Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Evidence for a Beneficial Effect of Oral N-acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke
Authors Sabetghadam M, Mazdeh M, Abolfathi P, Mohammadi Y ![]() , Mehrpooya M
, Mehrpooya M
Received 8 December 2019
Accepted for publication 1 May 2020
Published 18 May 2020 Volume 2020:16 Pages 1265—1278
DOI https://doi.org/10.2147/NDT.S241497
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Maryam Sabetghadam,1 Mehrdokht Mazdeh,2 Parnaz Abolfathi,1 Younes Mohammadi,3 Maryam Mehrpooya1
1Department of Clinical Pharmacy, School of Pharmacy, Medicinal Plants and Natural Products Research Center, Hamadan University of Medical Sciences, Hamadan, Iran; 2Department of Neurology, School of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran; 3Modeling of Noncommunicable Diseases Research Center, School of Public Health, Hamadan University of Medical Sciences, Hamadan, Iran
Correspondence: Maryam Mehrpooya
Department of Clinical Pharmacy, School of Pharmacy, Hamadan University of Medical Sciences, Shahid Fahmideh Ave, Hamadan 6517838678, Iran
Tel +9 881 382 1868
Fax +98 813 838 1591
Email [email protected]
Purpose: Numerous preclinical studies have demonstrated the potential neuroprotective effects of N-acetylcysteine (NAC) in the treatment of brain ischemia. Accordingly, the present study aimed to assess the potential therapeutic effects of oral NAC in patients with acute ischemic stroke.
Patients and Methods: In a randomized, double-blind, placebo-controlled trial study, 68 patients with acute ischemic stroke with the onset of symptoms less than 24 hours were randomly assigned to either the NAC-treated group or placebo-treated group. NAC and matched placebo were administrated by a 72-hour oral protocol (initially 4 grams loading dose and after on, 4 g in 4 equal divided doses for more 2 days). The primary outcomes were quantification of any neurologic deficit by the use of the National Institute of Health Stroke Scale (NIHSS) score and functional disability by the use of the modified Rankin scale (mRS) at 90 days after stroke. Additionally, serum levels of markers of oxidative stress and inflammation as a main mechanism of its action were assessed at baseline and the end of 3-day treatment protocol.
Results: NAC-treated patients in comparison with placebo-treated patients showed a significantly lower mean NIHSS scores at day 90 after stroke. A favorable functional outcome which was defined as an mRS score of 0 or 1, also in favor of NAC compared to placebo was noted on day 90 after stroke (57.6% in the NAC-treated group compared with 28.6% in the placebo-treated group). Further, compared to the placebo, NAC treatment significantly decreased serum levels of proinflammatory biomarkers such as interleukin 6 (IL-6), soluble intercellular cell adhesion molecule-1 (sICAM-1), nitric oxide (NO), malondialdehyde (MDA), and neuron-specific enolase (NSE) and significantly increased serum levels of anti-oxidant biomarkers such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and total thiol groups (TTG).
Conclusion: The pattern of results suggests that oral NAC administration early after an acute ischemic stroke is associated with a better outcome profile in terms of acute neurological deficit and disability grade compared to placebo. NAC may improve neurological outcomes of patients with stroke at least in part by its antioxidant and anti-inflammatory effects.
Keywords: ischemic stroke, oxidative stress, inflammation, N-acetylcysteine, antioxidant and anti-inflammatory compounds
Introduction
In addition to being one of the major causes of death,1 stroke is also the leading cause of long-term physical and intellectual disability particularly in the elderly worldwide.2 Ischemic stroke which represents about 80–85% of all strokes is mainly provoked by thrombosis, embolism, and systemic hypoperfusion. In any case, ischemic stroke eventually results in interruption or severe reduction of blood flow in cerebral arteries and consequently deprive neurons of necessary glucose and oxygen.3 Limited fuel reserves and high energy requirements render the brain particularly vulnerable to hypoxic conditions.4 Within seconds to minutes, brain ischemia activates a cascade of events including loss of adenine triphosphate (ATP), excitotoxicity of glutamate, oxidative stress, inflammation, reduced neurotrophic support, and multiple other metabolic stresses that all of which leads to neuronal death along with an irreversible loss of neuronal function.5,6 From a therapeutic point of view, although thrombolytic agents, antiplatelet and antithrombotic agents, and neuroprotective agents are currently the most effective type of therapy for acute ischemic stroke,7 current treatment of ischemic stroke is generally very limited and there is still room to improve efficacy and ameliorate outcomes associated with stroke.
There is increasing evidence that oxidative toxic stress (OTS) is an important mechanism in determining tissue injury following cerebral ischemia.8 An imbalance between the production of reactive oxygen/nitrogen species (ROS/RNS) and natural enzymatic/non-enzymatic antioxidant defense systems in favor of pro-oxidants leads to OTS which can cause damage to all cellular macromolecules including cellular proteins, membrane lipids, carbohydrates, and nucleic acids.9 In addition to its direct cellular damage, OTS also by activation of both the peripheral and brain resident immune pathways leads to exacerbation of cerebral ischemic injury.10 Lower plasma levels of nonenzymatic antioxidant like retinol, ascorbic acid, atocopherol, and carotenoids as well as antioxidant enzyme like superoxide dismutase (SOD) and glutathione peroxidase (GPx)11,12 and higher serum levels of OTS biomarkers like malondialdehyde (MDA), oxidized low-density lipoproteins (oxLDL), and DNA oxidative damage13–15 have been observed in patients with stroke compared to healthy controls. Moreover, it has been shown that plasma levels of antioxidants in the early hours post-stroke are inversely correlated with the infarct size and the severity of neurological damage.16–18
In addition to OTS, following ischemia, overproduction of pro-inflammatory cytokines and chemokines by activated microglia and macrophage also can result in an acute and potentially injurious inflammatory reaction.19,20 In this respect, both experimental and human stroke studies showed in the early stage of ischemic stroke loss in the balance between pro-inflammatory and anti-inflammatory cytokines in the periphery and in the brain play an important role in the development of ischemic damage and clinical prognosis.21 Further, some studies have reported that the magnitude of the serum pro-inflammatory cytokine levels such as tumor necrosis factor-alpha (TNF-α) and interleukine6 (IL-6) after acute ischemic stroke can predict brain infarct volume, stroke severity, and long-term outcomes.22,23 This is while inflammatory responses in the chronic phase have a beneficial effect in the post-stroke tissue healing process and functional recovery.21
Given the detrimental effects of inflammation and OTS pathways in the pathogenesis of ischemic stroke, pharmacological modulation of the inflammatory and oxidative stress responses may be an attractive therapeutic avenue for improving outcomes following acute stroke. One candidate molecule known to affect these mentioned pathological pathways is N-acetylcysteine (NAC). NAC is a cysteine pro-drug and glutathione (GSH) precursor that possesses potent anti-inflammatory and antioxidant properties. Apart from its direct scavenging activity of free radicals by providing sulfhydryl groups, NAC also through replenishment of intracellular GSH, exhibits potent antioxidant activity in the cell.24 Moreover, NAC through inhibitory effects on NF-κB activity (nuclear factor kappa-light-chain-enhancer of activated B cells) which plays a critical role in many aspects of the inflammation cascade and immune response, exhibits regulatory effects on pro-inflammatory cytokine production.25 Both animal and human studies have further confirmed the favorable influence of NAC treatment on inflammatory and OTS biomarkers.26–28 Due to its ability in modulating oxidative stress and inflammatory conditions, recently therapeutic effects of NAC over a wide range of disorders have been investigated.24 Previous preclinical studies have demonstrated the potential neuroprotective effects of NAC in treatment of various neurodegenerative diseases such as Huntington’s disease,29 Alzheimer’s disease,30 and Parkinson’s disease.31 Moreover, some experimental studies in stroke models have shown that NAC may exert beneficial effects on brain ischemia.32,33
To our best knowledge, no prior studies have addressed the influence of NAC supplementation in acute ischemic stroke. Accordingly, the primary aim of the present study was to assess the potential therapeutic effects of oral NAC on the clinical severity and functional ability in acute ischemic stroke patients. In addition, as a secondary outcome, we investigated the modulatory effects of oral NAC on serum levels of OTS and inflammatory biomarkers as the main mechanism of its action in these patients.
