Back to Journals » Drug Design, Development and Therapy » Volume 8
Evaluation of ceftiofur–PHBV microparticles in rats
Authors Vilos C, Constandil L, Rodas P, Cantin M, Zepeda K, Herrera N, Velasquez L
Received 10 January 2014
Accepted for publication 12 March 2014
Published 29 May 2014 Volume 2014:8 Pages 651—666
DOI https://doi.org/10.2147/DDDT.S60444
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Cristian Vilos,1,3 Luis Constandil,2,3 Paula I Rodas,1,3 Mario Cantin,4,5 Katherine Zepeda,1 Natalia Herrera,1 Luis A Velasquez1,3
1Center for Integrative Medicine and Innovative Science (CIMIS), Faculty of Medicine, Universidad Andres Bello, Santiago, 2Laboratory of Neurobiology, Department of Biology, Faculty of Chemistry and Biology, Universidad de Santiago de Chile, Santiago, 3Centro para el Desarrollo de la Nanociencia y Nanotecnología, Universidad de Santiago de Chile, Ecuador, Santiago, 4CIMA, Department of Integral Dentistry, Faculty of Dentistry, Universidad de La Frontera, Temuco, 5Center of Research in Biomedical Sciences, Universidad Autónoma de Chile, Temuco, Chile
Abstract: Despite the high number of antibiotics used for the treatment of infectious disease in animals, the development of slow release formulations presents a significant challenge, particularly in using novel biomaterials with low cost. In this report, we studied the pharmacokinetics, toxicity, and therapeutic activity of ceftiofur–PHBV (ceftiofur–poly(3-hydroxybutyrate-co-3-hydroxyvalerate)) in rats. The pharmacokinetic study demonstrated a sustained release of ceftiofur into the bloodstream, with detectable levels over the minimum inhibitory concentration for at least 17 days after a single intramuscular injection of ceftiofur–PHBV (10 mg/kg weight). In addition, the toxicological evaluation of biochemical, hematological, and coagulation blood parameters at the therapeutic dose demonstrated the safety of ceftiofur–PHBV, with no adverse effects. In addition, ceftiofur–PHBV exhibited a therapeutic effect for a longer time period than the nonencapsulated ceftiofur in rats challenged with Salmonella Typhimurium. The slow release of ceftiofur from the ceftiofur–PHBV, its low toxicity in the blood parameters evaluated, and the efficacy in the rats infected with Salmonella Typhimurium make ceftiofur–PHBV a strong candidate for biotechnological applications in the veterinary industry.
Keywords: microparticles, drug delivery, Salmonella Typhimurium, rat infection model, blood parameters
Introduction
Antibiotics are drugs used globally in both humans and animals.1 In the past 2 decades, numerous drug delivery systems based on biodegradable and noncytotoxic polymers have been developed to improve the effectiveness and availability of antibiotics, decreasing their degradation or inactivating and eliminating their side effects.2,3 Despite the advances in the field of polymeric microparticle formulation, in vivo activity of microparticles with antibiotics has not been widely addressed.
