Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Evidence of eosinophil extracellular trap cell death in COPD: does it represent the trigger that switches on the disease?
Authors Uribe Echevarría L, Leimgruber C, García González J, Nevado A, Álvarez R, García LN, Quintar AA, Maldonado CA
Received 28 June 2016
Accepted for publication 20 December 2016
Published 14 March 2017 Volume 2017:12 Pages 885—896
DOI https://doi.org/10.2147/COPD.S115969
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Video abstract presented by Loli Uribe Echevarría.
Views: 806
Loli Uribe Echevarría,1 Carolina Leimgruber,2 Jorge García González,1 Alberto Nevado,1 Ruth Álvarez,3 Luciana N García,2 Amado A Quintar,2 Cristina A Maldonado2
1Pneumonology Service, Sanatorio Allende (Nueva Córdoba), Córdoba, Argentina; 2Electronic Microscopy Center, Instituto de Investigaciones en Ciencias de la Salud (INICSA-CONICET), Facultad de Ciencias Médicas, Universidad Nacional de Córdoba, Córdoba, Argentina; 3Smoking Cessation Cente, Nuevo Hospital San Roque of Córdoba, Córdoba, Argentina
Abstract: In spite of the numerous studies on chronic obstructive pulmonary disease (COPD), the cellular and molecular basis of the disease’s development remain unclear. Neutrophils and eosinophils are known to be key players in COPD. Recently, neutrophil extracellular trap cell death (NETosis), a mechanism due to decondensation and extrusion of chromatin to form extracellular traps, has been demonstrated in COPD. However, there is limited knowledge about eosinophil extracellular trap cell death (EETosis) and its role in the pathogenesis of COPD. The aim of this study was to evaluate EETosis in stable COPD. Induced sputum obtained from healthy smokers and low exacerbation risk COPD A or B group patients or high exacerbation risk COPD C or D group patients were included. Samples were examined using electron microscopy and immunofluorescence. Healthy smokers (n=10) and COPD A (n=19) group exhibited neutrophilic or paucigranulocytic phenotypes, with NETosis being absent in these patients. In contrast, COPD B (n=29), with eosinophilic or mixed phenotypes, showed EETosis and incipient NETosis. COPD C (n=18) and COPD D groups (n=13) were differentiated from low exacerbation rate-COPD group by the abundant cellular debris, with COPD C group having an eosinophilic pattern and numerous cells undergoing EETosis. A hallmark of this group was the abundant released membranes that often appeared phagocytosed by neutrophils, which coincidentally exhibited early NETosis changes. The COPD D group included patients with a neutrophilic or mixed pattern, with abundant neutrophil extracellular trap-derived material. This study is the first to demonstrate EETosis at different stages of stable COPD. The results suggest a role for eosinophils in COPD pathophysiology, especially at the beginning and during the persistence of the disease, regardless of whether the patient quit smoking, with EETosis debris probably triggering uncontrolled NETosis. The main target of these findings should be young smokers with the potential to develop COPD.
Keywords: COPD, eosinophils, neutrophils, NETosis, EETosis, induced sputum
Introduction
Chronic obstructive pulmonary disease (COPD) is currently the third cause of death worldwide,1 as well as being a significant factor in terms of direct and indirect lost productivity. Although the current COPD epidemic is in part due to smoking, an important pathophysiological aspect is that not all smokers develop COPD, and the cellular and molecular basis to predict which smokers will develop the disease is completely unknown. Furthermore, the reason for the lack of achieving an attenuation of inflammatory signs, even years after having discontinued smoking, is still far from being clarified, thereby representing a real challenge for research related to this field.
COPD is characterized by the chronic inflammation of the airways as a result of exposure to inhaled irritants and the consequent epithelial damage. Neutrophils are considered to be the principal cell responsible for orchestrating COPD, with several reports having pointed out that excessive proteolytic activity of neutrophil elastase contributes to the tissue damage observed in the airways of COPD.2 However, this does not appear to be a major trigger of other clinical or pathophysiologic abnormalities in COPD.3 Furthermore, Singh et al4 observed a high prevalence of eosinophils in the blood and sputum of patients with COPD, but without any clinical relevance having been shown.
The death of inflammatory cells and their removal are critical events in affected tissues. Neutrophils usually die by apoptosis, a process that occurs by cleavage of DNA into oligonucleosomal size fragments, chromatin condensation, and the formation of apoptotic bodies, leading to phagocytosis by neighboring cells, thus preventing inflammation.5 Recently, an alternative mechanism of death was described for neutrophils,6,7 in which the cell releases its DNA forming neutrophil extracellular traps (NETs).8 This original neutrophil extracellular trap cell death (NETosis) process, referred to as “NETosis” by Steinberg and Grinstein, is distinct from apoptosis and necrosis9 and allows a complete neutrophil microbicidal function. NETs then need to be removed quickly to prevent further tissue damage or autoimmune phenomena, which have been described in pathologies that occur with alterations in the NETs resolution.10
The occurrence of NETosis has been described in many inflammatory conditions of the lung, including cystic fibrosis, which is an aggravating factor of airway obstruction.11,12 A recent study has demonstrated the presence of sterile NETs in the sputum of patients with stable and exacerbated COPD, which correlated with the severity of airflow limitation.13 As different cell types such as eosinophils, mast cells, and macrophages can also die by a similar mechanism, the process was renamed “ETosis,” meaning cell death with release of extracellular traps (ETs).14,15 However, there is no evidence demonstrating the occurrence of eosinophil extracellular trap cell death (EETosis) or its contribution in COPD. Therefore, the aim of this study was to evaluate the occurrence of eosinophil death by EETosis in stable COPD.