Patients and Methods
This is a prospective randomized, double-blind, placebo-controlled trial study that was conducted from June 2018 until September 2019 in the neurology Ward of Farshchian-Sina Hospital, affiliated to Hamadan University of Medical Sciences, Hamadan, Iran. Eligible patients or their representatives were fully informed about the study’s aims and the confidential and anonymous data handling. Participants or their representative signed the written informed consent and were randomly assigned either to the intervention or the control groups. The Ethics committee of Hamadan University of Medical Sciences has approved the study protocols, which was performed in accordance with the rules laid down in the Declaration of Helsinki and its later amendments. The study was registered at the Iranian Registry of Clinical Trials (IRCT20150629022965N16 and IRCT20120215009014N215; www.irct.ir).
Participants
All patients with acute ischemic stroke were screened for eligibility. Inclusion criteria at the screening visit were as follows: 1) males or females over 18 years; 2) diagnosed with first-ever ischemic stroke; 3) onset of symptoms less than 24 hours;and4) National Institute of Health Stroke Scale (NIHSS) score of >3 and <22. At baseline and during study period patients were excluded if any of the following exclusion criteria were met: 1) severe stroke (NIHSS>22); 2) hemorrhagic stroke; 3) evidence of other diseases of the Central Nervous System (CNS) including brain tumor, demyelinating diseases, craniotomy in the past, and severe brain injuries; 4) acute or chronic renal or hepatic failure (LFT’s>3× upper limit of normal, creatinine>2mg/dL); 5) acute or chronic infectious or inflammatory diseases; 6) patients who were candidate for thrombolysis and/or thrombectomy treatment; 7) consuming any antioxidant supplements or anti-inflammatory agents other than prescribed medications; 8) history of allergy or intolerance to NAC; 9) pregnancy or lactation or possibility of pregnancy; 10) medical, psychological, or pharmacological factors that interfere with the collection or interpretation of study data; 11) unlikely to be available for 90-day follow-up; and 12) presence of any adverse effects resulting in patients’ intolerance or complications.
Acute ischemic stroke was diagnosed by neurologic examination and head computed tomography (CT) and/or magnetic resonance imaging (MRI) on admission to the hospital. All patients were evaluated and treated according to the best accepted medical criteria and standard pharmacologic treatment (eg, dual antiplatelet therapy and antihypertensive agents).34
Intervention
Sixty-eight patients who met the inclusion/exclusion criteria were allocated into either the NAC group (intervention group; n=33) or Placebo group (control group; n=35) by block randomization method. The randomization was provided by an independent statistician to ensure that groups were matched for age, sex, and body mass index (BMI) where possible. Also, the study medication was blinded to both the study participants and the investigators. Required demographic and clinical variables of the study patients were taken from their medical records. They included demographic characteristics (age, sex, and BMI), smoking history, risk factors and other diseases, stroke etiology, prescribed medications, and mean time from stroke onset. Additionally, at baseline laboratory data including blood levels of total cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), triglyceride (TG), and fasting blood glucose (FBS), and also systolic and diastolic blood pressure were recorded for all the patients.
Drug Preparation and Administration
NAC and matched placebo were administrated by a 72-hour oral protocol according to Hoffer et al’s study which was conducted in the patients with mild traumatic brain injury.35 Based on this protocol, initially, 4 grams loading dose of NAC was given to the subjects, and 24 hours after the loading dose, 4 g NAC in 4 divided doses of 1 g were given to subjects for more 2 days (total protocol dose was 12 g). Because NAC’s ability to cross blood-brain barrier (BBB) is dependent on its dose and routes of administration, administration of such rather high doses of oral NAC was chosen in the percent study for increasing amount of NAC that crosses the BBB.36 NAC was administrated as liquid oral solution which was prepared from pure NAC powder in individual 50 mL bottle at a NAC concentration of 40 mg/mL, diluted in water. Placebo oral solution was similarly prepared by adding starch in water with identical taste and appearance. Each solution was freshly prepared by one nurse who was no further part of those patients’ care. The choice of 72-hour treatment duration of the study was based on previous evidence that most of the inflammatory molecules reach peak levels in the brain between 6 and 72 hours after the ischemic insult.37
Clinical Assessment
The primary efficacy variable was the quantification of any neurologic deficit 90 days after stroke in subjects treated with NAC and placebo with the use of the NIHSS. NIHSS consists of 15-item, each of which scores a specific ability between a 0 and 4. For each item, a score of 0 typically indicates a normal function in that specific ability, whereas a higher score is indicative of some level of impairment. The possible total NIHSS score 0 to 42, with higher values reflecting more severe neurologic impairment.38 Assessments were made at the time of admission and 90 days after onset of stroke. On admission time, the patients’ stroke severity according to the NIHSS score was categorized into mild stroke (the NIHSS score was ≤8), moderate stroke (NIHSS score 9 to 15), and severe stroke (the NIHSS score ≥16). The NIHSS findings on day 90 after the onset of stroke were categorized as a complete or nearly complete improvement (0 to 1), mild (2 to 7), moderate (8 to 14), and severe (≥15). A single experienced nurse, who was unaware of the treatment assignment, scored the NIHSS of each patient.
Additionally, as a secondary efficacy endpoint, functional disability at day 90 was evaluated by the modified Rankin scale (mRS).39 The mRS is a measure that is most widely used for assessment of functional outcomes in stroke trials. The mRS is a 6-point disability scale with possible scores ranging from 0 (no symptoms at all) to 5 (severe disability); a score of 6 indicates death. Favorable and unfavorable functional outcomes at day 90 were defined as an mRS score of 0 or 1 and 2 to 6, respectively. Three-month mRS outcomes were also obtained by a direct interview by a single experienced nurse.
To evaluate the adverse effects of medications, each patient was closely monitored for any possible adverse effects, and type and severity of adverse effects were recorded.
Measurement of Inflammatory and Oxidative Stress Biomarkers
Ten-milliliter fasting blood samples were taken from all recruited patients at the baseline (prior to treatment) and at the end of the 3-day treatment protocol. All blood samples were centrifuged at 3000 rpm for 10 minutes at 4°C. The serum was separated and stored at −70°C until analysis was performed. All samples were assessed in duplicate.
TAC, as a total antioxidant capacity, was measured by the ferric-reducing ability of plasma (FRAP method) and expressed as micromoles per liter (µmol/L).40 Total thiol groups (TTG), as a major portion of the total body antioxidants, were measured by spectrophotometric assay based on the Ellman’s method and were expressed as micromoles per liter (μmol/L).41 Superoxide dismutase (SOD), glutathione peroxidase (GPx), and Catalase (CAT) serum activities were determined using spectrophotometry according to commercially available kits (Zelbio co., Germany) and serum GPx and SOD levels are presented as Units per milliliter serum (U/mL) and CAT levels as milliUnits per milliliter serum (mU/mL). Malondialdehyde (MDA), as an indicator of final products of lipid peroxidation, was measured using the thiobarbituric acid-reactive substances method, as described by Botsoglou42 and was expressed as nmol/mL. Nitric oxide (NO), as the main reactive nitrogen species, was measured by using commercially available kits from Cayman Chemical Company based on determining the total nitrate/nitrite level, and results were expressed as μmol/L.
Furthermore, high-sensitivity sandwich enzyme-linked immunosorbent (ELISA) technique was exploited for the determination of serum levels of tumor necrosis factor-alfa (TNF-α), interleukin 6 (IL-6), and soluble intercellular cell adhesion molecule-1 (sICAM-1) as markers of immune-inflammatory (ZellBio GmbH, Germany) and neuron-specific enolase (NSE) as a specific indicator of brain neuronal damage (ZellBio GmbH, Germany). All of the inflammatory biomarkers were expressed as pg/mL.
