Back to Journals » Infection and Drug Resistance » Volume 12
Evaluation of treatment options for methicillin-resistant Staphylococcus aureus infections in the obese patient
Authors Narayanan N ![]() , Adams CD, Kubiak DW, Cheng S, Stoianovici R
, Adams CD, Kubiak DW, Cheng S, Stoianovici R ![]() , Kagan L, Brunetti L
, Kagan L, Brunetti L ![]()
Received 28 November 2018
Accepted for publication 12 February 2019
Published 17 April 2019 Volume 2019:12 Pages 877—891
DOI https://doi.org/10.2147/IDR.S196264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Suresh Antony
Navaneeth Narayanan,1,2 Christopher D Adams,1 David W Kubiak,3 Serena Cheng,4 Robyn Stoianovici,5 Leonid Kagan,1,6 Luigi Brunetti1,6
1Department of Pharmacy Practice, Rutgers University, Ernest Mario School of Pharmacy, Piscataway, NJ, USA; 2Division of Infectious Diseases, Department of Medicine, Rutgers Robert Wood Johnson Medical School, New Brunswick, NJ, USA; 3Department of Pharmacy, Brigham and Women’s Hospital, Boston, MA, USA; 4Department of Pharmacy, VA San Diego Healthcare System, San Diego, CA, USA; 5Department of Pharmacy, University of California, Davis Medical Center, Sacramento, CA, USA; 6Department of Pharmaceutics, Rutgers University, Ernest Mario School of Pharmacy, Piscataway, NJ, USA
Abstract: Methicillin-resistant Staphylococcus aureus (MRSA) has emerged as a major cause of infection in both the hospital and community setting. Obesity is a risk factor for infection, and the prevalence of this disease has reached epidemic proportions worldwide. Treatment of infections in this special population is a challenge given the lack of data on the optimal antibiotic choice and dosing strategies, particularly for treatment of MRSA infections. Obesity is associated with various physiological changes that may lead to altered pharmacokinetic parameters. These changes include altered drug biodistribution, elimination, and absorption. This review provides clinicians with a summary of the literature pertaining to the pharmacokinetic and pharmacodynamic considerations when selecting antibiotic therapy for the treatment of MRSA infections in obese patients.
Keywords: obesity, methicillin-resistant Staphylococcus aureus, antibiotics, pharmacokinetics
MRSA epidemiology
Methicillin-resistant Staphylococcus aureus (MRSA) was identified more than 50 years ago in 1961,1 shortly after the initial use of methicillin as a treatment option.2 Since then MRSA has emerged as a major cause of both healthcare-associated infections such as bacteremia or pneumonia and community-associated infections such as skin and soft tissue infections or osteomyelitis. According to data reported to the National Healthcare Safety Network, S. aureus accounted for 16% (8.5% MRSA) of the multidrug-resistant pathogens reported.3 From 1999 to 2005, the number of hospitalizations related to MRSA infections in the USA more than doubled, from approximately 127,000 to almost 280,000.4 Rates of hospitalizations related to MRSA continue to increase in the USA with estimates of approximately 460,000 MRSA-related hospitalizations in 2009.5 In a landmark report by the Centers for Disease Control and Prevention in 2013, MRSA is estimated to cause approximately 80,000 severe infections and over 11,000 deaths per year in the USA.2 The burden of MRSA is not limited to the USA, as 13–74% of S. aureus infections worldwide are methicillin resistant.6
Obesity is a significant risk factor for MRSA colonization and infection although mechanisms are yet to be conclusively defined.7–9 With the increased prevalence of MRSA infections and the growing presence of obesity, we need to understand the effects on treatment in this specific patient population. The estimated prevalence of overweight and obese individuals >20 years of age in the USA is 154.7 million and over 1.6 billion people are considered overweight or obese worldwide.10,11 Furthermore, by 2025 the global obesity prevalence will reach 18% in men and surpass 21% in women.12 Obesity is a risk factor for infection,13 antibiotic treatment failure,14 and antibiotic resistance due to antibiotic underdosing. Furthermore, treatment outcome for individuals affected with MRSA infection may be worse in the obese depending on treatment selection.15
Although the mechanisms of reduced treatment efficacy in patients with obesity are unknown, one can surmise that alterations in bio-distribution patterns may influence treatment success in obesity. While the number of individuals in the USA and worldwide with obesity has reached epidemic proportions, there is no requirement for the pharmaceutical industry to perform clinical studies in this population. Boyd et al performed a review of commonly used antibacterial agents in the UK and reported no advice was included in the manufacturer information for 83% of antibacterials evaluated.16 These data highlight the paucity of guidance available for drug dosing in this population. As a result, clinicians often dose therapies in obese patients without clear evidence. Much of the available literature that supports drug dosing in obesity makes the assumption that if adequate plasma concentration is achieved then this will translate into efficacy; however, this presumption does not account for adequacy at the target site. Typically, body mass index (BMI) is categorized as: underweight BMI<18.5 kg/m2; normal BMI 18.5–24.9 kg/m2; overweight BMI 25.0–29.9 kg/m2; and obese BMI>29.9 kg/m2. One of the challenges in the literature is that although BMI>29.9 kg/m2 is used to define obesity, many studies investigating treatment outcomes or drug pharmacokinetics use a variety of definitions. Despite widespread use of antibiotics, precise algorithms for dosing antibiotics in obese patients are not available, and the use of doses at the upper end of the approved dosage ranges is generally recommended.17 However, several studies reported that current dosing strategies result in subtherapeutic concentrations in target tissues.18–21 This review provides clinicians with a summary of the literature pertaining to the pharmacokinetic and pharmacodynamic considerations when selecting MRSA therapy in obese patients. In addition, recommendations for treating MRSA in the obese population are provided based on available clinical data.
Pharmacokinetic and pharmacodynamic alterations in obesity
Obesity is characterized by a significant alteration of body composition (disproportional increase in fat mass) and may affect other body functions. These physiological changes might lead to an alteration of pharmacokinetic parameters that would not be directly proportional to the increase in the actual body weight (ABW).22 In addition, describing drug pharmacokinetics using only the total volume of distribution (Vd) and total clearance (commonly derived using a non-compartmental approach from plasma data alone) may be too simplistic to describe the actual time course of exposure at the site of action. This consideration is particularly important for drugs that exhibit a time above the minimum inhibitory concentration (MIC) bactericidal effect (ie, beta-lactam antibiotics).
When considering obesity-induced changes in drug biodisposition, multiple factors should be considered. Drug physicochemical properties (such as lipophilicity and ionization state) and tissue composition (adipose vs other tissue) have been used for predicting steady-state tissue partition coefficients.23,24 Obese patients have a different adipose-to-lean tissue ratio compared to normal weight subjects, which may lead to alterations in relative drug exposure in different organs. Obesity does not appear to have an effect on drug binding to albumin, and reports regarding binding to alpha-1-acid glycoprotein are contradictory.25 The kinetics of tissue disposition can be also influenced by tissue blood flow, and an increase in total blood volume and cardiac output and a decrease in peripheral perfusion have been reported in obesity.26–30
Drug elimination is mostly dependent on liver and renal physiology, and obesity-induced changes of these systems including alterations in drug metabolizing enzymes and drug transporters have not been sufficiently studied. Increase in liver blood flow has been reported in obesity,30 which may be important for high extraction ratio drugs. Limited data regarding changes in metabolic function have usually been deduced from comparing the pharmacokinetic profiles of certain model drugs (that are known to be specifically metabolized by those cytochrome isoforms). For example, lower CYP3A4-mediated clearance has been reported for some probes (alfentanil, triazolam) but not others (trazodone, decetaxel).31 Estimated glomerular filtration rate was 62% higher in obese patients.32 On the other hand, a higher incidence of renal dysfunction has been also reported and is commonly attributed to various comorbidities (diabetes, hypertension).17,31
Potential changes in the absorption process should be also taken into consideration for antibiotics delivered by extravascular routes. Delayed gastric emptying in obese patients might affect oral bioavailability and the absorption rate.33,34 Increased subcutaneous fat content in obese patients can lead to inadvertent subcutaneous delivery for drugs intended for intramuscular injection.
Taken together, there are numerous changes that occur in drug pharmacokinetics in the obese population. Currently, we do not have adequate information to make definitive conclusions as to what alterations are clinically relevant and should be considered when selecting the drug or drug dosage. When evaluating drugs and dosing strategies one should consider both pharmacokinetic and outcome data to make the most informed decisions. Ultimately, a better understanding of pathophysiological changes in obese patients and their pharmacokinetic and pharmacodynamic consequences should facilitate selection of the most appropriate drug and dose optimization, including dose level and mode of administration (route, frequency, bolus vs infusion, immediate release vs controlled release). In each of the sections that follow, the available pharmacokinetic and outcome data related to specific drugs with activity for MRSA are reviewed. In addition, recommendations related to drug and dosage selection are provided.
