Back to Journals » International Journal of Nanomedicine » Volume 17
Everolimus-Loaded Reconstituted High-Density Lipoprotein Prepared by a Novel Dual Centrifugation Approach for Anti-Atherosclerotic Therapy
Authors Deuringer B, Härdtner C ![]() , Krebs K, Thomann R
, Krebs K, Thomann R ![]() , Holzer M, Hilgendorf I, Süss R
, Holzer M, Hilgendorf I, Süss R ![]()
Received 7 July 2022
Accepted for publication 3 October 2022
Published 31 October 2022 Volume 2022:17 Pages 5081—5097
DOI https://doi.org/10.2147/IJN.S381483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Benedikt Deuringer,1 Carmen Härdtner,2 Katja Krebs,2 Ralf Thomann,3 Martin Holzer,1 Ingo Hilgendorf,2,4,* Regine Süss1,*
1Department of Pharmaceutical Technology and Biopharmacy, Institute of Pharmaceutical Sciences, University of Freiburg, Freiburg, 79104, Germany; 2Department of Cardiology and Angiology, University Heart Center Freiburg-Bad Krozingen and Faculty of Medicine, University of Freiburg, Freiburg, 79106, Germany; 3FMF Materials Research Center, University of Freiburg, Freiburg, 79104, Germany; 4Institute for Experimental Cardiovascular Medicine, University Heart Center Freiburg-Bad Krozingen and Faculty of Medicine, University of Freiburg, Freiburg, 79110, Germany
*These authors contributed equally to this work
Correspondence: Benedikt Deuringer, Pharmaceutical Technology and Biopharmacy, Sonnenstraße 5, Freiburg, 79104, Germany, Tel +49 761 203 6329, Fax +49 761 203 6326, Email [email protected]
Purpose: The conventional techniques for the preparation of reconstituted high-density lipoprotein (rHDL) are hampered by long process times, the need for large amounts of starting material, and harsh preparation conditions. Here, we present a novel rHDL preparation method to overcome these challenges. Furthermore, we propose a dual mode of action for rHDL loaded with the immunosuppressant drug everolimus (Eve-rHDL) in the context of atherosclerosis and cardiovascular disease.
Methods: We use dual centrifugation for rHDL nanoparticle preparation and characterize the physicochemical properties by NS-TEM, N-PAGE, DLS, AF4, and HPLC. In addition, we determine the biological efficacy in human and murine cell culture with regard to cellular uptake, cholesterol efflux, and proliferation.
Results: We confirm the characteristic particle size of 10 nm, discoidal morphology, and chemical composition of the rHDL preparations and identify dual centrifugation as an ideal method for cost-effective aseptic rHDL manufacturing. rHDL can be prepared in approx. 1.5 h with batch sizes as little as 89 μL. Moreover, we demonstrate the cholesterol efflux capacity and anti-proliferative activity of Eve-rHDL in vitro. The anti-proliferative effects were comparable to free Eve, thus confirming the suitability of rHDL as a capable drug delivery vehicle.
Conclusion: Eve-rHDL shows great efficacy in vitro and may further be employed to target atherosclerotic plaques in vivo. Highly effective anti-atherosclerotic therapy might be feasible by reducing both inflammatory- and lipid burden of the plaques. Dual centrifugation is an ideal technique for the efficient application of the rHDL platform in cardiovascular disease and beyond.
Keywords: atherosclerosis, cholesterol efflux, drug delivery, everolimus, proliferation, protein-conjugate
Introduction
Cardiovascular disease (CVD) is a leading cause of morbidity and mortality worldwide. One of the underlying principles of CVD is the process of atherogenesis, which is associated with a retention of cholesterol-containing low-density lipoprotein in the intima layer of arteries and an inflammatory response.1 Standard treatment regimens comprise lifestyle management, control of vascular inflammation, and lipoprotein modification.2–4 High-density lipoprotein (HDL) can be employed to stabilize atherosclerotic plaques by the reverse cholesterol transport (RCT) mechanism. Cholesterol-efflux from foam cell macrophages is mediated by the interaction of HDL with cellular receptors such as scavenger receptor B1 and ATP-binding cassette transporters A1 and G1.5 The development of HDL-mimicking nanoparticles, ie, reconstituted HDL (rHDL), is therefore a promising option for plaque modification and the reduction of atherosclerotic burden.6
Small, discoidal rHDL is an 8 to 12 nm sized particle consisting of lipids and amphiphilic proteins such as apolipoprotein A1 (ApoA1). The proteins circumscribe the perimeter of the rHDL nanodiscs and stabilize the lipids in a planar bilayer.7 Due to the large capacity for hydrophobic cargo, rHDL can be exploited as a capable delivery vehicle for hydrophobic active pharmaceutical ingredients (APIs), which offers significant benefits in comparison to other delivery systems such as liposomes or polymeric nanoparticles. As rHDL resembles endogenous HDL, it can escape elimination by the mononuclear phagocytic system and protect incorporated APIs from degradation. Due to the extremely small size, the nanoparticles can accumulate in leaky microvasculature, congruent with the enhanced permeability and retention effect. Moreover, rHDL possesses an intrinsic targeting effect to atherosclerotic lesions where a direct exchange of hydrophobic cargo is possible.8 In this way, a dual mode of action is conceivable: reduction of plaque lipid content by RCT and targeted API delivery. This is of particular interest since plaque-destabilizing inflammation has been reported to be highly reliant on local macrophage proliferation.9 Inhibitors of the mammalian target of rapamycin (mTOR), such as everolimus (Eve), can reduce immune cell proliferation and even deplete atherosclerotic plaques of macrophages by autophagy.10 Thus, the loading of rHDL with Eve, ie, preparation of Eve-rHDL, has high therapeutic potential with the proposed synergistic effects of cardioprotective rHDL and immunosuppressive Eve. In addition, a high local concentration of Eve might be feasible without the dose-limiting side effects of systemic infusions.
The reconstitution of rHDL from lipid components by addition of ApoA1 was previously described by Jonas (1986).11 An inverse relationship exists between the lipid–ApoA1 reaction rates and the extent of lipid–lipid molecular interactions, ie, lipid vesicle stability. Thus, short-chain lipids such as DMPC (1,2-Dimyristoyl-sn-glycero-3-phosphocholine) react more readily than longer chain lipids due to less hydrophobic chain–chain interactions. The addition of MHPC (1-Myristoyl-2-hydroxy-sn-glycero-3-phosphocholine) might further increase reaction rates since lysolipids have been reported to form defect structures in vesicles at the gel to liquid-crystalline phase transition temperature (Tm).12 In general, defects in the lipid lattice occur most often at the Tm, and reconstitution methods are usually conducted at this temperature. While MHPC might affect the melting behavior of plain vesicles by altering the lipid packing, these effects might be resolved by complexation with protein. For example, Tall et al (1977) have shown that HDL does not display thermal transitions at the Tm of its lipid cargo.13 Changes in rHDL morphology as a function of temperature are thus unlikely. In fact, the morphology of the prepared rHDL particles depends rather on the lipid–protein ratio. A molar ratio of 100 typically leads to small, discoidal particles with a protein stoichiometry of 2, whereas higher amounts of lipid generate larger, vesicular structures with ≥3 protein molecules per particle.14
rHDL can be prepared by various published methods, such as incubation of preformed lipid vesicles with ApoA1, cosonication, detergent-based synthesis, and microfluidics.11,15–18 However, important aspects in formulation development are complicated in these procedures. Especially in early development stages, cost-effective screening is critical, and nanoparticle preparation has to be possible in both micro-scale and large quantities. With regard to the established techniques mentioned above, batch sizes usually comprise several hundred microliters, demanding high amounts of starting material or limiting product concentration. Furthermore, proteins or APIs are prone to degradation by harsh homogenization procedures such as cosonication and in vivo experiments usually demand aseptic manufacturing and extensive removal of potentially hemolytic detergents used in detergent-based synthesis. Energy transfer-based methods, such as cosonication, rely on the exact control of the process parameters for maximum efficiency and are thus difficult to scale-up. Consequently, rHDL currently investigated in clinical studies (CSL112) is prepared by the cholate dialysis method.6,19 Yet, despite extensive diafiltration, the amount of detergent remaining in CSL112 is still approx. 0.03 g per g of ApoA1.19 To overcome these process-related limitations, we present a novel, detergent-free approach for the preparation of rHDL or API-loaded rHDL based on dual centrifugation (DC). DC is an in-vial homogenization technique, which is effectively employed for the nanomilling of poorly soluble drugs or the preparation of nanoemulsions and liposomes.20–22 The DC device features a special rotor with a primary and secondary axis of rotation generating centrifugal force and a constant variation of vial orientation. Samples collide with the vial wall in high frequencies, which conveys efficient particle disruption. Ceramic ZY-beads can be employed to further increase the homogenization effect. During the preparation, the vesicles are especially fragile and should readily react with ApoA1. The process might therefore be categorized as facilitated self-assembly. Since the homogenization takes place in closed containers, aseptic manufacturing is easily achievable, and sample loss is minimized. Samples can be prepared in micro-scale with multiple samples per run, making DC a cost-effective screening tool with straightforward scale-up options. It can be regarded as a gentle and robust process, which, due to not requiring potentially hemolytic detergents, is applicable for in vivo applications and bench-to-bedside approaches.