Statistical Analyses
Data were analyzed using the SPSS for Windows (SPSS Inc., Chicago, IL, USA) version 16 software. The normality of variables was checked by the Kolmogorov–Smirnov test. Mean ± standard deviation (SD) and median (interquartile range [IQR]) were used to express normally- and non-normally distributed continuous variables. Categorical variables were reported as percentages. Mean (SD) and median (IQR) of continuous variables were compared between two groups using independent t-test and Mann–Whitney U-test, respectively. The distribution of categorical variables between the two groups was compared using Chi-square or Fisher exact test (if more than 20% of the categories were expected to have frequencies less than 5). The level of statistical significance was set to α=0.05. All analyses were performed on an Intention-to-Treat (ITT) analysis data set and missing data were replaced by the mean of the other group.
Results
Sample Characteristics
Figure 1 shows the flow diagram of the trial participants. Among 105 patients who admitted to our hospital with a diagnosis of acute ischemic stroke in the enrolment period, 37 patients were excluded (8 patients declined to participate and 29 patients did not meet inclusion and exclusion criteria). A total of 68 patients were enrolled in this study (NAC-treated group: 33 patients and placebo-treated group: 35 patients). All patients received at least one dose of the study medication. Of those, 11 patients discontinued participation in the study prematurely: 3 patients withdrew because of side effects (2 patients in the NAC-treated group and one patient in the placebo-treated group), 3 patients died in the acute phase due to stroke severity (one patient in the NAC-treated group and 2 patients in the placebo-treated group), and another 5 patients were lost to follow-up after discharge from the hospital (2 patients in the NAC-treated group and 3 patients in the placebo-treated group). As mentioned above, all analyses were performed on an Intention-to-Treat (ITT) analysis data set (on 68 patients). Because dropout rates of less than 20% and similar courses of disease in the comparison groups, missing data were replaced by the mean data of the other group.43
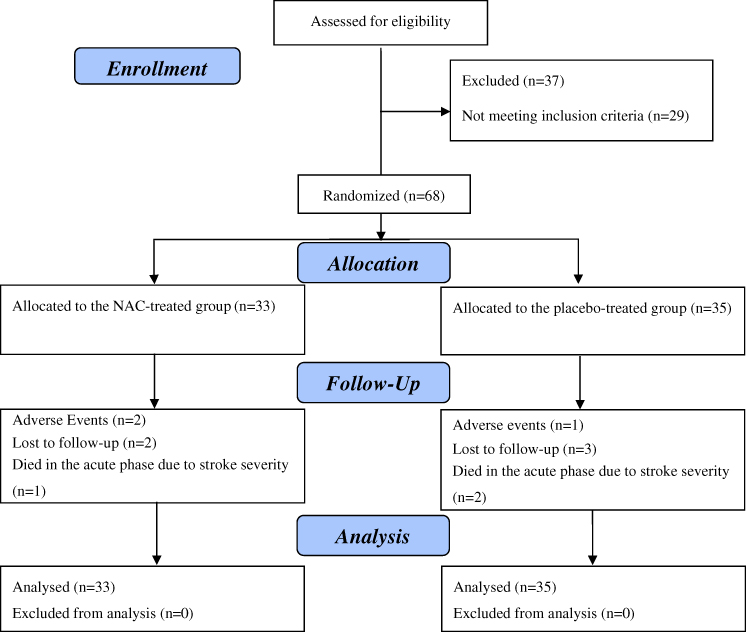 |
Figure 1 The flow diagram of the study. |
Table 1 shows the baseline demographic, laboratory, and clinical characteristics of the two groups. No significant intragroup differences in male/female ratio, mean age, and BMI were detected. No intragroup differences in laboratory test values including serum lipid profile and fasting blood sugar on admission time were noted. In respect of stroke risk factors, stroke etiology, and concomitant medications also no significant differences were observed between the 2 groups. No patients in either group used steroids or nonsteroidal anti-inflammatory drugs. Meantime to treatment after stroke onset for NAC- and placebo-treated subjects was 9.06±9.10 and 9.42±11.41 hours, respectively. No significant differences were observed between the 2 groups in this regard.
 |
Table 1 Demographics and Clinical Characteristics of the Intention-to-Treat Population at Baseline |
The study groups were similar with respect to the baseline median NIHSS scores (14 [IQR 16.5–9.5] in the NAC-treated group and 11 [IQR 16–9] in the placebo-treated group; P-value=0.59; Table 2). The distribution of the NIHSS score at baseline by 3-point categories also was similar in 2 groups (Figure 2A). On admission time, median mRS scores in the NAC- and placebo-treated groups were 3 (IQR 4–2) and 2 (IQR 3–1) respectively, and the group difference in the score of mRS did not reach statistical significance before drug intervention (Table 2). Distribution of mRS scores across 0–5 scores of the mRS also was similar in the 2 groups at admission time (Figure 2B).
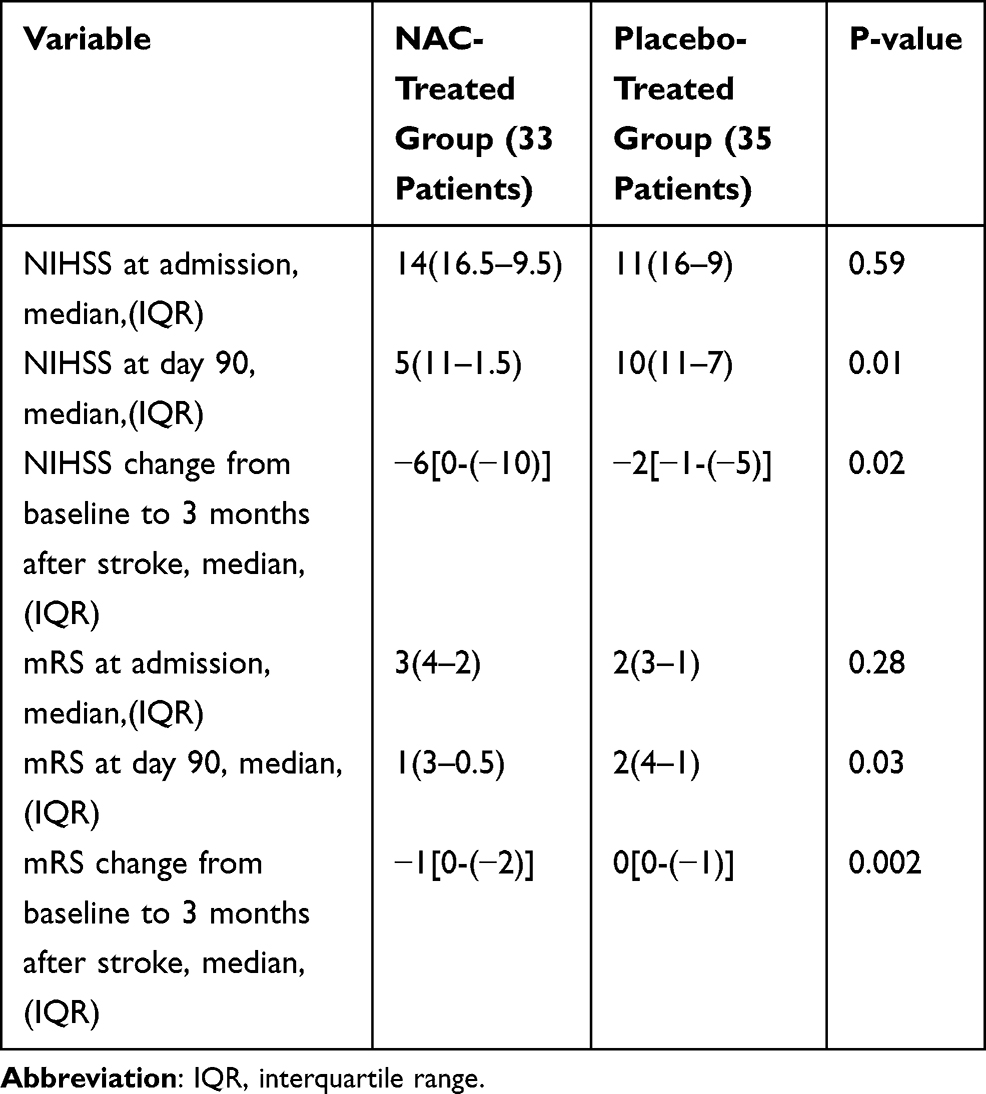 |
Table 2 National Institute of Health Stroke Scale (NIHSS) and Modified Rankin Scale (mRS) Scores at Baseline and at Day 90 |
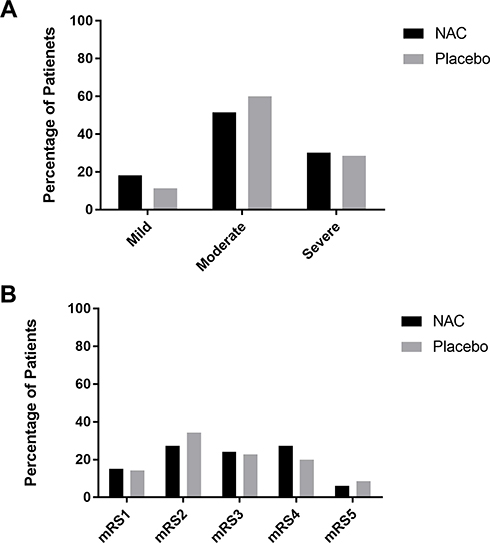 |
Figure 2 (A) Distribution of scores on the National Institute of Health Stroke Scale (NIHSS) and (B) modified Rankin scale (mRS) of 2 groups at baseline. Note: The patients’ stroke severity at baseline was categorized into mild stroke (the NIHSS score was ≤8), moderate stroke (NIHSS score 9 to 15), and severe stroke (the NIHSS score ≥16). |
Clinical Outcome Indicators Change at Day 90 After Stroke
On day 90, the median NIHSS scores decreased from 14 (IQR 16.5–9.5) at baseline to 5 (IQR11-1.50) in the NAC-treated group and from 11 (IQR 16-9) to 10 (IQR 11–7) in the placebo-treated group (Table 2). The median changes in the NIHSS scores at 90 days post-stroke compared to those at admission time were −6 [IQR 0-(−10)] in the NAC-treated subjects versus −2 [IQR −1-(−5)] in the placebo-treated subjects and difference between groups in the median changes in the NIHSS scores reached to the statistical significance level (P-value=0.02; Table 2). Distribution analysis by 4-point categories of NIHSS scores also showed that patients treated with NAC shifted towards a better outcome regarding NIHSS improvement by day 90 than did placebo-treated patients (P-value=0.02; Figure 3A).