Vancomycin
Vancomycin, a glycopeptide antibiotic, has been used for more than 50 years and remains the standard of care for treatment of MRSA infections.35 The bactericidal activity of vancomycin results from inhibition of bacterial wall synthesis.36 Vancomycin is predominantly eliminated by the kidneys, has a Vd ranging from 0.4 to 1 L/kg, and an elimination half-life ranging from 6 to 12 hours or longer depending on renal function.37 The pharmacodynamic target that has been associated with clinical efficacy is the ratio of area under the concentration–time curve to a minimum inhibitory concentration (AUC:MIC) of ≥400.36 There are variable data of the effects of obesity on vancomycin pharmacokinetics and conflicting recommendations on optimal dosing of vancomycin in obese patients.
Some of the changes in the pharmacokinetics of vancomycin can be attributed to physiologic alterations. Obese patients not only have increased adipose tissue but also muscle mass. Additionally, obesity is associated with an increase in plasma proteins and since vancomycin is ~55% (range 44–82%) protein-bound there may be less active free drug in the serum.38 Lastly, obesity is associated with an overall increase in cardiac output and blood volume, which leads to increases in renal blood flow and kidney mass; these may contribute to a larger Vd and increased systemic clearance of vancomycin resulting in lower serum concentrations.38
Current national guidelines for dosing and monitoring of vancomycin recommend using ABW to calculate initial vancomycin doses (15–20 mg/kg given via an intermittent infusion every 8–12 hours with normal renal function), adjusted to achieve a target serum trough concentration of 15–20 µg/mL.36 Adjustments range from pharmacy managed pharmacokinetic protocols to dose titration based on clinician judgment. The target serum trough recommendation is based on a pharmacokinetic study performed in 1982 on six healthy morbidly obese patients who received a single 1 g dose of vancomycin infusion (1 g/h) after gastric bypass surgery. These obese patients had a significantly larger Vd (43.0 L vs 28.9 L; P<0.005) but no significant difference in total body clearance (1.112±0.160 mL/min/kg vs 1.085±0.071 mL/min/kg of ABW) compared with normal weight control patients (n=4), which correlated most closely with ABW.39 However there have been several reports published after these guidelines which have challenged these recommendations and found that calculating vancomycin doses of 30–45 mg/kg/day on ABW often leads to supratherapeutic steady-state serum trough concentrations.40–45 Unfortunately, no study has been able to precisely determine the optimal dosing weight of vancomycin in obese and morbidly obese patients to achieve consistent serum trough concentrations of 15–20 µg/mL while minimizing nephrotoxicity. These efforts have been further hindered by confounding factors such as extremes of age, critical illness, and concomitantly administered nephrotoxic agents. Recent data indicate the minimal impact of serum trough concentration monitoring on clinical outcomes and in contrast, the safety advantage of AUC-based monitoring of vancomycin.44,46,47 Targeting an AUC:MIC ratio of 400–600 mg h/L may help maximize efficacy while reducing toxicity; however, this has not been well tested in a robust clinical trial. Methodical therapeutic drug monitoring coupled with patient-specific pharmacokinetic calculations is likely the optimal approach for dosing and monitoring systemic vancomycin in obese and morbidly obese patients.
In regards to empiric vancomycin dosing when patient-specific vancomycin serum concentrations are not yet available, Crass et al recently published a population pharmacokinetic study of obese patients treated with vancomycin.48 The analysis provides a dosing nomogram for empiric vancomycin dosing in obese and morbidly obese patients. The pharmacokinetic model for estimating vancomycin clearance was based on a linear combination of age, serum creatinine, sex, and weight. The targets of efficacy and toxicity in this study were developed utilizing AUC targets, and the dosing regimens were assessed using 1,000-subject Monte Carlo simulations. The nomogram, utilizing loading doses and AUC-targeted empiric dosing regimens, had a >90% probability of efficacy (AUC0–24≥400) for all vancomycin clearance levels analyzed and 0% probability of toxicity (AUC48–72≥700) for vancomycin clearance >2 L/h. Although not prospectively validated in a clinical study, the authors provide a practical empiric vancomycin dosing nomogram for obese patients that will likely maximize efficacy and minimize toxicity based on AUC targets.
Clindamycin
Clindamycin, first available in the late 1960s,49 is a bacteriostatic agent of the lincosamide class and works by blocking bacterial protein synthesis by inhibiting the peptidyltransferase reaction on the 50S subunit of the bacterial ribosome.49,50 Clindamycin is lipophilic and widely distributed in many fluids and tissues that can be affected by the larger than expected Vd of obese patients, thus potentially causing plasma drug concentrations to be low.51 It exhibits time-dependent killing and prolonged persistent effects, therefore maximizing the amount of drug is the most ideal approach to ensure efficacy.51
The usual dosing recommendation for clindamycin in MRSA-related infections is 300–450 mg orally three times daily for uncomplicated skin and soft tissue infection and 600 mg orally or intravenously three times daily for complicated skin and soft tissue infection, pneumonia, and osteomyelitis.35 Higher doses of 2,700–4,800 mg/day divided in 2, 3, or 4 equal doses are included in the drug labeling, but doses on the upper end (>3 g) of this range are seldom used clinically.10
A retrospective study of 50 adult patients who had received oral or intravenous clindamycin for treatment of osteomyelitis utilized a population pharmacokinetic model to determine the influence of covariates, including body weight. The body weight of the patients ranged from 23 to 133 kg, and the majority received 600 mg of clindamycin orally or intravenously three times a day, except for one patient who received 600 mg four times a day and two patients who received 600 mg once a day. The results showed that clindamycin clearance increased with body weight and concluded that 600 mg three times a day is effective for ABW less than 75 kg with the suggestion that the dose should be increased to 900 mg every 8 hours for >75 kg.50
A retrospective cohort study of 210 patients admitted for cellulitis/cutaneous abscess identified risk factors for clinical failure, including treatment with clindamycin. The population of the study was relatively obese with an average weight of 101 kg and a BMI of 34 kg/m2, including 21.9% morbidly obese patients (BMI≥40 kg/m2). Weight over 100 kg and BMI≥40 kg/m2 were identified as independent risk factors for clinical failure. A subgroup analysis demonstrated that morbidly obese patients were at a higher risk for clinical failure if they were discharged on a low dose of clindamycin (defined as 150–300 mg orally every 6–8 hours) versus high dose (defined as >300 mg every 8 hours).52 Given the findings of these two retrospective studies, it is evident that lower doses of clindamycin can place a patient at risk of treatment failure. When using higher doses of clindamycin, clinicians should be aware that gastrointestinal side effects are positively correlated to total dosage.53
Tetracyclines
Tetracyclines interfere with protein synthesis by reversibly binding to the bacterial 30S ribosomal subunit, subsequently preventing binding of tRNA to the mRNA-ribosome complex.54 Tetracycline antibiotics are highly lipophilic, and doxycycline and minocycline are approximately 3–5 times more lipophilic than tetracycline.54 As such, there is reason to suspect that the disposition of these antibiotics is altered in extremely high body weight. Despite this concern, there are limited data evaluating these antibiotics in obesity. Like other tetracycline derivatives, tigecycline has a large Vd resulting in wide tissue distribution and very low plasma drug concentrations. In a study including eight obese (median BMI 43.8 kg/m2) subjects, serum and urine pharmacokinetic parameters were similar to normal weight subjects.55 However, two population pharmacokinetic studies found that tigecyline clearance increased with increasing body weight.56,57 One option is to increase the dosage to 100 mg twice daily following a 200 mg loading dose (compared to the standard dose of 100 mg loading dose followed by 50 mg twice daily); however, this strategy is associated with an increased risk of nausea and vomiting.58 Further, evidence to support this strategy is inconclusive as different authors have reported contradicting conclusions on the influence of obesity on pharmacokinetics. Regardless of body habitus, tigecyline carries a black box warning for increased all-cause mortality versus comparators and therefore should be limited to last line treatment option.59 Further, in obese patients with invasive infections, tigecycline monotherapy is not recommended even as the last line due to rapid distribution into tissues and inadequate serum concentrations.