In this work, we use DC to prepare rHDL and Eve-rHDL and analyze the physicochemical properties of the nanoparticles. We show the resemblance to conventionally prepared rHDL and demonstrate excellent preparation efficiency. Furthermore, we evaluate the biological effectiveness of the formulation in regard to the encapsulated API and the rHDL cholesterol efflux capacity in macrophage cell culture.
Materials and Methods
Material
1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was obtained from Lipoid (Ludwigshafen, DE). 1-Myristoyl-2-hydroxy-sn-glycero-3-phosphocholine (MHPC) was purchased from Avanti Polar Lipids (Alabaster, US). Everolimus was purchased from MedChemExpress (Monmouth Junction, US). Ammonium peroxodisulfate (APS) and apolipoprotein A1 solution were purchased from Merck (Darmstadt, DE). Native HDL was purchased from Lee BioSolutions (Maryland Heights, US). Acetonitrile, ammonium carbonate, and methanol (HiPerSolv) were purchased from VWR (Darmstadt, DE). Bromophenol blue, glycine, HEPES, sodium chloride, sodium azide, sodium hydroxide, and trypan blue solution were purchased from Sigma-Aldrich (Steinheim, DE). Dimethyl sulfoxide, formic acid, glycerol, phosphotungstic acid hydrate, ROTI®Blue quick staining solution, TEMED, and TRIS were purchased from Carl Roth (Karlsruhe, DE). SiLibeads (0.3 to 0.4 mm, ZY-S) was purchased from Sigmund Lindner (Warmensteinach, DE). Two milliliter micro tubes for dual centrifugation were purchased from Sarstedt (Nümbrecht, DE). Four hundred mesh carbon film coated copper grids (5 to 6 nm) were purchased from Science Services (Munich, DE). Acrylamide 4K solution (40%) was purchased from AppliChem (Darmstadt, DE). DiI and DiO carbocyanine dyes, NativeMark™ protein standard, LysoTracker™ Deep Red, and Pierce™ formaldehyde solution (16%) were purchased from Thermo Fisher Scientific (Waltham, US). Vectashield® Vibrance™ Antifade mounting medium w/ DAPI was purchased from Vector Laboratories (Burlingame, US). Falcon® 8-well cell culture slides were purchased from Corning (Kaiserslautern, DE). Buffers and media for cell culture were purchased from Biochrom (Berlin, DE). Consumables for cell culture were purchased from Greiner Bio-One (Frickenhausen, DE).
Preparation of rHDL Nanoparticles
rHDL nanoparticles were prepared by dual centrifugation. Stock solutions of DMPC, MHPC, and Eve were prepared in methanol and added to 2 mL polypropylene reaction vials with 60 mg of 0.3 to 0.4 mm ZY-beads as homogenization aid for the dual centrifugation step. Mixing ratios were mMHPC/mDMPC=0.1 and mEve/mlipids=0.2 for Eve-rHDL with a lipid amount of mlipids=0.309 mg. If fluorescent labeling was desired, 1 mol% (in regard to the lipid amount) DiO or DiI were added to the mix. After removal of the organic solvent using an Eppendorf Concentrator plus evaporation centrifuge, the lipid film was incubated for 2 h in a high vacuum (approx. 15 mbar) and subsequently stored at −80 °C. The films were hydrated by addition of 89 µL ApoA1 in 10 mM ammonium bicarbonate buffer pH 7.4 in a mixing ratio of mApoA1/mlipids=0.4 (which corresponds to 108 lipid molecules per ApoA1 molecule) and a concentration of cApoA1=1.39 mg/mL. The reaction mix was subjected to a 30 min run in a Hettich ZentriMix 380R dual centrifuge at 2350 rpm and 15 °C, during which process temperatures of 24 °C, the Tm of DMPC, were reached. Afterwards, the samples were allowed to cool down during additional 30 min at 200 rpm and 4 °C. Potentially non-encapsulated, water-insoluble Eve was removed by centrifugation in an Eppendorf 5417R centrifuge at 10,000 × g and 4 °C for 15 min. The supernatant was collected and subsequently stored at −80 °C.
Negative-Stain Transmission Electron Microscopy (NS-TEM)
To introduce negative staining, the nanoparticles were mixed with an equal volume of a 0.22 µm filtered solution of 20 mg/mL phosphotungstic acid pH 7.4. Volumes of 3 µL of the mixture were cast on carbon-coated copper grids and allowed to attach for 20s. The samples were blotted with filter paper, air dried for 5 min, and the grids were stored at room temperature in the dark until evaluation using a Carl Zeiss LEO 912 Omega transmission electron microscope. The instrument was operated with an acceleration voltage of 120 kV, and images were taken with an Oxford Instruments Proscan HSC 2 camera. The maximum Feret diameter (dmax) was determined by manual evaluation of three micrographs (minimum particle count of 200 each) using the Carl Zeiss ZEN core 3.0 software.
Native Polyacrylamide Gel Electrophoresis (N-PAGE)
The samples were diluted with 10 mM ammonium carbonate buffer pH 7.4 to a concentration of 0.5 mg/mL (protein component mass) and were mixed 4+1 (v/v) with a sample buffer containing 312.5 mM TRIS, 50% (v/v) glycerol, and 0.5 g/l bromophenol blue, pH 6.8. Volumes of 5 µL were loaded onto native 8% polyacrylamide gels with 3.75% stacking gels, which were prepared in a Bio-Rad multicasting chamber using a standard gel casting protocol (consecutive combination of TRIS buffer, acrylamide solution, APS, and TEMED). Electrophoresis was performed for 135 min at 80 V using a Bio-Rad Mini-PROTEAN™ Tetra Cell electrophoresis system with a 25 mM TRIS, 192 mM glycine running buffer. Afterwards, the gels were stained with Coomassie®-staining solution. A calibration curve (2nd order polynomial fit) was generated from the size of each NativeMark™ protein standard (size according to Holzer et al (2017)23) and the respective distance from the bromophenol blue band. The analyte size was then calculated from the band distance accordingly.
Dynamic Light Scattering (DLS)
Size analysis was performed on a Malvern Zetasizer Nano ZS ZEN3600 using a ZEN2112 ultra low volume quartz batch cuvette. All measurements were carried out at a 173° scattering angle and 25 °C after a 2 min equilibration phase. For the sample optical properties, the preset values for protein samples with a refractive index of 1.450 and absorption of 0.001 were used. For the dispersant, the preset values for water with a viscosity of 0.8872 mPa∙s and a refractive index of 1.330 were selected. The sample viscosity was approximated accordingly to 0.8872 mPa∙s. The measurement position and attenuation settings were determined automatically by the Zetasizer software, and the distribution fit of the preset general-purpose analysis model was used to conduct size analysis.
Asymmetric-Flow Field-Flow Fractionation (AF4)
For rHDL separation and analysis, a Wyatt Eclipse AF4 system with Wyatt DAWN HELEOS II MALS detector (LS), Wyatt Optilab T-rEX refractive index detector (dRI), and Agilent 1260 Infinity variable wavelength detector (UV) set to 278 nm was employed. Additional hardware consisted of Agilent 1260 Infinity autosampler and quaternary pump. Separation was performed on a Wyatt Eclipse short channel with 490 µm wide-type spacer and 10 kDa regenerated cellulose membrane using a 0.1 µm filtered solution of 10 mM HEPES, 140 mM sodium chloride, 0.2 g/l sodium azide, pH 7.4 for sample elution.
All AF4 experiments were conducted at 25 °C with 10 µL injection volume, an inject flow (Vinj) of 0.2 mL/min, and detector flow (Vd) of 1.0 mL/min. After several focusing and injection steps with a focusing position at 30% channel length, sample elution was initiated after 21 min (Table 1). A linear cross-flow (Vx) gradient of 3.0 mL/min to 0.0 mL/min over 60 min with additional 30 min at 0.0 mL/min was used for sample fractionation.
 |
Table 1 AF4 Instrument Method |
High-Performance Liquid Chromatography (HPLC)
Lipids and Eve were analyzed on a Thermo Fisher UltiMate 3000 HPLC system with DAD-3000 diode array detector (UV) set to 278 nm and ESA 542 charged aerosol detector (CAD). A Merck Purospher STAR RP-18 endcapped (5 μm) 250–4.6 column was used as stationary phase. Separation was conducted at 35 °C. Mobile phase A consisted of water, B consisted of acetonitrile, and C consisted of methanol containing 0.3% (v/v) formic acid. A 6-point calibration was generated by injecting mixtures of analytes in methanol in known concentrations ranging from 20 to 500 µg/mL for DMPC and 2 to 50 µg/mL for MHPC and Eve. The lipids were quantified using a 2nd order polynomial fit on the CAD data. Eve was quantified using linear regression on the UV data. Typical injection volumes were 10 to 20 µL. Eve-rHDL samples were mixed with a 9-fold excess of methanol to precipitate protein components, centrifuged at 10,000 × g and 4 °C for 15 min, and the supernatant was injected into the HPLC system. The HPLC method is displayed in Table 2.