Materials and methods
Subjects
Ex-smokers who had quit smoking for 6 months or longer, COPD and healthy patients with a smoking history of ≥20 pack/year, of both genders and of age over 60 years, were enrolled for this study after being admitted to the Inpatient Service of Pneumonology of the Sanatorio Allende and the Centre of Smoking Cessation of the Nuevo Hospital San Roque of Córdoba, Argentina. To be included, COPD patients had to fulfill requirements of a forced expiratory volume in one second (FEV1) <80% and FEV1/forced vital capacity (FVC) <70% following inhalation of a bronchodilator. In addition, only healthy ex-smokers with no respiratory symptoms and normal lung function tests (FEV1/FVC ≥70%) were included. The COPD patients were classified as low exacerbation risk (LER) or high exacerbation risk (HER) groups according to the criteria and classification established by the Global Initiative for Chronic Obstructive Lung Disease (GOLD).1,16
Patients with a history of infectious disease during the six weeks prior to the enrollment and those with bronchial asthma in childhood and/or adolescence were excluded. In addition, subjects with chronic respiratory diseases other than COPD, gastroesophageal reflux diseases, obesity (body mass index ≥30), cardiovascular diseases, cancer, collagen diseases, drugs abuse or known pneumotoxic or pulmonary occupational exposure risks, and unconscious patients or those who declined to participate were excluded. Patients who had had COPD exacerbations treated with systemic or inhaled corticosteroids during the past six weeks were also excluded. HER COPD patients were treated with a maintenance therapy according to the GOLD guidelines for the management of stable COPD.1 Written informed consent was obtained from all subjects, and the study and protocol were approved by the Medical Ethics Committee of Sanatorio Allende (Nueva Córdoba).
Study design
This was a cross-sectional study. After admission, individual patients were subjected to lung function examinations,17 and samples of induced sputum (IS) were obtained.
IS and sample processing
To obtain an IS sample, 200 mg of salbutamol by inhalation with a metered-dose dispenser was administered to the patients ten minutes before sputum induction. Nebulization was carried out with NaCl saline solutions in increasing concentrations (3%, 4%, and 5%) for 7 minutes, with a 5 minute interval between each dose. Before attempting expectoration, patients were asked to remove nasal secretions and rinse the mouth and throat with running water; they were then asked to cough energetically to expectorate bronchial secretions. Before and after each nebulization, the peak expiratory flow (PEF) was measured; if it decreased to less than 10%, patients were exposed to the next salt concentration. Conversely, if PEF fall was 10%–20%, nebulization was prolonged for 7 minutes before continuing with the next hypertonic concentration. The protocol was interrupted if the patient evidenced symptoms of respiratory distress or demonstrated a PEF less than 20% of baseline.
Expectorated sputum was processed immediately or kept at 4°C for a period no longer than 2 hours. The total volume of sputum obtained was weighed, and the mucous fraction different from the saliva was selected. This portion was dissolved in a volume of 1% dithiothreitol (DTT) (Sputolysin® Reagent Cat No 560000 Calbiochem, EMD Millipore, Billerica, MA, USA) equivalent to four times the weight of the selected fraction. The mixture was stirred for 15 seconds and transferred to a water bath for 15 minutes with continuous stirring. Then, 0.1 M phosphate-buffered saline (PBS) was added in equal volume to DTT used and filtered with 48 mm thick gauze. The filtrate was centrifuged at 1,500 rpm for 10 minutes and the supernatant stored in Eppendorf tubes at −70°C for future biochemical analysis. For the cytological exam, the cellular pellet was suspended in 0.5 mL of PBS. The viability of the cells was determined with the Trypan blue exclusion technique. A sample quality was considered acceptable if cell viability was equal to or greater than 60%. Cells were counted in a Neubauer chamber and resuspended in PBS to a final concentration of 0.75×106–1×106 cells/mL. Cell monolayers (cytospins) were prepared with 50 μL aliquots on glass slides, and these were centrifuged at 500 rpm for 4 minutes in a cytocentrifuge (Giumelli®; Giumelli, Buenos Aires, Argentina).
After air drying, cytospins were stained with May Grünwald-Giemsa stain to enable differential cell counts, including a count of neutrophils, eosinophils, and macrophages, with the percentages based on a minimum of 200 nonbronchial squamous cells. In order to characterize the cellular inflammatory pattern, we further classified patients according to the predominant cells in the cytospins of the IS as eosinophilic (≥3%), neutrophilic (≥67%), mixed (Eosinophilic and neutrophilic predominance), or pausigranulocytic (neither eosinophilic nor neutrophilic).18
Electron microscopy
Fresh sputum plugs were fixed in a mixture of 4% formaldehyde and 2% glutaraldehyde, omitting the DTT treatment, and embedded in Araldite (Electron Microscopy Sciences, Hatfield, PA, USA) for ultrastructural analysis. Thin sections were then mounted on nickel grids for observation in a Zeiss LEO 906 E Electron Microscope (Carl Zeiss, Oberkochen, Germany). Some samples were also embedded in LR-White (Electron Microscopy Science), a hydrophilic resin, in order to perform DAPI staining to evaluate the extracellular DNA traps.
Immunofluorescence
Immunofluorescence to analyze NETosis/EETosis occurrence was performed on unprocessed freshly collected plugs of IS that had been fixed for 5–10 minutes to slides without poly-l-lysine and then fixed in 4% paraformaldehyde. After permeabilization with 0.25% Triton X-100, slides were treated for 1 h with 5% PBS-BSA to block nonspecific binding and incubated overnight at 4°C in a humidified chamber with a rabbit polyclonal anti-neutrophil elastase antibody (1/100; Abcam, Cambridge, MA) or a mouse monoclonal anti-human eosinophil major basic protein antibody (1/100; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Afterwards, the slides were washed three times with PBS and further incubated with Alexa 594-conjugated goat anti-rabbit polyclonal antibody or conjugated to rabbit anti-mouse polyclonal antibody (Thermo Scientific, Waltham, MA, USA) for 1 h and mounted using a fluoromount containing DAPI (4′, 6-diamidino-2-phenylindole). Images were then obtained using an inverted confocal laser scanning microscope, FluoView FV 1000 (Olympus; Tokyo, Japan).
Extracellular traps quantification at epifluorescence microscopy level
In order to quantify extranuclear DNA, 0.5–1 μm semithin LR-White-embedded IS sections were obtained at three different levels, and at least 10 pictures were taken at each level after DAPI staining. ImageJ (NHI, Bethesda MD, USA) was used to detect and quantify DNA utilizing home-built plugins. The amount of extranuclear/extracellular DNA was obtained by calculating the difference between the total DAPI stain and the well-recognized nuclear DAPI signal.
Extracellular trap quantification at electron microscopy level
Thin sections (80 nm) from three different regions of araldite-embedded IS samples were mounted on copper grids; to avoid repeated counting, only one section per grid was mounted. A total of 20 photographs, taken at 4,600×, were evaluated from each region. The mean area of extracellular DNA was calculated using the ImageJ software in each picture. Extracellular DNA originating from eosinophils (EETosis) was recognized as compact structures with sharp borders and punctiform content, frequently associated with preserved eosinophil secretory granules or adhered debris. Another distinctive feature was the leftover condensed regions at the periphery or in the interior of the cells, with the presence of abundant cell membrane debris near the extracellular DNA material being the most distinct evidence of EETosis.