Sulfamethoxazole-trimethoprim
Trimethoprim (TMP)/sulfamethoxazole (SMX) is a broad spectrum antimicrobial combination that has been utilized in the outpatient setting for over 50 years. TMP competitively inhibits the production of dihydro-folate reductase and SMX interferes with the production of dihydrofolate.60 The co-administration of these agents are synergistic and the combination is bactericidal against most organisms in vitro.61 Both components are well distributed (1.4–1.8 L/kg, TMP and 0.43 L/kg, SMX) after administration.62 TMP is primarily cleared through the kidneys (excreted 75–85% unchanged in urine) while sulfamethoxazole is metabolized by liver microsomes (cytochrome P450 2C9).63,64 The dosing of this agent has primarily been extrapolated from pediatric cohorts and has displayed significant interindividual variability in the adult population.65–68 The recommended dosing, based on small pharmacokinetic studies, has been capped at 100 mg/kg/day for SMX and 20 mg/kg/day for TMP, based on patient’s ABW.69,70 Intravenous and oral dosing are essentially equivalent due to the high oral bioavailability of the agent. Dosing and concentrations are organism dependent, with varying target concentrations existing for each specific organism as well as the location of the infection.71,72 Clinical decision-making is imperative to balance exposure versus safety profile in each patient.71 Target concentrations for SMX at the site of infection have been observed to be 100–150 µg/mL with increased toxicities occurring with concentrations greater than 200 µg/mL, yet the limited data linking the relationship between target SMX concentrations and outcomes deters routine therapeutic drug monitoring.73
TMP/SMX is a lipophilic, highly protein-bound (44% for TMP, 70% for SMX) agent that is predominantly excreted through the kidneys. A pharmacokinetic study in healthy subjects determined the Vd of each agent; with TMP having a larger distribution at 1.4 L/kg versus 0.4 L/kg for SMX.65,74 Data are lacking on the variables affecting the pharmacokinetics of TMP/SMX; critical illness and trauma have been shown to increase the Vd, but other factors such as shock, fluid resuscitation and obesity are not well studied.71,75 There is a concerning lack of data on the appropriate weight metrics for dosing of TMP/SMX. A small study by Garrett et al found no difference in the Vd of sulfisoxazole (a sulfonamide with similar pharmacokinetic profile) in morbidly obese patients over time after jejunal-ileal bypass surgery resulting in up to a 44% reduction in body mass.76 Hall et al, on the other hand, demonstrated a decrease in maximum plasma concentration (Cmax) and AUCs in overweight patients.70
The lack of knowledge of target concentrations and appropriate dosing for TMP/SMX, especially in the setting of MRSA infections, is concerning, and more information is required in this arena.77–81 In a recent retrospective analysis, Dao et al attempted to link therapeutic drug monitoring and dosage optimization and were unable to determine appropriate dosing regimens with target concentration attainment.73 Small studies have shown that TMP/SMX is successful in eradicating MRSA infections but the heterogeneity of drug dosing and lack of available drug concentrations make determination of optimal dosing strategies difficult.79,80 In conclusion, ABW should be utilized clinically to ensure adequate concentrations are being reached, while monitoring closely for adverse effects associated with higher doses.
Linezolid/tedizolid
Linezolid (FDA approval April 2000) and tedizolid (FDA approval May 2014) are part of the oxazolidinone class of antibiotics. The mechanism of action of the oxazolidinones is through binding of the 50S bacterial ribosomal subunit.82 That prevents the formation of the 70S initiation complex that leads to protein synthesis. This class is effective for the treatment of skin and soft tissue infections and lung infections caused by Gram-positive organisms, particularly MRSA.83 Linezolid and tedizolid are available in intravenous and oral formulations with high bioavailability (linezolid, 100% and tedizolid, >80%).84,85 Linezolid Vd approximates the total body water compartment (40–50 L), is cleared by renal and non-renal mechanisms, and elimination half-life ranges from 3.4 to 7.4 hours.85 Tedizolid Vd ranges from 67 to 80 L, is predominantly eliminated hepatically although not a substrate of cytochrome P450 and half-life is approximately 9 hours.86
The first of the oxazolidinones was linezolid. It has time-dependent killing with both percentage of time over the MIC (T>MIC) and the AUC:MIC.82 T>MIC of 82% was correlated to bacterial eradication. In a murine model, a 24-hour AUC:MIC ratio of ~83 (range of 39–167) is required for bacteriostatic activity against staphylococci.87 In healthy volunteers, linezolid was found to have a Vd of 40–50 L and plasma protein binding of 31%. Its elimination half-life is 3.4–7.4 hours, and 65% is cleared by non-renal mechanisms.85 In vivo, linezolid undergoes slow non-enzymatic oxidation by ubiquitous reactive species into inactive metabolites.85,88 Linezolid demonstrated good penetration into bone, joints, and soft tissues, making it an attractive option for treatment of skin and soft tissue infections and staphylococcal pneumonia.85,89
The standard dose for linezolid is 600 mg every 12 hours with no dose adjustment for moderate renal or hepatic dysfunction. Due to its hydrophobic properties, linezolid is less susceptible to changes of the extracellular fluid volume.83 However, studies have shown that linezolid is cleared more quickly in critically ill patients, indicating other hypermetabolic factors are resulting in a lower serum concentration of linezolid.82,90,91 Stein et al obtained serum samples from 7 patients with an actual body weight >50% of their ideal body weight (IBW) after administration of linezolid 600 mg by mouth every 12 hours for treatment of cellulitis.87 Although all patients had achieved clinical cure after 12 days of therapy, the serum concentrations measured were diminished compared to those in non-obese patients; the mean linezolid serum concentration at one hour was 12.3 µg/mL compared to 16.3–24 µg/mL in previous studies. This reduction in AUC:MIC resulted in prolonged activity in S. aureus with a MIC of 1.0 µg/mL and a lack of activity to those with a MIC>2 µg/mL.87 In healthy adults, linezolid reached tissue concentrations that were active against pathogens with a MIC up to 4 µg/mL.85 Additionally, the decrease in serum concentrations can be related to the variability in Vd of linezolid due to an increased amount of adipose tissue in obese patients. Adipose tissues of obese mice have also been shown to have a higher production of reactive oxygen species, thus could increase the non-enzymatic oxidation of linezolid.83
One approach is to optimize its time-dependent killing with increased dosing frequency. Corcione et al reported variability in linezolid serum concentrations of two patients with BMI≥40 kg/m2 who received linezolid 600 mg intravenously every 8 hours, suggesting that increasing the dosing frequency may not achieve desired targets. Despite an increased frequency, the patient with a BMI of 71 kg/m2 could only achieve an AUC0–24 of 55.05 mg h/L.83 Also, in contrast to the potential pharmacodynamic benefits of increased dosing frequency, 600 mg every 8 hours is likely associated with an increased risk of toxicity (ie thrombocytopenia) due to higher trough concentrations.92 De Pascale et al compared linezolid concentrations in the plasma and epithelial lining fluids when administered by intermittent or continuous infusion to 22 obese (median BMI of 33.2 kg/m2) critically ill patients for treatment of ventilator-associated pneumonia.93 Although the T>MIC was higher in the continuous infusion group, the AUC:MIC and measured penetration into the lungs were not statistically different. Additionally, there was no detectable added benefit with the use of continuous infusion and no difference in clinical improvement on day 4.93
Tedizolid was the second oxazolidinone that was FDA approved for the treatment of acute bacterial skin and skin structure infections caused by MRSA. It is administered in its prodrug form, tedizolid phosphate, requiring activation by phosphatases into the active form, tedizolid.94 The standard dose is 200 mg daily without the need for dose adjustment in patients with hepatic or renal impairment. It also distributes well into the skin and soft tissue. The efficacy of tedizolid is dependent on the AUC:MIC ratio.84 Few data are available evaluating this drug in obesity. One study evaluated 18 patients (9 obese and 9 normal weight-matched control subjects) with either a BMI of ≥40 kg/m2 or of 18.5–29.9 kg/m2. The tedizolid median Cmax and AUC were not significantly different compared to non-obese controls.94
Overall, caution is required as altered pharmacokinetics and pharmacodynamics in obesity has shown to affect the serum concentrations of linezolid and tedizolid. Obese patients will have a larger Vd and more reactive oxygen species, which results in lower serum concentrations of linezolid and possibly tedizolid. That can ultimately prevent linezolid and tedizolid obtaining the optimal AUC:MIC ratio to inhibit the growth of MRSA with MIC≥2. However, there is no clear guidance for dose adjustments as the clinical significance of these pharmacokinetic and pharmacodynamic alterations are limited due to the limited number of studies in obese patients and small sample sizes.
Daptomycin
Daptomycin is a cyclic lipopeptide antibiotic FDA approved (September 2003) for the treatment of complicated skin and skin structure infections caused by susceptible Gram-positive bacteria and staphylococcal bacteremia including infective endocarditis. Daptomycin exerts its bactericidal effects through various mechanisms, including insertion into and disruption of the bacterial cytoplasmic membrane.95 Daptomycin also has the ability to inhibit bacterial protein, DNA, RNA, and lipoteichoic acid synthesis.95,96 Unlike beta-lactams that are effective primarily during bacterial replication, daptomycin is active at all bacterial growth phases including the stationary phase.95 These qualities are desirable for treating deep-seated and indolent serious infections that involve MRSA.95 Pharmacokinetics of daptomycin are independent of time and linear up to doses of 12 mg/kg administered for 14 days.97 Daptomycin is eliminated primarily by the kidneys and requires dose adjustment in patients with a creatinine clearance <30 mL/min. The Vd is approximately 0.1 L/kg and limited primarily to the extracellular fluid. Approximately 92% of the administered dose is protein bound.