Ceftiofur is a third generation cephalosporin exclusively used in animals. It has a broad spectrum of activity against Gram-positive and Gram-negative bacteria such as Pasteurella multocida, Pasteurella haemolytica, Haemophilus somnus and Escherichia coli.4–6 This antibiotic has been globally approved for the treatment of infectious diseases in pigs,7 cattle,8 sheep,9 goats, and horses,10,11 and for the treatment of metritis and infections of the extremities in cattle.12
Ceftiofur is a β-lactam antibiotic, and therefore exerts its bactericidal effect by inhibiting cell wall synthesis through binding to the penicillin binding proteins, and disrupting the production of peptidoglycan leading to cell lysis in an isotonic environment.13 Similar to the bactericidal activities of other β-lactam antibiotics, the bactericidal activity of ceftiofur is time-dependent, meaning that it exhibits antimicrobial activity when the free concentration of antibiotic exceeds the minimum inhibitory concentration (MIC) in time (t>MIC).14 Studies in rats, cattle and swine have demonstrated that ceftiofur is metabolized in the liver to desfuroylceftiofur (primary metabolite), which has similar antimicrobial activity because it conserves its β-lactam structure.15–17 Due to the biotransformation of ceftiofur in the liver, ceftiofur cannot be directly measured; however the plasma concentration of ceftiofur can be quantified indirectly as desfuroylceftiofur acetamide after the derivatization process as described in previous reports.16,18
In the last decade, the biosynthesis of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) and other polyhydroxyalkanoates from large-scale bacterial systems has enabled the development of several biodegradable polymers with low production costs; making feasible the development of drug delivery technology for veterinary applications.19–21
Injectable polymeric microparticles have shown high effectiveness as a sustained-release drug delivery system in humans, and represent an excellent candidate for use in the release of antibiotics in veterinary applications.22 The main advantage of these microparticles is their biodegradability, as it is not necessary to remove them through surgery even when the therapy is completed. The microparticles not only protect the drug from degradation and/or inactivation, but also do not require the use of solvents for their administration.23,24
In the previous report, we described the synthesis and in vitro characterization of ceftiofur–PHBV microparticles, which exhibited a size in the range of 1.65–2.37 μm with a spherical shape and smooth surface. The encapsulation efficiency obtained was 39.5%±1.1% weight/weight (w/w) and the experimental drug loading was 6.6%±0.2% w/w. In addition, ceftiofur–PHBV exhibited an antibacterial activity in E. coli and a low cytotoxicity in Hep G2 cultures with a half maximal inhibitory concentration (IC50) of >10 mg/mL, and under in vitro conditions ceftiofur–PHBV released ceftiofur over 7 days.25
In this report, we present the pharmacological, therapeutic, and toxicological activity of ceftiofur–PHBV microparticles in rats, with a hope to progress to drug delivery systems for application in the veterinary industry.
Materials and methods
Animals
This investigation was performed by following the guidelines on ethical standards detailed in the Guide for the Care and Use of Laboratory Animals of National Institutes of Health (NIH),26 and protocols were approved by the Bioethics Committee of the Universidad de Santiago de Chile. Experiments were performed in 80 healthy male Sprague Dawley rats weighing 280–320 g. They were obtained from the Animal Facility of the Universidad de Santiago de Chile and were separated into randomized groups to evaluate the pharmacological, therapeutic, and toxicological activity of microparticles. Every effort was made to minimize the number and the suffering of animals used in the experiments. On arrival at the facility, the animals were clinically examined, weighed, and housed (six animals per cage). Animals were kept in an environment where the temperature ranged from 19°C to 21°C and the light was controlled (12 hour light/dark cycle; light from 8 am to 8 pm). Rats had ad libitum access to food and water. Rats were allowed to habituate to the housing facility for 3 days before commencing the experiments, and these housing conditions remained constant throughout the experiments. The animals were euthanized at the end of the experiment via CO2 overdose followed by a bilateral thoracotomy.
Synthesis of ceftiofur–PHBV
Ceftiofur–PHBV were formulated by the double emulsion method (w1/o1/w2 [water/oil/water]) according to our previous work.25 Briefly, 15 mg of ceftiofur was dissolved in 450 μL of a 2:1 mixture of water:methanol solution, and added to 1 mL of PHBV (25 mg/mL) dissolved in dichloromethane. The first emulsion (w1/o1) was prepared with a handheld homogenizer (Tisssue-Tearor™; Biospec Products, Bartlesville, OK, USA) at 35,000 rpm for 40 seconds. Afterwards, the water-in-oil emulsion was further emulsified under the same conditions in 4 mL of polyvinyl alcohol solution (0.5% w/v [weight/volume]) (w2). This w1/o1/w2 emulsion was immediately poured into a beaker containing 25 mL of polyvinyl alcohol (0.1% w/v) solution and was stirred in an orbital shaker at 120 rpm for 12 hours to evaporate the dichloromethane and methanol. The ceftiofur–PHBV was washed three times and harvested by centrifugation at 5,000 rpm for 10 minutes. The microparticles were either stored at 4°C for immediate use, or lyophilized for storage at −80°C for later use.