By contrast, extracellular nets originating from neutrophils were distinguishable by the presence of small secretory granules, organelles, lipid droplets, and small membranous vesicles associated with the chromatin material. In COPD D patients, the abundant extracellular nets were almost devoid of organelles; in this case, they were characterized by their irregular shapes and baggy borders. Neutrophils undergoing NETosis (NETotic) could appear homogenously decondensed or could still preserve thin linear condensations without associated secretory granules (Figures 1, 2 and 3 for representative images).
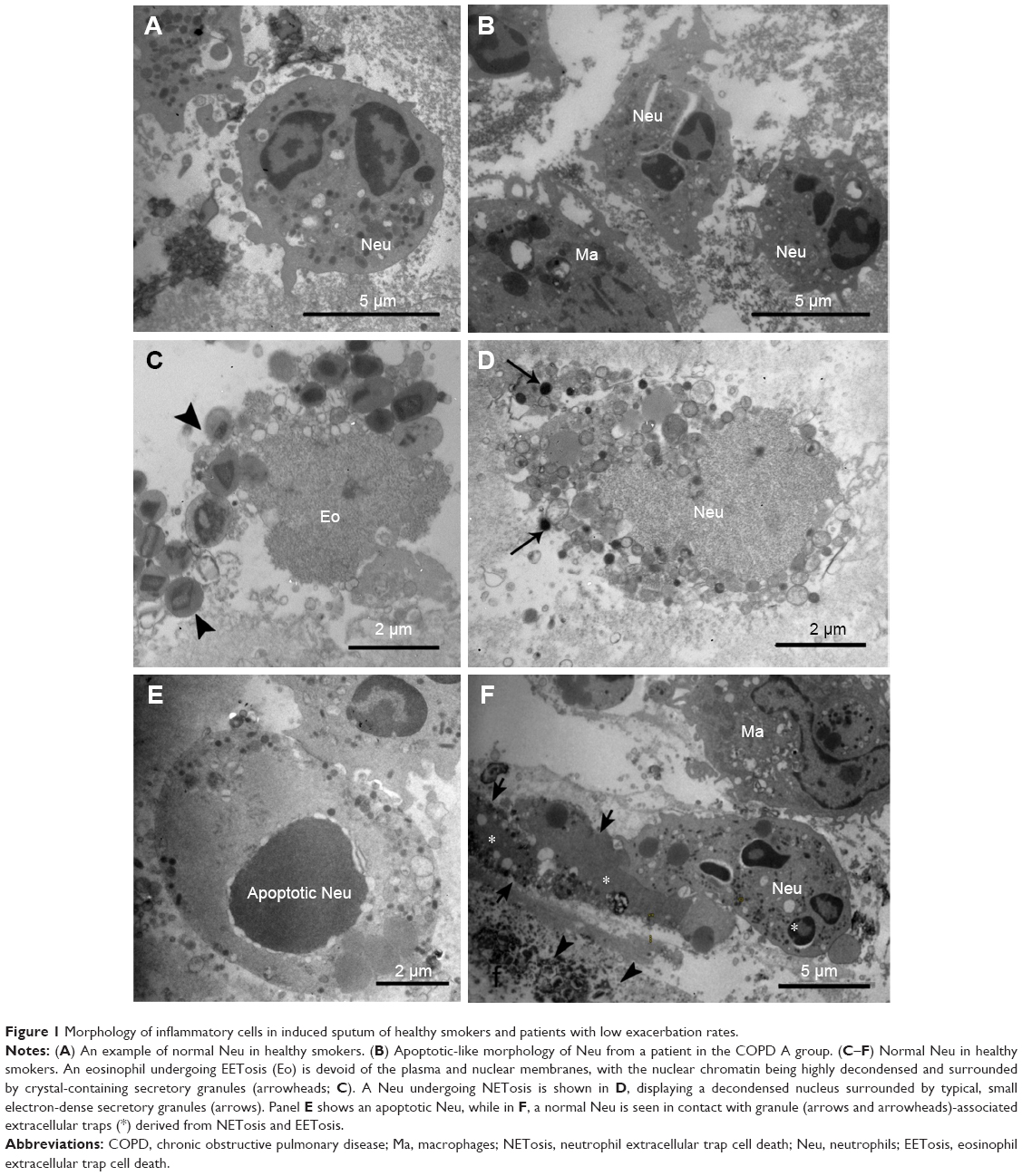 | Figure 1 Morphology of inflammatory cells in induced sputum of healthy smokers and patients with low exacerbation rates. |