There have been several pharmacokinetic studies evaluating daptomycin in obese and non-obese individuals. The importance of considering ABW when dosing daptomycin is underscored by a population pharmacokinetic analysis which included 29 extremely obese (BMI>40 kg/m2), 333 moderately obese (BMI 25–40 kg/m2), and 255 non-obese subjects (BMI<25 kg/m2).98 This analysis determined that ABW was a potential factor influencing clearance and Vd. While both moderately and extremely obese subjects had a 28% and 42% higher AUC, respectively, these values were within the coefficient of variation (55%) seen in normal weight subjects. Dvorchik et al evaluated daptomycin 4 mg/kg based on ABW as a single dose in 6 morbidly obese (BMI>40 kg/m2), 6 moderately obese (BMI 25–39.9 kg/m2) and 2 non-obese healthy subjects.99 Compared to the non-obese subjects, Vd was increased by 58% and 24% in the moderately and morbidly obese subjects, respectively. AUC and Cmax were increased by 25% and 30%, respectively, compared to the non-obese subjects. Similarly, Pai et al compared the pharmacokinetics of a single daptomycin 4 mg/kg dose based on ABW in 7 morbidly obese versus 7 non-obese female subjects.100 AUC was significantly increased by approximately 61% and Cmax by 59% in the morbidly obese subjects compared to non-obese control subjects. The difference in Vd and clearance were increased in morbidly obese subjects but failed to reach statistical significance. Furthermore, the Vd was strongly correlated to ABW and not IBW. In addition to pharmacokinetic studies, Ng et al described an institution-wide protocol that switched from an ABW to IBW dosing strategy for daptomycin.101 They reported no significant differences in microbiological outcomes, length of stay, mortality, or adverse effects. Importantly, the mean body mass index in this analysis was approximately 31 kg/m2 and treatment success rates range from 79% to 100% depending on the etiology of infection. While these data suggest IBW dosing may be appropriate, the external validity of the findings is limited by the small sample size (n=117), single institution, and dosing strategy of 4–6 mg/kg.
Pharmacokinetic studies completed to date suggest ABW as the appropriate descriptor to use when selecting the dosage. Although these studies used doses of 4–6 mg/kg, it is common to prescribe daptomycin doses of up to 8–10 mg/kg in the treatment of severe infections. Pharmacokinetics have been reported as linear up to 12 mg/kg in normal weight patients.95 The primary safety concern with daptomycin is myositis and elevated creatinine phosphokinase. This adverse effect typically occurs after 2 weeks of exposure, and the probability of an elevated creatinine phosphokinase with a minimum plasma concentration (Cmin) of ≥24.3 mg/L is 50% compared to 2.9% when the Cmin is below this value.102 Based on Monte Carlo simulations, using IBW in patients weighing more than 111 kg would decrease the probability of achieving a Cmin of ≥24.3 mg/L and therefore decrease the probability of creatinine phosphokinase elevations. Nonetheless, in patients with extreme obesity clinicians should weigh the risks and benefits of dosing based on ABW when high dose daptomycin is considered. In many cases, the risks of underdosing and treatment failure are greater than the risk of toxicity from using ABW. In obese patients treated with high dose daptomycin, it is prudent to monitor for signs of toxicity and check creatinine phosphokinase at least weekly and perhaps more frequently if the patient has other risk factors for elevated creatinine phosphokinase or muscle toxicity.103,104
Recently, a population pharmacokinetic study evaluated daptomycin exposure and safety of fixed versus weight-based dosing in morbidly obese and nonobese healthy subjects.105 Monte Carlo simulations were performed to compare pharmacokinetic parameters (Cmax, Cmin, and AUC) of daptomycin 6 mg/kg/day or 500 mg daily. As compared to nonobese subjects, morbidly obese subjects who received 6 mg/kg/day had an approximately two times higher AUC, Cmax, and Cmin. Subjects given a fixed dose of 500 mg daily demonstrated relatively isometric exposure measures between the two groups. Also, there was a higher proportion of morbidly obese subjects given 6 mg/kg/day that had a Cmin associated with creatinine phosphokinase elevations (Cmin>24.3 mg/L) as compared to those that received 500 mg daily. Further clinical studies are needed in order to evaluate the comparative clinical effectiveness of fixed-dose regimens, particularly in patients with severe MRSA infections (ie bacteremia) where daptomycin is generally used for treatment.
Ceftaroline
Ceftaroline fosamil, the prodrug of ceftaroline, is a broad spectrum fifth-generation cephalosporin FDA approved (October 2010) for the treatment of acute bacterial skin and skin structure infections and community-acquired pneumonia. Ceftaroline exhibits potent activity against Streptococcus pyogenes, Streptococcus agalactiae, and Streptococcus pneumoniae, Escherichia coli, Klebsiella pneumoniae, and β-lactamase-positive and negative isolates of Haemophilus influenzae.106 In addition, in vitro studies support the efficacy of ceftaroline against methicillin-susceptible and methicillin-resistant isolates of S. aureus as well as isolates with reduced susceptibility to vancomycin or linezolid.107 These data have been corroborated in randomized controlled trials where subsets of patients with MRSA skin and skin structure infections achieved clinical success with ceftaroline.108–110 Ceftaroline exerts its bactericidal effect by binding to the penicillin-binding proteins (PBP) 1–3 and inhibiting bacterial cell wall synthesis.
Ceftaroline has a low Vd (20.3 L) and is not extensively protein bound (approximately 20%).106 Both of these attributes are favorable in the setting of obesity since ceftaroline distribution is primarily in the total body water compartment and protein binding may be altered in obesity. The primary route of elimination is through the kidneys (88%).106 The pharmacokinetics of ceftaroline have been evaluated in 32 healthy normal weight and obese volunteer subjects.111 Subjects were evenly assigned to one of four groups (normal to overweight; BMI, 18.5–29.9 kg/m2; obese class I; BMI, 30–34.9 kg/m2; obese class II; BMI, 35–39.9 kg/m2; obese class III; BMI, >39.9 kg/m2). Mean ceftaroline Cmax was 30% lower in subjects with a BMI≥40 kg/m2 compared to subjects with a BMI<30 kg/m2. Despite the lower concentration, Monte Carlo simulations performed suggested that when the MIC is ≤1 µg/mL, the probability of target attainment is ≥90%. Additionally, outcomes in obese patients (defined as BMI≥30 kg/m2) were evaluated in The Clinical Assessment Program and TEFLARO Utilization Registry (CAPTURE) multicentre retrospective cohort study.112 Data were collected from 261 normal BMI patients and 690 patients with a BMI≥30 kg/m2 (of which 239 [34.6%] had a BMI≥40 kg/m2) receiving ceftaroline for acute bacterial skin and skin structure infections. Outcomes in the obese patients were similar to normal BMI patients (clinical success rates ranging from 85.1% to 89.0%). Collectively these data suggest that no dosage adjustment is necessary for ceftaroline in obese patients and this treatment represents a viable option when there is a concern for inadequate antibiotic exposure due to extreme weight. Higher dosing (ie, every 8-hour regimen) may be considered for severe MRSA infections although further studies are needed to assess any clinical advantage to higher dosing in this scenario.113
Telavancin/dalbavancin/oritavancin
Telavancin, dalbavancin, and oritavancin are the newest of the glycopeptide antibiotic class with the latter two FDA approved in 2014 for the treatment of acute bacterial skin and skin structure infections in adult patients. Telavancin was initially approved in 2009 for treatment of complicated skin and skin structure infections and more recently approved in 2013 for hospital-acquired and ventilator-associated bacterial pneumonia.114 Compared to previously discussed antibiotics, pharmacokinetic and clinical data regarding the newer glycopeptides are limited.