Pharmacokinetic analysis
The pharmacokinetic parameters were evaluated in 12 animals randomly separated into four experimental groups (n=3 animals per group), which were injected with a disposable tuberculin syringe and 23G needle in the right gastrocnemius muscle. The groups were injected with sterile vehicle solution (physiological saline); empty microparticles (PHBV–Ø), 10 mg/kg weight; ceftiofur–PHBV, 10 mg/kg weight; and 700 μg/kg ceftiofur equivalent amount encapsulated (ceftiofur). One hundred microliter blood samples were obtained from the tip of the tail of the animals via a small incision at 0, 0.3, 1, 3, 7, 14, and 20 days. To prevent any discomfort and pain, all the animals were kept under gaseous anesthesia (isoflurane) and the tails were first cleaned with alcohol (95%) and then locally anesthetized with lidocaine (1%).
Pharmacokinetic parameters were calculated based on the plasma concentration of desfuroylceftiofur acetamide (DCA), and adjusting to a two-compartment pharmacokinetic model according to the work described by Tang in 2010.14 The maximum plasma concentration (Cmax) and time to maximum concentration (Tmax) were obtained from the semilogarithmic curve of concentration versus time, and the area under the curve (AUC) was determined by the linear trapezoidal method.
The quantification of DCA was performed by ultra-performance liquid chromatography (UPLC) using an Acquity system (Waters, Milford, MA, USA) equipped with a binary solvent delivery pump, an auto sampler, and a tunable ultraviolet detector. The chromatographic separation was performed in a Waters Acquity BEH 50×2.1 mm, 1.7 μm C18 column. The mobile phase was a 78:22 (volume/volume) mixture of 20 mM disodium hydrogen phosphate dihydrate buffer (pH 6.0, the pH was adjusted by the addition of 85% ortho-phosphoric acid) and acetonitrile loaded at a flow rate of 0.6 mL/min. The peak detection was carried out at a wavelength of 292 nm. The injection volume of samples was 0.5 μL, and the mobile phase was used as a diluent, while the column was maintained at 27°C.
The plasma samples were quantified after the serum derivatization according to protocol described by Jaglan et al.27 In this protocol, ceftiofur is treated with dithioerythritol and iodoacetamide generating the complex DCA that can be measured at 292 nm.27 Experimentally, blood samples were centrifuged for 5 minutes at 5,000 rpm to collect the supernatant. The proteins were precipitated with 500 μL of 20% trichloroacetic acid, and treated with 7 mL of 0.4% dithioerythritol solution prepared in pH 9 borate buffer. Afterwards the samples were placed in a water bath at 50°C for 15 minutes, and the derivatization was carried out by adding 500 μL of 14% iodoacetamide solution prepared in phosphate-buffered saline (PBS; 25 mM phosphate at pH 7.4). Finally, 75 mL of 20% phosphoric acid was added and centrifuged for 10 minutes at 300 rpm. The supernatant was collected and the concentration of DCA was quantified by UPLC. Calibration curves were made following the same derivatization procedure using ceftiofur standard solutions (0.25, 50, 100, and 150 μg/mL) prepared in fresh human serum.
In vivo toxicological evaluation
Changes in biochemical, hematological and hemostasis parameters were studied in animals that received an intramuscular (im) injection of ceftiofur–PHBV (10 mg/kg) or saline, at 0.3, 1, 3, and 7 days postadministration of microparticles. The biochemistry, hematology and hemostasis parameters were analyzed in a veterinary clinical laboratory (CAMPVSVet, Santiago, Chile). The data were compared intragroup and intergroup during the 7 days.
