Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Evaluation of CYP2C19 Gene Polymorphisms in Patients with Acid Peptic Disorders Treated with Esomeprazole
Authors Díaz-Ordóñez L ![]() , Ramírez-Montaño D
, Ramírez-Montaño D ![]() , Candelo E
, Candelo E ![]() , González-Restrepo C, Silva-Peña S
, González-Restrepo C, Silva-Peña S ![]() , Rojas CA, Sepulveda Copete M, Echavarria HR, Pachajoa H
, Rojas CA, Sepulveda Copete M, Echavarria HR, Pachajoa H ![]()
Received 20 October 2020
Accepted for publication 25 January 2021
Published 29 April 2021 Volume 2021:14 Pages 509—520
DOI https://doi.org/10.2147/PGPM.S285144
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Lorena Díaz-Ordóñez,1– 3 Diana Ramírez-Montaño,1– 3 Estephania Candelo,2– 4 Carolina González-Restrepo,1 Sebastián Silva-Peña,1 Carlos Arturo Rojas,5 Mario Sepulveda Copete,5 Hector Raul Echavarria,6 Harry Pachajoa1– 3
1Basic Medical Science Department, Faculty of Health Sciences, Universidad Icesi, Cali, Colombia; 2Clinical Genetic Department, Fundación Valle del Lili, Cali, Colombia; 3Research Centre in Rare Diseases and Congenital Abnormalities (CIACER), Universidad Icesi, Cali, Colombia; 4Research Centre, Fundación Valle de Lili, Cali, Colombia; 5Gastroenterology Department, Fundacion Valle del Lili, Cali, Colombia; 6Centro Medico Imbanaco, Cali, Colombia
Correspondence: Estephania Candelo Calle 18 122-135, Cali, 76003, Colombia
Tel +57 2 5552334
Fax + 57 2 555 1441
Email [email protected]
Background: CYP2C19 is a highly polymorphic gene that encodes an enzyme with the same name and whose function is associated with the metabolism of many important drugs, such as proton pump inhibitors (such as esomeprazole, which is used for the treatment of acid peptic disease). Genetic variants in CYP2C19 alter protein function and affect drug metabolism. This study aims to genotypically and phenotypically characterize the genetic variants in the CYP2C19 gene in 12 patients with acid peptic disorders and different therapeutic profiles to proton pump inhibitor (PPI) drugs. The patients were randomly selected from a controlled, randomized and blinded clinical pilot trial of 33 patients. We determined the presence and frequency of single nucleotide polymorphisms (SNPs) within exons 1– 5 and 9, the intron-exon junctions, and a fragment in the 3ʹ UTR region of the CYP2C19 gene using Sanger sequencing. Undescribed polymorphisms were analyzed by free online bioinformatics tools to evaluate the potential molecular effects of these genetic variants.
Results: We identified nine polymorphisms, six of which had no reported functions. One of these genetic variants, with a functional impact, not yet reported (p.Arg132Trp) was predicted by bioinformatic tools as potentially pathogenic. This finding suggests that p.Arg132Trp could be related to poor metabolizers of drugs metabolized by CYP2C19.
Conclusion: We identified the genotype spectrum of variants in CYP2C19. The genotype spectrum of variants in CYP2C19 could predict the treatment response and could support to evaluate clinical efficacy in patients treated with esomeprazole.
Keywords: single nucleotide polymorphism, cytochrome P450 CYP2C19, pharmacogenetics, computational biology, treatment failure, proton pump inhibitors
Background
Acid peptic disorders result from an imbalance at the gastroduodenal level between aggressive factors, such as the concentration of acid and pepsin in the gastric lumen, and the defensive factors of the mucous membrane.1 Proton pump inhibitors (PPIs) are the first-line treatment for these disorders. PPIs modify chloride acid production by irreversibly inhibiting H/K-ATPase on parietal cells.2 There are currently five PPI drugs commercially available: omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole, which have very similar pharmacological properties.
The enzymatic superfamily cytochrome P450 (P450) regulates the metabolic transformation of a broad spectrum of drugs, such as PPIs, via oxidative reactions.3 There are approximately 2740 CYP450 sequences in animals, 57 known genes, and 59 pseudogenes, which are divided among 57 families and 43 subfamilies.4 Of the 57 known families, families 1, 2, and 3 are the most relevant in clinical studies because they catalyze a critical step in biotransformation reactions in humans.5 Within family 2, the most important subfamily is CYP2C, which in the case of humans is integrated by four enzymes: CYP2C8, CYP2C9, CYP2C18, and CYP2C19.6 This subfamily has an important role in the metabolism of nearly 25% of clinically important drugs.7 The enzyme CYP2C19 is encoded by the gene with the same name at locus 10q23.23. Although its expression represents only 1% of the total hepatic CYP enzymes, the CYP2C19 enzyme is involved in the metabolism of approximately 10% of drugs8 and is the main enzyme implicated in the metabolic pathway of PPIs. Therefore, variants in this gene significantly impact drug metabolism, specifically affecting their pharmacokinetics and clinical efficiency.9
To date, more than 40 polymorphic variants of CYP2C19 have been identified, and they are related to nearly 35 enzyme isoforms.10 Allelic variants are associated with defined metabolizer phenotypes. The poor metabolizer (PM) phenotype is characterized by the presence of both alleles, either nonfunctional or null (CYP2C19*2, CYP2C19*3). The intermediate metabolizer phenotype presents one null allele and one functional allele. The normal metabolizer (NM) phenotype refers to the wild-type phenotype, in which both alleles are functional the ultrarapid metabolizer (UM) phenotype has been characterized in homozygous promoter region variants that potentiate gene expression, increasing the enzymatic activity of the protein.11,12 Recently, has been described a rapid metabolizer (RM) phenotype such as an individual carrying one normal function allele and one increased function allele.13 It has been shown that in drugs such as omeprazole, lansoprazole, and rabeprazole, the ratio of the mean values between the plasma concentration and intragastric pH is higher in PMs than in EMs. These differences in pharmacokinetic properties imply that the clinical effects of the drug are altered.14 Thus, it can be assumed at a general level that polymorphisms affecting drug metabolism increase the risk of drug side effects and the rate of therapeutic failure unless an alternative metabolic route is available.15–19
Methodologies based on pharmacokinetic studies are usually used to phenotypically determine the metabolizer type. However, these types of tests disregard the genotypic impact on an enzyme’s capacity to generate a clinical response.19,20 Therefore, the use of molecular biology techniques, such as Sanger sequencing, should be considered to optimally determine the genotype and to predict the metabolizer phenotype.21,22 In Colombia, only two reports have characterized the CYP2C19 polymorphisms of the population, and the results revealed that only the allele variants CYP2C19*1 and CYP2C19*2 were prevalent in the Colombian population.23,24
Because PPIs are commonly administered for acid peptic disease treatment, the genotypic and phenotypic characterization of CYP2C19 is important to determine the therapeutic approach.25 Furthermore, self-medication and the high rates of therapeutic failure are currently increasing.26 The results of this study could be applied to diverse scenarios with other drugs that are metabolized by this enzyme. For example, antidepressants and antiplatelet drugs, such as clopidogrel,48 and the methodology could be replicated with other CYP enzymes used in personalized medicine. This study aimed to perform genotypic and phenotypic characterizations of a population with acid peptic disorder and different physiological responses to esomeprazole. To date, this is the first study to characterize the population frequency of CYP2C19 enzyme polymorphisms related to the use of PPIs in the Colombian population.