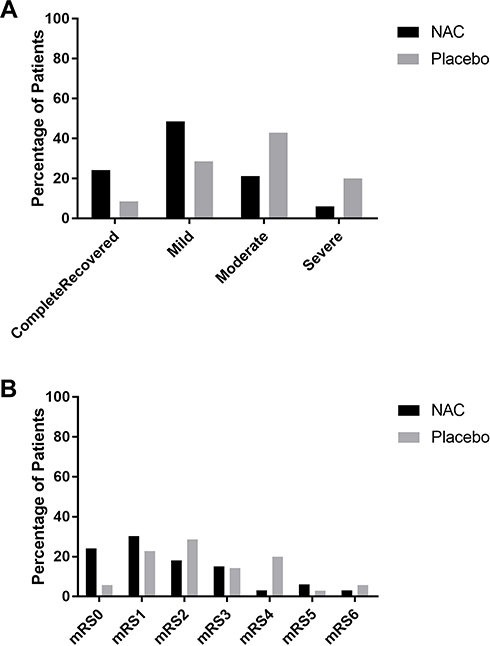 |
Figure 3 (A) Distribution of scores on the National Institute of Health Stroke Scale (NIHSS) and (B) modified Rankin scale (mRS) of 2 groups on day 90. Note: the NIHSS findings on day 90 were categorized as a complete or nearly complete improvement (0 to 1), mild (2 to 7), moderate (8 to 14), and severe (≥15). |
Similar to the results for the NIHSS, concerning mRS, the stroke patients treated with NAC at 90 days post-stroke, showed a significantly lower median mRS score compared with the stroke subjects treated with placebo (1[IQR3-0.5] vs 2[IQR4-1]; P-value=0.03; Table 2).The median changes of mRS scores from baseline to day 90 post-stroke also were significantly greater in the NAC-treated subjects compared with the placebo-treated subjects (−1[0-(−2)] vs 0[0-(−1)]; P-value=0.002; Table 2). Although distribution analysis across all scores of the mRS on day 90 post-stroke did not show statistically significant difference in the NAC-treated subjects compared to the placebo-treated subjects (P-value=0.09) (Figure 3B), a favorable functional outcome which was defined as an mRS score of 0 or 1, in favor of NAC were noted, so that a favorable mRS was reported in 57.6% of the patients in the NAC-treated group (19 out of 33 patients) compared with 28.6% of those in the placebo-treated group (10 out 0f 35 patients) (P-value=0.02).
Safety and Tolerability
Results regarding the occurrence of adverse effects have been shown in Table 3. The most frequently reported side effects by the study patients regardless of treatment group were nausea, vomiting, dyspepsia, and headache. As shown in Table 3, although the incidence of adverse effects such as nausea, dyspepsia, and headache was significantly higher in the NAC-treated patients compared to the placebo-treated patients, most of the adverse effects were rated as mild to moderate in severity and none of the reported adverse effects was serious or caused any complication for the patients.
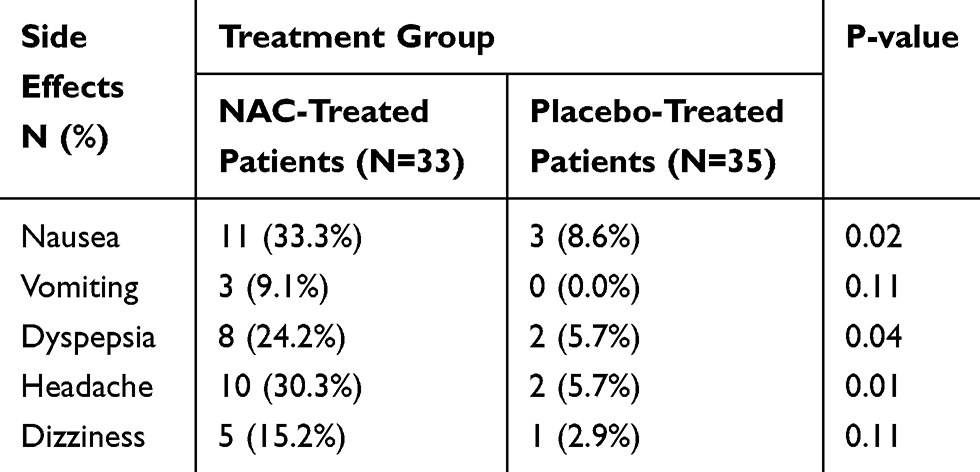 |
Table 3 Frequency of Drug-Related Adverse Effects Among Patients in Each Group |
Changes of Oxidative Stress and Inflammatory Biomarkers: From Baseline to Day 3 After Treatment and Between the Groups
As shown in Table 4, with regard to OTS and inflammatory biomarkers at baseline, there were no significant differences between 2 groups. Seventy-two hours after treatment, the serum levels of GPx, TTG, and SOD (as enzymatic antioxidants which catalyze ROS/RNS conversion to less reactive or inert species) were significantly increased in the NAC-treated subjects compared to the placebo-treated subjects (P-value = 0.01, 0.01, and <0.001, respectively). Also, 72 hours after treatment, levels of the MDA (as final products of lipid peroxidation) and IL-6 (as a pro-inflammatory cytokine) were significantly decreased in the NAC group compared to the placebo group (P-value = 0.04 and <0.001, respectively). Additionally, mean changes in the serum levels of factors including IL-6, ICAM-1, TTG, MDA, NO, NSE, and SOD from baseline to day 3 after treatment showed a significant difference between two groups, with favoring NAC treatment group. Although 72 hours after treatment, serum levels of CAT, TAC, and TNF-α were more favorable in the NAC-treated subjects compared to the placebo-treated subjects, serum values of these factors did not display significant differences in inter- or intra-group comparisons over time.