Animals were randomly separated into three groups, which received sterile vehicle solution (saline, n=3); PHBV–Ø, (n=3), and ceftiofur–PHBV (n=3). Investigators were double-blinded to the treatment groups. Clinical observations were recorded daily to determine the presence of signs of toxicity or adverse effects caused by the microparticles. Also, body weight, food consumption and temperature were measured daily. The blood samples were taken by decapitation under isoflurane anesthesia.
At 7 days postadministration of microparticles, we studied the toxicological effect of high concentrations of PHBV–Ø and ceftiofur–PHBV on blood parameters and the general health condition of animals such as food intake, weight gain, and response to handling.
The rats were separated into three groups, each treated with sterile vehicle solution (saline, n=3), PHBV–Ø (n=3), and ceftiofur–PHBV (n=3), respectively. The doses injected were 20, 30, and 50 mg/kg of weight. The blood parameters of animals that received saline in the previous set of toxicological experiments were used as control.
Acute systemic infection in rat model
Salmonella enterica serovar Typhimurium (ATCC 14028) is a reference strain widely used in a rat model of acute systemic salmonellosis infection.28–30 Bacteria was grown aerobically in Luria-Bertani broth overnight at 37°C. Bacterial subcultures were grown until log phase, then the cultures were harvested, washed, and suspended in PBS. Serial ten-fold dilutions were made, plated out in nutrient agar, and incubated overnight at 37°C. The colonies were counted after incubation and colony forming units (CFU) were calculated.
The in vivo efficacy of ceftiofur–PHBV microparticles was evaluated using the acute systemic salmonellosis infection rat model as previously described,28–30 in 14 animals divided into four groups: infected (n=3) intramuscularly injected with 0.3 mL of PBS, and 2.0×107 bacteria via intraperitoneal injection; noninfected (n=3) injected with 0.3 mL of PBS intramuscularly, and 1 mL of PBS via intraperitoneal injection; ceftiofur–PHBV (n=4) injected with 10 mg/kg ceftiofur–PHBV intramuscularly in 0.3 mL of PBS, and 2.0×107 bacteria via intraperitoneal injection; ceftiofur (n=4) injected with 700 μg/kg of ceftiofur intramuscularly in 0.3 mL of PBS and 2.0×107 bacteria via intraperitoneal injection. The bacterial inoculum was prepared in 1 mL of sterile PBS and injected 2 days postadministration of ceftiofur, ceftiofur–PHBV, and PBS.
Daily blinded clinical observations were made on the general health of the animals during 6 days: weight, body temperature, food consumption, consistency of feces and behavior changes. The feces were microbiologically tested by the plating of homogenates prepared in PBS onto Salmonella-Shigella agar (SS agar) in order to detect the presence of bacteria. On day 6, the animals were euthanized and the liver, colon, spleen, and intestine of each animal were removed and homogenized in 3 mL of PBS aseptically. The homogenates were serially diluted and the 0.01 mL dilutions were plated onto SS agar plates. The plates were incubated overnight, and the number of viable bacteria (CFU/mg tissue) was counted. To analyze hematological changes generated by S. Typhimurium infection, postmortem blood samples were taken and the hematological parameters were registered.
Statistical analysis
The results were expressed as mean ± standard deviation (SD) for each parameter analyzed. A nonparametric Kruskal–Wallis statistical analysis was performed to determine significance among the different experimental groups. The significance was set at P-value less than or equal to 0.05, and for those cases in which there were significant differences, the Mann–Whitney post hoc test was applied. All statistical analyses were performed with the Prism 3.0 software (GraphPad Software, Inc., San Diego CA, USA).
Results and discussion
Pharmacokinetics of ceftiofur–PHBV microparticles
Like many other drugs that undergo biotransformation in the body, ceftiofur metabolizes to desfuroylceftiofur and related metabolites within an hour of injection. Since this is the case, direct quantification of ceftiofur in vivo is virtually impossible. For our pharmacokinetic study, we have quantified DCA in rats treated with saline, PHBV–Ø, ceftiofur–PHBV, and ceftiofur. The plasma measurements of DCA were carried out with samples taken from the tail tip of rats at 0, 0.3, 1, 3, 7, 14, and 20 days. After the single im injection of ceftiofur, the plasma levels of DCA reached 0.3±0.04 μg/mL after 8 hours (0.3 day) and the maximum (36.6±2.8 μg/mL) after 24 hours (1 day). These levels quickly decreased until they were undetectable after 72 hours (3 days postinjection).