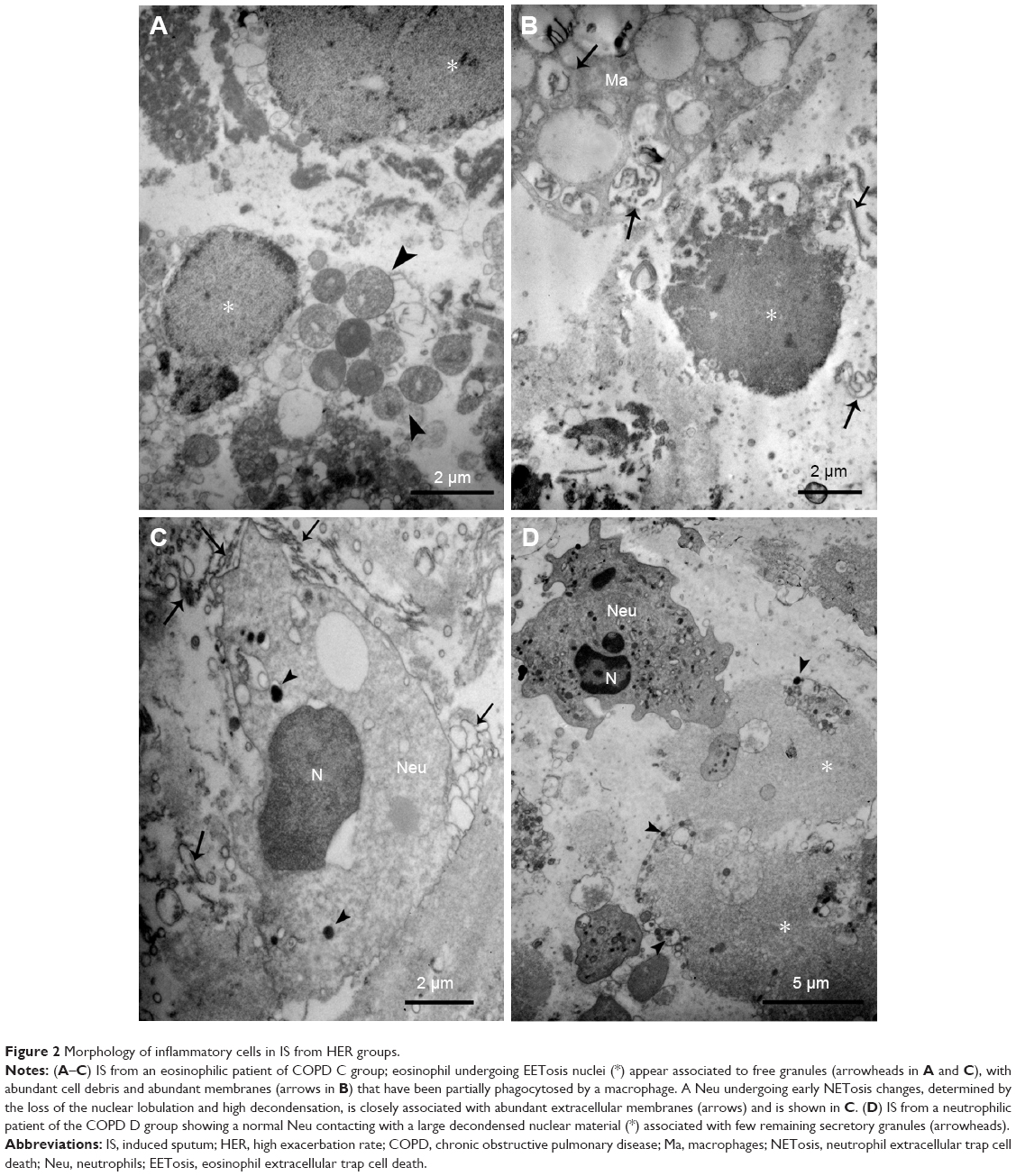 | Figure 2 Morphology of inflammatory cells in IS from HER groups. |
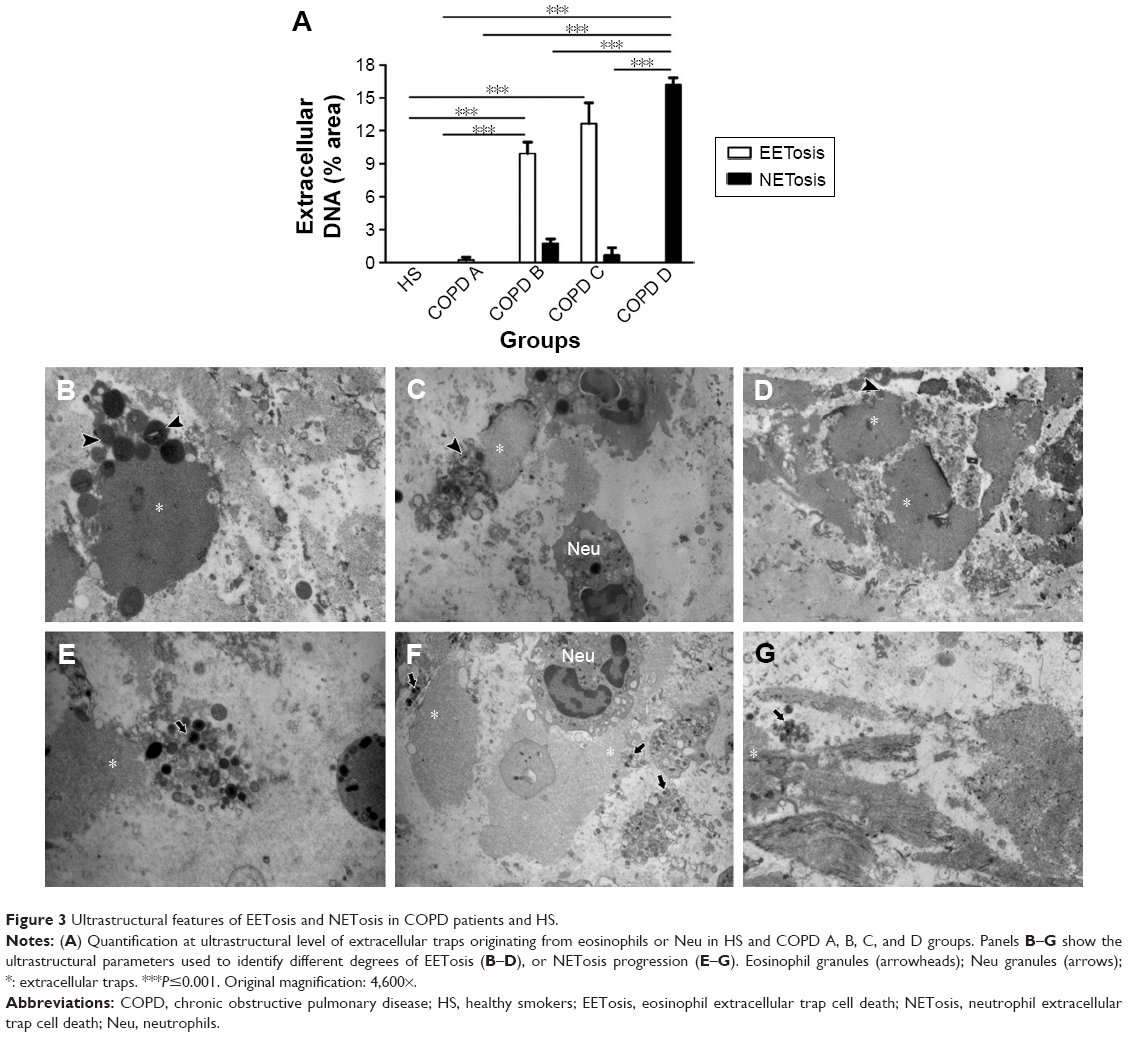 | Figure 3 Ultrastructural features of EETosis and NETosis in COPD patients and HS. |
Statistical analysis
Results are expressed as mean ± standard error of the mean. The data are presented as relative frequency differences between groups, examined using the Mann–Whitney U-test and analysis of variance-Tukey’s test for nonnormality and the independent unpaired t-test for normally distributed data. The Infostat statistical software package (Infostat, FCA-UNC, Córdoba, Argentina) was used for the analysis, and a P-value <0.05 was considered statistically significant.