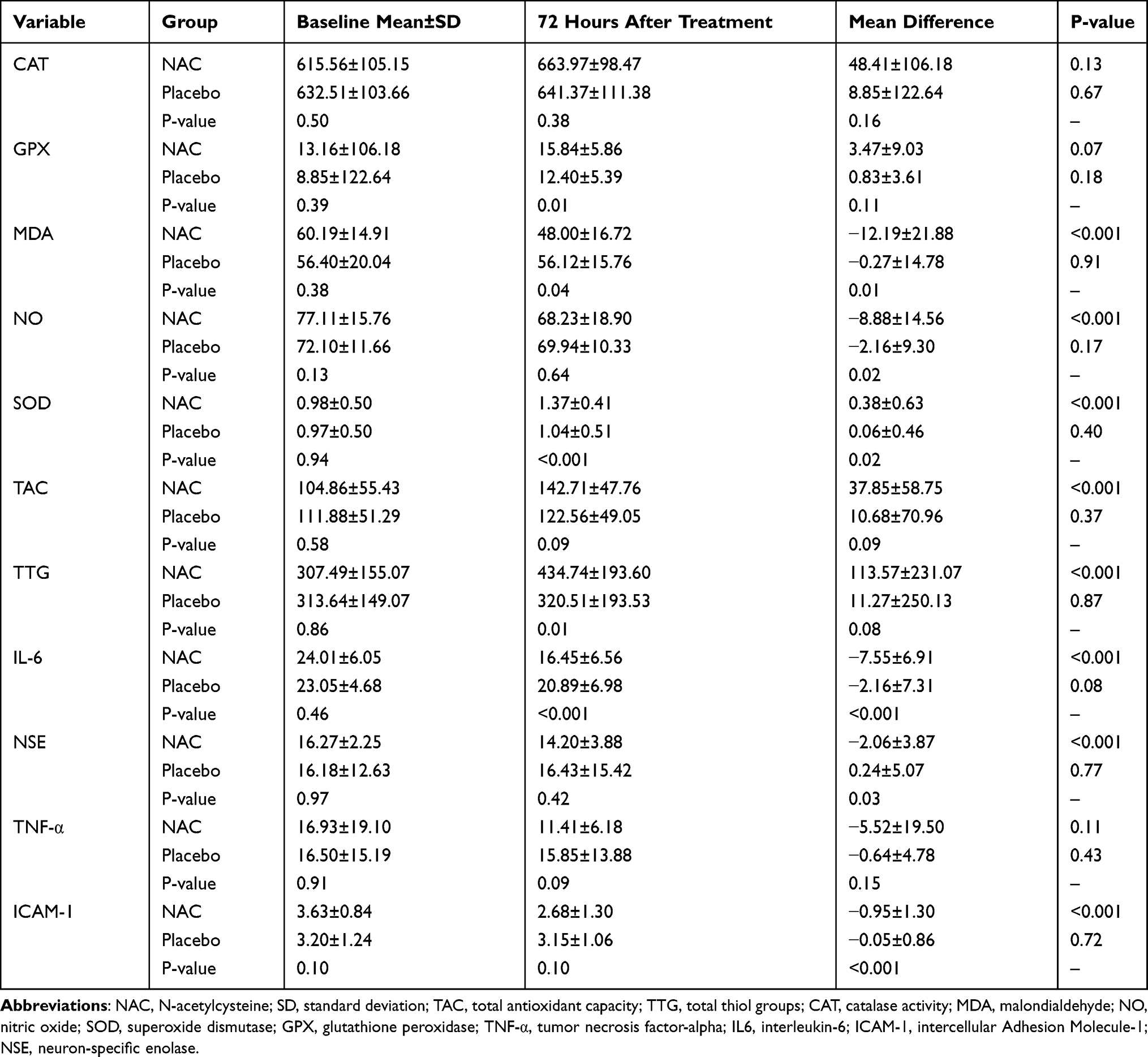 |
Table 4 The Serum Levels of Oxidative Stress and Inflammatory Biomarkers of Two Groups, at Baseline and 72 Hours After Treatment |
To summarize, compared to the placebo group, adjuvant NAC significantly decreased serum levels of MDA and, ICAM-1, NSE, NO, and IL-6 and significantly increased serum levels of SOD, GPx, and TTG.
Discussion
Our study for the first time evaluated the influence of oral NAC treatment on clinical outcomes of patients with acute ischemic stroke in a randomized, double-blind, and placebo-controlled study. The key findings of the present study were as follows: oral NAC administered early after an acute ischemic stroke is associated with a better outcome profile in terms of acute neurological deficit and disability grade compared to placebo. Next, ameliorative effects of NAC on OTS and inflammatory status of the acute phase of stroke demonstrated NAC may improve the neurological outcomes of patients with stroke at least in part by its antioxidant and anti-inflammatory effects.
Although reperfusion therapy with recombinant tissue plasminogen activator (rt-PA) is considered the cornerstone of treatment for acute ischemic stroke, due to its narrow treatment time window (within 4.5 h of the onset of ischemia)44 and the number of contraindications for thrombolysis, only a limited fraction of all stroke patients are treated with this treatment.45 Thus, in the past several years, attempts have been made to develop new pharmacological treatment modalities for acute ischemic stroke.
One candidate molecule that may be effective in the treatment of stroke is NAC. Previously, in several experimental studies, potential neuroprotective effects of NAC in cerebral ischemia have been investigated. In this respect, in a study that was conducted in a rat model of experimental stroke, intraperitoneally administration of NAC (150 mg/kg) at the time of reperfusion followed by another dose of NAC 6 hr later resulted in a significant reduction in an infarct area and an improvement in neurologic scores.46 In another study, the influence of individual and combination treatment with pramipexole and NAC, as two neuroprotective agents, on global cerebral ischemia/reperfusion injury model in rats was investigated. According to the results of this study, the combination treatment was more able to improve the histopathological parameter and the oxidative stress parameters when compared to the individual treatment.47 Sekhon et al also in their study showed preadministration of NAC by protecting cells against free radical damage can attenuate focal cerebral ischemia in an animal model of transient focal cerebral ischemia.48 Similar to these findings, results of another experimental study revealed administration of ceftriaxone (200 mg/kg) or NAC (150 mg/kg) for 5 days before focal cerebral ischemia by modulating of glutamate transporter expression and normalizing extracellular glutamate concentrations can significantly limit stroke-related damage and increase brain tolerance to ischemia.49 In a similar vein, Cuzzocrea et al in an experimental study in Mongolian gerbils subjected to cerebral ischemia showed early treatment with NAC (20 mg/kg 30 min before reperfusion and 1, 2, and 6 h after reperfusion) can reduce brain injury induced by transient cerebral ischemia.33 However, despite promising results with NAC treatment in animal models of cerebral ischemia, no data are yet available on the use of NAC in acute ischemic stroke patients.
