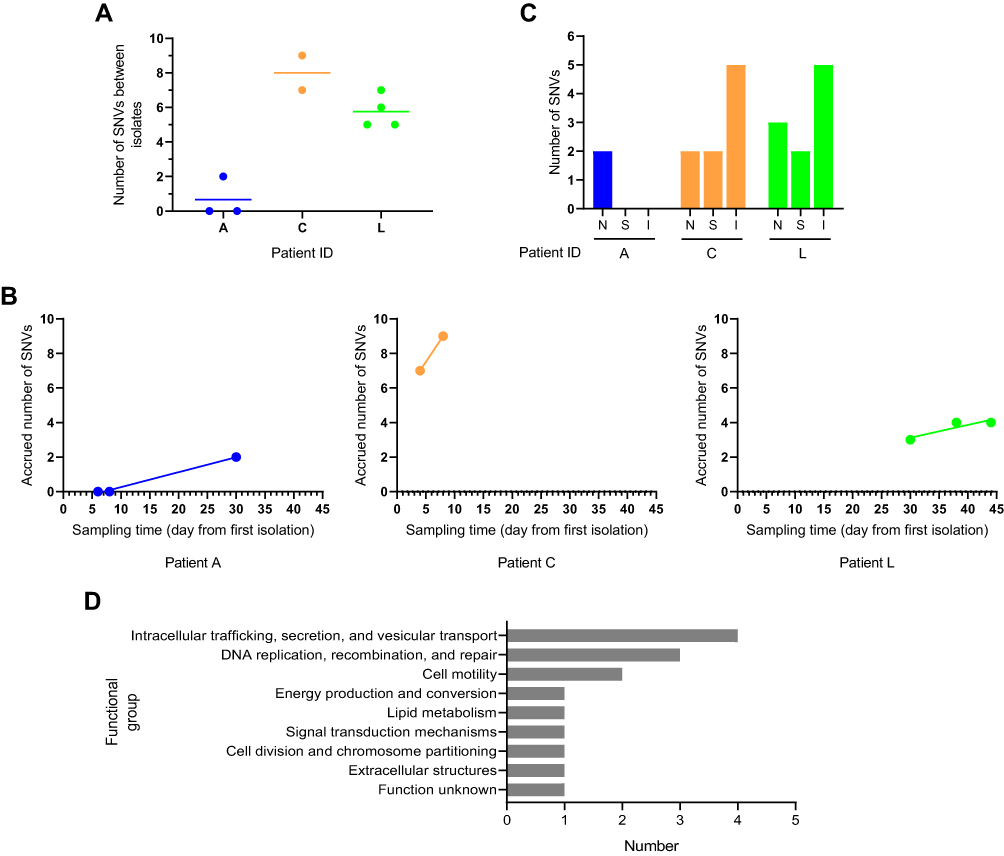 |
Figure 2 Characteristics of single nucleotide variants (SNVs) from respiratory tract carriage to bacteremia in three A. baumannii bacteremia patients. SNVs of the genomes were identified by mapping sequence reads for each isolate against the first isolates in each patient (A1 for patient A, C2 for patient C, and L6 for patient L). (A) Within-host genetic diversity. The plots showing the number of SNVs calculated between isolates within the same patient. (B) Within-host mutation rates. Linear relationship between the number of accrued SNVs and sampling time was assessed using linear regression. (C) The genic and intergenic SNVs identified during within-host evolution. The letters N, S and I stand for non-synonymous, synonymous and intergenic SNVs respectively. (D) Functional classification of genes with SNVs. The genes were analysed clusters of orthologous groups functional categories. |
In two of three patients, intergenic SNVs occurred more frequently than synonymous and non-synonymous SNVs (Figure 2C). Five intergenic SNVs were identified in patient C, including 2 upstream gene variants and 3 intergenic variants. In Patient L, we discovered a total of 5 intergenic SNVs, comprising 3 upstream gene variants and 2 intergenic variants. Non-synonymous mutations mostly occurred during the transition from respiratory tract carriage to bacteremia. In Patient A and C, non-synonymous variants were only detected during the transition. In Patient L, we discovered a total of 3 missense variants among the respiratory tract isolates. Functional analysis suggested that most of the mutations were in genes associated intracellular trafficking, secretion, and vesicular transport, DNA replication, recombination, and repair and cell motility (Figure 2D).