When the animals were injected with ceftiofur–PHBV, the plasma concentration of DCA reached 0.04 μg/mL after 24 hours (1 day), and a maximum (6.3±0.1 μg/mL) after 72 hours (3 days). The plasma concentration of DCA in the group injected with ceftiofur–PHBV was maintained over the MIC (>1 μg/mL) until the end of the experiment (day 20 postadministration). On the other hand, the PHBV–Ø and saline groups did not show measurable levels of antibiotic during the experiment (Figure 1).
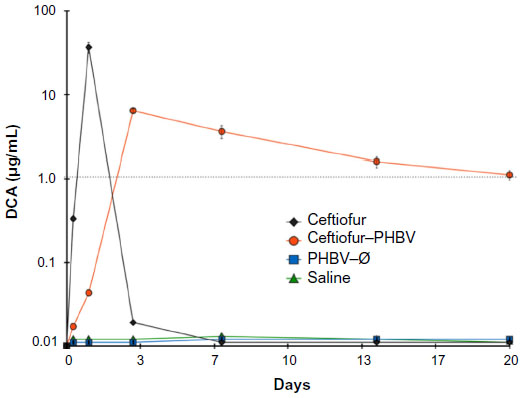 | Figure 1 Semilogarithmic curve of the plasma concentration of DCA in rats that received an intramuscular injection of nonencapsulated ceftiofur (700 μg/kg body weight), ceftiofur–PHBV (10 mg/kg body weight), PHBV–Ø (10 mg/kg body weight), and saline (n=3 for each group). |
The pharmacokinetic properties presented in Table 1 were calculated under a two-compartment pharmacokinetic model from a semilogarithmic curve of plasma concentration of drug over time, as described in previous reports.14 The Cmax of DCA in animals treated with ceftiofur–PHBV (6.31±0.3 μg/mL) was 5.9 times lower than the Cmax of animals which received nonencapsulated ceftiofur (36.6±2.8 μg/mL).
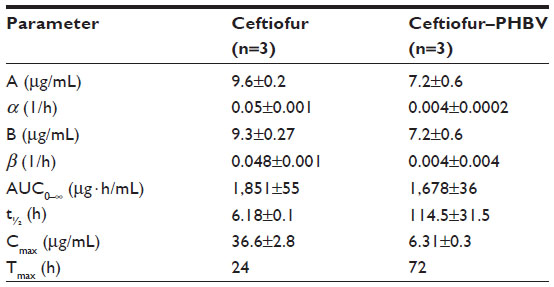 | Table 1 Pharmacokinetic parameters calculated from biexponential curve of the average plasma concentration of DCA in rats |
The Cmax of DCA obtained in the group treated with ceftiofur–PHBV was 1.9 times lower than the Cmax (11.8±1.6 μg/mL) described in the study of Brown et al in pigs that received 3 mg/kg weight via im injection.18 Also, the Cmax of DCA in the group of ceftiofur–PHBV was 1.6 times lower than the Cmax (10.3±1.2 μg/mL) reported by Goudah in camels treated with 2.2 mg/kg weight of ceftiofur using the same route of administration.31 Lower Cmax implies reduced potential of adverse effects related to high doses of antibiotics.