NAC via multiple pathways may act in the brain following stroke. Unlike other organs, the brain for many reasons including, high concentrations of peroxidizable lipids, low levels of protective antioxidants, high oxygen consumption, and high levels of iron is vulnerable to ROS increases. Excessive ROS generation after stroke whose effects cannot be balanced by endogenous antioxidants can cause excessive neuronal cell damage in ischemia.50 So, it might be suspected that supplementation with exogenous antioxidants acts as a possible way to protect brain tissue against oxidative damage. One of the key mechanisms of NAC is its role in oxidative homeostasis. NAC through at least three different mechanisms can exert antioxidant activity. First, NAC as a precursor of cysteine increases plasma cysteine levels that finally involve a subsequent increase in intracellular glutathione. Second, as a thiol compound, NAC shows direct antioxidant properties toward some radical and non-radical oxidants. Third, by breaking thiolated proteins and releasing free thiols, NAC can regulate the redox state.51 Beneficial effects of NAC on oxidative stress biomarkers in various pathological conditions such as pneumonia,26 rheumatoid arthritis,27 diabetes mellitus,52 and myocardial infarction53 have been reported in previous studies. NAC’s influence on oxidative stress biomarkers during brain injury also has been investigated in some experimental studies. In this regard, in one study in an animal model of Alzheimer’s disease, NAC administration inhibited lipid peroxidation and increased the activity of glutathione reductase in brain tissue.54 Similarly, in another study treatment with NAC was associated with increased glutathione levels in the brain tissue of animals.55
Elevated ROS production through depletion of intra-platelet antioxidant content and reduced synthesis and bioavailability of anti-thrombotic nitric oxide (NO) contributes to the hyperaggregability of platelets.56 Moreover, since the production of endothelium-derived NO is an essential component of the maintenance of vascular tone, oxidative stress through alterations of the NO metabolism can exacerbate cerebral vascular ischemia.57 In vitro, NAC at therapeutically relevant concentrations (10–100 micro-molar) by increasing intraplatelet glutathione levels has exhibited inhibitory effects on thrombin and adenosine diphosphate-induced platelet aggregation.58 In this regard, the previous study in a mouse model of diabetes showed NAC treatment can attenuate platelet activation and thrombus formation.32 In a similar vein, results of one study in diabetic patients showed oral NAC therapy probably by normalization of intraplatelet GSH, coupled with a reduction in platelet–monocyte conjugation can reduce atherothrombotic risk in type 2 diabetes.59 The beneficial effects of NAC treatment on improving endothelial function in some animal and human studies have been also reported.60,61 Moreover, recently some preliminary evidence has been raised on the thrombolytic effects of NAC. In this regard, in experimental models of thrombotic stroke, NAC administration promoted lysis of arterial thrombi that were resistant to conventional approaches such as recombinant tissue-type plasminogen activator, direct thrombin inhibitors, and antiplatelet treatments.62 It is hypothesized that NAC by reducing the disulfide bonds inside multimeric proteins, such as von Willebrand factor (VWF), inhibited vWF-dependent platelet aggregation and collagen binding in thrombotic conditions in which excessive vWF-mediated cell adhesion plays a causative role.63
Extensive research has identified inflammatory mechanisms as another key contributory pathway underlying brain injury soon after the onset of ischemia. Once neuro-inflammation happens, it enhances the release of a number of pro-inflammatory mediators such as cytokines, chemokines, cell adhesion molecules (CAMs), and matrix metalloproteinases (MMPs) in the brain that can exacerbate neuronal damage and death.21 Thus, antagonizing the activity of these pro-inflammatory mediators can be an attractive therapeutic avenue in the pharmacological treatment of ischemic stroke. Another potential mechanism of action of NAC in stroke treatment stems from its anti-inflammatory properties. It is found that NAC through inhibitory effects on NF-κB which has a cardinal role in regulation and expression of stress response genes under inflammatory and oxidative conditions can exert its anti-inflammatory effects. Indeed, since oxidative stress is an important inducer of NF-κB, NAC by modulating oxidative stress can suppress NF-κB activation and subsequent cytokine production.64 Ameliorative effects of NAC treatment on plasma levels of pro-inflammatory cytokines, such as TNFα, IL-6, and IL-1β have been reported in humans subjected to hemodialysis or septic shock.65,66 Further, NAC treatment following traumatic brain injury or focal cerebral ischemia in rodents has demonstrated ameliorative effects on pro-inflammatory molecules such as TNF-α, IL-1β, and NF-κB.46,67 Inhibitory effects of NAC on the expression of adhesion molecule such as ICAM-1 (Intercellular Adhesion Molecule 1) and VCAM-1 (Vascular Cell Adhesion Molecule 1) that by recruitment of circulating leukocytes into the areas of inflammation play an important role in inflammation and progression of ischemic injury after acute stroke68 has been also reported in some experimental studies. It seems inhibitory effects of NAC on adhesion molecule are mediated via suppression of NF-κB expression which has an important role in the regulation of redox-sensitive inflammation-related cytokines responsible for the expression of these molecules.69,70
The mitochondria-protective effect is another mechanism that NAC through it can protect brain tissue from oxidative insults during cerebral ischemia.71 NAC probably by protecting mitochondrial respiratory chain proteins from oxidative damage can maintain mitochondrial bioenergetic capacity.72 Such findings are in accord with those experimental studies which showed NAC treatment can effectively restore mitochondrial dysfunction in brain tissue.73,74
Interruption or severe reduction of blood flow in the affected region of the brain results in an excessive release of excitatory amino acids particularly glutamate to the extracellular compartment that by over-activation of glutamate receptors induce or aggravate neuronal damage.75 Preliminary research indicates NAC probably via the cysteine-glutamate exchange (Xc-system) that is essential components of glutamate homeostasis can show regulatory effects on the amount of glutamate present in the extracellular space.76,77
During stroke attacks, over-activation and expression of MMPs, a group of metal-dependent endopeptidases that degrade all components of the extracellular matrix, in ischemic stroke have been associated with various complications including excitotoxicity, neuronal damage, apoptosis, BBB opening leading to cerebral edema, and hemorrhagic transformation.78 Therefore, MMP inhibitors may be also considered as a potential therapeutic strategy in the management of acute ischemic stroke. Given that activation of MMPs is dependent on the modification of the cysteine residue and reaction of ROS with thiol groups can activate these protolithic proteins,79 it is thought NAC with abundant cysteine residues in its structures may have inhibitory effects on MMP-9 production and activation.80,81
Taken together, this evidence explains NAC at least in theory via multiple mechanisms of action can exert protective effects on brain tissue during ischemic attacks. Our data also support that early use of NAC in stroke pationts is able to improve the clinical outcomes of these patients. Moreover, our results raise the intriguing possibility that that these beneficial effects of NAC are mediated at least in part by its antioxidant and anti-inflammatory effects. One aspect that has made NAC as a particularly attractive drug for this purpose is its documented safety profile. NAC at doses as high as 500 mg/kg has been employed to treat acute acetaminophen toxicity.82 Although severe and in some instances, life-threatening anaphylactoid reactions have been reported by intravenous administration of NAC, its oral administration is relatively safe. Mild gastrointestinal disturbance such as nausea, vomiting, and heartburn are the only adverse effects that have been reported by oral administration of NAC.83 Therefore, this margin of safety and excellent efficacy make oral NAC a potentially useful therapy in the management of ischemic stroke.
Despite the novelty of the findings, several limitations warn against the overgeneralization of the results. The first limitation of this study is the relatively small number of subjects. More studies with larger sample sizes are warranted. The second limitation of the present study is that only two samples were taken from each patient at baseline and 72 h after treatment. Therefore, the quality of the data does not allow a deeper inspection and understanding of the biochemical changes of biomarkers during acute ischemic stroke. Third, the patients in our study received NAC treatment orally; IV NAC treatment may have been different and be more effective. Last but not least, the time window of treatment in our study was less than 24 hours after onset of symptoms post-stroke; earlier treatment may have different efficacy.
Conclusion
In conclusion, our study adds to the limited clinical database that NAC, a natural supplement with proven antioxidant and anti-inflammatory properties, can improve the clinical outcomes after ischemic stroke at least in part by improving oxidative balance and suppressing inflammatory biomarkers. We expect that the result of this study may stimulate further investigation of NAC use as a therapy for ischemic stroke.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request up to 2 years after publication.