Results
Patient’s characteristics
Table 1 exhibits the demographic features of 89 patients. A significant difference in group age was found, with the severe HER group being the oldest compared to the remaining groups. The percentage of the eosinophils and macrophages as well as the inflammatory patterns (eosinophilic, neutrophilic, mixed, or pausigranulocytic) were significantly different among patient subgroups. As shown in Figure 4, group A was the only one that did not include patients with an eosinophilic phenotype. Surprisingly, HS group included similar proportions of patients with paucigranulocytic and eosinophilic patterns. On the other hand, in the COPD B group, all patients exhibited elevated percentages of eosinophilic and mixed patterns. The HER groups also showed eosinophilic patients, but in contrast with LER group B, the neutrophilic phenotype was strong.
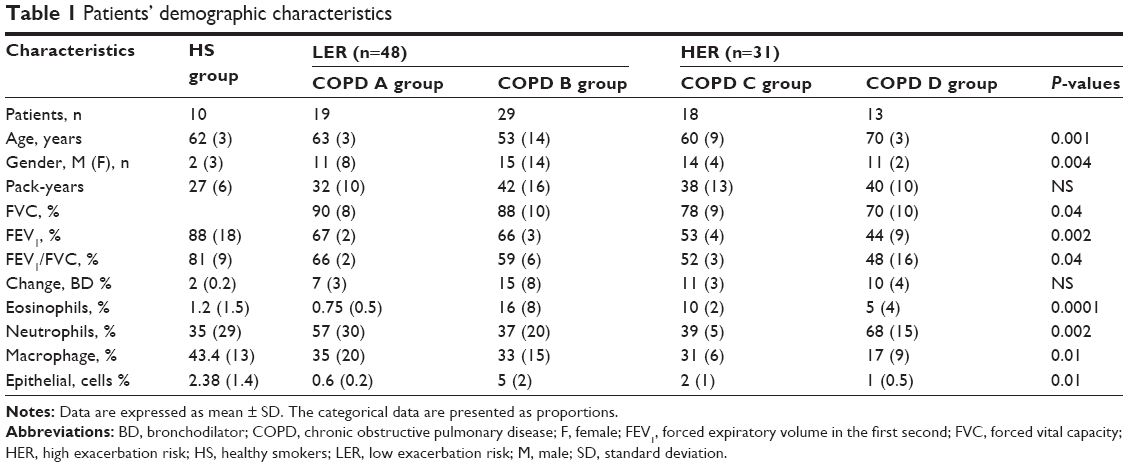 | Table 1 Patients’ demographic characteristics |
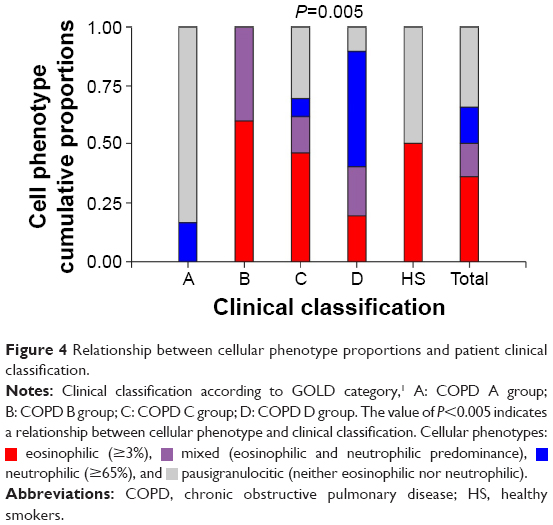 | Figure 4 Relationship between cellular phenotype proportions and patient clinical classification. |
 eosinophilic (≥3%),
eosinophilic (≥3%),  mixed (eosinophilic and neutrophilic predominance),
mixed (eosinophilic and neutrophilic predominance),  neutrophilic (≥65%), and
neutrophilic (≥65%), and  pausigranulocitic (neither eosinophilic nor neutrophilic).
pausigranulocitic (neither eosinophilic nor neutrophilic).Morphological analysis by electron microscopy and DNA labeling
In order to study the incidence of NETosis or EETosis in the different groups of COPD patients, an ultrastructural study of plugs isolated from IS without DTT treatment was performed. Neutrophils and eosinophils from HS mostly revealed a normal morphology when evaluated by immunofluorescence (Figure 5). On electron microscopy, neutrophils exhibited normal ultrastructure with numerous secretory granules (Figure 1A). In LER patients, most neutrophils of the COPD A group exhibited nuclear characteristics compatible with apoptosis, including margination of dense masses of chromatin beneath the nuclear membrane and sharp areas separated from the diffuse chromatin (Figure 1B); no evidence of NETosis was found. DAPI staining on semithin sections showed that neither HS nor COPD A patients exhibited extracellular DNA (Figure 6A and B); some apoptotic-like neutrophils were clearly visualized, which was in agreement with the ultrastructural findings in COPD A (Figure 6B).