Homologous Recombination During Within-Host Evolution
Homologous recombination is the major driver of evolution in bacterial pathogens. To identify or rule out the occurrence of recombination, we aligned genomes of the isolates from each patient to assess whether we could identify genomic regions with high density of SNVs, a well-known signature for recombination. We found evidence of within-host recombination in one patient (Patient L). The range of recombination blocks was 1–3, while the average number of SNVs within each block was 14 (8–36) per recombination block. We then assessed the ratio of imported SNVs via recombination relative to random substitutions (r/m) and total recombination blocks relative to random substitutions (ρ/θ), which are widely used statistics for quantifying the contribution of recombination to genomic diversification. The r/m and ρ/θ averaged across all phylogenetic branches where recombination had occurred were 3.02 (0–26) and 0.35 (0–3), respectively.
Patient Specific Gene Content During Within-Host Evolution
Gene content showed patient specificity. Only 1.2–9.4% of the A. baumannii gene clusters observed in a specific patient (ie, the subject-specific pan-genome), and 88.5–92.9% of gene clusters observed in all isolates of a specific patient (ie, the subject-specific core-genome) were shared between all three patients, and 0.6–6.4% of core gene clusters were entirely unique to a patient (Figure 3A and B). A total of 259 genes were found to be gained or lost during the within-host evolution, and 231 genes were only detected in one patient (Figure 3C, Table S3). Gene function annotation results suggested that most genes (71/259) were related to replication, recombination, and repair. Five genes were altered in all patients, including three putative membrane proteins, one transcriptional regulator, and one type I secretion C-terminal target domain-containing protein. Eighteen unique genes were found to be gained or lost in the bloodstream isolates of three patients, but most of the genes had unknown functions (Table S3). The number of bloodstream specific genes varied among three patients (2–13). Universal bloodstream specific genes were not found in all three patients, and only one putative membrane protein related gene was lost in two patients (Patient A and L).
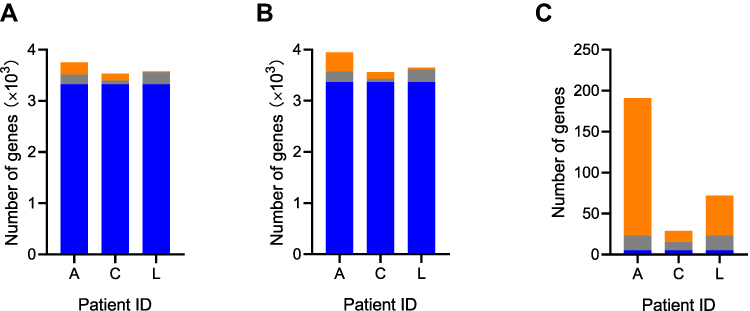 |
Figure 3 Characteristics of gene content in three A. baumannii bacteremia patients. Shared versus unique patient-specific core genes (A), pan genes (B), and gained or lost genes (C). The blue, grey, and orange colours indicated genes shared among all patients, shared between two patients, and unique to one patient, respectively. |
Antibiotic resistant gene and virulence gene profiling showed that most genes were consistent in each patient (Figure S1). Only one isolate A2 contained the fosfomycin resistance gene fosB4. The gene armA was only absent in isolate L7. For virulence genes, only the bap gene showed difference within isolates with the same ST. Isolates C4 and C6 had two biofilm-associated bap genes, while C2 had only one. These findings suggested a personalization of gene content at the population level.
Discussion
A. baumannii bacteremia is a devastating public health threat, with high mortality in vulnerable populations. The rate of antimicrobial resistance in A. baumannii is increasing alarmingly.31 Previous colonization with causative microorganisms is significantly associated with bacteremia.12 Several studies have suggested that colonization of the respiratory tract or respiratory tract infection is a risk factor for A. baumannii bloodstream infection.32,33 However, there are few studies on the phylogenomic analysis of respiratory tract and bloodstream A. baumannii isolates from the same host.