Acknowledgments
This study was a part of a greater study that was supported by Vice-Chancellor of Research and Technology of Hamadan University of Medical Sciences, Hamadan, Iran (No: 9705022453 and 970211698). The authors thank all patients for helping and participating in the study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zandieh A, Kahaki ZZ, Sadeghian H, et al. A simple risk score for early ischemic stroke mortality derived from National Institutes of Health Stroke Scale: a discriminant analysis. Clin Neurol Neurosurg. 2013;115(7):1036–1039. doi:10.1016/j.clineuro.2012.10.034
2. Caplan LR, Hon FK. Clinical diagnosis of patients with cerebrovascular disease. Prim Care. 2004;31(1):95–109. doi:10.1016/S0095-4543(03)00118-0
3. Caplan L. basic pathology, anatomy, and pathophysiology of stroke. Caplan stroke. 2009:22–63.
4. Kunz A, Iadecola C. Cerebral vascular dysregulation in the ischemic brain. Handb Clin Neurol. 2009;92:283–305.
5. Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: an overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 2010;17(3):197–218. doi:10.1016/j.pathophys.2009.12.001
6. Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22(9):391–397. doi:10.1016/S0166-2236(99)01401-0
7. Bansal S, Sangha KS, Khatri P. Drug treatment of acute ischemic stroke. Am J Cardiovasc Drugs. 2013;13(1):57–69. doi:10.1007/s40256-013-0007-6
8. Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int. 2013;62(5):712–718. doi:10.1016/j.neuint.2012.11.009
9. Hepel M, Andreescu S. Oxidative Stress and Human Health. Oxidative Stress: Diagnostics, Prevention, and Therapy Volume 2. ACS Symposium Series. 1200:American Chemical Society; 2015.p.1–33. doi:10.1021/bk-2015-1200.ch001
10. Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27(6):1124–1129. doi:10.1161/01.STR.27.6.1124
11. Chang CY, Lai YC, Cheng TJ, Lau MT, Hu ML. Plasma levels of antioxidant vitamins, selenium, total sulfhydryl groups and oxidative products in ischemic-stroke patients as compared to matched controls in Taiwan. Free Radic Res. 1998;28(1):15–24. doi:10.3109/10715769809097872
12. Cherubini A, Polidori Maria C, Bregnocchi M, et al. Antioxidant profile and early outcome in stroke patients. Stroke. 2000;31(10):2295–2300. doi:10.1161/01.STR.31.10.2295
13. Polidori MC, Cherubini A, Stahl W, Senin U, Sies H, Mecocci P. Plasma carotenoid and malondialdehyde levels in ischemic stroke patients: relationship to early outcome. Free Radic Res. 2002;36(3):265–268. doi:10.1080/10715760290019273
14. Uno M, Kitazato KT, Nishi K, Itabe H, Nagahiro S. Raised plasma oxidised LDL in acute cerebral infarction. J Neurol Neurosurg Psychiatry. 2003;74(3):312. doi:10.1136/jnnp.74.3.312
15. Liu H, Uno M, Kitazato KT, et al. Peripheral oxidative biomarkers constitute a valuable indicator of the severity of oxidative brain damage in acute cerebral infarction. Brain Res. 2004;1025(1–2):43–50. doi:10.1016/j.brainres.2004.07.071
16. De Keyser J, De Klippel N, Merkx H, Vervaeck M, Herroelen L. Serum concentrations of vitamins A and E and early outcome after ischaemic stroke. Lancet. 1992;339(8809):1562–1565. doi:10.1016/0140-6736(92)91830-2
17. Spranger M, Krempien S, Schwab S, Donneberg S, Hacke W. Superoxide dismutase activity in serum of patients with acute cerebral ischemic injury. Stroke. 1997;28(12):2425–2428.
18. Gariballa SE, Hutchin TP, Sinclair AJ. Antioxidant capacity after acute ischaemic stroke. QJM. 2002;95(10):685–690. doi:10.1093/qjmed/95.10.685
19. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17(7):796–808. doi:10.1038/nm.2399
20. Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62(2):127–136. doi:10.1093/jnen/62.2.127
21. Jin R, Liu L, Zhang S, Nanda A, Li G. Role of inflammation and its mediators in acute ischemic stroke. J Cardiovasc Transl Res. 2013;6(5):834–851. doi:10.1007/s12265-013-9508-6
22. Smith CJ, Emsley HC, Gavin CM, et al. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi:10.1186/1471-2377-4-2
23. Intiso D, Zarrelli MM, Lagioia G, et al. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke patients. Neurol Sci. 2004;24(6):390–396. doi:10.1007/s10072-003-0194-z
24. Pei Y, Liu H, Yang Y, Yang Y, Jiao Y. Biological activities and potential oral applications of n-acetylcysteine: progress and prospects. Stroke. 2018;2018:2835787.
25. Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107(2):135–142. doi:10.1172/JCI11914
26. Zhang Q, Ju Y, Ma Y, Wang T. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia: a randomized controlled trial. Medicine (Baltimore). 2018;97(45):e13087. doi:10.1097/MD.0000000000013087
27. Hashemi G, Mirjalili M, Basiri Z, et al. A pilot study to evaluate the effects of oral N-Acetyl cysteine on inflammatory and oxidative stress biomarkers in rheumatoid arthritis. Curr Rheumatol Rev. 2019;15(3):246–253. doi:10.2174/1573403X14666180926100811
28. Kilciksiz S, Demirel C, Erdal N, et al. The effect of N-acetylcysteine on biomarkers for radiation-induced oxidative damage in a rat model. Acta Med Okayama. 2008;62(6):403–409. doi:10.18926/AMO/30946
29. Sandhir R, Sood A, Mehrotra A, Kamboj SS. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener Dis. 2012;9(3):145–157. doi:10.1159/000334273
30. Tchantchou F, Graves M, Rogers E, Ortiz D, Shea TB. N-acetyl cysteine alleviates oxidative damage to central nervous system of ApoE-deficient mice following folate and vitamin E-deficiency. J Alzheimers Dis. 2005;7(2):
31. Pan J, Xiao Q, Sheng CY, et al. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem Int. 2009;54(7):418–425. doi:10.1016/j.neuint.2009.01.013
32. Wang B, Yee AT, Stokes KY. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox Biol. 2018;14:218–228. doi:10.1016/j.redox.2017.09.005
33. Cuzzocrea S, Mazzon E, Costantino G, et al. Beneficial effects of n-acetylcysteine on ischaemic brain injury. Br J Pharmacol. 2000;130(6):1219–1226. doi:10.1038/sj.bjp.0703421
34. Powers William J, Rabinstein Alejandro A, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 Update to the 2018 guidelines for the early management of acute ischemic stroke: a Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2019;50(12):e344–e418. doi:10.1161/STR.0000000000000211
35. Hoffer M, Balaban CD, Slade M, Tsao J, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by n-acetyl cysteine: a double-blind, placebo controlled study. PLoS One. 2013;8:e54163. doi:10.1371/journal.pone.0054163
36. Bavarsad Shahripour R, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4(2):108–122. doi:10.1002/brb3.208
37. Perera M, Ma H, Arakawa S, et al. Inflammation following stroke. J Clin Neurosci. 2006;13:1–8. doi:10.1016/j.jocn.2005.07.005
38. Muir KW, Weir CJ, Murray GD, Povey C, Lees KR. Comparison of neurological scales and scoring systems for acute stroke prognosis. Stroke. 1996;27(10):1817–1820. doi:10.1161/01.STR.27.10.1817
39. van Swieten JC, Koudstaal PJ, Visser MC, Schouten HJ, van Gijn J. Interobserver agreement for the assessment of handicap in stroke patients. Stroke. 1988;19(5):604–607. doi:10.1161/01.STR.19.5.604
40. Benzie IF, Szeto YT. Total antioxidant capacity of teas by the ferric reducing/antioxidant power assay. J Agric Food Chem. 1999;47(2):633–636. doi:10.1021/jf9807768
41. Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. doi:10.1016/0003-9861(59)90090-6
42. Botsoglou NA, Fletouris DJ, Papageorgiou GE, Vassilopoulos VN, Mantis AJ, Trakatellis AG. Rapid, sensitive, and specific thiobarbituric acid method for measuring lipid peroxidation in animal tissue, food, and feedstuff samples. J Agric Food Chem. 1994;42(9):1931–1937. doi:10.1021/jf00045a019
43. Unnebrink K, Windeler J. Intention-to-treat: methods for dealing with missing values in clinical trials of progressively deteriorating diseases. Stat Med. 2001;20(24):3931–3946. doi:10.1002/sim.1149
44. Li B-H, Ding X, Yin Y-W, et al. Meta-analysis of clinical outcomes of intravenous recombinant tissue plasminogen activator for acute ischemic stroke: within 3 hours versus 3–4.5 hours. Curr Med Res Opin. 2013;29(9):1105–1114. doi:10.1185/03007995.2013.818533
45. Adeoye O, Hornung R, Khatri P, Kleindorfer D. Recombinant tissue-type plasminogen activator use for ischemic stroke in the United States: a doubling of treatment rates over the course of 5 years. Stroke. 2011;42(7):1952–1955. doi:10.1161/STROKEAHA.110.612358
46. Khan M, Sekhon B, Jatana M, et al. Administration of N‐acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res. 2004;76(4):519–527. doi:10.1002/jnr.20087
47. Danduga RCSR, Reddy DS, Seshadri SM, Has KSS, Kumar KP. Effect of combination therapy with pramipexole and n-acetylcysteine on global cerebral ischemic reperfusion injury in rats. Iran J Basic Med Sci. 2018;21(6):569.