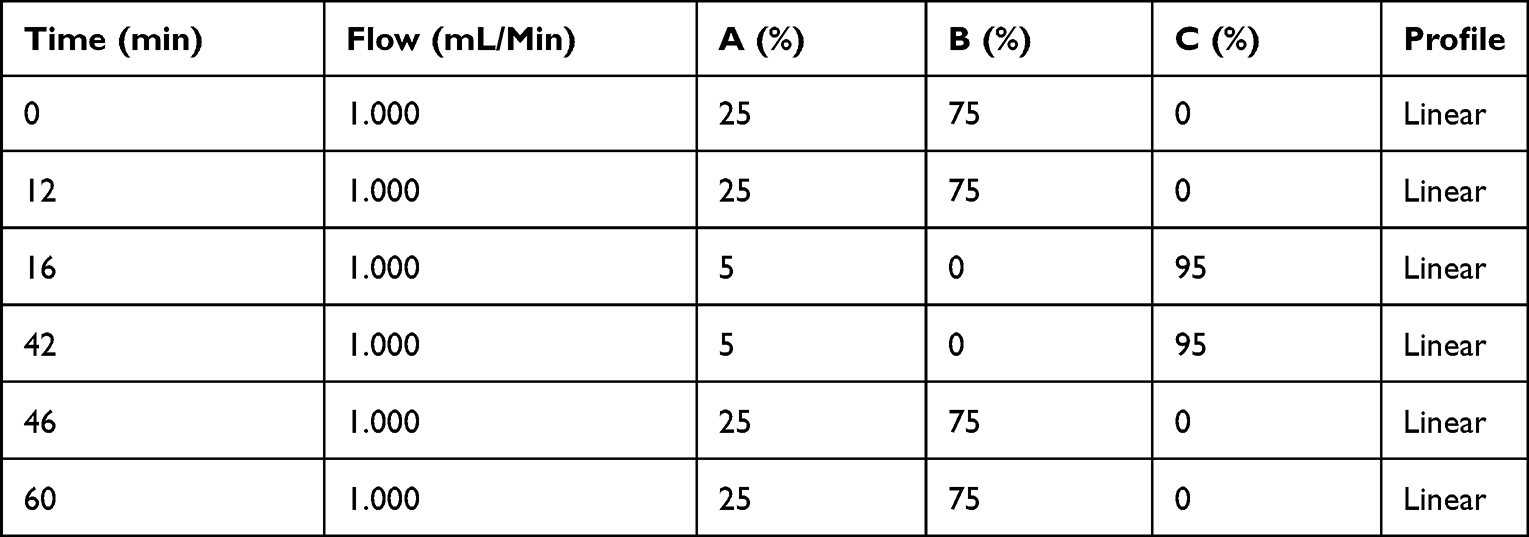 |
Table 2 HPLC Instrument Method |
In vitro Cell Assays
J774A.1 murine macrophages (DSMZ, Braunschweig, DE) were cultivated in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% (v/v) fetal calf serum (FCS) in Greiner CELLSTAR 100 × 20 mm cell culture dishes. The cultures were split twice per week at 80 to 90% confluence using a cell scraper.
Murine bone marrow-derived macrophages (BMDMs) were generated from murine bone marrow cells in accordance with ethical approval from the animal care committee and regional council Freiburg, DE. Human peripheral blood mononuclear cells (PBMCs) were retrieved from human blood samples collected and processed in accordance with ethical approval from the institutional review board University Hospital Freiburg, DE.
In brief, for BMDM generation, murine bone marrow cells (C57Bl6/J, Janvier Labs, FR) were isolated from femur, hip, and tibia. The cell suspension was processed through a 40 µm cell strainer. About 100,000 bone marrow cells per well of a 48-well plate were cultivated in 1 mL RPMI w/ 10% (v/v) FCS w/ 30 ng/mL murine M-CSF (PeproTech, Hamburg, DE) for 5 to 7 days. The media with supplements was changed every two days.
For PBMC isolation, 34 mL of human whole blood was anticoagulated with 6 mL sodium citrate, diluted 1+1 (v/v) with PBS, and distributed on four SepMate50 tubes (STEMCELL Technologies, Cologne, DE), lined with Bicoll. The samples were further processed according to the manufacturer’s instructions. After collection, the PBMC samples were directly placed in a 4 °C prechilled freezing container and stored at −80 °C. The cells were thawed quickly in a 37 °C prewarmed water bath, diluted with warm PBS, centrifuged, and the pellet was resuspended in RPMI w/ 10% (v/v) FCS w/ 100 ng/mL human M-CSF (PeproTech, Hamburg, DE). About 1,200,000 cells per well were cultivated in 1 mL RPMI w/ 10% (v/v) FCS w/ 100 ng/mL human M-CSF for 5 to 7 days in a 48-well plate. The media with supplements was changed every two days.
All cultures were incubated at 37 °C in a 5% CO2 atmosphere.
Cytotoxicity and Cell Viability
Cytotoxicity and cell viability were determined using Promega CellTox Green and CellTiter-Glo assays in a multiplex setup. J774A.1 cells were harvested by scraping, and about 3000 cells in 100 µL culture medium were seeded into Greiner opaque 96-well cell culture microplates. After a preincubation of 24 h, the cells were treated with 100 µL of culture medium containing various concentrations of Eve-rHDL or free Eve, dissolved in DMSO (Eve-DMSO). The amount of DMSO in the final dilutions was <0.25% (v/v). Several types of controls were used to substantiate the assay results: vehicle controls treated with the same concentration of rHDL and DMSO as in the highest Eve-rHDL and Eve-DMSO treatments, untreated and toxicity controls treated with culture medium, and blank medium controls containing no cells. After 48 h of incubation, the assays were conducted following the manufacturer’s instructions. After readout of the CellTox Green fluorescence with a BioTek Instruments FLx800 microplate reader (ex. 485 to 500 nm, em. 528 ± 20 nm), the CellTiter-Glo assay was performed, and the luminescence was analyzed. Each readout was blank-corrected and normalized to toxicity and untreated controls, respectively.
Proliferative Activity
Proliferative activity was determined using a Roche bromodeoxyuridine (BrdU) colorimetric assay. About 3000 J774A.1 cells in 100 µL culture medium were seeded into Greiner clear 96-well cell culture microplates as described above. An additional background control was established. After 48 h of incubation, the assay was conducted following the manufacturer’s instructions. Dual-wavelength absorption was recorded at 450 nm and 750 nm reference wavelength with a Packard SpectraCount™ BS10001 microplate reader. Each readout was blank-corrected and normalized to the untreated controls.
Prior to cell processing, images of the well center positions were taken with a Zeiss Axiovert 40 CFL microscope with 5× objective magnification and an Axiocam 305 camera.
Cholesterol Efflux Capacity
Human PBMCs and murine BMDMs were incubated for 90 min with 0.025 mM TopFluor® Cholesterol (Avanti Polar Lipids, Alabaster, US) in cell culture medium. The labeling medium was removed, and the cells were incubated overnight with 2 µg/mL acyl-coenzyme A:cholesterol O-acyltransferase (ACAT)-inhibitor (Tocris Bioscience, Wiesbaden, DE) in cell culture medium w/ 2 g/l bovine serum albumin (BSA). Cholesterol efflux was initiated by incubation for 4 h with 25 µg/mL cholesterol acceptors (native HDL, rHDL, and Eve-rHDL; protein component mass) in cell culture medium. Untreated controls w/o labeled cholesterol and controls w/ labeled cholesterol but w/o cholesterol acceptor treatment were prepared accordingly.
The supernatant was collected, centrifuged, and analyzed with a Molecular Devices SpectraMax M2 plate reader with 490 nm excitation filter, 520 nm emission filter, and 515 nm cutoff. The amount of cholesterol efflux was quantified using linear regression (5-point calibration on concentrations ranging from 1 to 25 µM TopFluor® Cholesterol).
Flow Cytometry
About 60,000 J774A.1 cells in 400 µL culture medium were seeded into clear 24-well cell culture multiwell plates. After a 24 h preincubation, the cells were treated for 1 h with 100 µL of culture medium containing rHDL or Eve-rHDL, which had been labeled with DiO. Concentrations of 0.15 mM (lipid component amount) were used, which translated to 1.5 µM DiO and 18 µM Eve for Eve-rHDL. After preparation for flow cytometry, the samples were mixed with a trypan blue solution in a final concentration of 0.04% to quench external DiO fluorescence. Flow cytometry was performed on a BD LSRFortessa™. FSC and SSC were recorded to establish a live gate with a minimum of 10,000 events. The DiO fluorescence intensity of the live gate was recorded using an excitation laser at 488 nm and a 530 ± 30 nm emission bandpass filter.