Through pulsed-field gel electrophoresis, previous studies have shown that nasal carriage is a source of Staphylococcus aureus bacteremia.34 In recent decades, high-throughput sequencing has facilitated understanding of bacterial molecular evolution of bacteremia within the host. The genomic correlation between respiratory tract isolates and bloodstream isolates has been reported in Staphylococcus aureus. Invasive bloodstream S. aureus has been shown to emerge from a nasal population of methicillin-sensitive S. aureus.35 By applying comparative genomics to S. aureus bacteremia with sequential invasive S. aureus isolates or paired colonizing isolates, previous studies have described specific patterns of S. aureus diversity within the host. Isolates from persistent S. aureus bacteremia have a distinctive molecular signature, characterised by a low mutation frequency and high proportion of non-silent mutations.36 One previous research has used comparative and functional genomics to identify and characterise signals of vancomycin-resistant Enterococcus faecium adaptation during gastrointestinal tract colonization and bloodstream infection in immunocompromised paediatric hosts. Variants have been shown to be enriched in genes involved in carbohydrate metabolism, and phenotypic analysis has revealed associated differences in carbohydrate utilisation among isolates.37 Another research has utilised whole-genome sequencing to characterise E. faecium associated with stool carriage and bloodstream infection in patients. A comparative genomic analysis of related isolates in stool and blood of the same patient has failed to reveal adaptive mutations associated with invasive disease.38 However, there are very few studies on the origin and evolution of A. baumannii bacteremia within the host. Therefore, we conducted this study to assess the correlation between A. baumannii isolates from the respiratory tract and bloodstream within the host. In our study, the genome of bloodstream isolates was highly homologous to that of respiratory isolates, suggesting that A. baumannii bloodstream infection may originate from the respiratory tract. Our results indicated that within-host evolution in A. baumannii bacteremia was driven by genic and intergenic mutations, gene content changes and limited effect of recombination. Blood stream specific genes varied among three patients in our study, which indicated patient-specific evolution of A. baumannii bacteremia. Only one putative membrane protein related gene was lost in two of three patients’ bloodstream isolates, and biofilm formation assay showed decreased biofilm formation ability in bloodstream isolates (data not shown). Functions and mechanisms of the altered genes identified in this study need to be further explored. In our study, within host diversity was identified, which suggested interplay of both host and pathogen factors in within-host genetic diversity. We found genic mutations in twelve genes. Genic evolution occurred particularly in secretion system related genes. Four genes were related with secretion system, including srpA, gspJ and two fimV genes. Besides, one type I secretion C-terminal target domain-containing protein changed in all three patients and was absent in two bloodstream isolates. Protein secretion is a key virulence mechanism of pathogenic bacteria. Secretion systems are required to transport proteins across the cell membrane and play a role in virulence and fitness.39,40 SrpA plays versatile regulatory functions by binding directly to the promoter region of the type III secretion system and type VI secretion system.41 GspJ is general secretion pathway protein of type II secretion system (T2SS). In the bacteria Burkholderia cenocepacia, gspJ is required for multihost pathogenicity in nematode Caenorhabditis elegans and onions.42 There were few studies that have been conducted on gspJ in A. baumannii. However, another T2SS gene gspD has been found to play an important role in multihost pathogenicity. GspD is required for optimal virulence of A. baumannii in Galleria mellonella larvae. Percent survival of larvae infected with gspD deleted strain was significantly higher than that in gspD wild type group.43 In another study, mice were infected intranasally with the wild type and gspD mutant to generate a murine pneumonia infection model, and the mice infected with gspD mutant showed lower lung colonization and proliferation advantage.44 FimV has been shown to interact with T2SS protein Vfr by a mechanism that acts as a modulator of the Vfr level.45 Recent studies have shown that the T2SS supported survival of A. baumannii in a bacteremia mouse model through extracellular secretion of a variety of enzymes, including the glycan-specific metalloprotease CpaA. CpaA interferes with blood coagulation through cleavage of factor XII of the contact activation system. Preventing factor XII-mediated generation of fibrin may allow A. baumannii to escape containment by intravascular clots and to disseminate.46 T4SSs was involved in Helicobacter pylori adaptation to the hostile environment in the human stomach.47 Further research is needed to clarify functions and mechanisms of secretion system in A. baumannii during within-host evolution.