For the elimination half-life (t½), ceftiofur–PHBV showed a longer plasma circulation time than nonencapsulated ceftiofur, and also longer t½ than others studies in pigs (16.7±2.3 hours) injected with 3 mg/kg weight (im) of ceftiofur, llamas (8.1±1.7 hours) injected with 2.2 mg/kg (im), and alpacas (8.2±1.3 hours) that received doses in the range of 1.30 to 1.51 mg/kg via im injection.18,32 In addition, the sustained release of ceftiofur from ceftiofur–PHBV was longer (17 days) than that of other long-acting drug delivery systems based on liposomes (2 days) and crystalline-free acid sterile suspension (10 days).33,34 The pharmacokinetic study demonstrated that animals treated with ceftiofur–PHBV exhibited t>MIC for at least 17 days, suggesting ceftiofur–PHBV can be clinically effective. Advantages of using ceftiofur–PHBV include the reduction of the administration frequency of the drug into a single dose, and generation of a more uniform and constant therapeutic effect by minimizing the fluctuations in plasma concentration. Despite the existence of other reports regarding formulations of PHBV microparticles with other antibiotics such as rifampicin,35 gentamicin,36 tetracycline,2 this finding is of significance because it is the very first to present the pharmacokinetic properties of ceftiofur and evaluation of this antibiotic in animals.
Toxicological evaluation of ceftiofur–PHBV microparticles
Given that blood clinical parameters such as biochemistry, hematology and coagulation, provide a comprehensive assessment of health status, we evaluated the blood clinical parameters of rats treated with therapeutic and high doses of microparticles.