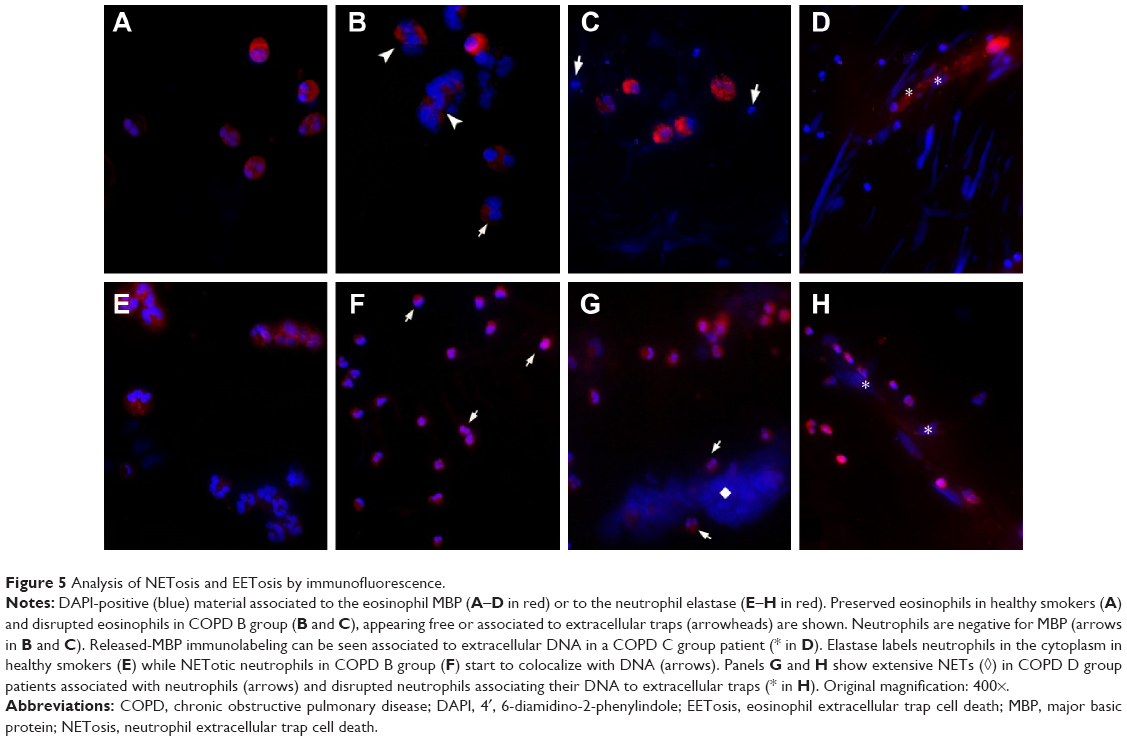 | Figure 5 Analysis of NETosis and EETosis by immunofluorescence. |
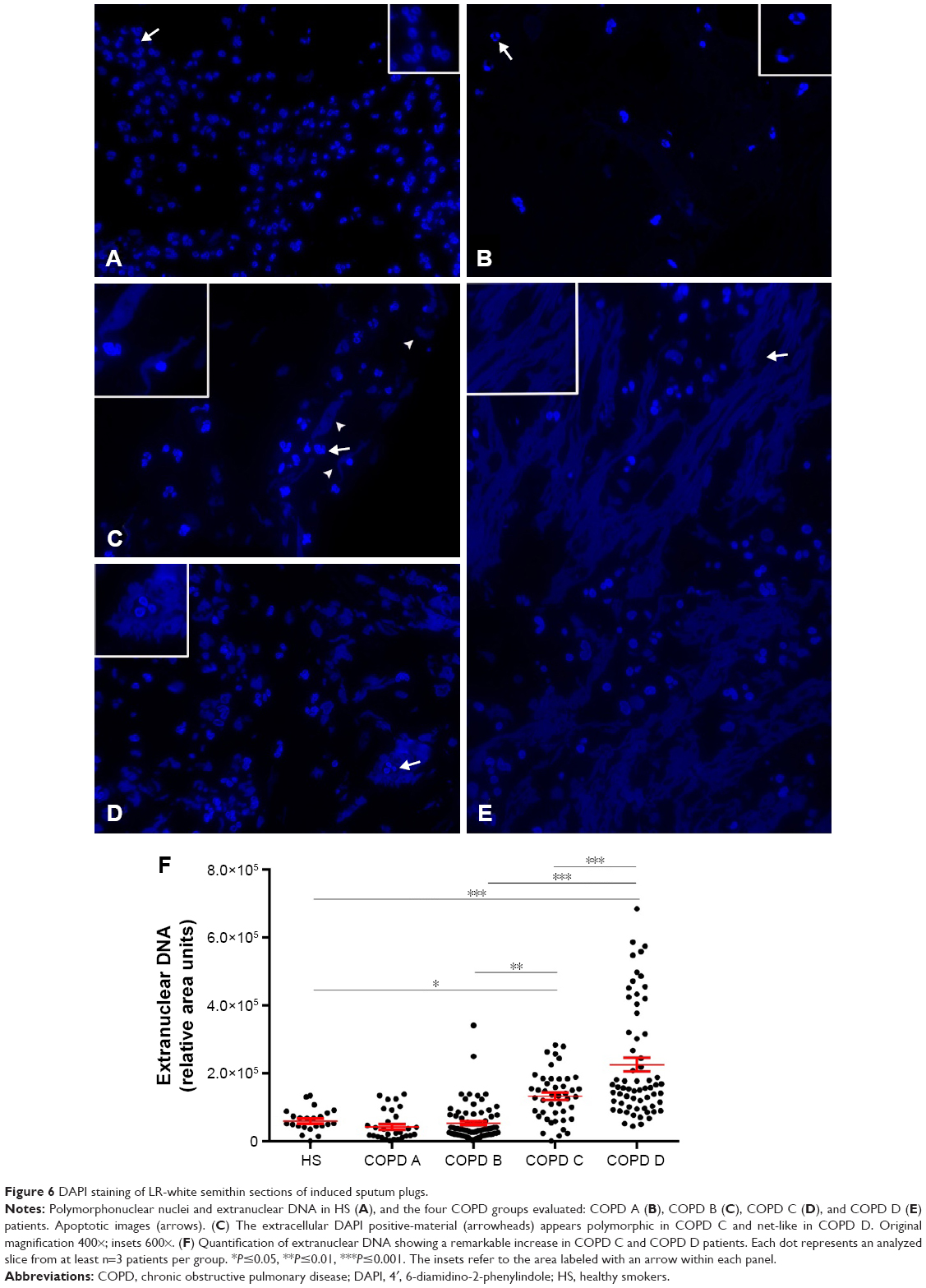 | Figure 6 DAPI staining of LR-white semithin sections of induced sputum plugs. |
An interesting observation in the LER-COPD B group, which exhibited the eosinophilic phenotype, was the presence of EETosis. This could be identified by the presence of eosinophil-like secretory granules associated with the nucleus, which exhibited decondensed chromatin, and it was already denudated of the nuclear membrane (Figure 1C). EETosis occurrence was confirmed by immunofluorescence, with typical images of extruded DNA, associated in part with the major basic protein (MBP) (Figure 5B and C). The typical morphology of NETosis could also be detected in the IS of COPD B, clearly recognized by the presence of neutrophil secretory granules associated with decondensed nuclear material (Figure 1D). By immunofluorescence, neutrophil elastase was found to be colocalized with DNA in some pre-NETotic or NETotic neutrophils (Figure 5E), together with the presence of some extracellular DNA intermingled with inflammatory cells (Figure 6C). However, extracellular traps forming classical NETs were rarely seen in the COPD B group. However, this group appeared to be quite a heterogeneous group with regard to its neutrophil morphology and functional state. Some patients of this group mainly exhibited numerous apoptotic cells (Figure 1E), while others displayed highly preserved and active neutrophils, coexisting with the typical morphology of cell death by NETosis and EETosis (Figure 1F).