Materials and Methods
Participants
A group of 33 patients were recruited from a clinical trial pilot study, this group was doubled blinded and randomized for different esomeprazole presentations (modified-release esomeprazole, Tecnoquimicas vs NEXIUM-MUPS®, AstraZeneca). They were selected by the presence of unstudied dyspepsia, were previously evaluated by digestive endoscopy and biopsy and gastric pH with Lab pH meter inoLab pH 7110 (Xylem, Germany); Congo red stain were used to excluded patients with organic cause of dyspepsia and pH>4 suggesting hypochloridria. Table 1 describes the inclusion and exclusion criterias for the study. Patients with a diagnosis of non-study gastritis were subjected to a treatment of 40 mg/day of esomeprazole uptake for 28 days, the follow-up of the subjects was performed by esophageal 24-hour pH/impedance monitoring at 14 days of treatment, followed by a validated clinical scale for symptoms and quality of life (SODA and QoL-PEI) (Figure 2 and 3) at 2 and 4 weeks post treatment. All the methodologies for the patient samples are provided in this previously published study (Rojas et al 2019)44 and Table 1. Then, a randomly selected subsample of 12 (36.3%) subjects from the aforementioned group was randomly selected according to different physiological responses, which was based on a spectrum from poor metabolizer phenotypes to ultrarapid metabolizer phenotypes using randomization software. The physiological response was determined by the number of hours during 24-hour pH/impedance reflux monitoring that the subject showed pH >4 (this showed the effect of the PPIs on stomach acid production). The genotypes of the subsamples were blinded to the phenotypes (physiological response). All participants provided written informed consent before enrollment in the study. All the patients have not been previously studied for dyspepsia; they do not present alarming signs such as previous gastric surgery, gastric neoplasia, gastric and duodenum ulcers or erosive esophagitis. None of the patients have received other proton-pump inhibitors or histamine 2 antagonist and non-steroidal anti-inflammatory drugs before the treatment started. Only one participant presented a minor adverse effect consisting of nausea. Other clinical and demographic characteristics of the participants are presented in Table 2.
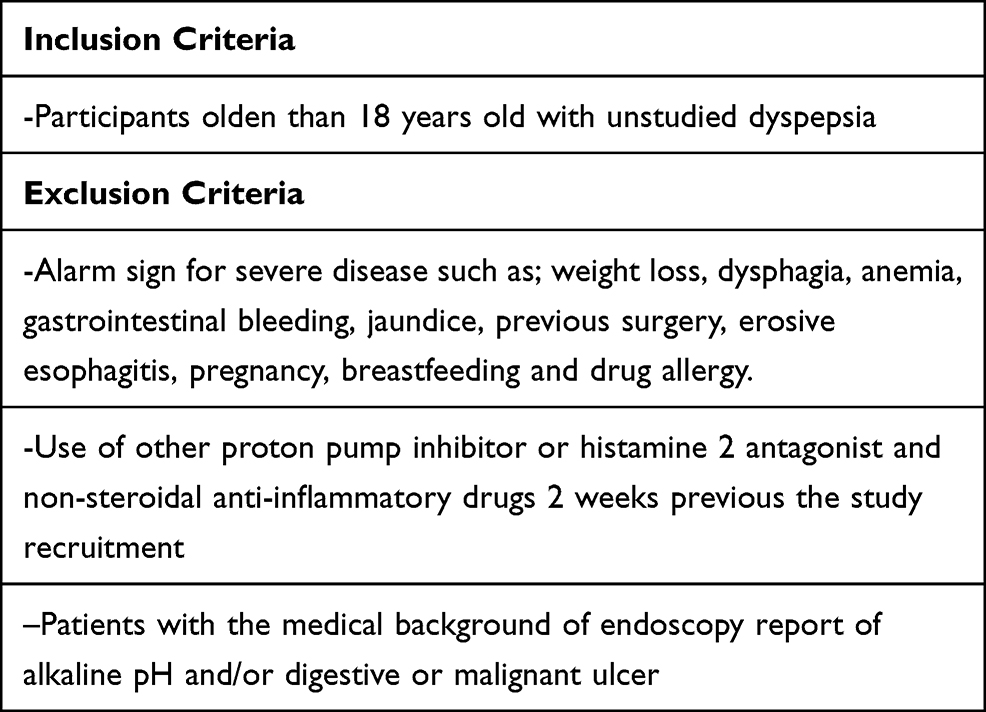 |
Table 1 Inclusion and Exclusion Criteria for the Present Study |
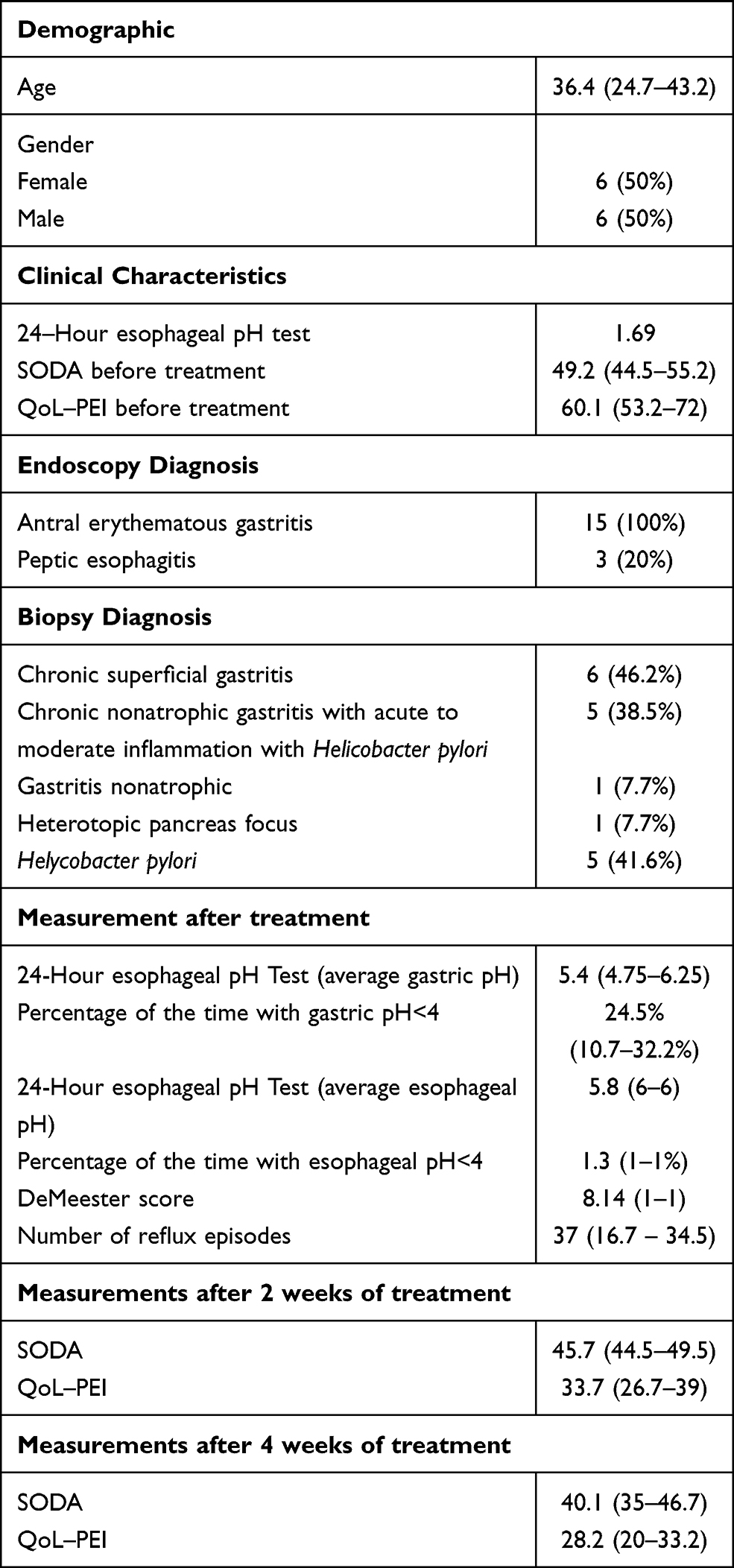 |
Table 2 Demographic, Clinical, Endoscopy and Biopsy Characteristics of the Study Sample as Well as Clinical Scores After 2 and 4 Weeks Treatment Exposure |
Genotyping CYP2C19
A blood sample (5 mL) was taken from each subject in an EDTA tube, and genomic DNA was extracted using the automated EZ1 system (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Exons 1 to 5 and 9 and the promoter region of the CYP2C19 gene were separately amplified by conventional PCR in a final volume of 25 µL per reaction, containing 1X buffer, 2 mM MgCl2, 0.4 mM each primer, 0.2 mM dNTPs, 0.5 U of Taq polymerase (Invitrogen, California, USA), and 100 ng DNA. Primer sequences and their annealing temperatures are listed in Table 3. The thermal cycling conditions were as follows: denaturation at 95°C for 5 min; followed by 38 cycles of denaturation at 95°C for 30 sec, annealing at 59–61°C for 30 sec and extension at 72°C for 1 min; and a final extension at 72°C for 5 min.