Telavancin is a highly protein-bound drug (90%) that is primarily excreted in the urine (~75%) with an elimination half-life of approximately 8 hours.114 In obese patients defined as BMI≥35 kg/m2, the mean AUC is approximately 26–35% higher in comparison to non-obese patients (BMI<35 kg/m2). Also in obese patients, the drug clearance (L/h) and Cmax is 20% and 10% higher, respectively, versus non-obese patients.115 In a population pharmacokinetic study that included both healthy subjects and infected patients, AUC changes were minimal despite increases in doses for obese individuals. For the complicated skin and skin structure infections model, a 50% dose increase in obese (BMI≥35 kg/m2) patients only resulted in a 34% higher median AUC for obese compared to non-obese patients. This was similar in the hospital-acquired pneumonia model in which a 55% dose increase in obese patients resulted in an 18% increase in the median AUC.116 Dosing for patients in clinical trials is based on both ABW and renal function as measured by CrCl.117 Clinical data for use of telavancin in obese patients is primarily derived from post hoc analyses from two phase 3 clinical trials evaluating treatment of skin infections (ATLAS studies). Clinical cure rates were similar for telavancin versus the comparator vancomycin in the obese (BMI≥35 kg/m2) subgroup (72% and 73%, respectively).118 The specific pathogen (ie, MRSA vs other bacteria) was not delineated in the post hoc analysis. Based on limited post hoc data analysis, pharmacokinetic changes in obese patients were minimal, which is consistent with clinical findings in which clinical cure rates for skin infections were similar. Also important to note is that there is growing evidence that telavancin flat dosing at 750 mg daily performs similarly to weight-based dosing.119 Recently, a study was completed assessing single-dose pharmacokinetics of weight-stratified fixed dose telavancin in obese and non-obese healthy subjects. A fixed dose in obese patients of 750 mg (ABW 90–99.9 kg) or 1,000 mg (ABW≥100 kg; maximum dose) was compared to weight-based dosing for the probability of AUC target attainment. The fixed-dose strategy resulted in more uniform AUC measures as compared to the projected AUC measures from the current standard, ABW-based dosing. The authors concluded that a fixed dose of 750 mg is a potentially safe and effective alternative to weight-based (total or adjusted) dosing for telavancin in obese patients with normal renal function.120
Dalbavancin is the first approved long-acting glycopeptide antibiotic with currently two dosing regimens. The first is a two-dose regimen of 1,000 mg followed by 500 mg one week later. More recently, a regimen of 1,500 mg as a single dose was approved for the treatment of acute bacterial skin and skin structure infections. It has a half-life of 346 hours with approximately 33% of the drug excreted unchanged in urine.121 Data with dalbavancin in obese patients is limited to post hoc analyses of two phase 3 trials of the two-dose regimen versus vancomycin. Overall efficacy of dalbavancin was similar to vancomycin regardless of BMI. In patients with BMI≥30 kg/m2 vs 25–<30 kg/m2 vs <25 kg/m2, there were similar clinical cure rates for dalbavancin as well.122 There is no available data for clinical efficacy specifically in obese patients with MRSA infections. For acute bacterial skin and skin structure infections, cure rates seem to be unaffected by BMI and require no dose adjustments.
Oritavancin is the second FDA approved long-acting glycopeptide antibiotic given as a single dose regimen (1,200 mg). It has a long half-life of approximately 245 hours.123 In an efficacy analysis of subgroups from two pivotal phase 3 clinical trials, there were similar rates of the primary efficacy outcome in patients with BMI≥30 kg/m2 in the oritavancin and vancomycin treatment arms (77.3% and 74.9%, respectively).124 In a separate analysis of the same acute bacterial skin and skin structure infections clinical trial data, there was not a statistically significant difference of clinical success for BMI≥29.1 kg/m2 in comparison to BMI<24.8 kg/m2 specifically for patients with S. aureus (odds ratio 0.377 [95% CI, 0.087–1.63]).125 Additionally, in a population pharmacokinetic analyses of the same phase 3 clinical trial patients (mean weight 79.9 kg, range 42.7–178 kg), covariate analyses concluded no dose adjustment is needed for weight or BMI given the lack of relation to the interindividual variability in oritavancin pharmacokinetics. The standard dose (1,200 mg) is appropriate regardless of variations in body size based on this pharmacokinetic analysis.126 Based on limited pharmacokinetic and clinical data in the context of acute bacterial skin and skin structure infections, there appears to be no significant difference in outcomes for oritavancin in subgroups of obese patients although conclusions must be drawn cautiously, given the absence of high-quality evidence.
Summary and conclusions
There are a variety of agents available to treat MRSA infections. As the obesity epidemic continues to reach new heights, clinicians are frequently faced with decisions related to drug selection and dosing strategy when managing these infections. The general recommendation is to dose at the higher end of the dosing range and use therapeutic drug monitoring if available. Another important consideration is the source of infection as different antibiotics may be preferred depending on the setting. Table 1 provides a summary of the anti-MRSA antibiotics discussed in this manuscript and includes pertinent antibiotic information to help clinicians select optimal therapy and choose the most appropriate dose.
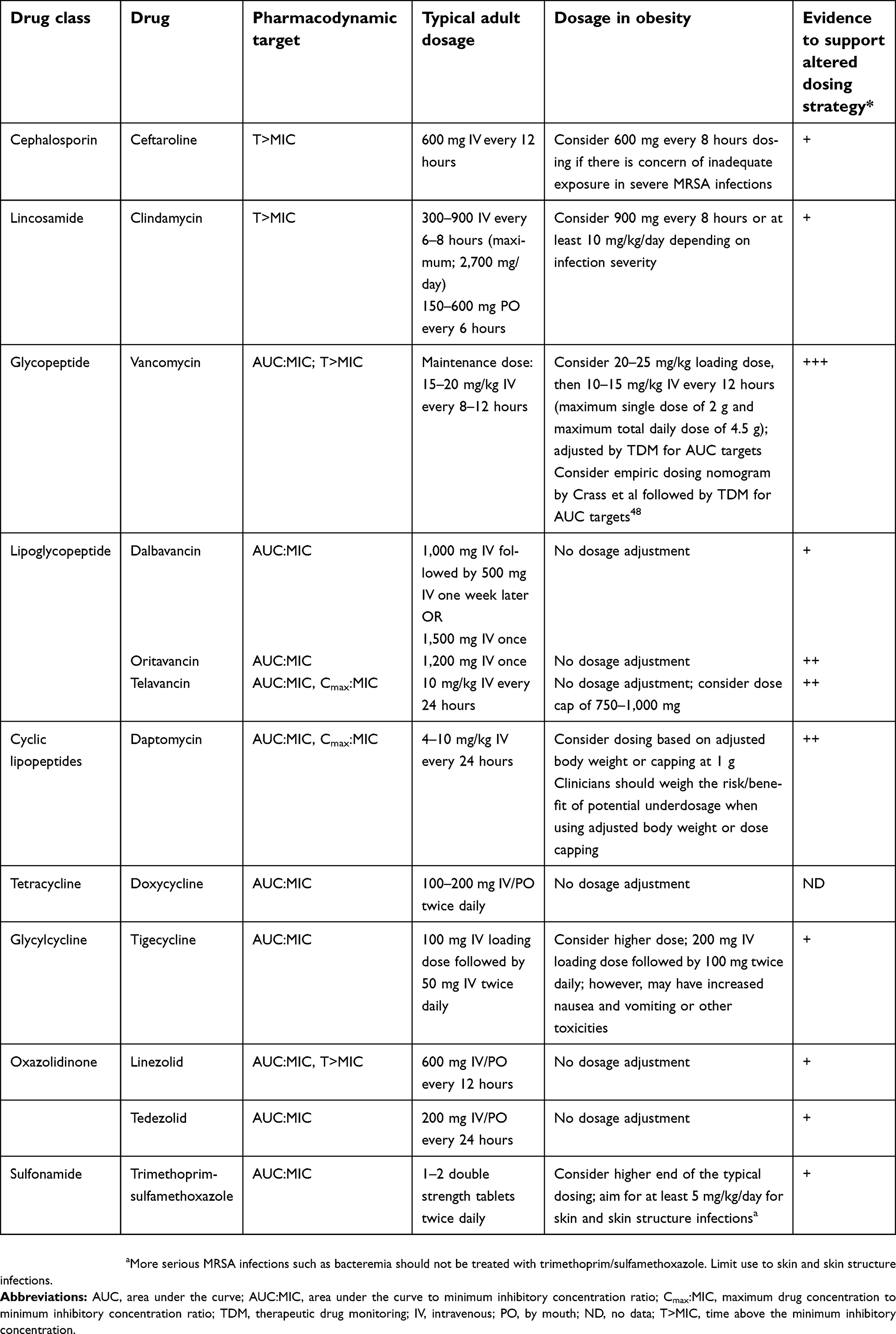 | Table 1 Summary of antibiotic characteristics for agents used to treat methicillin-resistant Staphylococcus aureus infections |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jevons MP. “Celbenin” - resistant Staphylococci. Br Med J. 1961;1(5219):124–125. doi:10.1136/bmj.1.5219.124-a
2.