There were clear differences between HER and LER patients regarding the presence of abundant cellular debris in their IS, with some of these variations depending on the cellularity. In the HER COPD C group that included patients with an eosinophilic inflammatory pattern (Table 1), numerous burst eosinophils were observed with signs of EETosis as expelled nuclei still associated with eosinophil secretory granules. However, in contrast with NETosis, nuclei did not seem decondensed to the same extent as in neutrophils and instead appeared as round masses (Figure 2A) or as irregular DAPI-stained structures in semithin sections (Figure 6D). A significant observation was that the higher the number of eosinophils that underwent EETosis, the higher the presence of cell debris, especially in the form of numerous free membranes and some detached cilia (Figure 2B), with macrophages being observed to phagocyte small portions of nuclear decondensated material and free membranes (Figure 2B). The neutrophils in these patients frequently seemed to be pre-NETotic as indicated by the chromatin decondensation and the loss of nuclear poly-lobulation, both events that precede the extrusion of the nuclear material, but still preserving their nuclear and plasma membranes (Figure 2C). Also, pre-NETotic neutrophils could often be seen incorporating cell debris (chromatin and membranes) by phagocytosis.
In HER COPD D patients with a neutrophilic inflammatory pattern, normal neutrophils frequently appeared associated with NET-like structures that could be identified by the presence of abundant decondensed nuclear material, with a few remaining electron-dense granules and small membrane-limited vesicles (Figures 2D and 5G and H). We next quantified the proportion of EETosis and NETosis at the ultrastructural level. The analysis confirmed that EETosis is predominant in COPD B and C, whereas NETosis is particularly abundant in COPD D (Figure 3).
These findings were then confirmed by immunofluorescence (Figure 5F). DAPI staining revealed that extracellular traps appeared to gather as fibers with a NET-like distribution (Figure 6E), with the analysis of IS by DAPI staining making it possible to identify the LER and HER groups based on the different quality–quantity patterns of the extracellular nuclear material. Quantification of the DAPI signal revealed a significant increase in COPD C and D patients (Figure 6F).
Discussion
This study focuses on aspects of inflammation that could be associated with the early symptoms and disease progression of COPD. Here, we demonstrated the occurrence of eosinophil death by EETosis in stable LER symptomatic COPD B and in HER patients with eosinophilic or mixed patterns. Furthermore, in high-exacerbation COPD patients, abundant extracellular traps were observed, thereby indicating an increase in NETosis and EETosis occurrence.
To date, most of the research has addressed the neutrophil in COPD. For instance, the ECLIPSE study has evaluated the relationship between sputum neutrophils and FEV1, but only found a weak association.1 This is in agreement with our study in that the neutrophil percentage did not change significantly among different groups of patients. Recently, Grabcanovic-Musija et al13 concluded that the severity of lung functions in COPD was associated to neutrophil death by NETosis. However, they only evaluated neutrophils and included patients with both exacerbated and stable stages of the disease.
Our study confirms the occurrence of NETosis but suggests that eosinophils might also play an important role in COPD. This was evidenced by the fact that the asymptomatic COPD A group, which lacked eosinophils, did not exhibit NETosis. Conversely, all LER symptomatic COPD B patients with eosinophilic or mixed cellular phenotypes showed an early presence of EETosis, thus revealing a factor that probably contributes to triggering the disease symptoms. In agreement, Singh et al4 claimed that eosinophils are a common cause of chronic cough in middle-aged patients, associated with variable airflow obstruction or airway hyperresponsiveness, accelerated decline in FEV1 and COPD development.
The HER COPD subset exhibited either combined eosinophil and neutrophil extracellular traps or only those derived from neutrophils. However, when eosinophils and EETosis coexisted in HER patients, IS showed abundant free organelles and membranes, indicative of greater cellular damage.
Several reports have focused on clarifying the beginning of the “burning” process of COPD.19 Our observations have led us to place eosinophils not only at the triggering of the COPD disease, but probably in its maintenance as well, in spite of smoking cessation, as a consequence of the prolonged accumulation of eosinophils and their debris in the airways.16,20 Therefore, cell debris of eosinophils undergoing EETosis, especially chromatin, accumulates in airways and could become a chemotactic stimulus for sterile inflammation by recruiting neutrophils to phagocytose cell debris. In this way, the process of NETosis could be started and self-perpetuated over the long time period of disease evolution in the COPD D subset, even if EETosis is no longer present, as occurs in many other chronic diseases.21,22 Even though a longitudinal study is needed to demonstrate such a progression, it is supported by Agusti et al,23 who concluded, based on the cohort study ECLIPSE, that severity of disease progressed in COPD B and C patients.
Although patients with less than 10 years history of smoking were reported to have normal lung function,24 the presence of eosinophils in their IS samples (shown in the present study) indicates the importance of evaluating inflammatory cells in the IS of HS and LER patients in order to initiate a specific anti-inflammatory treatment in the early stages of the disease. Therefore, we hypothesize that eosinophils may be implicated in the early development of COPD and that the presence of eosinophils might indicate the smokers who are most at risk of developing the disease.
It is well known that the smoke induces a complex microenvironment containing over 4,000 toxins and leading to proinflammatory cytokine induction in macrophages. In addition, thousands of reactive oxygen species (ROS) are produced in the burning cigarette, and these are able to damage the epithelial cells lining the airways.25 ROS also activate intracellular signaling cascades leading to inflammatory gene activation (IL-8 and TNF-α) that promote chronic immune cell recruitment and inflammation. Therefore, EETosis induction in COPD patients, as described in this study, probably is a secondary feature in genetically predisposed patients to have eosinophils in airways. According to our observations, eosinophils would be very sensitive to these toxins and respond widely by triggering EETosis, as indicated by the lack of preserved eosinophils.