We have some limitations in this study. Since patients were recruited after blood cultures were found to be positive for A. baumannii, colonizing A. baumannii isolates were available only for a very small proportion of patients. Additionally, because this study was retrospective study, sample collection interval varied among different patients, which might miss some information about genomic evolution from respiratory tract carriage to bacteremia. A larger dataset and more comprehensive screening are required to determine whether the findings described here can be more widely generalized. Considering that we predominantly sequenced single colonies, these might have failed to capture temporal dynamics of co-colonizing strains, especially those present at low frequency. Therefore, future prospective studies sequencing either multiple colonies or better yet the entire culture will be required to fully unravel within-sample genetic diversity and temporal dynamics of the wild-type and recombinant variants. Intergenic mutations and replication, recombination and repair related gene content variations occupied a large proportion at all stages, which indicated potential transcription or translation regulation during within-host evolution of A. baumannii. Transcription level research should be carried out in future work, since regulatory networks are important for survival of A. baumannii within the host.48 Our study found genomic variation characteristics in the evolution of the host, which provided a theoretical basis for elucidating the occurrence of A. baumannii bacteremia. However, in the process of interaction between bacteria and host, the evolution of bacteria within the host exhibited patient specificity. This showed that only focusing on bacteria was not sufficient. The role of the host in the evolution of bacteria cannot be ignored. This also requires an in-depth research in the future.
Conclusion
Our findings showed within-host evolution of A. baumannii from respiratory tract carriage to bacteremia with evidence of adaptations through mutations in intergenic and genic regions association with secretion system, DNA recombination, and cell motility proteins. Gene content diversity between different patients was identified, which suggested interplay of both the host and pathogen factors on within-host genetic diversity. Replication, recombination, and repair related genes were found to be gained or lost more frequently. Our findings enhanced our understanding of within-host evolution of A. baumannii bacteremia and provided a framework for discovering novel genomic changes and pathogenicity genes important for bacteremia that will be validated in future studies. We hope that these findings can ultimately be leveraged to develop more effective strategies for the treatment and control of A. baumannii bacteremia in vulnerable patient populations.
Acknowledgments
This research was funded by National Natural Science Foundation of China (grant number 81871693 and 81991533) and Peking University People’s Hospital Research and Development Funds (grant number RS2018-03).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Antunes LC, Visca P, Towner KJ. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis. 2014;71(3):292–301. doi:10.1111/2049-632X.12125
2. Jin L, Zhao C, Li H, Wang R, Wang Q, Wang H. Clinical profile, prognostic factors, and outcome prediction in hospitalized patients with bloodstream infection: results from a 10-Year Prospective Multicenter Study. Front Med (Lausanne). 2021;8:629671. doi:10.3389/fmed.2021.629671
3. Ballouz T, Aridi J, Afif C, et al. Risk factors, clinical presentation, and outcome of Acinetobacter baumannii bacteremia. Front Cell Infect Microbiol. 2017;7:156. doi:10.3389/fcimb.2017.00156
4. Wang X, Zhang L, Sun A, et al. Acinetobacter baumannii bacteraemia in patients with haematological malignancy: a multicentre retrospective study from the infection working party of Jiangsu society of hematology. Eur J Clin Microbiol Infect Dis. 2017;36(7):1073–1081. doi:10.1007/s10096-016-2895-2
5. Di Franco S, Alfieri A, Pace MC, et al. Blood stream infections from MDR bacteria. Life (Basel). 2021;11(6):575. doi:10.3390/life11060575