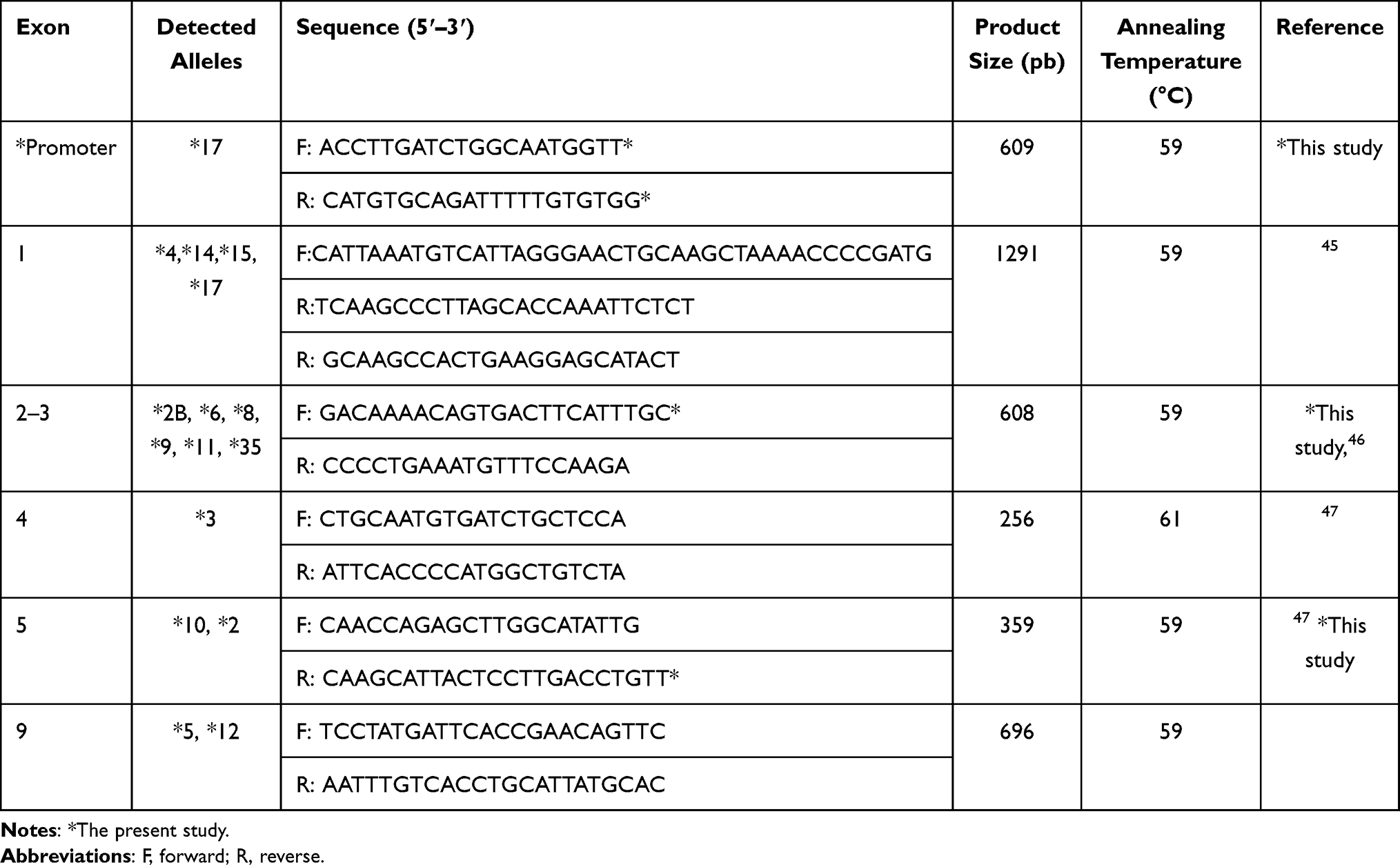 |
Table 3 Primers Used in the PCR Experiments to Detect CYP2C19 Alleles |
Sanger Sequencing Method
Polymerase chain reaction (PCR) products were separated by agarose gel electrophoresis to ensure proper amplification and were subsequently purified using the E.Z.N.A.® Cycle Pure Kit (Omega Bio-Tek, USA) according to the manufacturer’s recommendations. Sanger sequencing was performed using the BigDye Terminator 3.1 Kit (Thermo Fisher Scientific, USA). The cycle sequencing reaction consisted of 1 µL of BigDye Terminator, 1.5 µL of 5X sequencing buffer, 3.2 pmol primers, and 1 µL of PCR product. The reaction consisted of pre-denaturation at 96°C for 1 min, followed by 35 cycles of denaturing at 96°C for 15 sec, annealing at 55°C for 15 sec, and extension at 72°C for 1 min, and a final extension at 72°C for 5 min. Excess BigDye terminators were removed using the BigDye XTerminator™ Purification Kit (Thermo Fisher Scientific, USA) according to manufacturer recommendations, and samples were analyzed on the 3500 DNA Analyzer (Thermo Fisher Scientific, USA). Sequence data were analyzed using SeqScape software (Thermo Fisher Scientific, USA) with the GenBank CYP2C19 genomic reference sequence NG_008384. To confirm the presence of SNPs, we performed additional SNP genotyping by Snapshot chemistry (Applied Biosystems, CA, USA) according to the manufacturer’s recommendations. Probe sequences and PCR conditions are available upon request.
Bioinformatic Analysis
All genetic variants were searched for in four different sources: dbSNP, ClinVar, PubMed and PharmVar to obtain all available information about the variants. The effects of missense variants were predicted by SIFT (Sorting Intolerant From Tolerant), PolyPhen-2 (Polymorphism Phenotyping v2), Provean (Protein Variation Effect Analyzer), Mutation Taster, FATHMM (Functional Analysis through Hidden Markov Models), CADD v1.3 (Combined Annotation–Dependent Depletion), and DANN (Deleterious Annotation of genetic variants using Neural Networks).
Finally, the effect of promoter SNPs was predicted using rSNPBase 3.1 (SNP-related regulatory elements, element-gene pairs and SNP-based regulatory networks) and PROMO v3.0.2, which is a virtual laboratory for the identification of putative transcription factor binding sites (TFBS) in DNA sequences from a species or groups of species of interest. For each promoter SNP, both sequences (wild-type and mutant) were loaded to search for potential transcription factor binding sites. The prediction was carried out considering only transcription factor binding sites and only human transcription factors.
Results
These results were confirmed by snapshot chemistry. CYP2C19 allele frequencies in this study are summarized in Table 4. Among the CYP2C19 variant alleles analyzed, CYP2D19*1 was the most common allele (62.5%) of this population, followed by CYP2D19*2, CYP2D19*17 and CYP2D19*35. These polymorphisms have not been previously described, and their frequency in our study is summarized in Table 4.