Microscopy
About 60,000 J774A.1 cells in 300 µL culture medium were seeded into 8-well cell culture slides. After a 24 h preincubation, the cells were treated for 1 h with 100 µL of culture medium containing rHDL or Eve-rHDL, which had been labeled with DiI. Concentrations of 0.15 mM (lipid component amount) were used, which translated to 1.5 µM DiI and 18 µM Eve for Eve-rHDL. Concentrations of 50 nM LysoTracker™ Deep Red were used to stain acidic cell organelles. Multiple controls for each individual dye and the respective dye combinations were established to assess cellular autofluorescence and spectral overlap.
The cells were washed and incubated for 15 min with a 3% Pierce™ formaldehyde solution in PBS at room temperature in the dark. The samples were mounted with Vectashield® Vibrance™ Antifade mounting medium w/ DAPI and stored overnight at room temperature in the dark. Imaging was performed on a Leica TCS SP8 X inverted confocal microscope with 63×/1.40 oil objective magnification. DAPI fluorescence was recorded with a PMT detector at 419 to 484 nm, DiI fluorescence was recorded with a HyD SMD detector at 561 to 606 nm, and the lysotracker fluorescence was recorded with a HyD SMD detector at 657 to 764 nm. For sample excitation, lasers at 405, 553, and 647 nm were used. The illumination settings were kept fixed during the analysis.
Statistical Analysis and Software
If applicable, all results are reported as mean±SD. Statistical significance was determined by GraphPad Prism 8.01 using one-way ANOVA and Dunnett’s test: ***P≤0.001, **P≤0.01, *P≤0.05. Microsoft Excel 2016 was used for all further calculations.
Results and Discussion
In this study, rHDL was prepared from DMPC, MHPC, and ApoA1 via a novel dual centrifugation method, as depicted in Scheme 1. The loading with Eve was achieved by integration of the hydrophobic API into the lipid film. Following the lipid film preparation, the dual centrifugation process was completed after one hour. After the reconstitution process, potentially non-encapsulated Eve was removed by conventional centrifugation, which should promote the sedimentation of free API. The supernatant containing the API-loaded nanoparticles was recovered. Given the low aqueous solubility (<0.01%) and high hydrophobicity (log n-octanol-water partition coefficient of 4) of Eve,24 the remaining amount of non-encapsulated API was considered negligible with regard to the concentration in the rHDL particles. Considering the profound instability of Eve in aqueous solution, the samples were immediately frozen at −80 °C after preparation. For all experiments, the samples were thawed at room temperature for less than 30 min, vortex-mixed, and immediately subjected to the analysis.
 |
Scheme 1 Nanoparticle preparation by dual centrifugation. (A) The preparation of the lipid film is finished after approx. 3 h. The films may be prepared in large batch sizes and stored at −80 °C for convenience. (B) The preparation of Eve-rHDL is completed after approx. 1.5 h, requiring only a minimum amount of manual labor. |
rHDL is a Discoidal Particle in the 10 nm Size Range
We used NS-TEM to analyze the morphology of the prepared rHDL and Eve-rHDL nanoparticles. Figure 1A and B show micrographs of rHDL and Eve-rHDL and reveal excellent nanoparticle homogeneity. The stacking of nanoparticles was in line with the characteristic formation of stain-based rouleau artifacts in lipoprotein micrographs (see Zhang et al (2013)25 for further information). Since rouleaux formation is based on interactions with the staining agent, no stacking should be present in solution. Despite rouleaux, no further aggregation was identified in the micrographs. The discoidal morphology of the nanoparticles could be demonstrated through observation of both a nanoparticle side-view in the rouleaux and a top-view of individual nanoparticles. The dmax was determined to 13.3 ± 2.5 nm for rHDL and 11.0 ± 1.4 nm for Eve-rHDL by manual evaluation of three individual micrographs (n=648).
 |
Figure 1 Nanoparticle size measurement. NS-TEM micrographs of (A) rHDL and (B) Eve-rHDL revealed aparticle morphology congruent with discoidal HDL; 100 nm scale bar. The preparations were highly homogenous, and both single particles and particle stacks (rouleaux) could be identified. dmax,rHDL=13.3±2.5 nm, dmax,Eve-rHDL=11.0±1.4 nm (n=648). The (C) N-PAGE bands for rHDL (3, 5, 7) and Eve-rHDL (4, 6, 8) were clearly distinct from an ApoA1 starting material standard. rHDL was found at higher positions on the gel than Eve-rHDL, indicating a larger hydrodynamic size: dh,rHDL=11.5±0.3 nm, dh,Eve-rHDL=9.7±0.1 nm (n=3). Different storage durations did not convey a change in migration behavior or nanoparticle integrity. In contrast to the single bands of rHDL, native HDL presented a sharp band at approx. 66 kDa and an additional band in the rHDL region, which was embedded in a diffuse smear. The hydrodynamic size could furthermore be retrieved using (D) DLS distribution analysis: dh,rHDL=10.9±0.8 nm. Interestingly, in contrast to rHDL, no size peak in the 10 nm region could be observed for Eve-rHDL. |
The excellent preparation efficiency and homogeneity was further demonstrated by N-PAGE. The electrophoresis was conducted under native conditions to preserve the nanoparticle integrity. As presented in Figure 1C, the rHDL and Eve-rHDL bands were clearly separated from the ApoA1 band, which indicated a complete reaction of the starting material. The nanoparticles were stable for at least 14 weeks since no difference in the migration behavior in the gel could be observed for different storage durations at −80 °C. Using a native protein standard as reference, average sizes of 11.5 ± 0.3 nm for rHDL and 9.7 ± 0.1 nm for Eve-rHDL (n=3) were determined. However, this method presented limitations in regard to molecular weight analysis since the specific mass of the lipid-based rHDL nanoparticles differed from the plain-protein standards. Moreover, the molecular weight of ApoA1 (approx. 28 kDa) was overestimated by the protein standard, which suggested self-association of ApoA1.
We further investigated DLS as a rapid method for rHDL size determination. The use of cumulant analysis for the measurements was unsuccessful and reported large z-average and PDI values. Potentially, this finding was attributed to the presence of large vesicular remnants, which had not undergone complete transition to discoidal rHDL during the DC process. Due to the exponential size–scattering intensity relationship, these large particles may have dominated the analysis even though the actual particle amounts were likely to be negligible. Particle size was therefore analyzed using distribution analysis, as reported in Figure 1D. A hydrodynamic diameter (dh) of 10.9 ± 0.8 nm was retrieved for rHDL (n=3). However, the size of Eve-rHDL could not be conclusively determined by distribution analysis, suggesting a slightly larger number of large particles or aggregates in Eve-rHDL, which were overrepresented in the analysis.
A summary of the size measurements is displayed in Table 3.
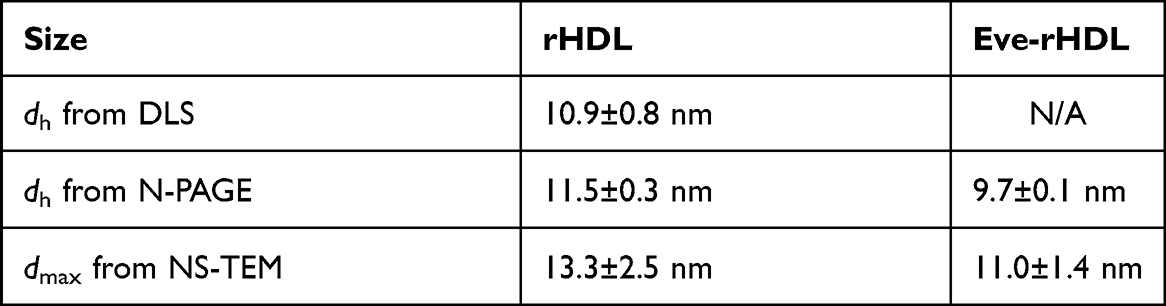 |
Table 3 Nanoparticle Size |
Analysis of rHDL Homogeneity and Composition Can Be Realized in Detail Using AF4
We used AF4 analysis to gain further insight into the preparation homogeneity and to investigate the analyte molar mass and composition. AF4 is a separation technique for macromolecules and colloids. The samples are retained on the trapezoid separation channel by a separation field, which is generated by a cross flow (Vx). Sample elution by the orthogonal detector flow (Vd) is controlled by the analyte diffusion coefficient and thus macromolecule size. While the analysis was conducted at 25 °C and thus close to the Tm of the lipids, no effect on the fractionation behavior was expected due to the stabilizing effects of ApoA1 on the lipid packing.13 The AF4 chromatograms (Figure 2) showed a sharp signal peak at low retention times. The signals in the concentration detectors (UV and dRI) were clearly distinct from the ApoA1 starting material standard peak, indicating successful reconstitution. The LS detectors indicated the elution of larger particles at late retention times. Corresponding to the DLS experiments, a larger number of aggregates or vesicular remnants could be determined in Eve-rHDL samples by the highly sensitive LS detectors. However, the actual amount of large particles present could be considered negligible with regard to the concentration detectors, which do not suffer a signal bias by analyte size.
 |
Figure 2 AF4 chromatograms. The signal peaks for rHDL and Eve-rHDL were clearly distinct from an ApoA1 starting material standard peak. Only a minor amount of nanoparticle signal was present in the ApoA1 region. Additional peaks at late retention times were only visible in the LS detectors. |
To gain deeper insight into the rHDL physicochemical properties, protein–conjugate analysis was conducted. To obtain the necessary coefficients, the ApoA1 amino acid sequence was retrieved from the neXtProt knowledgebase (NX_P02647)26 and used to calculate the ApoA1 mass attenuation coefficient and dn/dc via the Sedfit software.27
For the lipids, the attenuation coefficient was estimated to be zero, and the lipid dn/dc standard value of 0.160 mL/g was used.28 The rHDL attenuation coefficient, dn/dc, molar mass, as well as the individual protein and lipid component molar masses were then determined by the Wyatt Astra 7 software from the detector data. The amounts of single protein and lipid molecules per rHDL particle were determined via division of the component molar masses by the single molecule molar masses. For ApoA1, the single molecule molar mass was determined to 26.7 kDa by AF4. For the lipids, Equation 1 is employed to calculate the average single molecule molar mass of 651 g/mol from the HPLC quantification data of the individual lipids.