48. Sekhon B, Sekhon C, Khan M, et al. N‐acetyl cysteine protects against injury in animal model of transient focal cerebral ischemia. J Neurochem. 2002;81:101–105. doi:10.1046/j.1471-4159.81.s1.40_12.x
49. Krzyżanowska W, Pomierny B, Bystrowska B, et al. Ceftriaxone-and N-acetylcysteine-induced brain tolerance to ischemia: influence on glutamate levels in focal cerebral ischemia. PLoS One. 2017;12:10. doi:10.1371/journal.pone.0186243
50. Rodrigo R, Fernández-Gajardo R, Gutiérrez R, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12(5):698–714.
51. Aldini G, Altomare A, Baron G, et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52(7):751–762. doi:10.1080/10715762.2018.1468564
52. Ribeiro G, Roehrs M, Bairros A, et al. N-acetylcysteine on oxidative damage in diabetic rats. Drug Chem Toxicol. 2011;34(4):467–474. doi:10.3109/01480545.2011.564179
53. Yesilbursa D, Serdar A, Senturk T, Serdar Z, Sağ S, Cordan J. Effect of N-acetylcysteine on oxidative stress and ventricular function in patients with myocardial infarction. Heart Vessels. 2006;21(1):33–37. doi:10.1007/s00380-005-0854-4
54. Huang Q, Aluise CD, Joshi G, et al. Potential in vivo amelioration by N‐acetyl‐L‐cysteine of oxidative stress in brain in human double mutant APP/PS‐1 knock‐in mice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010;88(12):2618–2629. doi:10.1002/jnr.22422
55. Yin J, Ren W, Yang G, et al. l-Cysteine metabolism and its nutritional implications. Mol Nutr Food Res. 2016;60(1):134–146. doi:10.1002/mnfr.201500031
56. Freedman JE. Oxidative stress and platelets. Arterioscler Thromb Vasc Biol. 2008;28(3):s11–s16. doi:10.1161/ATVBAHA.107.159178
57. Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci (Landmark Ed). 2011;16:1733–1745. doi:10.2741/3816
58. Gibson KR, Winterburn TJ, Barrett F, Sharma S, MacRury SM, Megson IL. Therapeutic potential of N-acetylcysteine as an antiplatelet agent in patients with type-2 diabetes. Cardiovasc Diabetol. 2011;10(1):43. doi:10.1186/1475-2840-10-43
59. Treweeke A, Winterburn T, Mackenzie I, et al. N-Acetylcysteine inhibits platelet–monocyte conjugation in patients with type 2 diabetes with depleted intraplatelet glutathione: a randomised controlled trial. Diabetologia. 2012;55(11):2920–2928. doi:10.1007/s00125-012-2685-z
60. Pieper GM, Siebeneich W. Oral administration of the antioxidant, N-acetylcysteine, abrogates diabetes-induced endothelial dysfunction. J Cardiovasc Pharmacol. 1998;32(1):101–105. doi:10.1097/00005344-199807000-00016
61. Sahin G, Yalcin AU, Akcar N. Effect of N-acetylcysteine on endothelial dysfunction in dialysis patients. Blood Purif. 2007;25(4):309–315. doi:10.1159/000106103
62. Martinez de Lizarrondo S, Gakuba C, Herbig BA, et al. Potent thrombolytic effect of N-acetylcysteine on arterial thrombi. Circulation. 2017;136(7):646–660. doi:10.1161/CIRCULATIONAHA.117.027290
63. Chen J, Reheman A, Gushiken FC, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. 2011;121(2):593–603. doi:10.1172/JCI41062
64. Samuni Y, Goldstein S, Dean OM, Berk M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830(8):4117–4129. doi:10.1016/j.bbagen.2013.04.016
65. Emet S, Memiş D, Pamukçu Z. The influence of N-acetyl-L-cystein infusion on cytokine levels and gastric intramucosal pH during severe sepsis. Crit Care. 2004;8(4):R172. doi:10.1186/cc2866
66. Nascimento MM, Suliman ME, Silva M, et al. Effect of oral N-acetylcysteine treatment on plasma inflammatory and oxidative stress markers in peritoneal dialysis patients: a placebo-controlled study. Perit Dial Int. 2010;30(3):336–342. doi:10.3747/pdi.2009.00073
67. Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm. 2008;2008:1–8. doi:10.1155/2008/716458
68. Frijns C, Kappelle L. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33(8):2115–2122. doi:10.1161/01.STR.0000021902.33129.69
69. Spiecker M, Darius H, Kaboth K, Hübner F, Liao JK. Differential regulation of endothelial cell adhesion molecule expression by nitric oxide donors and antioxidants. J Leukoc Biol. 1998;63(6):732–739. doi:10.1002/jlb.63.6.732
70. Radomska-Lesniewska D, Sadowska A, Van Overveld F, Demkow U, Zielinski J, De Backer W. Influence of** N**-acetylcysteine on ICAM-1 expression and il-8 release from endothelial and epithelial cells. J Physiol Pharmacol. 2007;57(S4):325–334.
71. Fries GR, Kapczinski F. N-acetylcysteine as a mitochondrial enhancer: a new class of psychoactive drugs? Braz J Psychiatry. 2011;33(4):321–322. doi:10.1590/S1516-44462011000400003
72. Basha RH, Priscilla DH. An in vivo and in vitro study on the protective effects of N-acetylcysteine on mitochondrial dysfunction in isoproterenol treated myocardial infarcted rats. Exp Toxicol Pathol. 2013;65(1–2):7–14. doi:10.1016/j.etp.2011.05.002
73. Xiong Y, Peterson P, Lee C. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma. 1999;16(11):1067–1082. doi:10.1089/neu.1999.16.1067
74. Cocco T, Sgobbo P, Clemente M, et al. Tissue-specific changes of mitochondrial functions in aged rats: effect of a long-term dietary treatment with N-acetylcysteine. Free Radic Biol Med. 2005;38(6):796–805. doi:10.1016/j.freeradbiomed.2004.11.034
75. Lai TW, Zhang S, Wang YT. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol. 2014;115:157–188. doi:10.1016/j.pneurobio.2013.11.006
76. Dean O, Giorlando F, Berk M. N-acetylcysteine in psychiatry: current therapeutic evidence and potential mechanisms of action. J Psychiatry Neurosci. 2011;36(2):78. doi:10.1503/jpn.100057
77. Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25(27):6389–6393. doi:10.1523/JNEUROSCI.1007-05.2005
78. Lakhan SE, Kirchgessner A, Tepper D, Aidan L. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. doi:10.3389/fneur.2013.00032
79. Yu F, Kamada H, Niizuma K, Endo H, Chan PH. Induction of mmp-9 expression and endothelial injury by oxidative stress after spinal cord injury. J Neurotrauma. 2008;25(3):184–195. doi:10.1089/neu.2007.0438
80. Ramaesh T, Ramaesh K, Riley S, West J, Dhillon B. Effects of N-acetylcysteine on matrix metalloproteinase-9 secretion and cell migration of human corneal epithelial cells. Eye. 2012;26(8):1138–1144. doi:10.1038/eye.2012.135
81. Voronkina I, Vakhromova E, Kirpichnikova K, Smagina L, Gamaley I. Matrix metalloproteinase activity in transformed cells exposed to an antioxidant. Cell Tissue Biol. 2015;9(1):16–23. doi:10.1134/S1990519X15010113
82. Heard KJ. Acetylcysteine for acetaminophen poisoning. New Engl J Med. 2008;359(3):285–292. doi:10.1056/NEJMct0708278
83. De Rosa S, Zaretsky M, Dubs J, et al. N‐acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest. 2000;30(10):915–929. doi:10.1046/j.1365-2362.2000.00736.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.