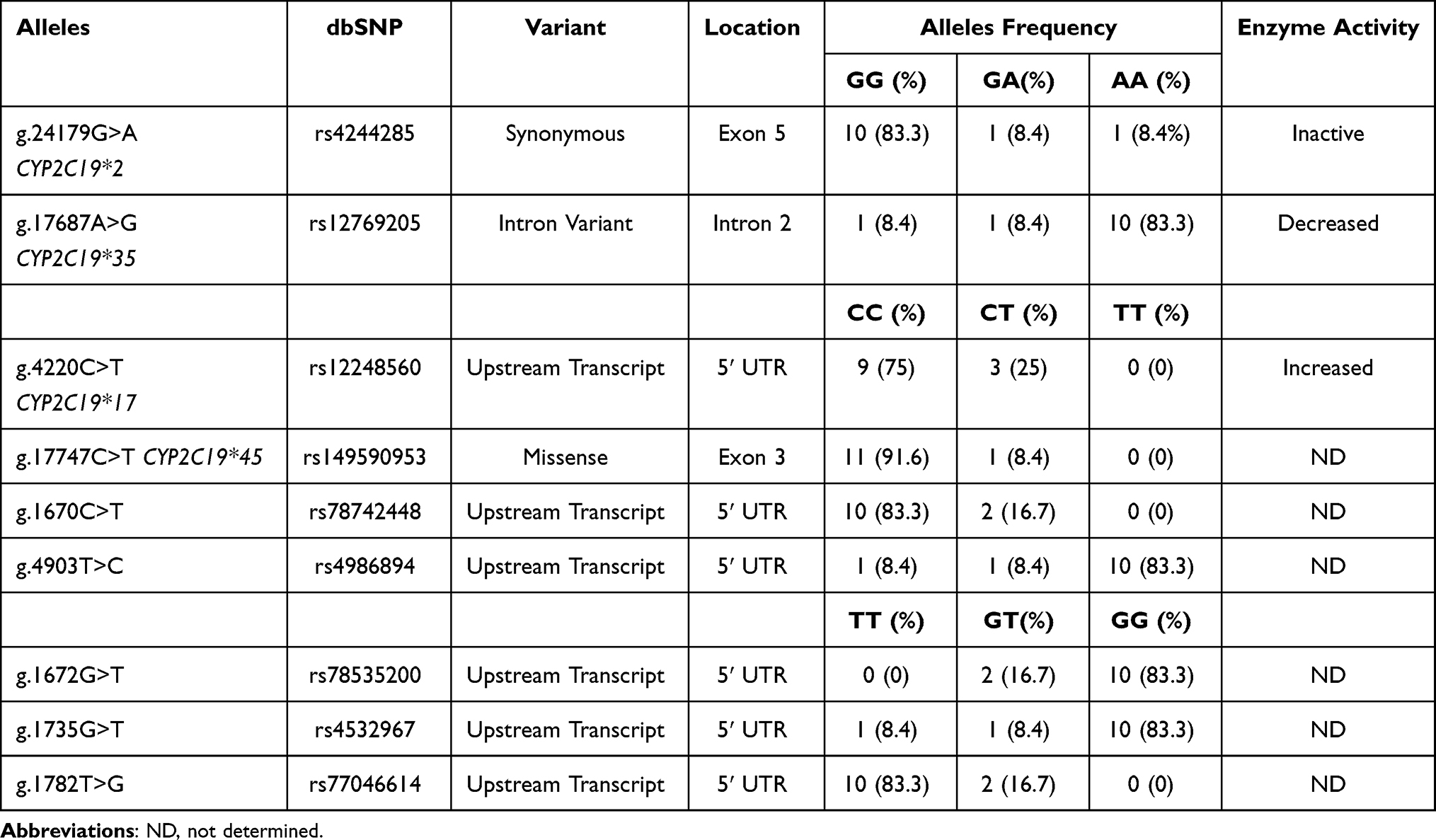 |
Table 4 Frequencies of the CYP2C19 Alleles Identified |
As detailed in Table 4, one patient was heterozygous for rs149590953 (g.17747C>T; p.Arg132Trp; CYP2C19*45), a missense variant that has not been previously reported. Six different in silico tools were used to predict the potential pathogenicity of this variant. The prediction by SIFT indicated that p.Arg132Trp is not a tolerable variant. The prediction conducted by PolyPhen-2 demonstrated that this variant is probably damaging in terms of conservation with a score of 0.994 and possibly damaging in terms of variation, with a score of 0.735. The prediction by PROVEAN and CADD 1.3 classifies this as a deleterious variant. The DANN score of this variant was 0.9974, and the value range was 0 to 1, with 1 indicating that the variants are predicted to be the most damaging. Finally, MutationTaster predicted that this variant is a polymorphism.
The genetic variants localized in the 5ʹ untranslated regions (5ʹ UTR) were analyzed by three different in silico tools, and the prediction results are described in Table 5. According to reference sequence NG_008384, PROMO data reveal that the wild-type nucleotide cytosine, in genomic placement 1695 (rs78742448), is located in a binding site of three transcription factors: 1) general transcription factor II–I (TFII-I), 2) signal transducer and activator of transcription 4 (STAT4) and 3) ETS Proto-Oncogene 1, Transcription Factor (c-Ets-1). Meanwhile, the alternative nucleotide (thymine) in the same position (g.1695C>T) binds two additional transcription factors: glucocorticoid receptor isoform β (GR-beta) and CCAAT enhancer binding protein beta (C/EBP beta) (Figure 1). In addition, when the rs77046614 T allele was replaced by the G allele, only one additional transcription factor (GR-beta) was able to bind to it (Figure 1). The other analyzed polymorphisms did not reveal changes in transcription factor binding sites. Finally, rs4986894 was predicted by rSNPBase 3.1 to be related to disease because the genetic variant g.4903T>C is located in a circular RNA (circRNA) region.