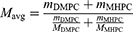
A summary of the analysis results is provided in Table 4. The rHDL protein-to-lipid ratio was very close to the employed ratio of starting materials, indicating great reconstitution efficiency. The amount of protein molecules per rHDL particle was in excellent agreement with the reported ApoA1 stoichiometry of 2 for small, discoidal rHDL.14 However, Eve-rHDL could not be characterized by protein–conjugate analysis since the Eve attenuation coefficient and dn/dc were not available. As an alternative, the Eve-rHDL molar mass was determined using the rHDL dn/dc, which was determined by the protein–conjugate analysis. All experiments were conducted using three independent replicates.
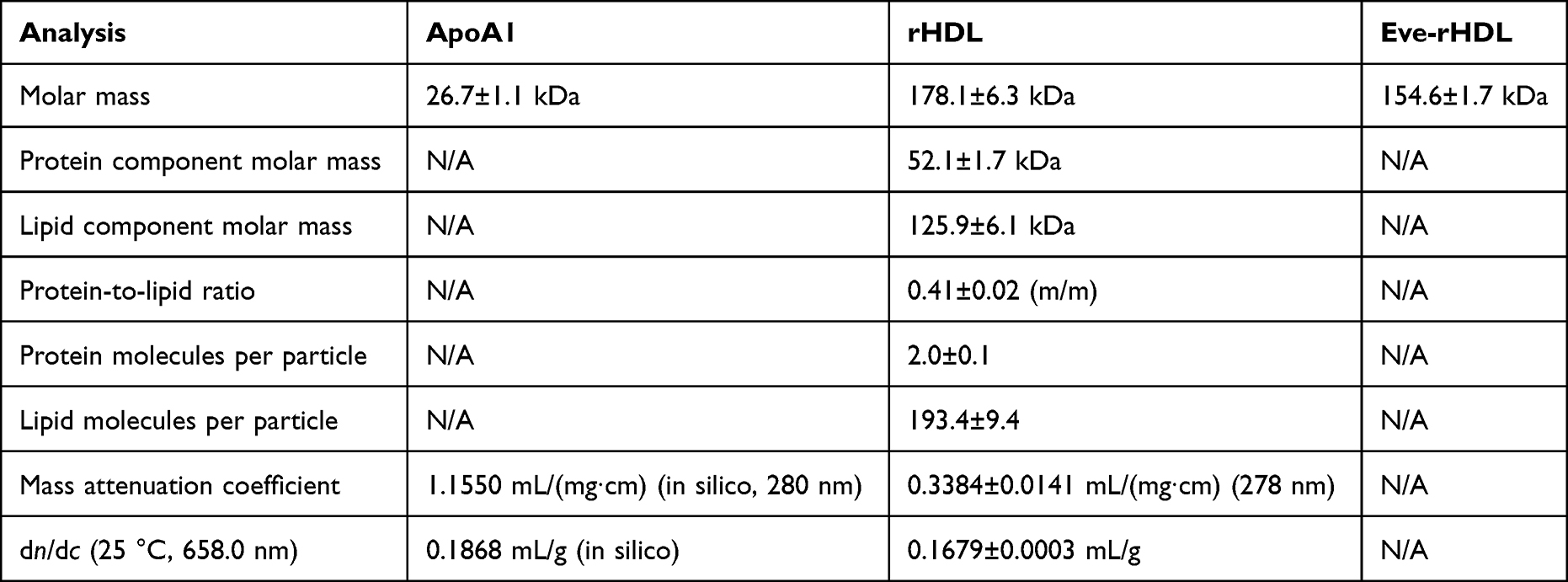 |
Table 4 Results of AF4 Analysis |
Considering the size- and AF4 experiments, Eve-rHDL seemed to be smaller than rHDL. Potentially, this might have been due to a disruption of the lipid packing by Eve and a displacement of lipids. The theory was further substantiated by the slightly elevated amount of large particles in Eve-rHDL, as determined by DLS and AF4.
Eve-rHDL Stability is Preserved During DC and Frozen Storage
We used HPLC to determine the concentrations of lipid and Eve and thus the Eve-rHDL drug loading. An exemplary HPLC chromatogram is displayed in Figure 3A. The total lipid amount was determined to 321.9 ± 13.2 µg (291.9 ± 12.1 µg DMPC and 30.0 ± 2.2 µg MHPC) per 89 µL batch (104.1 ± 4.3%, n=8). The Eve amount was determined to 56.1 ± 3.1 µg per 89 µL batch (90.7 ± 5.0%, n=8), and a drug-to-lipid molar ratio of 0.12 ± 0.01 was calculated. The Eve loading was highly efficient, and no considerable degradation could be detected in the HPLC chromatograms, given immediate freezing after preparation via DC.
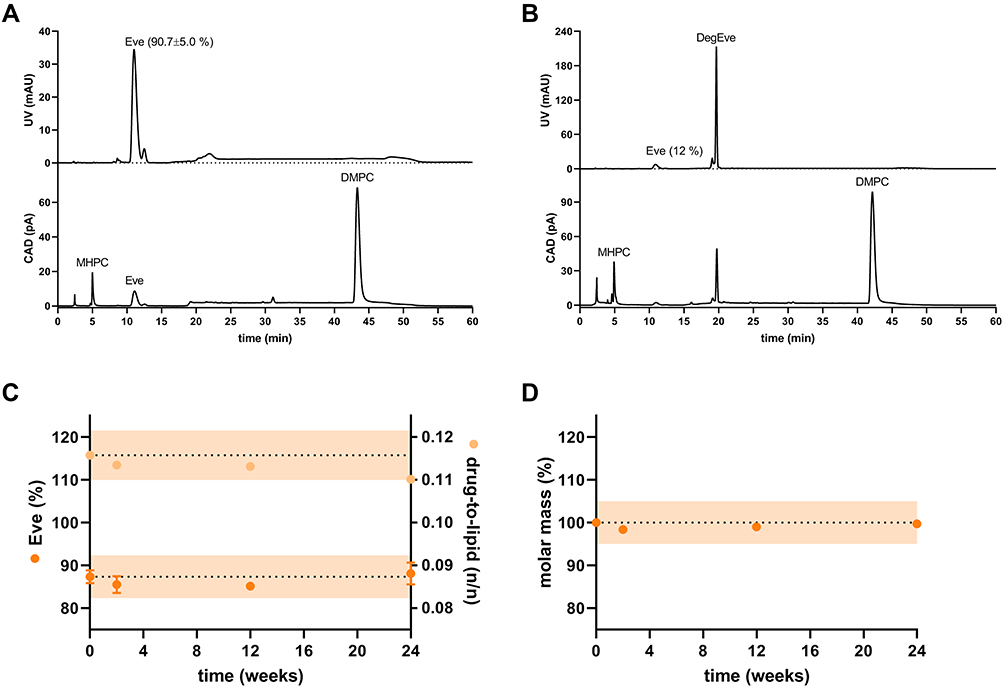 |
Figure 3 HPLC chromatograms and stability studies on Eve-rHDL. The samples were either (A) frozen immediately after preparation (drug-to-lipid molar ratio of 0.12±0.01, mEve=90.7±5.0% of theoretical value, n=8) or (B) stored at 24 °C for 24 h (mEve=12% of theoretical value). The baseline noise at approx. 20 min and 50 min was due to the mobile phase gradients. For the stability studies, the nanoparticles were stored at −80 °C for up to 24 weeks. (C) HPLC measurements. The initial Eve content and drug-to-lipid ratio after preparation is indicated by the dashed line with an orange ±5% interval of the starting value. The content of Eve did not considerably decline during storage; n=2. (D) AF4 measurements: the Eve-rHDL molar mass remained constant during the course of the experiment; n=1. |
If Eve-rHDL was stored at 24 °C, only 12% of Eve remained after 24 h, and an Eve degradation product (DegEve) signal was observed in the chromatograms (Figure 3B). A potential degradation mechanism of the macrolide API might have been the hydrolytic cleavage of the lactone moiety as proposed by Il’ichev et al (2007).29 Eve degradation is most likely related to the aqueous media and temperature stress, ie, activation energy, rather than being related to the rHDL vehicle. The short DC processing time and gentle homogenization mechanism were thus ideally suited for retaining the chemical integrity of the fragile API since only 1.5 h of water exposure and a small amount of thermal energy were introduced by DC. The stability of Eve in Eve-rHDL after immediate freezing at −80 °C was further investigated over 24 weeks (Figure 3C). No considerable degradation could be observed in the frozen state, which was most likely due to a reduction in activation energy and immobilization of water molecules in a solid phase. Moreover, the nanoparticle molar mass remained constant, as determined by AF4 (Figure 3D). This indicated excellent storage stability in regard to the particle integrity, a finding which had also been demonstrated by the N-PAGE experiments.
rHDL is Taken Up by Murine Macrophages
To analyze the cellular uptake of the rHDL nanoparticles, we performed flow cytometry experiments. For this purpose, J774A.1 cells were incubated with DiO-labeled rHDL and Eve-rHDL, and the cellular fluorescence was analyzed. The external fluorescence of attached but not ingested particles was quenched by trypan blue, which absorbs light in the emission spectrum of DiO.30 The results are displayed in Figure 4A. After 1 h of incubation, the cellular DiO fluorescence was significantly increased in regard to untreated controls, indicating the uptake of the nanoparticles.