The cytotoxic effects of eosinophils may favor a more symptomatic disease and also contribute to increased exacerbations, and therefore faster progression. Free destroyed eosinophil secretory granules can be aggressive for the epithelium and endothelium as the proteins they contain, contribute to tissue damage and dysfunction.26,27 In addition, eosinophil histone release, either from nuclei or from mitochondrial DNA, has been demonstrated to have a killing activity on pathogens and mammalian cells.28–32 Extracellular DNA extrusion from eosinophils was verified in Crohn disease, where IL-5 or IFNγ, followed by lipopolysaccharide, induced these cells to release mitochondrial DNA independently of cell death.32 In our study, a different methodology was used and eosinophil death was observed with typical ultrastructural changes that precede EETosis, as has been described before by many authors (for example, Fuchs et al7) for neutrophils. Cigarette-smoking-induced microenvironment in COPD airways may explain the difference as it does trigger cell death.
Although the inhaled corticosteroid is the recommended treatment for HER, we showed that a significant number of HER patients still had elevated percentages of eosinophils in spite of corticosteroids. In line with this, other authors reported that inhaled corticosteroids failed to suppress inflammation in COPD patients.33 Our observations by electron microscopy raise doubts about the efficiency of corticosteroids to induce eosinophil control once they are destroyed by EETosis, thereby strengthening the importance of early pharmacological intervention.
In summary, we demonstrated, for the first time, the presence of EETosis, in parallel with NETosis, in COPD at different stages in subjects at risk and with stable disease status. EETosis was useful to explain the eosinophil role in the pathophysiology, especially at the beginning and during COPD progression, regardless of whether the patient quit smoking. It is not necessary, however, to evaluate its presence in the clinical practice, as it is sufficient to study the occurrence of eosinophils in the IS of young healthy smokers without any spirometric changes since this might predict the development of COPD and be a guide for effective early treatment. Finally, our findings contribute by characterizing clear differences among groups, which is in agreement with their clinical manifestations. These results could contribute to prevention of the development of COPD in young smokers with a predisposition to develop the disease, with novel therapeutic techniques still being needed to treat the advanced disease.
Acknowledgments
The authors want to acknowledge Elena Pereyra, Lucía Artino, and Soledad Santa Cruz for the excellent technical assistance, and Nestor Boetto for his assistance with the electron microscopy. They would also like to thank Dr Paul Hobson for English language revision of the manuscript. This work was supported by research grants from Ministry of Science and Tehcnology of Córdoba, National Council for Scientific Technical Research (CONICET) and from the Fund for Scientific and Technological Research-National Agency for Science and Technology Promotion (FONCyT-ANPCyT), Ministry of Science, Technology and Productive Innovation, Argentina.
Disclosure
The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Shapiro SD. The pathogenesis of emphysema: the elastase:antielastase hypothesis 30 years later. Proc Assoc Am Physicians. 1995;107(3):346–352. | ||
Singh D, Edwards L, Tal-Singer R, Rennard S. Sputum neutrophils as a biomarker in COPD: findings from the ECLIPSE study. Respir Res. 2010;11:77. | ||
Singh D, Kolsum U, Brightling CE, Locantore N, Agusti A, Tal-Singer R. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697–1700. | ||
Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2(3):216–227. | ||
Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. | ||
Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241. | ||
Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007(379):pe11. | ||
Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Koutsopoulos A, Siafakas NM. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):161–171. | ||
Steen VD, Powell DL, Medsger TA Jr. Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 1988;31(2):196–203. | ||
Marcos V, Zhou Z, Yildirim AO, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med. 2010;16(9):1018–1023. | ||
Obermayer A, Stoiber W, Krautgartner WD, et al. New aspects on the structure of neutrophil extracellular traps from chronic obstructive pulmonary disease and in vitro generation. PLoS One. 2014;9(5):e97784. | ||
Grabcanovic-Musija F, Obermayer A, Stoiber W, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. 2015;16:59. | ||
Guimaraes-Costa AB, Nascimento MT, Wardini AB, Pinto-da-Silva LH, Saraiva EM. ETosis: a microbicidal mechanism beyond cell death. J Parasitol Res. 2014;2012:929743. | ||
Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil extracellular DNA trap cell death mediates lytic release of free secretion-competent eosinophil granules in humans. Blood. 2013;121(11):2074–2083. | ||
Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Front Immunol. 2013;3:420. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardization of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Nair P. Update on clinical inflammometry for the management of airway diseases. Can Respir J. 2013;20(2):117–120. | ||
Siafakas NM. “In the Beginning” of COPD: is evolution important? Am J Respir Crit Care Med. 2007;175(5):423–424. | ||
Eltboli O, Bafadhel M, Hollins F, et al. COPD exacerbation severity and frequency is associated with impaired macrophage efferocytosis of eosinophils. BMC Pulm Med. 2014;14:112. | ||
Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. | ||
Sayah DM, Mallavia B, Liu F, et al. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med. 2015;191(4):455–463. | ||
Agusti A, Hurd S, Jones P, et al. FAQs about the GOLD 2011 assessment proposal of COPD: a comparative analysis of four different cohorts. Eur Respir J. 2013;42(5):1391–1401. | ||
Kohansal R, Martinez-Camblor P, Agusti A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10. | ||
Lee J, Taneja V, Vassallo R. Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res. 2012;91(2):142–149. | ||
Ueki S, Tokunaga T, Fujieda S, et al. Eosinophil ETosis and DNA traps: a new look at eosinophilic inflammation. Curr Allergy Asthma Rep. 2016;16(8):54. | ||
Schorn C, Janko C, Latzko M, Chaurio R, Schett G, Herrmann M. Monosodium urate crystals induce extracellular DNA traps in neutrophils, eosinophils, and basophils but not in mononuclear cells. Front Immunol. 2012;3:277. | ||
Ueki S, Konno Y, Takeda M, et al. Eosinophil extracellular trap cell death-derived DNA traps: their presence in secretions and functional attributes. J Allergy Clin Immunol. 2016;137(1):258–267. | ||
Wang Y, Chen Y, Xin L, et al. Differential microbicidal effects of human histone proteins H2A and H2B on Leishmania promastigotes and amastigotes. Infect Immun. 2011;79(3):1124–1133. | ||
Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15(11):1318–1321. | ||
von Kockritz-Blickwede M, Goldmann O, Thulin P, et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood. 2008;111(6):3070–3080. | ||
Yousefi S, Gold JA, Andina N, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. 2008;14(9):949–953. | ||
Barnes PJ. Inhaled corticosteroids in COPD: a controversy. Respiration. 2010;80(2):89–95. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.