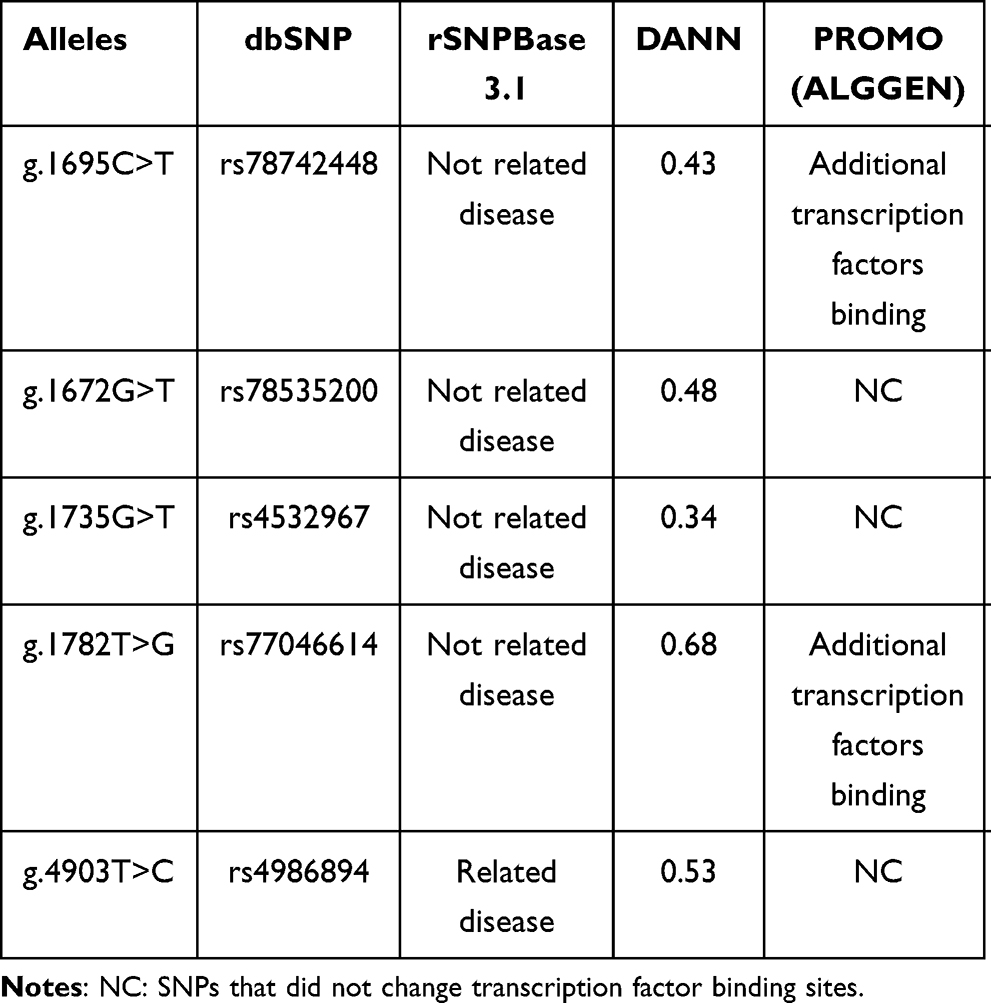 |
Table 5 Bioinformatic Analysis of 5ʹ UTR Variants |
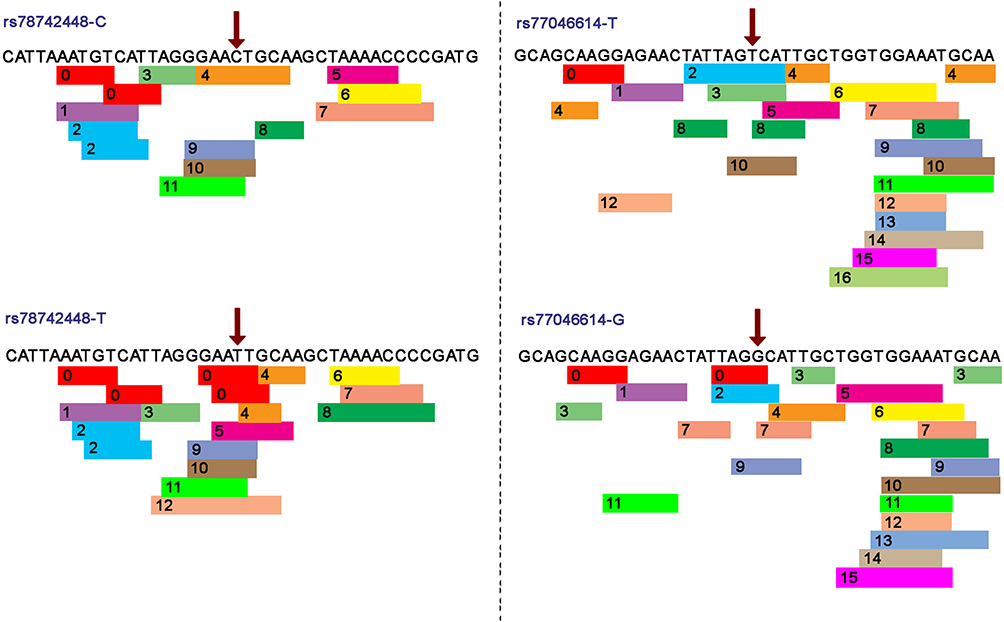 |
Figure 1 Transcription factor binding site prediction at rs78742448 and rs77046614 in the CYP2C19 promoter. The red arrow indicates the SNP position. 0: GR-beta (T01920); 1: c-Jun (T00133); 2: XBP-1 (T00902); 3: GR-alpha (T00337); 4: FOXP3 (T04280); 5: RXR-alpha (T01345); 6: RAR-beta (T00721); 7: C/EBPbeta (T00581); 8: TFII-I (T00824); 9: STAT4 (T01577); 10:c-Ets-1 (T00112). |
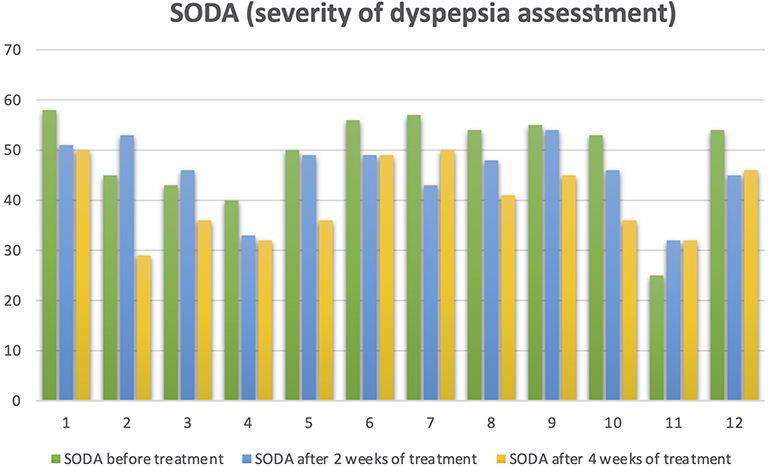 |
Figure 2 Trend of SODA (Severity Dyspepsia Assessment) before and after treatment. |
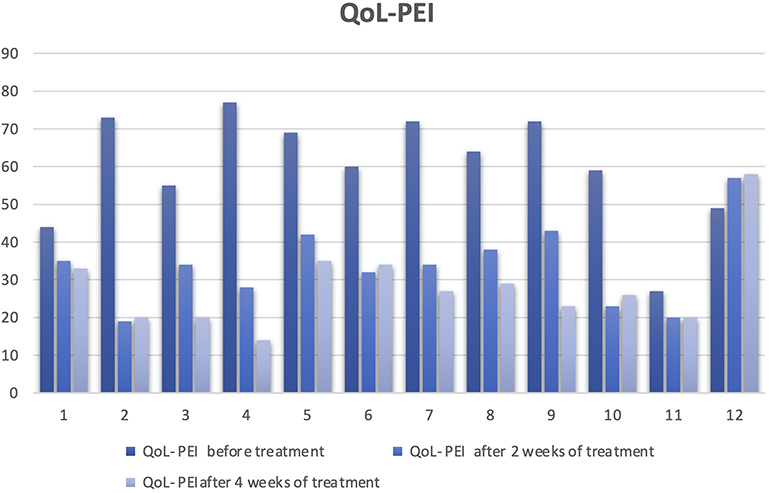 |
Figure 3 Trend of quality of life, resilience, perception and illness behaviour in patients with functional dyspepsia (QoL-PEI) before and after treatment. |
Discussion
CYP2C19 is a highly polymorphic gene that plays a substantial role in the metabolism of proton pump inhibitors such as esomeprazol.27 Studies have demonstrated that CYP2C19 genetic polymorphisms can result in a significant alteration in enzyme activity and modified drug responses.28 We investigated the frequency of genetic variants in the CYP2C19 gene in 12 patients with different physiological responses to PPIs. The relationship between the genotype and phenotype is summarized in Table 6. Using Sanger sequencing, we identified nine genetic variants of the CYP2C19 gene. Of these, three had been previously characterized and included the CYP2C19*2, CYP2C19*17 and CYP2C19*35 haplotypes. Among these variants, CYP2C19*2 and CYP2C19*35 have been categorized as PM variants, and the haplotype CYP2C19*17 is considered an UM variant.