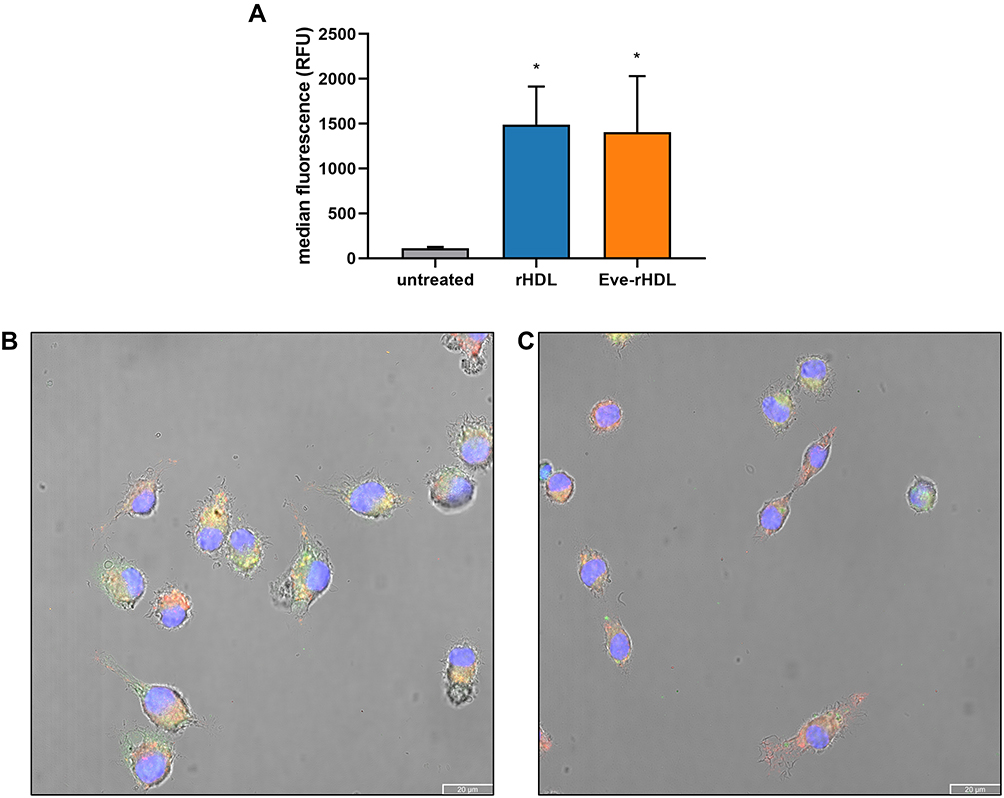 |
Figure 4 Cellular uptake experiments. (A) Flow cytometry analysis of cellular uptake into J774A.1 cells. The cellular fluorescence intensity after 1 h of incubation with DiO-labeled nanoparticles was significantly increased in regard to untreated control cells; n=3, statistical significance determined by one-way ANOVA and Dunnett’s test: *P≤0.05. Confocal micrographs of J774A.1 treated with DiI-labeled (B) rHDL and (C) Eve-rHDL for 1 h; 20 µm scale bar. Blue, green, and red colors represent DAPI-, DiI-, and lysotracker staining, respectively. Yellow coloring could be observed due to colocalization of DiI and lysotracker fluorescence. |
To substantiate the results and provide a visual representation of the nanoparticle localization in the cells, confocal fluorescence microscopy was performed. To this end, DiI-labeled rHDL and Eve-rHDL were prepared to exploit the superior distinction to cellular autofluorescence in regard to DiO. The cells were incubated with the nanoparticles for 1 h, fixed with Pierce™ methanol-free formaldehyde solution to avoid dye extraction by organic solvents, and mounted to glass slides. Additional staining was performed with LysoTracker™ Deep Red to stain acidic cell organelles and DAPI to stain the cell nucleus. Exemplary micrographs are displayed in Figure 4B and C. A partial colocalization of DiI and the lysotracker signal suggested the processing of the nanoparticles into late endosomes or lysosomes. However, the DiI staining also appeared spread throughout the cytosol, which may be explained by a lateral diffusion of the dye or direct cytosolic delivery by the rHDL vehicle.
rHDL Promotes Cholesterol Efflux
To assess the biological effects of the rHDL vehicle, we conducted a cholesterol efflux assay. The assay was performed on human PBMCs and murine BMDMs, which had been fed a fluorescent cholesterol derivative and were subsequently incubated with the cholesterol acceptors. The results of the supernatant fluorescence readout are displayed in Figure 5. The supernatant fluorescence intensity and associated cholesterol efflux from the cells was highest for a human plasma HDL control. This might have been due to a broader arsenal of HDL subpopulations in regard to the homogeneous rHDL preparations (see Figure 1C), each contributing to the overall cholesterol efflux capacity. Additional plasma proteins present in the HDL sample (as indicated by the N-PAGE band at approx. 66 kDa) may also have contributed to the elevated cholesterol efflux capacity (see Phillips (2014)5 for further information). However, the results for rHDL and Eve-rHDL were still significant in regard to cells which had been incubated without cholesterol acceptors. Thus, the ability of rHDL to provoke cholesterol efflux was clearly demonstrated.
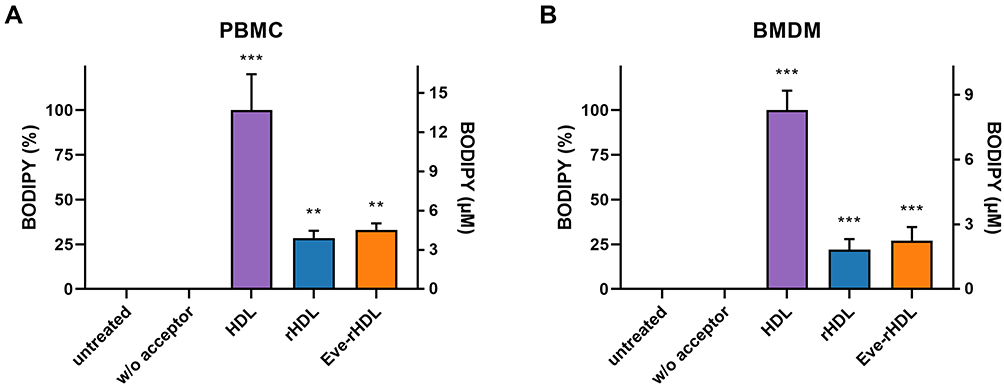 |
Figure 5 Cholesterol efflux of (A) human PBMCs and (B) murine BMDMs treated with cholesterol acceptors for 4 h. The data was normalized to the cholesterol efflux capacity of a native HDL standard. The efflux capacity of rHDL and Eve-rHDL was lower than that of native HDL but significantly distinct from controls w/o cholesterol acceptor treatment; n=5 technical replicates, statistical significance determined by one-way ANOVA and Dunnett’s test: ***P≤0.001, **P≤0.01. |
Eve-rHDL is a Potent Proliferation Inhibitor
The antiproliferative effect of Eve-rHDL and free Eve was investigated using J774A.1 cells. After incubation of the cells with the compounds for 48 h, bright-field microscopy was conducted to visualize cell growth, and a BrdU proliferation assay was performed. The results are displayed in Figure 6A. The cellular proliferative activity was reduced over a wide concentration range. The microscopy images of the treated specimen suggested a reduced number of cells in regard to vehicle controls. For 50 µM of free Eve, no remaining proliferative activity could be detected, and a change in cell morphology was observed.