The blood parameters were determined at 0.3, 1, 3, and 7 days after administration of a single therapeutic dose of ceftiofur–PHBV, and the AUC was compared both within and between groups (versus saline). Daily clinical observations of the animals showed no symptoms of toxicity or adverse effects. In addition, no changes were reported in the fur or appearance of eyes. All the groups showed normal response at handling, and no deaths occurred in this test. Furthermore, animals showed an increase in weight similar to the control group (Figure S1).
The examination of biochemical parameters did not show significant changes in the experimental groups compared with saline control, which suggests that the microparticles or their degradation products did not cause alterations in metabolic parameters, nor did they affect the liver and kidney function, as described in Figure 2. In addition, the quantitative analyses of hematological parameters showed no significant differences in blood counts (hematocrit, hemoglobin, leukocytes), as illustrated in Figure 3. On the other hand, the blinded morphological examination of blood smears stained with May-Grünwald/Giemsa exhibited normal parameters, including slight anisocytosis of red blood cells with the absence of pathological granulations in polymorphonuclear cells (neutrophils, eosinophils and basophils), and a normal morphology of mononuclear leukocytes (lymphocytes and monocytes), as shown in Figure 4. The study of the prothrombin time (PT) and the partial thromboplastin time (PTT) showed no abnormalities of hemostasis, ruling out any pro-thrombotic activity of microparticles administered at therapeutic doses, as described in Figure 5 (full data report of blood parameters in Tables S1–S3).
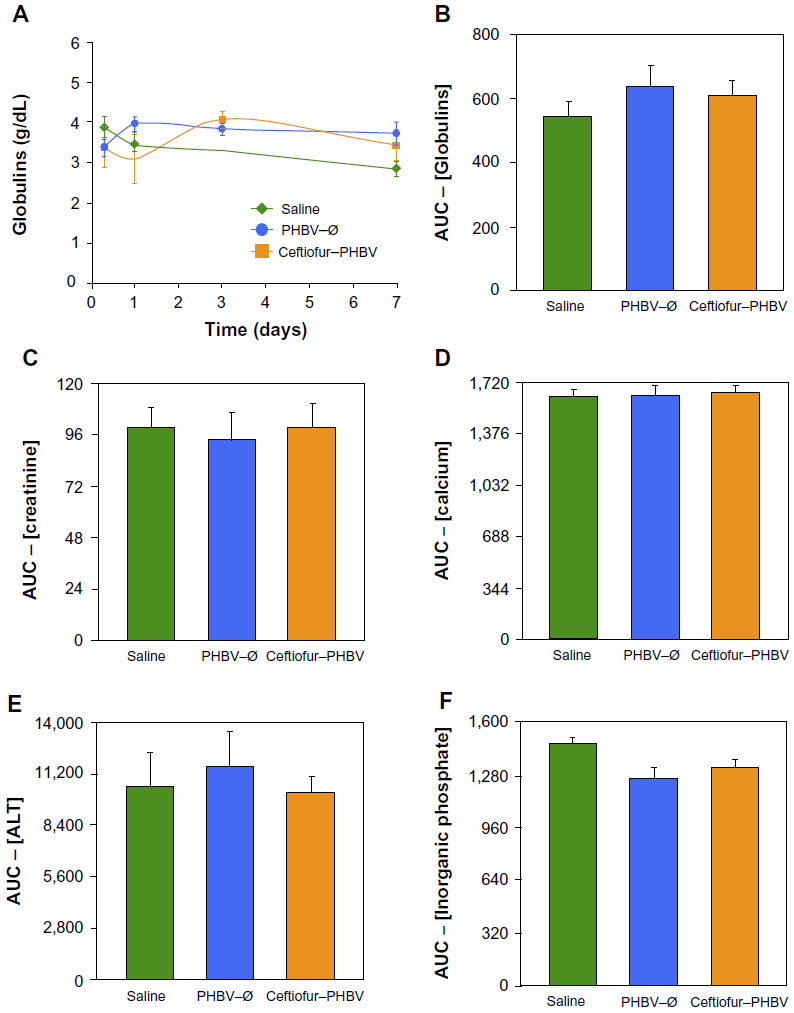 | Figure 2 (A) Blood levels (g/dL) of globulins at 0, 1, 3, and 7 days of the different study groups (saline, PHBV–Ø and ceftiofur–PHBV). (B) Comparison of area under the curve (AUC) of the plasma globulins at 0, 1, 3, and 7 days. (C–F) Other biochemical blood parameters determined in this study, expressed as the area under the curve of the plasma concentrations obtained in 7 days (P>0.05, Kruskal–Wallis). |