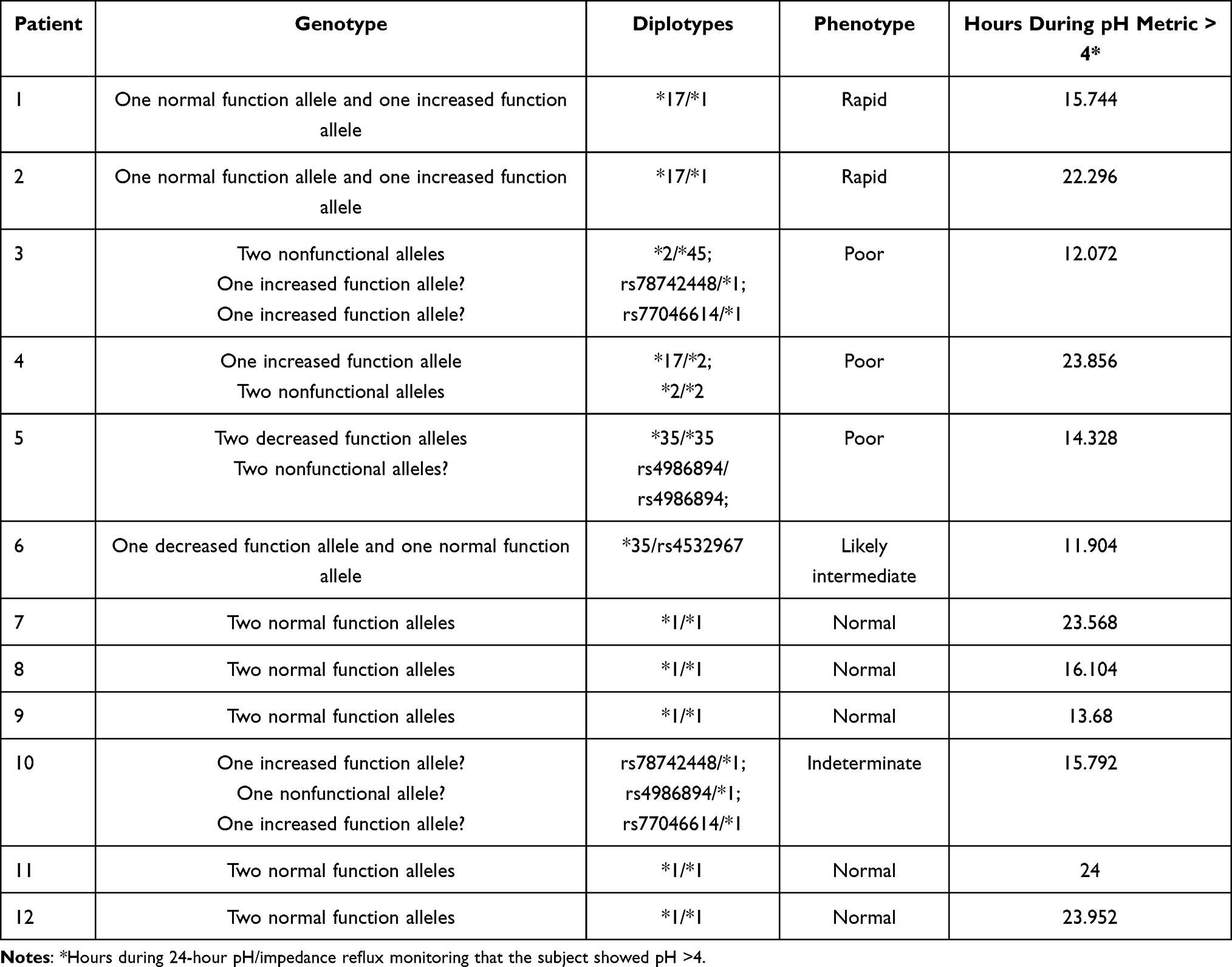 |
Table 6 Genotype–Phenotype Relationship of the Study Population |
The haplotype CYP2C19*2 results in aberrant splicing and loss of enzyme activity in the PM phenotype.29 The disruption in the branch site in CYP2C19 intron 2 creates a novel exon 2B. This alternative CYP2C19 mRNA will generate a nonfunctional protein since the insertion of exon 2B creates an out-of-frame protein with 87 novel amino acid residues followed by a premature termination codon, resulting in a truncated 197 amino acid protein.30 However, CYP2C19*2 was only present in 3 of the 24 alleles, with a total frequency of 12.5%. In one of the subjects presented in this study, the deleterious effect of this variant had more influence than the UM CYP2C19*17 variant and this subject showed a PM physiological response (Table 6). In Colombia, only two reports have characterized CYP2C19 polymorphisms in the population, and the results revealed that only the allele variants CYP2C19*1 and CYP2C19*2 were prevalent in the evaluated Colombian population.23,24
The variant CYP2C19*35 is commonly associated with a nonfunctional phenotype. It is an intronic polymorphism whose clinical relevance is linked with drug response, generating therapeutic failure of the antiplatelet clopidogrel and antidepressants such as escitalopram, citalopram, and sertraline (WEB: https://www.ncbi.nlm.nih.gov/snp/rs12769205#clinical_significance). The reported patients with this variant have a poor metabolic activity of CYP2C19, and in patients with homozygous conditions, this variant generates a complete loss of the protein activity of CYP2C19.30 This variant was found in only 2 of 12 patients, representing approximately 16.6% of the study population.
CYP2C19*17 is the polymorphism in which the C>T transition in the intronic region, specifically the promoter, creates a consensus binding site for the GATA transcription factor family, generating a UM phenotype; thus, the CYP2C19*17 gene has increased expression and activity (https://www.pharmgkb.org/vip/PA166169770). The CYP2C19*19 allele is present in 3 of 12 patients, representing 12.5% of the allele frequency and explains the decrease in hours with a pH metric >4 of 2 cases. The reported frequencies of variants in CYP2C19*2, CYP2C19*35, and CYP2C19*17 were approximately 8%, 8% and 25%, respectively, showing similarities with the European (range 18–27%)31 and Brazilian populations (range 15.8– 26.3%) in the case of CYP2C19*17,32 and with CYP2C19*2 and CYP2C19*35, the Asian (range 24–27%)33 and African-American (19.4–24%)34 populations are the most similar in terms of allelic frequency.
The rs12769205 and rs4244285 variants are closely related, and they mapped to the same haplotype block on chromosome 10 at positions 96535124 and 96541616, respectively. Although these SNPs have been reported in linkage disequilibrium (LD) in a Colombian population (Medellin – Colombia),35 they were both included in the analysis for the following reasons: 1) small sample size (n=148) can affect the LD estimate for overrepresentation of the D’36 and 2) accessing the functional consequences of each genetic variant is fundamental due to the recombination events during the time, possibly creating movement of the variants from linkage disequilibrium to linkage equilibrium.37 Finally, Sanger sequencing allowed us to obtain both variants without an additional test.
The presence of the diplotype *2/*17 in sample 4 (Table 6) is one of the most remarkable features identified in this study. According to a previous report,38 this diplotype could generate an intermediate phenotype. However, in our sample patient 4 genotype was identified as homozygous for CYP2C19*2 and heterozygous for CYP2C19*17. The heterozygous status to CYP2C19*17 leads to a rapid metabolizer phenotype due to g.4220C>T being associated with increased transcription of CYP2C19 gene. Although, the presence of the homozygous status of CYP2C19*2 leads to a PM phenotype due to g.24179G>A being associated with truncated and nonfunctional protein. For those reasons, we might assume that even increased transcription did produce non-functional CYP2C19.39 Furthermore, the intronic location of isoform 17 and the fact that isoform 2 may be a synonymous variant, which highlights the importance of the characterization and evaluation of these types of variants that are normally considered to exert a low impact. This is particularly important because of the 10 polymorphisms found, 6 were located in an intronic region. Only one polymorphism was located in a codifying region, but it resulted in a synonymous variation.
In this study, the use of reliable bioinformatic predictors that have been utilized by many authors for in silico predictions has enabled us to establish the impact of the allele CYP2C19*45 located in an exonic region, whose effect had been predicted as a damaging SNP.40 As reported in Table 5, we also found that this variant has a high probability of damaging protein folding and function. Additionally, this variant changes the free energy of the molecule, and the amino acid chemical properties of this variant are different from those of the wild-type protein. This prediction supports a correlation with the PM phenotype because in this case the protein is considerably damaged, generating a possible nonfunctional product. The predictor PROMO also provided information about the transcription factor binding sites (TFBS) in DNA sequences, which is useful to analyze SNPs that are located in the intronic regions, allowing us to predict the possible impact of SNPs on gene expression. In that sense, when the predictor shows a response of additional transcription factor binding sites, this implies that the final product could be overexpressed; alternatively, the product could not be expressed, or the transcription process could stop early. With this program, we compared the sequence of the SNPs rs78724448, rs78535200, rs4532967, rs77046614, and rs4986894 with the sequence of the wild-type gene (Table 5). We found that the SNPs rs78742448 and rs77046614 present additional transcription factor binding sites (Figure 1). However, in vivo tests are necessary to confirm these results.