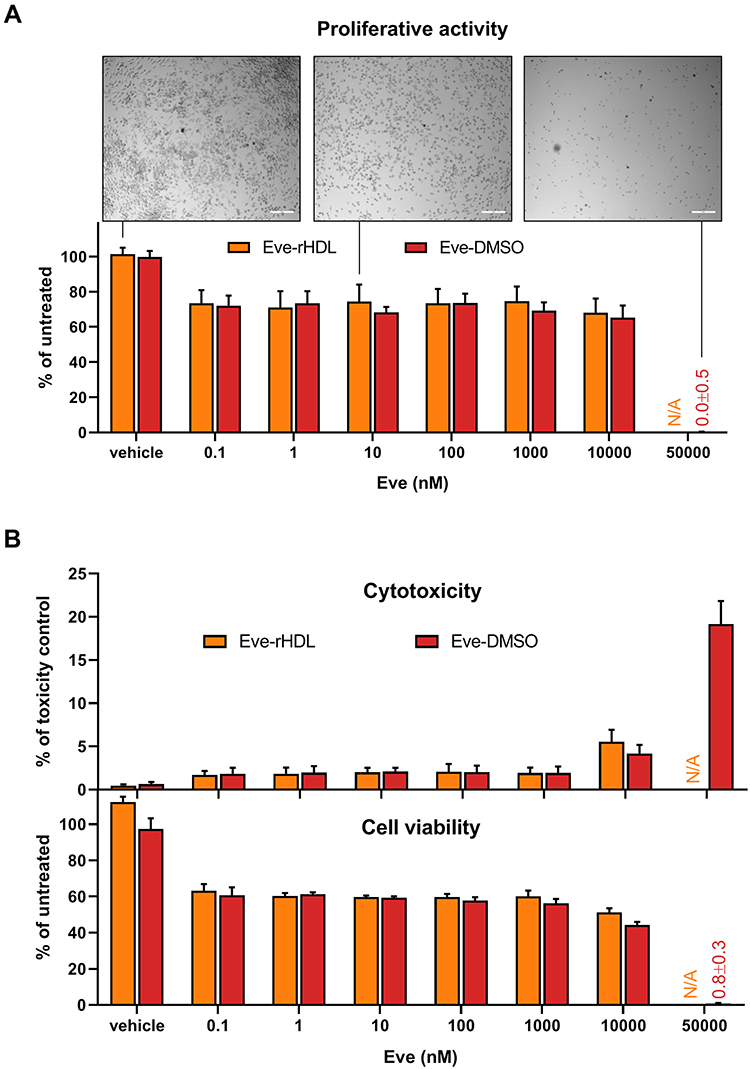 |
Figure 6 Effects on cell viability. (A) Proliferative activity of J774A.1 after 48 h of treatment. Bright-field microscopy images revealed a reduced cell growth for treated cells and a change in cell morphology for very high Eve concentrations; 200 µm scale bar. The images were taken of the well center positions prior to cell processing (Zeiss Axiovert 40 CFL microscope, 5× objective magnification, Axiocam 305 camera). A BrdU proliferation assay reported a reduced proliferative activity in regard to untreated controls and a complete proliferation halt for very high Eve concentrations; n=3. (B) Cytotoxicity and cell viability of J774A.1 after 48 h of treatment. The cell viability was diminished over a wide concentration range and further reduced for Eve concentrations >1000 nM. Correspondingly, cytotoxicity measurements revealed an increase in membrane permeability at this concentration. For very high Eve concentrations, the cell viability was completely lost, and a considerable increase in membrane permeability could be observed; n=3. |
To further substantiate these findings, the cytotoxicity and viability of the cells were investigated using a CellTox Green–CellTiter-Glo multiplex assay (Figure 6B). While BrdU is actively incorporated into the genome of proliferating cells and is thus a specific marker for cellular proliferation, the decline in cell viability (determined as a function of cellular ATP content by CellTiter-Glo) might also be related to cytotoxic events. To establish the connection between proliferation inhibition and viability loss, cytotoxicity also has to be analyzed (for example, via determination of cell membrane permeability by CellTox Green). After 48 h of incubation with the nanoparticles or with free Eve, the cell viability was reduced over a wide concentration range, corresponding to a reduced cellular proliferative activity. All cell viability was lost for the treatment with 50 µM free Eve, which indicated a toxic effect of the high Eve concentrations on top of the antiproliferative effect. The finding was substantiated by the cytotoxicity measurements. However, relevant cytotoxicity was only observed at Eve concentrations of >1 µM with an inverse relationship between the cell viability data and cytotoxicity.
Taken together, the assays suggested a mainly anti-proliferative effect of both Eve-rHDL and free Eve at concentrations ranging from 0.1 nM to 1 µM and cytotoxic effects at higher concentrations. It is noteworthy that high concentrations of the plain rHDL vehicle induced a slight cell viability increase. Considering the flow cytometry data discussed before, cellular uptake was in effect after 1 h of particle exposure enabling the anti-proliferative effects of the formulation well before the decomposition of Eve in the aqueous media at 37 °C. In regard to the nearly identical performance of Eve-rHDL and free Eve, the suitability of rHDL as a drug delivery vehicle was clearly demonstrated.
While the application of free drug may be justified in some cases, API-loaded rHDL offers substantial benefits over the application of free API in anti-atherosclerotic therapy. For example, a desirably reduced plaque macrophage proliferation has been detected after oral administration of statins in mice and patients.31 However, the concentration of orally administered API in the atherosclerotic plaque was negligible, and the reduction of macrophage proliferation was presumably due to effects on the overall lipid metabolism rather than direct inhibition. Targeting plaque macrophages directly through API-loaded rHDL offers the benefit of elevated local API concentration and associated higher efficacy. Correspondingly, injectable, statin-loaded rHDL has been proven to accumulate in the affected blood vessel walls and decrease the macrophage-positive plaque area.32 Similarly, plaque macrophage activity has been successfully reduced by inhibition of the CD40–TRAF6 interaction using TRAF-STOP-loaded rHDL, preventing atherosclerotic plaque progression.33 Regarding the potent anti-proliferative effects of Eve-rHDL in vitro, we expect a similar effect in vivo and substantial plaque stabilization. For this evaluation and the required preparation of Eve-rHDL, dual centrifugation is an ideal manufacturing technique. Eve-rHDL preparation would not have been successful using the cosonication approach which, for example, has been applied to generate statin-loaded rHDL and TRAF-STOP-loaded rHDL. The high temperatures during the sonication process would lead to rapid API degradation, as suggested by the detection of DegEve via HPLC after Eve-rHDL storage at 24 °C. Correspondingly, the long process times of detergent-based synthesis would lead to compound hydrolysis. The dual centrifugation process is highly scalable from miniature screening batch sizes to larger scale. Dual centrifugation retains sterility during manufacture and, considering the robust homogenization process, is easily transferable to different manufacturing sites. These features may also be achieved using a microfluidic approach, but the technique requires expensive single-use chip consumables containing the microfluidic channel. The limitations of dual centrifugation lie in the preparation of ultra-large batch sizes. The physical dimensions of the centrifuge are naturally limited, and continuous manufacturing is not possible using the dual centrifugation approach. Therefore, the technique might not be ideally suited to produce the amounts of product required for full commercialization.
However, researchers will benefit from the economic way of rHDL manufacturing and dual centrifugation will be a valuable tool to bring rHDL from bench to bedside.
Conclusion
In this work, we present a novel approach for the aseptic preparation of rHDL and drug-loaded rHDL. The dual centrifugation process requires only minimal time and manual labor. Dual centrifugation is suited for micro-scale manufacturing and thus efficient formulation screening. Here, we incorporated everolimus into rHDL and demonstrated the successful nanoparticle reconstitution in regard to particle size, morphology, and chemical composition.
Considering the biological efficacy, we demonstrated that everolimus-loaded rHDL both diminishes cellular proliferation and provokes cholesterol efflux from human and murine macrophages. It may therefore be employed to target atherosclerotic plaques in vivo and reduce the plaque inflammatory- and lipid burden. With a dual mode of action, the formulation is a promising candidate for further clinical development, and dual centrifugation may enable the efficient application of the rHDL platform in cardiovascular disease and beyond.
Abbreviations
ACAT, Acyl-coenzyme A:cholesterol O-acyltransferase; AF4, Asymmetric-flow field-flow fractionation; API, Active pharmaceutical ingredient; ApoA1, Apolipoprotein A1; APS, Ammonium peroxodisulfate; BMDM, Bone marrow-derived macrophages; BrdU, Bromodeoxyuridine; BSA, Bovine serum albumin; CAD, Charged aerosol detector; CVD, Cardiovascular disease; DC, Dual centrifugation; DegEve, Everolimus degradation product; dh, Hydrodynamic diameter; DLS, Dynamic light scattering; dmax, Maximum Feret diameter; DMEM, Dulbecco’s modified eagle’s medium; DMPC, 1,2-Dimyristoyl-sn-glycero-3-phosphocholine; dRI, Differential refractometer; Eve, Everolimus; Eve-DMSO, Free Eve, dissolved in DMSO; Eve-rHDL, Reconstituted high-density lipoprotein, loaded with Everolimus; FCS, Fetal calf serum; HDL, High-density lipoprotein; HPLC, High-performance liquid chromatography; LS, Light scattering; MHPC, 1-Myristoyl-2-hydroxy-sn-glycero-3-phosphocholine; mTOR, Mammalian target of rapamycin; N/A, not applicable; N-PAGE, Native polyacrylamide gel electrophoresis; NS-TEM, Negative-stain transmission electron microscopy; PBMC, Peripheral blood mononuclear cells; RCT, Reverse cholesterol transport; rHDL, Reconstituted high-density lipoprotein; Tm, Phase transition temperature; UV, Ultraviolet; Vd, Detector flow; Vinj, Inject flow; Vx, Cross flow.