6. Hu F, Zhu D, Wang F, Wang M. Current status and trends of antibacterial resistance in China. Clin Infect Dis. 2018;67(suppl_2):S128–S134. doi:10.1093/cid/ciy657
7. Lemos EV, de la Hoz FP, Einarson TR, et al. Carbapenem resistance and mortality in patients with Acinetobacter baumannii infection: systematic review and meta-analysis. Clin Microbiol Infect. 2014;20(5):416–423. doi:10.1111/1469-0691.12363
8. Marchaim D, Navon-Venezia S, Schwartz D, et al. Surveillance cultures and duration of carriage of multidrug-resistant Acinetobacter baumannii. J Clin Microbiol. 2007;45(5):1551–1555. doi:10.1128/JCM.02424-06
9. Chen YP, Liang CC, Chang R, et al. Detection and colonization of multidrug resistant organisms in a regional teaching hospital of Taiwan. Int J Environ Res Public Health. 2019;16(7):1104. doi:10.3390/ijerph16071104
10. Ng D, Marimuthu K, Lee JJ, et al. Environmental colonization and onward clonal transmission of carbapenem-resistant Acinetobacter baumannii (CRAB) in a medical intensive care unit: the case for environmental hygiene. Antimicrob Resist Infect Control. 2018;7(1):51. doi:10.1186/s13756-018-0343-z
11. Didelot X, Walker AS, Peto TE, Crook DW, Wilson DJ. Within-host evolution of bacterial pathogens. Nat Rev Microbiol. 2016;14(3):150–162. doi:10.1038/nrmicro.2015.13
12. Lee JY, Kang CI, Ko JH, et al. Clinical features and risk factors for development of breakthrough gram-negative bacteremia during carbapenem therapy. Antimicrob Agents Chemother. 2016;60(11):6673–6678. doi:10.1128/AAC.00984-16
13. Kim SY, Cho SI, Bang JH. Risk factors associated with bloodstream infection among patients colonized by multidrug-resistant Acinetobacter baumannii: a 7-Year Observational Study in a general hospital. Am J Infect Control. 2020;48(5):581–583. doi:10.1016/j.ajic.2019.07.025
14. Wen H, Wang K, Liu Y, et al. Population dynamics of an Acinetobacter baumannii clonal complex during colonization of patients. J Clin Microbiol. 2014;52(9):3200–3208. doi:10.1128/JCM.00921-14
15. Kim SJ, Kim YJ, Ko KS. Genomic analysis of consecutive Acinetobacter baumannii strains from a single patient. Front Microbiol. 2018;9:2840. doi:10.3389/fmicb.2018.02840
16. Wright MS, Iovleva A, Jacobs MR, Bonomo RA, Adams MD. Genome dynamics of multidrug-resistant Acinetobacter baumannii during infection and treatment. Genome Med. 2016;8(1):26. doi:10.1186/s13073-016-0279-y
17. Hua X, Zhou Z, Yang Q, et al. Evolution of Acinetobacter baumannii in vivo: international clone II, more resistance to ceftazidime, mutation in ptk. Front Microbiol. 2017;8:1256. doi:10.3389/fmicb.2017.01256
18. Gerson S, Betts JW, Lucassen K, et al. Investigation of novel pmrB and eptA mutations in isogenic Acinetobacter baumannii isolates associated with colistin resistance and increased virulence in vivo. Antimicrob Agents Chemother. 2019;63(3):e01586–18. doi:10.1128/AAC.01586-18
19. Marano V, Marascio N, Pavia G, et al. Identification of pmrB mutations as putative mechanism for colistin resistance in A. baumannii strains isolated after in vivo colistin exposure. Microb Pathog. 2020;142:104058. doi:10.1016/j.micpath.2020.104058
20. Clinicaland Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: Twenty-Ninth Informational Supplement. M100S-S29. Wayne, PA: Clinical and Laboratory Standards Institute; 2019.
21. Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi:10.1101/gr.074492.107
22. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi:10.1093/bioinformatics/btu153
23. Page AJ, Cummins CA, Hunt M, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi:10.1093/bioinformatics/btv421
24. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi:10.1093/bioinformatics/btu033
25. Larsen MV, Cosentino S, Rasmussen S, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50(4):1355–1361. doi:10.1128/JCM.06094-11
26. Darling AE, Mau B, Perna NT, Stajich JE. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi:10.1371/journal.pone.0011147
27. Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using gubbins. Nucleic Acids Res. 2015;43(3):e15. doi:10.1093/nar/gku1196
28. Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi:10.1093/nar/gky1085
29. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. doi:10.4161/fly.19695