Patient’s response to esomeprazole treatment was correlated with the genotype in Table 6. However, there was a poor correlation between genotype and phenotype. For example, the RM carrying CYP2C19*1/*17 displayed different results in 24-hour pH/impedance reflux monitoring with pH >4. While patient #1 showed 15.7 hours, patient #2 showed 22.2 hours. Similarly, the NM carrying CYP2C19*1/*1 displayed long-term and short-term reduction of acid production (between 13.68 and 24 hours). These findings suggest that although approximately 70% of the esomeprazole metabolism depends on CYP2C19 enzyme activity, the remaining 30% of the metabolism depends on CYP3A4 enzyme activity and might be influencing the phenotype.41 Even though patient #4 was PM, the test shows a reduction of acid production of 23.8 hours. This finding suggests that other mechanisms may be involved in the metabolism of esomeprazole, as has been reported for other drugs such as Cytarabine42 that could have a compensatory effect. Finally, we expected patient #5 to display no CYP2C19 activity, but his test shows a reduction of acid production of 14.3 hours, which might be related to higher CYP3A4 enzyme activity or the influence of regulatory mechanisms. However, additional studies are required to clarify these hypotheses.
A genotype-phenotype correlation was generated starting from the premise that all 12 samples were obtained from a broad physiological response to PPIs, and all test results for each patient suggested that the difference in the clinical response was probably a genetic polymorphism on the enzyme facilitator (CYP2C19). In this sense, this study was driven to determine the most likely type of metabolizer that is related to this response, showing an approximation of the possible effects that the presence of each polymorphism causes on the metabolism of drugs that are metabolized by CYP2C19. As in other previously published studies, our results support the use of genotype-guided prescriptions in clinical practice. It is necessary to define the optimal approach to implement clinical testing in diverse health care practices, including the timing and methodology of genotyping in clinical settings.43 Furthermore, for chronic therapy (ie, >12 weeks) in patients who are classified as having the CYP2C19 PM phenotype, a reduction in the daily dose by 50% is recommended due to an increase in the plasma concentration of PPI and the chance of potential toxicity compared to those who are classified as having the NM phenotype.
In the future, the use of a panel-based preventive approach before prescribing medications will require genotype information on hundreds of pharmacogenes readily available in the patient’s medical history to guide drug therapy.43 In our population, it is necessary to determine the frequency of the polymorphisms in these pharmacogenes previously reported in the literature before implementing these approaches. This will elucidate the impact of a polymorphism on the metabolizing phenotype and even decrease the influence of other genetic or nongenetic factors, since a correct genotype and metabolizer phenotype correlation was not possible in all patients. This is because our population is characterized by a heterogeneous genetic background of three parental populations (European, African, and Native Amerindians) with a wide degree and diverse patterns of genetic admixture. Knowing these data will allow the selection of the correct therapeutic approach and determine, in the long term, which types of drugs might pose a risk to some patients and which types and correct dosages of drugs are the safe alternatives for them.
Finally, the limitations of the study include the lack of a control group and the small sample size. Further studies are required to check whether there are significant differences between controls and patients.
Conclusion
Our results allow us to characterize in silico genetic variants in CYP2C19 with functional effects not yet described, which improves our understanding of the effect of polymorphisms located in unusual locations such as in the intronic and promoter regions. It will be necessary to conduct studies with a larger sample that determine the importance of our findings at the population level and define which polymorphisms should be tested in our patients before the use of pharmacological therapies. Furthermore, our results contribute to the improvement of the tools currently used in personalized medicine.
Abbreviations
PPIs, proton pump inhibitors; CYP, cytochrome P450; PCR, polymerase chain reaction; PM, poor metabolizer; NM, normal metabolizer; UM, ultrarapid metabolizer; RM, rapid metabolizer; TFBS, transcription factor binding site; SODA, severity of dyspepsia assessment; QoL-PEI, Quality of life in relation to Stomach and Intestinal Problems questionnaire; rSNPBase 3.1, SNP-related regulatory elements, element-gene pairs and SNP-based regulatory network; DANN, Deleterious Annotation of genetic variants using Neural Networks; CADD v1.3, Combined Annotation–Dependent Depletion; FATHMM, Functional Analysis through Hidden Markov Models; Provean, Protein Variation Effect Analyzer; PolyPhen-2, Polymorphism Phenotyping v2; SIFT, Sorting Intolerant from Tolerant; 5ʹ UTR, 5ʹ untranslated region.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This research was conducted in accordance with the Declaration of Helsinki. The subjects provided written informed consent to the reporting of data, and the study was approved by the Fundación Valle de Lili ethics committee with the reference number. Information revealing the subjects’ identities was not included in the manuscript. The patients were identified by number and not by his/her real name.
Consent for Publication
All the images and patient material presented in this study have been approved for publication, and they are available upon request.
Acknowledgment
The authors thank Fundación Valle del Lili and Universidad ICESI for providing the samples, facilities and support for this work.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
All forms of support and financial involvement were provided by the employers of the authors (Universidad Icesi and Fundación Valle del Lili).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mejia A, Kraft WK. Acid peptic diseases: pharmacological approach to treatment. Expert Rev Clin Pharmacol. 2009;2(3):295–314. doi:10.1586/ecp.09.8
2. Cardona-Ospina JA, Medina-Morales DA, Rodr?guez-Morales AJ, Machado-Alba JE. Efectos adversos a largo plazo de los inhibidores de la bomba de protones. Perspectiva desde la medicina basada en la evidencia. Rev Colomb Gastroenterol. 2016;31:403–408. doi:10.22516/25007440.115
3. Donato MT
4. Hunt SE, McLaren W, Gil L, et al. Ensembl variation resources. Database. 2018;2018.
5. Guengerich FP, Rendic S. Update information on drug metabolism systems— 2009, part I. Curr Drug Metab. 2010;11:1–3. doi:10.2174/138920010791110908
6. Zhang H-F, Wang -H-H, Gao N, et al. Physiological content and intrinsic activities of 10 cytochrome P450 isoforms in human normal liver microsomess. J Pharmacol Exp Ther. 2016;358(1):83–93. doi:10.1124/jpet.116.233635
7. Ph. D. Lemke TL, Ph. D. Williams DA, Ph. D. Roche VF, Ph. D. Zito SW, eds. Foye’s Principles of Medicinal Chemistry. Vol. 106. 2013. doi:10.1023/B:PRES.0000030927.38007.36
8. Hiratsuka M. Genetic polymorphisms and in vitro functional characterization of CYP2C8, CYP2C9, and CYP2C19 allelic variants. Biol Pharm Bull. 2016;39(11):1748–1759. doi:10.1248/bpb.b16-00605
9. Samer CF, Lorenzini KI, Rollason V, Daali Y, Desmeules JA. Applications of CYP450 testing in the clinical setting. Mol Diagn Ther. 2013;17(3):165–184. doi:10.1007/s40291-013-0028-5
10. Gaedigk A, Ingelman-Sundberg M, Miller NA, et al. The Pharmacogene Variation (PharmVar) consortium: incorporation of the human Cytochrome P450 (CYP) allele nomenclature database. Clin Pharmacol Ther. 2018;103(3):399–401. doi:10.1002/cpt.910
11. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141.