Acknowledgments
We thank Lipoid (Ludwigshafen, DE) for the kind support with lipid reagents. Furthermore, we acknowledge support by the Open Access Publication Fund of the University of Freiburg. We also want to express our gratitude to Ulrich Massing for the expert advice on the dual centrifugation process and to Monika Köll-Weber for the technical assistance with cell culture experiments.
Funding
This research was supported by grants to I.H. (German Research Foundation grant HI1573/2; I.H. is a member of the Collaborative Research Center SFB1425 grant 42268184).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bäck M, Hansson G. Basic mechanisms of atherosclerosis. In: de Lemos JA, Omland T editors. Chronic Coronary Artery Disease. Elsevier; 2018:45–54. doi:10.1016/B978-0-323-42880-4.00004-2.
2. Lechner K, von Schacky C, McKenzie AL, et al. Lifestyle factors and high-risk atherosclerosis: pathways and mechanisms beyond traditional risk factors. Eur J Prev Cardiol. 2020;27(4):394–406. doi:10.1177/2047487319869400
3. Bertrand MJ, Tardif JC. Inflammation and beyond: new directions and emerging drugs for treating atherosclerosis. Expert Opin Emerg Drugs. 2017;22(1):1–26. doi:10.1080/14728214.2017.1269743
4. Bergheanu SC, Bodde MC, Jukema JW. Pathophysiology and treatment of atherosclerosis. Neth Heart J. 2017;25(4):231–242. doi:10.1007/s12471-017-0959-2
5. Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. 2014;289(35):24020–24029. doi:10.1074/jbc.R114.583658
6. Huang J, Wang D, Huang LH, Huang H. Roles of reconstituted high-density lipoprotein nanoparticles in cardiovascular disease: a new paradigm for drug discovery. Int J Mol Sci. 2020;21(3):739. doi:10.3390/ijms21030739
7. Martin DDO, Budamagunta MS, Ryan RO, Voss JC, Oda MN. Apolipoprotein A-I assumes a “looped belt” conformation on reconstituted high density lipoprotein. J Biol Chem. 2006;281(29):20418–20426. doi:10.1074/jbc.M602077200
8. Kornmueller K, Vidakovic I, Prassl R. Artificial high density lipoprotein nanoparticles in cardiovascular research. Molecules. 2019;24(15):2829. doi:10.3390/molecules24152829
9. Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19(9):1166–1172. doi:10.1038/nm.3258
10. Kurdi A, De Meyer GRY, Martinet W. Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br J Clin Pharmacol. 2016;82(5):1267–1279. doi:10.1111/bcp.12820
11. Jonas A. Reconstitution of high-density lipoproteins. In: Segrest JP, Albers JJ editors. Methods in Enzymology. Vol 128. Plasma Lipoproteins Part A: Preparation, Structure, and Molecular Biology. Academic Press; 1986:553–582. doi:10.1016/0076-6879(86).
12. Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta BBA. 2005;1716(2):77–96. doi:10.1016/j.bbamem.2005.08.007
13. Tall AR, Deckelbaum RJ, Small DM, Shipley GG; Tall AR, Deckelbaum RJ, Small DM, Shipley GG. Thermal behavior of human plasma high density lipoprotein. Biochimica et Biophysica Acta. 1977;487(1):145–153. doi:10.1016/0005-2760(77)
14. Brouillette CG, Jones JL, Ng TC, Kercret H, Chung BH, Segrest JP. Structural studies of apolipoprotein A-I/phosphatidylcholine recombinants by high-field proton NMR, nondenaturing gradient gel electrophoresis, and electron microscopy. Biochemistry. 1984;23(2):359–367. doi:10.1021/bi00297a027
15. Bricarello DA, Mills EJ, Petrlova J, Voss JC, Parikh AN. Ganglioside embedded in reconstituted lipoprotein binds cholera toxin with elevated affinity. J Lipid Res. 2010;51(9):2731–2738. doi:10.1194/jlr.M007401
16. Ramos-Cabrer P, Fay F, Sanchez-Gaytan BL, et al. Conformational changes in high-density lipoprotein nanoparticles induced by high payloads of paramagnetic lipids. ACS Omega. 2016;1(3):470–475. doi:10.1021/acsomega.6b00108
17. Berney E, Sabnis N, Panchoo M, Raut S, Dickerman R, Lacko AG. The SR-B1 receptor as a potential target for treating glioblastoma. J Oncol. 2019;2019:1–10. doi:10.1155/2019/1805841
18. Kim Y, Fay F, Cormode DP, et al. Single step reconstitution of multifunctional high-density lipoprotein-derived nanomaterials using microfluidics. ACS Nano. 2013;7(11):9975–9983. doi:10.1021/nn4039063
19. Diditchenko S, Gille A, Pragst I, et al. Novel formulation of a reconstituted high-density lipoprotein (CSL112) dramatically enhances ABCA1-dependent cholesterol efflux. ATVB. 2013;33(9):2202–2211. doi:10.1161/ATVBAHA.113.301981
20. Hagedorn M, Bögershausen A, Rischer M, Schubert R, Massing U. Dual centrifugation – a new technique for nanomilling of poorly soluble drugs and formulation screening by an DoE-approach. Int J Pharm. 2017;530(1):79–88. doi:10.1016/j.ijpharm.2017.07.047
21. Krämer W, Schubert R, Massing U. Small-scale preparation of perfluorocarbon-nanoemulsions utilizing dual centrifugation. Int J Pharm. 2019;572:118753. doi:10.1016/j.ijpharm.2019.118753
22. Massing U, Ingebrigtsen SG, Škalko-Basnet N, Holsæter AM. Dual centrifugation - A novel “in-vial” liposome processing technique. In: Catala A editor. Liposomes. IntechOpen; 2017:3–28. doi:10.5772/intechopen.68523.
23. Holzer M, Kern S, Trieb M, Trakaki A, Marsche G. HDL structure and function is profoundly affected when stored frozen in the absence of cryoprotectants. J Lipid Res. 2017;58(11):2220–2228. doi:10.1194/jlr.D075366
24. European Medicines Agency. Afinitor CHMP Assessment Report EMEA/533232/2009; 2009. https://www.ema.europa.eu/en/medicines/human/EPAR/afinitor.
25. Zhang L, Tong H, Garewal M, Ren G. Optimized negative-staining electron microscopy for lipoprotein studies. Biochim Biophys Acta BBA. 2013;1830(1):2150–2159. doi:10.1016/j.bbagen.2012.09.016
26. Zahn-Zabal M, Michel PA, Gateau A, et al. The neXtProt knowledgebase in 2020: data, tools and usability improvements. Nucleic Acids Res. 2020;48(D1):D328–D334. doi:10.1093/nar/gkz995
27. Zhao H, Brown PH, Schuck P. On the distribution of protein refractive index increments. Biophys J. 2011;100(9):2309–2317. doi:10.1016/j.bpj.2011.03.004
28. Theisen A, Johann C, Deacon MP, Harding SE. Refractive Increment Data-Book for Polymer and Biomolecular Scientists. Nottingham University Press; 2000.
29. Il’ichev YV, Alquier L, Maryanoff CA. Degradation of rapamycin and its ring-opened isomer: role of base catalysis. Arkivoc. 2007;2007(12):110–131. doi:10.3998/ark.5550190.0008.c09
30. Sahlin S, Hed J, Runfquist I. Differentiation between attached and ingested immune complexes by a fluorescence quenching cytofluorometric assay. J Immunol Methods. 1983;60(1):115–124. doi:10.1016/0022-1759(83)
31. Härdtner C, Kornemann J, Krebs K, et al. Inhibition of macrophage proliferation dominates plaque regression in response to cholesterol lowering. Basic Res Cardiol. 2020;115(6):78. doi:10.1007/s00395-020-00838-4
32. Duivenvoorden R, Tang J, Cormode DP, et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi:10.1038/ncomms4065
33. Seijkens TTP, van Tiel CM, Kusters PJH, et al. Targeting CD40-induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol. 2018;71(5):527–542. doi:10.1016/j.jacc.2017.11.055
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Peptide-Based HDL as an Effective Delivery System for Lipophilic Drugs to Restrain Atherosclerosis Development

Gao J, Li Z, Li J, Song P, Yang J, Xiao W, Li N, Xu R
International Journal of Nanomedicine 2022, 17:3877-3892
Published Date: 6 September 2022
Recent Advances in Nanozymes for the Treatment of Atherosclerosis
Zou Z, Bi X, Ma H, Chen L, Jin H, Gong S, Li X, Liu X
International Journal of Nanomedicine 2025, 20:9447-9472
Published Date: 29 July 2025