30. Letunic I, Bork P. Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–W259. doi:10.1093/nar/gkz239
31. Holmes CL, Anderson MT, Mobley H, Bachman MA. Pathogenesis of gram-negative bacteremia. Clin Microbiol Rev. 2021;34(2):e00234–20. doi:10.1128/CMR.00234-20
32. Liu CP, Shih SC, Wang NY, et al. Risk factors of mortality in patients with carbapenem-resistant Acinetobacter baumannii bacteremia. J Microbiol Immunol Infect. 2016;49(6):934–940. doi:10.1016/j.jmii.2014.10.006
33. Liu Y, Wang Q, Zhao C, et al. Prospective multi-center evaluation on risk factors, clinical characteristics and outcomes due to carbapenem resistance in Acinetobacter baumannii complex bacteraemia: experience from the Chinese Antimicrobial Resistance Surveillance of Nosocomial Infections (CARES) network. J Med Microbiol. 2020;69(7):949–959. doi:10.1099/jmm.0.001222
34. von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group. N Engl J Med. 2001;344(1):11–16. doi:10.1056/NEJM200101043440102
35. Young BC, Golubchik T, Batty EM, et al. Evolutionary dynamics of Staphylococcus aureus during progression from carriage to disease. Proc Natl Acad Sci U S A. 2012;109(12):4550–4555. doi:10.1073/pnas.1113219109
36. Giulieri SG, Baines SL, Guerillot R, et al. Genomic exploration of sequential clinical isolates reveals a distinctive molecular signature of persistent Staphylococcus aureus bacteraemia. Genome Med. 2018;10(1):65. doi:10.1186/s13073-018-0574-x
37. Chilambi GS, Nordstrom HR, Evans DR, et al. Evolution of vancomycin-resistant Enterococcus faecium during colonization and infection in immunocompromised pediatric patients. Proc Natl Acad Sci U S A. 2020;117(21):11703–11714. doi:10.1073/pnas.1917130117
38. Moradigaravand D, Gouliouris T, Blane B, et al. Within-host evolution of Enterococcus faecium during longitudinal carriage and transition to bloodstream infection in immunocompromised patients. Genome Med. 2017;9(1):119. doi:10.1186/s13073-017-0507-0
39. Weber BS, Kinsella RL, Harding CM, Feldman MF. The secrets of Acinetobacter secretion. Trends Microbiol. 2017;25(7):532–545. doi:10.1016/j.tim.2017.01.005
40. Harding CM, Hennon SW, Feldman MF. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol. 2018;16(2):91–102. doi:10.1038/nrmicro.2017.148
41. You J, Sun L, Yang X, et al. Regulatory protein SrpA controls phage infection and core cellular processes in Pseudomonas aeruginosa. Nat Commun. 2018;9(1):1846. doi:10.1038/s41467-018-04232-6
42. Somvanshi VS, Viswanathan P, Jacobs JL, Mulks MH, Sundin GW, Ciche TA. The type 2 secretion Pseudopilin, gspJ, is required for multihost pathogenicity of Burkholderia cenocepacia AU1054. Infect Immun. 2010;78(10):4110–4121. doi:10.1128/IAI.00558-10
43. Kinsella RL, Lopez J, Palmer LD, et al. Defining the interaction of the protease CpaA with its type II secretion chaperone CpaB and its contribution to virulence in Acinetobacter species. J Biol Chem. 2017;292(48):19628–19638. doi:10.1074/jbc.M117.808394
44. Elhosseiny NM, El-Tayeb OM, Yassin AS, Lory S, Attia AS. The secretome of Acinetobacter baumannii ATCC 17978 type II secretion system reveals a novel plasmid encoded phospholipase that could be implicated in lung colonization. Int J Med Microbiol. 2016;306(8):633–641. doi:10.1016/j.ijmm.2016.09.006
45. Michel G, Aguzzi A, Ball G, Soscia C, Bleves S, Voulhoux R. Role of fimV in type II secretion system-dependent protein secretion of Pseudomonas aeruginosa on solid medium. Microbiology (Reading). 2011;157(Pt 7):1945–1954. doi:10.1099/mic.0.045849-0
46. Waack U, Warnock M, Yee A, et al. CpaA is a glycan-specific adamalysin-like protease secreted by Acinetobacter baumannii that inactivates coagulation factor XII. Mbio. 2018;9(6):e01606–18. doi:10.1128/mBio.01606-18
47. Fischer W, Tegtmeyer N, Stingl K, Backert S. Four chromosomal type IV secretion systems in Helicobacter pylori: composition, structure and function. Front Microbiol. 2020;11:1592. doi:10.3389/fmicb.2020.01592
48. Allen JL, Tomlinson BR, Casella LG, Shaw LN. Regulatory networks important for survival of Acinetobacter baumannii within the host. Curr Opin Microbiol. 2020;55:74–80. doi:10.1016/j.mib.2020.03.001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.