12. Marjani A. Genetic variations in cytochrome P450 2C9 and 2C19: a review. Curr Pharmacogenomics Person Med. 2016;14:18–28. doi:10.2174/1875692115666161214152223
13. Lima JJ, Thomas CD, Barbarino J, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for CYP2C19 and proton pump inhibitor dosing. Clin Pharmacol Ther. 2020. doi:10.1002/cpt.2015
14. Klotz U, Schwab M, Treiber G. CYP2C19 polymorphism and proton pump inhibitors. Basic Clin Pharmacol Toxicol. 2004;95(1):2–8. doi:10.1111/j.1600-0773.2004.pto950102.x
15. Guengerich FP. Cytochrome P450 and chemical toxicology. Chem Res Toxicol. 2008;21:70–83. doi:10.1021/tx700079z
16. Kamiya C, Inui N, Hakamata A, et al. Effect of co-administered inducer or inhibitor on omeprazole pharmacokinetics based on CYP2C19 genotype. J Pharmacol Sci. 2019;139:361–366. doi:10.1016/j.jphs.2019.03.001
17. Velazquez MNR, Parween S, Udhane SS, Pandey AV. Variability in human drug metabolizing cytochrome P450 CYP2C9, CYP2C19 and CYP3A5 activities caused by genetic variations in cytochrome P450 oxidoreductase. Biochem Biophys Res Commun. 2019;515(1):133–138. doi:10.1016/j.bbrc.2019.05.127
18. Tunthong R, Sukasem C, Puangpetch A, et al. Pharmacogenetic study of CYP2C19 variation and clopidogrel dose adjustment according to platelet reactivity monitoring in atherothromboticrisk patients in Thailand. Curr Pharmacogenomics Person Med. 2013;11:154–161. doi:10.2174/1875692111311020007
19. Cattaneo D, Perico N, Remuzzi G. From pharmacokinetics to pharmacogenomics: a new approach to tailor immunosuppressive therapy. Am J Transplant. 2004;4:299–310. doi:10.1111/j.1600-6143.2004.00312.x
20. Daly AK, Cascorbi I. Opportunities and limitations: the value of pharmacogenetics in clinical practice. Br J Clin Pharmacol. 2014;77:583–586. doi:10.1111/bcp.12354
21. Ur Rehman K, Akhtar T, Sabar MF, Tariq MA. Allele frequency distribution of CYP2C19*2 allelic variants associated with clopidogrel resistance in cardiac patients. Exp Ther Med. 2015;10:309–315. doi:10.1124/dmd.32.8.821
22. Zhong Z, Hou J, Li B, et al. Analysis of CYP2C19 genetic polymorphism in a large ethnic hakka population in southern China. Med Sci Monit. 2017;23:6186–6192. doi:10.12659/MSM.905337
23. Heredia MM, Araujo D, Rodríguez M, Sierra E. Genotypic and fenotypic characterization of CYP2C19 from a mestiza population in Colombia. Clin Chem Lab Med. 2015;53:S770.
24. Isaza C, Henao J, Isaza Martínez JH, Sepúlveda Arias JC, Beltrán L. Phenotype-genotype analysis of CYP2C19 in Colombian mestizo nidividuals. BMC Clin Pharmacol. 2007;7. doi:10.1186/1472-6904-7-6
25. Serrano D, Torrado S, Torrado-Santiago S, Gisbert JP. The influence of CYP2C19 genetic polymorphism on the pharmacokinetics/- pharmacodynamics of proton pump inhibitor-containing Helicobacter pylori treatments. Curr Drug Metab. 2012;13:1303–1312. doi:10.2174/138920012803341393
26. Bennadi D. Self-medication: a current challenge. J Basic Clin Pharm. 2013;5:19–23. doi:10.4103/0976-0105.128253
27. Scott SA, Martis S, Peter I, et al. Identification of CYP2C19*4B: pharmacogenetic implications for drug metabolism including clopidogrel responsiveness. Pharmacogenomics J. 2012;12(4):297–305. doi:10.1038/tpj.2011.5
28. Li XQ, Andersson TB, Ahlström M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole, and rabeprazole on human cytochrome P450 activities. Drug Metab Dispos. 2004;32:821–827.
29. Aquilante CL, Niemi M, Gong L, Altman RB, Klein TE. PharmGKB summary: very important pharmacogene information for cytochrome P450, family 2, subfamily C, polypeptide 8. Pharmacogenet Genomics. 2013;23:721–728. doi:10.1097/FPC.0b013e3283653b27
30. De Morais SMF, Wilkinson GR, Blaisdell J, et al. The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem. 1994;269(22):15419–15422. doi:10.1016/S0021-9258(17)40694-6
31. Chaudhry AS, Prasad B, Shirasaka Y, et al. The CYP2C19 intron 2 branch point SNP is the ancestral polymorphism contributing to the poor metabolizer phenotype in livers with CYP2C19*35 and CYP2C19*2 alleles. Drug Metab Dispos. 2015;43(8):1226–1235. doi:10.1124/dmd.115.064428
32. Scott SA, Sangkuhl K, Gardner EE, et al. Clinical pharmacogenetics implementation consortium guidelines for cytochrome P450-2C19 (CYP2C19) genotype and clopidogrel therapy. Clin Pharmacol Ther. 2011;90:328–332. doi:10.1038/clpt.2011.132
33. Favela-Mendoza AF, Martinez-cortes G, Hernandez-zaragoza M, et al. Genetic variability of CYP2C19 in a Mexican population: contribution to the knowledge of the inheritance pattern of CYP2C19*17 to develop the ultrarapid metabolizer phenotype. J Genet. 2015;94(1):3–7. doi:10.1007/s12041-015-0477-1
34. Sugimoto K, Uno T, Yamazaki H, Tateishi T. Limited frequency of the CYP2C19*17 allele and its minor role in a Japanese population. Br J Clin Pharmacol. 2008;65:437–439. doi:10.1111/j.1365-2125.2007.03057.x
35. Martis S, Peter I, Hulot J-S, et al. Multi-ethnic distribution of clinically relevant CYP2C genotypes and haplotypes. Pharmacogenomics J. 2013;13(4):369–377. doi:10.1038/tpj.2012.10
36. CLM. IGSR population. Availbale from: https://www.internationalgenome.org/data-portal/population/CLM.
37. Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. doi:10.1038/nrg777
38. Bush WS, Moore JH, Lewitter F, Kann M. Chapter 11: genome-wide association studies. PLoS Comput Biol. 2012;8(12):e1002822. doi:10.1371/journal.pcbi.1002822
39. Dean L. Diazepam therapy and CYP2C19 genotype. In: Pratt VM, editor. Medical Genetics Summaries. US: National Center for Biotechnology Information; 2012.
40. Langaee TY, Zhu H-J, Wang X, et al. The influence of the CYP2C19*10 allele on clopidogrel activation and CYP2C19*2 genotyping. Pharmacogenet Genomics. 2014;24(8):381–386. doi:10.1097/FPC.0000000000000068
41. Zhang L, Sarangi V, Moon I, et al. CYP2C9 and CYP2C19: deep mutational scanning and functional characterization of genomic missense variants. Clin Transl Sci. 2020;13(4):727–742. doi:10.1111/cts.12758
42. El Rouby N, Lima JJ, Johnson JA. Proton pump inhibitors: from CYP2C19 pharmacogenetics to precision medicine. Expert Opin Drug Metab Toxicol. 2018;14(4):447–460. doi:10.1080/17425255.2018.1461835
43. Bhise NS, Elsayed AH, Cao X, Pounds S, Lamba JK. MicroRNAs mediated regulation of expression of nucleoside analog pathway genes in acute myeloid leukemia. Genes. 2019;10(4):319. doi:10.3390/genes10040319
44. Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–350. doi:10.1038/nature15817
45. Rojas CA, Sepúlveda Copete M, García Abadía JA, et al. Un ensayo clínico piloto de la efectividad clínica de dos presentaciones de esomeprazol. Rev Colomb Gastroenterol. 2019;34(3):261–268. doi:10.22516/25007440.335
46. Dong Y, Xiao H, Wang Q, et al. Analysis of genetic variations in CYP2C9, CYP2C19, CYP2D6 and CYP3A5 genes using oligonucleotide microarray. Int J Clin Exp Med. 2015;8(10):18917–18926.
47. Ye C, Jin H, Zhang R, et al. Variability of warfarin dose response associated with CYP2C9 and VKORC1 gene polymorphisms in Chinese patients. J Int Med Res. 2014;42(1):67–76. doi:10.1177/0300060513499094
48. Al-Jenoobi FI, Alkharfy KM, Alghamdi AM, et al. CYP2C19 genetic polymorphism in Saudi Arabians. Basic Clin Pharmacol Toxicol. 2013;112(1):50–54. doi:10.1111/j.1742-7843.2012.00919.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.