3. Sievert DM, Ricks P, Edwards JR, et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol. 2013;34(1):1–14. doi:10.1086/668770
4. Klein E, Smith DL, Laxminarayan R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis. 2007;13(12):1840–1846. doi:10.3201/eid1312.070629
5. Klein EY, Sun L, Smith DL, Laxminarayan R. The changing epidemiology of methicillin-resistant Staphylococcus aureus in the United States: a national observational study. Am J Epidemiol. 2013;177(7):666–674. doi:10.1093/aje/kws273
6. Kock R, Becker K, Cookson B, et al. Methicillin-resistant Staphylococcus aureus (MRSA): burden of disease and control challenges in Europe. Euro Surveill. 2010;15(41):19688. doi:10.2807/ese.15.41.19688-en
7. Dodds Ashley ES, Carroll DN, Engemann JJ, et al. Risk factors for postoperative mediastinitis due to methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2004;38(11):1555–1560. doi:10.1086/420819
8. Early GJ, Seifried SE. Risk factors for community-associated Staphylococcus aureus skin infection in children of Maui. Hawaii J Med Public Health. 2012;71(8):218–223.
9. Khawcharoenporn T, Tice AD, Grandinetti A, Chow D. Risk factors for community-associated methicillin-resistant Staphylococcus aureus cellulitis–and the value of recognition. Hawaii Med J. 2010;69(10):232–236.
10. Wilborn C, Beckham J, Campbell B, et al. Obesity: prevalence, theories, medical consequences, management, and research directions. J Int Soc Sports Nutr. 2005;2:4–31. doi:10.1186/1550-2783-2-2-4
11. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi:10.1161/01.cir.0000441139.02102.80
12. Cesare MD, Bentham J, Stevens GA, et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet. 2016;387(10026):1377–1396. doi:10.1016/S0140-6736(16)30054-X
13. Huttunen R, Syrjanen J. Obesity and the risk and outcome of infection. Int J Obes (Lond). 2013;37(3):333–340. doi:10.1038/ijo.2012.62
14. Longo C, Bartlett G, Macgibbon B, et al. The effect of obesity on antibiotic treatment failure: a historical cohort study. Pharmacoepidemiol Drug Saf. 2013;22(9):970–976. doi:10.1002/pds.3461
15. Caffrey AR, Noh E, Morrill HJ, LaPlante KL. The effects of obesity on the comparative effectiveness of linezolid and vancomycin in suspected methicillin-resistant Staphylococcus aureus pneumonia. Adv Pharmacoepidemiol Drug Saf. 2015;4:176. doi:10.4172/2167-1052.1000176
16. Boyd SE, Charani E, Lyons T, Frost G, Holmes AH. Information provision for antibacterial dosing in the obese patient: a sizeable absence? J Antimicrob Chemother. 2016;71(12):3588–3592. doi:10.1093/jac/dkw324
17. Janson B, Thursky K. Dosing of antibiotics in obesity. Curr Opin Infect Dis. 2012;25(6):634–649. doi:10.1097/QCO.0b013e328359a4c1
18. Toma O, Suntrup P, Stefanescu A, London A, Mutch M, Kharasch E. Pharmacokinetics and tissue penetration of cefoxitin in obesity: implications for risk of surgical site infection. Anesth Analg. 2011;113(4):730–737. doi:10.1213/ANE.0b013e31821fff74
19. Brill MJ, Houwink AP, Schmidt S, et al. Reduced subcutaneous tissue distribution of cefazolin in morbidly obese versus non-obese patients determined using clinical microdialysis. J Antimicrob Chemother. 2014;69(3):715–723. doi:10.1093/jac/dkt444
20. Barbour A, Schmidt S, Rout WR, Ben-David K, Burkhardt O, Derendorf H. Soft tissue penetration of cefuroxime determined by clinical microdialysis in morbidly obese patients undergoing abdominal surgery. Int J Antimicrob Agents. 2009;34(3):231–235. doi:10.1016/j.ijantimicag.2009.03.019
21.
22. Al-Dorzi HM, Al Harbi SA, Arabi YM. Antibiotic therapy of pneumonia in the obese patient: dosing and delivery. Curr Opin Infect Dis. 2014;27(2):165–173. doi:10.1097/QCO.0000000000000045
23. Poulin P, Schoenlein K, Theil FP. Prediction of adipose tissue: plasma partition coefficients for structurally unrelated drugs. J Pharm Sci. 2001;90(4):436–447.
24. Poulin P, Theil FP. A priori prediction of tissue: plasmapartition coefficients of drugs to facilitate the use of physiologically-based pharmacokinetic models in drug discovery. J Pharm Sci. 2000;89(1):16–35. doi:10.1002/(SICI)1520-6017(200001)89:1<16::AID-JPS3>3.0.CO;2-E
25. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71–87. doi:10.2165/11318100-000000000-00000
26. Morrish GA, Pai MP, Green B. The effects of obesity on drug pharmacokinetics in humans. Expert Opin Drug Metab Toxicol. 2011;7(6):697–706. doi:10.1517/17425255.2011.570331
27. Leykin Y, Miotto L, Pellis T. Pharmacokinetic considerations in the obese. Best Pract Res Clin Anaesthesiol. 2011;25(1):27–36.
28. Alexander JK, Dennis EW, Smith WG, Amad KH, Duncan WC, Austin RC. Blood volume, cardiac output, and distribution of systemic blood flow in extreme obesity. Cardiovasc Res Cent Bull. 1962;1:39–44.
29. Zavorsky GS. Cardiopulmonary aspects of obesity in women. Obstet Gynecol Clin North Am. 2009;36(2):267–284, viii. doi:10.1016/j.ogc.2009.03.006
30. Casati A, Putzu M. Anesthesia in the obese patient: pharmacokinetic considerations. J Clin Anesth. 2005;17(2):134–145. doi:10.1016/j.jclinane.2004.01.009
31. Brill MJ, Diepstraten J, van Rongen A, van Kralingen S, van Den Anker JN, Knibbe CA. Impact of obesity on drug metabolism and elimination in adults and children. Clin Pharmacokinet. 2012;51(5):277–304. doi:10.2165/11599410-000000000-00000
32. Pai MP. Estimating the glomerular filtration rate in obese adult patients for drug dosing. Adv Chronic Kidney Dis. 2010;17(5):e53–e62. doi:10.1053/j.ackd.2010.05.010
33. Maddox A, Horowitz M, Wishart J, Collins P. Gastric and oesophageal emptying in obesity. Scand J Gastroenterol. 1989;24(5):593–598.
34. Jackson SJ, Leahy FE, McGowan AA, Bluck LJ, Coward WA, Jebb SA. Delayed gastric emptying in the obese: an assessment using the non-invasive (13)C-octanoic acid breath test. Diabetes Obes Metab. 2004;6(4):264–270. doi:10.1111/j.1462-8902.2004.0344.x
35. Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–55. doi:10.1093/cid/ciq146
36. Rybak M, Lomaestro B, Rotschafer JC, et al. therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. am j health syst pharm. 2009;66(1):82–98. doi:10.2146/ajhp080434
37. Rybak MJ. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin Infect Dis. 2006;42(Suppl 1):S35–39. doi:10.1086/491712
38. Grace E. Altered vancomycin pharmacokinetics in obese and morbidly obese patients: what we have learned over the past 30 years. J Antimicrob Chemother. 2012;67(6):1305–1310. doi:10.1093/jac/dks066
39. Blouin RA, Bauer LA, Miller DD, Record KE, Griffen WO
40. Richardson J, Scheetz M, O’Donnell EP. The association of elevated trough serum vancomycin concentrations with obesity. J Infect Chemother. 2015;21:507–511. doi:10.1016/j.jiac.2015.03.007
41. Morrill HJ, Caffrey AR, Noh E, LaPlante KL. Vancomycin dosing considerations in a real-world cohort of obese and extremely obese patients. Pharmacotherapy. 2015;35(9):869–875. doi:10.1002/phar.1625
42. Kubiak DW, Alquwaizani M, Sansonetti D, Barra ME, Calderwood MS. An evaluation of systemic vancomycin dosing in obese patients. Open Forum Infect Dis. 2015;2(4):ofv176. doi:10.1093/ofid/ofv176
43. Kosmisky DE, Griffiths CL, Templin MA, Norton J, Martin KE. Evaluation of a new vancomycin dosing protocol in morbidly obese patients. Hosp Pharm. 2015;50(9):789–797. doi:10.1310/hpj5009-789
44. Adane ED, Herald M, Koura F. Pharmacokinetics of vancomycin in extremely obese patients with suspected or confirmed Staphylococcus aureus infections. Pharmacotherapy. 2015;35(2):127–139. doi:10.1002/phar.1531
45. Reynolds DC, Waite LH, Alexander DP, DeRyke CA. Performance of a vancomycin dosage regimen developed for obese patients. Am J Health Syst Pharm. 2012;69(11):944–950. doi:10.2146/ajhp110324
46. Prybylski JP. Vancomycin trough concentration as a predictor of clinical outcomes in patients with Staphylococcus aureus bacteremia: a meta-analysis of observational studies. Pharmacotherapy. 2015;35(10):889–898. doi:10.1002/phar.1638
47. Finch NA, Zasowski EJ, Murray KP, et al. A quasi-experiment to study the impact of vancomycin area under the concentration-time curve-guided dosing on vancomycin-associated nephrotoxicity. Antimicrob Agents Chemother. 2017;61(12). doi:10.1128/AAC.01293-17.
48. Crass RL, Dunn R, Hong J, Krop LC, Pai MP. Dosing vancomycin in the super obese: less is more. J Antimicrob Chemother. 2018;73(11):3081–3086.
49. Smieja M. Current indications for the use of clindamycin: a critical review. Can J Infect Dis. 1998;9(1):22–28.
50. Bouazza N, Pestre V, Jullien V, et al. Population pharmacokinetics of clindamycin orally and intravenously administered in patients with osteomyelitis. Br J Clin Pharmacol. 2012;74(6):971–977. doi:10.1111/j.1365-2125.2012.04292.x
51. Levison ME, Levison JH. Pharmacokinetics and pharmacodynamics of antibacterial agents. Infect Dis Clin North Am. 2009;23(4):791–815, vii. doi:10.1016/j.idc.2009.06.008
52. Halilovic J, Heintz BH, Brown J. Risk factors for clinical failure in patients hospitalized with cellulitis and cutaneous abscess. J Infect. 2012;65(2):128–134. doi:10.1016/j.jinf.2012.03.013
53. Lusk RH, Fekety FR
54. Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother. 2006;58(2):256–265. doi:10.1093/jac/dkl224
55. Pai MP. Serum and urine pharmacokinetics of tigecycline in obese class III and normal weight adults. J Antimicrob Chemother. 2014;69(1):190–199. doi:10.1093/jac/dkt299
56. Xie J, Roberts JA, Alobaid AS, et al. Population pharmacokinetics of tigecycline in critically ill patients with severe infections. Antimicrob Agents Chemother. 2017;61(8). doi:10.1128/AAC.00345-17.
57. Van Wart SA, Owen JS, Ludwig EA, Meagher AK, Korth-Bradley JM, Cirincione BB. Population pharmacokinetics of tigecycline in patients with complicated intra-abdominal or skin and skin structure infections. Antimicrob Agents Chemother. 2006;50(11):3701–3707. doi:10.1128/AAC.01636-05
58. Falagas ME, Vardakas KZ, Tsiveriotis KP, Triarides NA, Tansarli GS. Effectiveness and safety of high-dose tigecycline-containing regimens for the treatment of severe bacterial infections. Int J Antimicrob Agents. 2014;44(1):1–7. doi:10.1016/j.ijantimicag.2014.01.006
59. Yahav D, Lador A, Paul M, Leibovici L. Efficacy and safety of tigecycline: a systematic review and meta-analysis. J Antimicrob Chemother. 2011;66(9):1963–1971. doi:10.1093/jac/dkr242
60. Burman LG. The antimicrobial activities of trimethoprim and sulfonamides. Scand J Infect Dis. 1986;18(1):3–13.
61. Kielhofner MA. Trimethoprim- sulfamethoxazole: pharmacokinetics, clinical uses, and adverse reactions. Tex Heart Inst J. 1990;17(2):86–93.
62.
63. Siber GR, Gorham CC, Ericson JF, Smith AL. Pharmacokinetics of intravenous trimethoprim-sulfamethoxazole in children and adults with normal and impaired renal function. Rev Infect Dis. 1982;4(2):566–578.
64. Kremers P, Duvivier J, Heusghem C. Pharmacokinetic studies of co-trimoxazole in man after single and repeated doses. J Clin Pharmacol. 1974;14(2):112–117.
65. Chin TW, Vandenbroucke A, Fong IW. Pharmacokinetics of trimethoprim-sulfamethoxazole in critically ill and non-critically ill AIDS patients. Antimicrob Agents Chemother. 1995;39(1):28–33.
66. Blaser J, Joos B, Opravil M, Luthy R. Variability of serum concentrations of trimethoprim and sulfamethoxazole during high dose therapy. Infection. 1993;21(4):206–209.
67. Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. A prospective randomized trial. Ann Intern Med. 1986;105(1):37–44.
68. Bowden FJ, Harman PJ, Lucas CR. Serum trimethoprim and sulphamethoxazole levels in AIDS. Lancet. 1986;1(8485):853. doi:10.1016/S0140-6736(86)90958-X
69. Stevens RC, Laizure SC, Williams CL, Stein DS. Pharmacokinetics and adverse effects of 20-mg/kg/day trimethoprim and 100-mg/kg/day sulfamethoxazole in healthy adult subjects. Antimicrob Agents Chemother. 1991;35(9):1884–1890.
70. Hall RGN, Pasipanodya JG, Meek C, Leff RD, Swancutt M, Gumbo T. Fractal geometry-based decrease in trimethoprim-sulfamethoxazole concentrations in overweight and obese people. CPT Pharmacometrics Syst Pharmacol. 2016;5(12):674–681. doi:10.1002/psp4.12146
71. Brown GR. Cotrimoxazole - optimal dosing in the critically ill. Ann Intensive Care. 2014;4:13. doi:10.1186/2110-5820-4-13
72. Dudley MN, Levitz RE, Quintiliani R, Hickingbotham JM, Nightingale CH. Pharmacokinetics of trimethoprim and sulfamethoxazole in serum and cerebrospinal fluid of adult patients with normal meninges. Antimicrob Agents Chemother. 1984;26(6):811–814.
73. Dao BD, Barreto JN, Wolf RC, Dierkhising RA, Plevak MF, Tosh PK. Serum peak sulfamethoxazole concentrations demonstrate difficulty in achieving a target range: a retrospective cohort study. Curr Ther Res Clin Exp. 2014;76:104–109. doi:10.1016/j.curtheres.2014.08.003
74.
75. Hess MM, Boucher BA, Laizure SC, et al. Trimethoprim-sulfamethoxazole pharmacokinetics in trauma patients. Pharmacotherapy. 1993;13(6):602–606.
76. Garrett ER, Suverkrup RS, Eberst K, Yost RL, O’Leary JP. Surgically affected sulfisoxazole pharmacokinetics in the morbidly obese. Biopharm Drug Dispos. 1981;2(4):329–365.
77. Polso AK, Lassiter JL, Nagel JL. Impact of hospital guideline for weight-based antimicrobial dosing in morbidly obese adults and comprehensive literature review. J Clin Pharm Ther. 2014;39(6):584–608. doi:10.1111/jcpt.12200
78. Goldberg E, Bishara J. Contemporary unconventional clinical use of co-trimoxazole. Clin Microbiol Infect. 2012;18(1):8–17. doi:10.1111/j.1469-0691.2011.03613.x
79. Goldberg E, Paul M, Talker O, et al. Co-trimoxazole versus vancomycin for the treatment of methicillin-resistant Staphylococcus aureus bacteraemia: a retrospective cohort study. J Antimicrob Chemother. 2010;65(8):1779–1783. doi:10.1093/jac/dkq179
80. Campbell ML, Marchaim D, Pogue JM, et al. Treatment of methicillin-resistant Staphylococcus aureus infections with a minimal inhibitory concentration of 2 mug/mL to vancomycin: old (trimethoprim/sulfamethoxazole) versus new (daptomycin or linezolid) agents. Ann Pharmacother. 2012;46(12):1587–1597. doi:10.1345/aph.1R211
81. Lemaire S, Kosowska-Shick K, Appelbaum PC, Glupczynski Y, Van Bambeke F, Tulkens PM. Activity of moxifloxacin against intracellular community-acquired methicillin-resistant Staphylococcus aureus: comparison with clindamycin, linezolid and co-trimoxazole and attempt at defining an intracellular susceptibility breakpoint. J Antimicrob Chemother. 2011;66(3):596–607. doi:10.1093/jac/dkq478
82. Morata L, Cuesta M, Rojas JF, et al. Risk factors for a low linezolid trough plasma concentration in acute infections. Antimicrob Agents Chemother. 2013;54(4):1913–1917. doi:10.1128/AAC.01694-12
83. Corcione S, Pagani N, Baietto L, et al. Pharmacokinetics of high dosage of linezolid in two morbidly obese patients. J Antimicrob Chemother. 2015;70(8):2417–2418. doi:10.1093/jac/dkv126
84. Flanagan S, Passarell J, Lu Q, Fiedler-Kelly J, Ludwig E, Prokocimer P. Tedizolid population pharmacokinetics, exposure response, and target attainment. Antimicrob Agents Chemother. 2014;58(11):6462–6470.
85. Dryden MS. Linezolid pharmacokinetics and pharmacodynamics in clinical treatment. J Antimicrob Chemother. 2011;66(Suppl 4):iv7–iv15. doi:10.1093/jac/dkr072
86.
87. Stein GE, Schooley SL, Peloquin CA, et al. Pharmacokinetics and pharmacodynamics of linezolid in obese patients with cellulitis. Ann Pharmacother. 2005;39(3):427–432. doi:10.1345/aph.1E484
88. Tsuji Y, Hiraki Y, Matsumoto K, et al. Evaluation of the pharmacokinetics of linezolid in an obese Japanese patient. Scand J Infect Dis. 2012;44(8):626–629. doi:10.3109/00365548.2011.652164
89. Lovering AM, Zhang J, Bannister GC, et al. Penetration of linezolid into bone, fat, muscle and haematoma of patients undergoing routine hip replacement. J Antimicrob Chemother. 2002;50(1):73–77.
90. Adembri C, Fallani S, Cassetta MI, et al. Linezolid pharmacokinetic/pharmacodynamic profile in critically ill septic patients: intermittent versus continuous infusion. Int J Antimicrob Agents. 2008;31(2):122–129. doi:10.1016/j.ijantimicag.2007.09.009
91. Thallinger C, Buerger C, Plock N, et al. Effect of severity of sepsis on tissue concentrations of linezolid. J Antimicrob Chemother. 2008;61(1):173–176. doi:10.1093/jac/dkm431
92. Cojutti P, Pai MP, Pea F. Population pharmacokinetics and dosing considerations for the use of linezolid in overweight and obese adult patients. Clin Pharmacokinet. 2018;57(8):989–1000. doi:10.1007/s40262-017-0606-5
93. De Pascale G, Fortuna S, Tumbarello M, et al. Linezolid plasma and intrapulmonary concentrations in critically ill obese patients with ventilator-associated pneumonia: intermittent vs continuous administration. Intensive Care Med. 2015;41(1):103–110. doi:10.1007/s00134-014-3550-y
94. Pai MP. Pharmacokinetics of tedizolid in morbidly obese and covariate-matched nonobese adults. Antimicrob Agents Chemother. 2016;60(8):4585–4589. doi:10.1128/AAC.00682-16
95. Hancock RE. Mechanisms of action of newer antibiotics for Gram-positive pathogens. Lancet Infect Dis. 2005;5(4):209–218. doi:10.1016/S1473-3099(05)70051-7
96. LaPlante KL, Rybak MJ. Daptomycin - a novel antibiotic against Gram-positive pathogens. Expert Opin Pharmacother. 2004;5(11):2321–2331. doi:10.1517/14656566.5.11.2321
97. Benvenuto M, Benziger DP, Yankelev S, Vigliani G. Pharmacokinetics and tolerability of daptomycin at doses up to 12 milligrams per kilogram of body weight once daily in healthy volunteers. Antimicrob Agents Chemother. 2006;50(10):3245–3249. doi:10.1128/AAC.00247-06
98.
99. Dvorchik BH, Damphousse D. The pharmacokinetics of daptomycin in moderately obese, morbidly obese, and matched nonobese subjects. J Clin Pharmacol. 2005;45(1):48–56. doi:10.1177/0091270004269562
100. Pai MP, Norenberg JP, Anderson T, et al. Influence of morbid obesity on the single-dose pharmacokinetics of daptomycin. Antimicrob Agents Chemother. 2007;51(8):2741–2747. doi:10.1128/AAC.00059-07
101. Ng JK, Schulz LT, Rose WE, et al. Daptomycin dosing based on ideal body weight versus actual body weight: comparison of clinical outcomes. Antimicrob Agents Chemother. 2014;58(1):88–93. doi:10.1128/AAC.01018-13
102. Bhavnani SM, Rubino CM, Ambrose PG, Drusano GL. Daptomycin exposure and the probability of elevations in the creatine phosphokinase level: data from a randomized trial of patients with bacteremia and endocarditis. Clin Infect Dis. 2010;50(12):1568–1574. doi:10.1086/652767
103. Parra-Ruiz J, Duenas-Gutierrez C, Tomas-Jimenez C, Linares-Palomino JP, Garrido-Gomez J, Hernandez-Quero J. Safety analysis of high dose (>6 mg/kg/day) daptomycin in patients with concomitant statin therapy. Eur J Clin Microbiol Infect Dis. 2012;31(8):1771–1774. doi:10.1007/s10096-011-1500-y
104. Hair PI, Keam SJ. Daptomycin: a review of its use in the management of complicated skin and soft-tissue infections and Staphylococcus aureus bacteraemia. Drugs. 2007;67(10):1483–1512. doi:10.2165/00003495-200767100-00008
105. Butterfield-Cowper JM, Lodise TP
106. Biek D, Critchley IA, Riccobene TA, Thye DA. Ceftaroline fosamil: a novel broad-spectrum cephalosporin with expanded anti-Gram-positive activity. J Antimicrob Chemother. 2010;65(Suppl 4):iv9–16. doi:10.1093/jac/dkq251
107. Sader HS, Fritsche TR, Kaniga K, Ge Y, Jones RN. Antimicrobial activity and spectrum of PPI-0903M (T-91825), a novel cephalosporin, tested against a worldwide collection of clinical strains. Antimicrob Agents Chemother. 2005;49(8):3501–3512. doi:10.1128/AAC.49.8.3501-3512.2005
108. Corey GR, Wilcox M, Talbot GH, et al. Integrated analysis of CANVAS 1 and 2: phase 3, multicenter, randomized, double-blind studies to evaluate the safety and efficacy of ceftaroline versus vancomycin plus aztreonam in complicated skin and skin-structure infection. Clin Infect Dis. 2010;51(6):641–650. doi:10.1086/655827
109. Corey GR, Wilcox MH, Talbot GH, Thye D, Friedland D, Baculik T. CANVAS 1: the first Phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother. 2010;65(Suppl 4):iv41–51. doi:10.1093/jac/dkq254
110. Wilcox MH, Corey GR, Talbot GH, Thye D, Friedland D, Baculik T. CANVAS 2: the second Phase III, randomized, double-blind study evaluating ceftaroline fosamil for the treatment of patients with complicated skin and skin structure infections. J Antimicrob Chemother. 2010;65(Suppl 4):iv53–iv65. doi:10.1093/jac/dkq255
111. Justo JA, Mayer SM, Pai MP, et al. Pharmacokinetics of ceftaroline in normal body weight and obese (classes I, II, and III) healthy adult subjects. Antimicrob Agents Chemother. 2015;59(7):3956–3965. doi:10.1128/AAC.00498-15
112. Evans JD, Udeani G, Cole P, Friedland HD. Ceftaroline fosamil for the treatment of acute bacterial skin and skin structure infections in obese patients. Postgrad Med. 2014;126(5):128–134. doi:10.3810/pgm.2014.09.2807
113. Burnett YJ, Echevarria K, Traugott KA. Ceftaroline as salvage monotherapy for persistent MRSA bacteremia. Ann Pharmacother. 2016;50(12):1051–1059. doi:10.1177/1060028016664361
114.
115. Worboys P, Barriere S Population pharmacokinetic analysis suggests minimal imp\act of obesity on telavancin (TLV) exposure.
116. Samara E, Shaw JP, Barriere SL, Wong SL, Worboys P. Population pharmacokinetics of telavancin in healthy subjects and patients with infections. Antimicrob Agents Chemother. 2012;56(4):2067–2073. doi:10.1128/AAC.05915-11
117. Stryjewski ME, Graham DR, Wilson SE, et al. Telavancin versus vancomycin for the treatment of complicated skin and skin-structure infections caused by gram-positive organisms. Clin Infect Dis. 2008;46(11):1683–1693. doi:10.1086/587896
118. Slover CM, Azie N, Barriere S, Lu Q. Telavancin (TLV) for treatment of complicated skin and skin structure infections (cSSI) in obese patients.
119. Hassoun A, Edwards J Evaluation of telavancin dose capping in a large community hospital.
120. Bunnell KL, Pai MP, Sikka M, et al. Pharmacokinetics of telavancin at fixed doses in normal-body-weight and obese (Classes I, II, and III) adult subjects. Antimicrob Agents Chemother. 2018;62(4). doi:10.1128/AAC.02475-17.
121.
122. Puttagunta S, Dunne M Dalbavancin for the treatment of acute bacterial skin and skin structure infections in obese patients.
123.
124. Corey GR, Good S, Jiang H, et al. Efficacy and safety outcomes by subgroups in studies of a single dose of oritavancin for the treatment of acute bacterial skin and skin structure infections (ABSSSI).
125. Bhavnani SM, Hammel JP, Rubino CM, et al. Oritavancin pharmacokinetic-pharmacodynamic analyses for efficacy based on data from patients with acute bacterial skin and skin structure infections enrolled in SOLO I and II.
126. Rubino CM, Bhavnani SM, Moeck G, Bellibas SE, Ambrose PG. Population pharmacokinetic analysis for a single 1,200-milligram dose of oritavancin using data from two pivotal phase 3 clinical trials. Antimicrob Agents Chemother. 2015;59(6):3365–3372. doi:10.1128/AAC.00176-15
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.