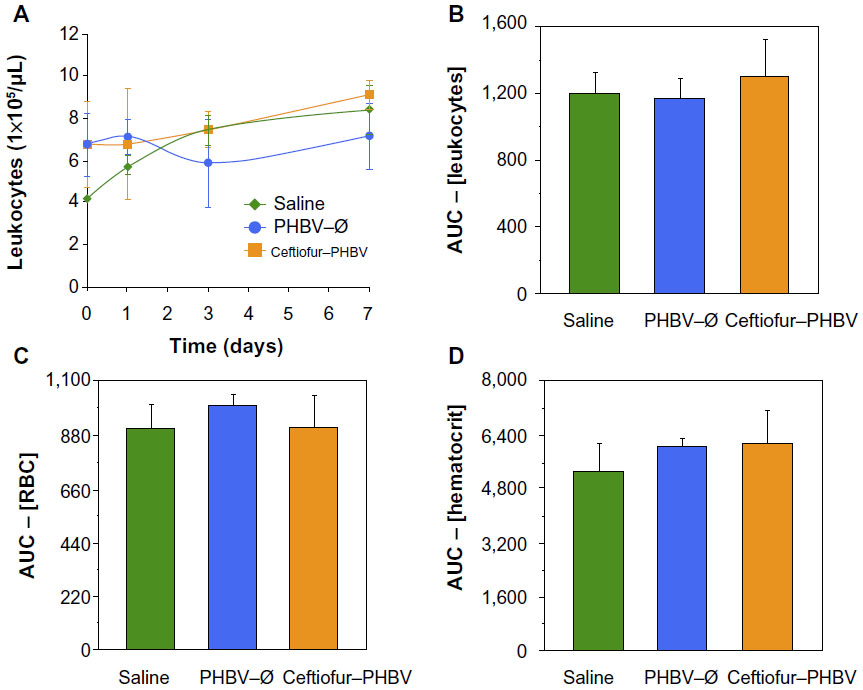 | Figure 3 (A) Leukocyte count at 0, 1, 3, and 7 days in the different study groups (saline, PHBV–Ø and ceftiofur–PHBV). (B) Comparison of the area under the curve (AUC) the values of the leucocyte counts 0, 1, 3, and 7 days. (C and D) Other hematological blood parameters evaluated in this study (P>0.05, Kruskal–Wallis). |
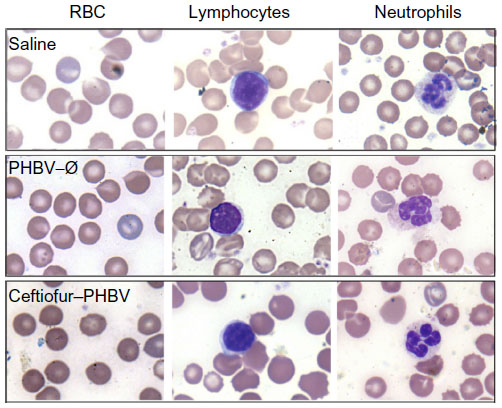 | Figure 4 Images of peripheral blood smear of experimental groups analyzed by optical microscopy (100×). |
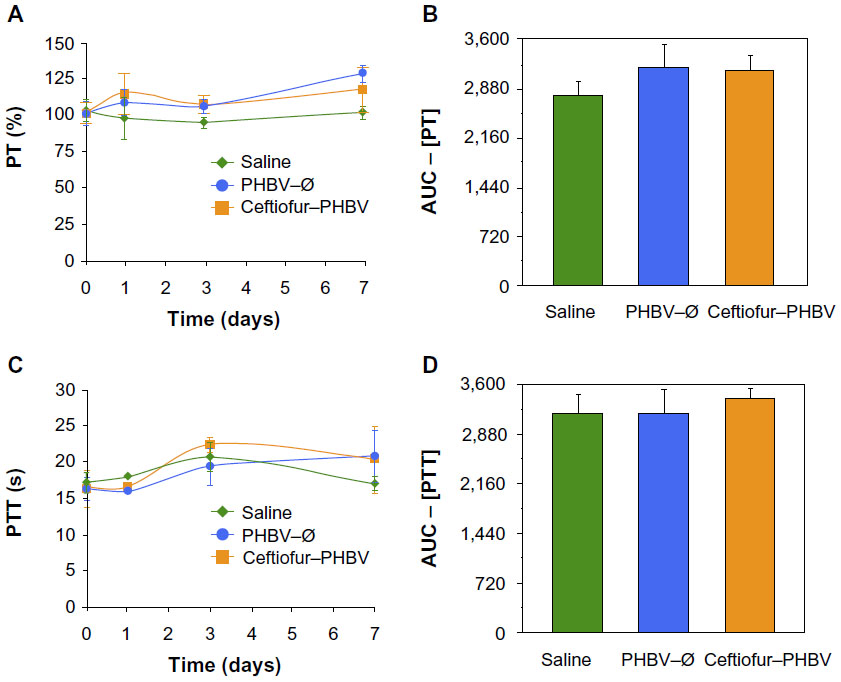 | Figure 5 (A) Determination of prothrombin time (PT) and (C) partial thromboplastin time (PTT) at 0, 1, 3, and 7 days in the animals at therapeutic dose (saline, PHBV–Ø and ceftiofur–PHBV). (B and D) represent the area under the curve (AUC) of the PT and PTT respectively (P>0.05, Kruskal–Wallis). |
In order to evaluate the toxicological effects of microparticles administered in higher doses after 7 days, blood parameters were analyzed. The daily observations showed no signs of toxicity or adverse effects, and all groups showed an increment of weight equivalent to saline control (7.5%±0.8%) (Figure S2). During the study no deaths occurred under experimental conditions.
Blood biochemical measurements of animals that received high doses of microparticles displayed a significant decrease of inorganic phosphate in all experimental groups compared to the saline group (Figure 6). In addition, quantification of hematological parameters revealed no significant differences between the doses administered, as shown in Figure 7. On the other hand, the hemostasis evaluation showed a significant decrease of activated PTT compared to that of saline control in all groups as shown in (Figure 8; Tables S4–S6).
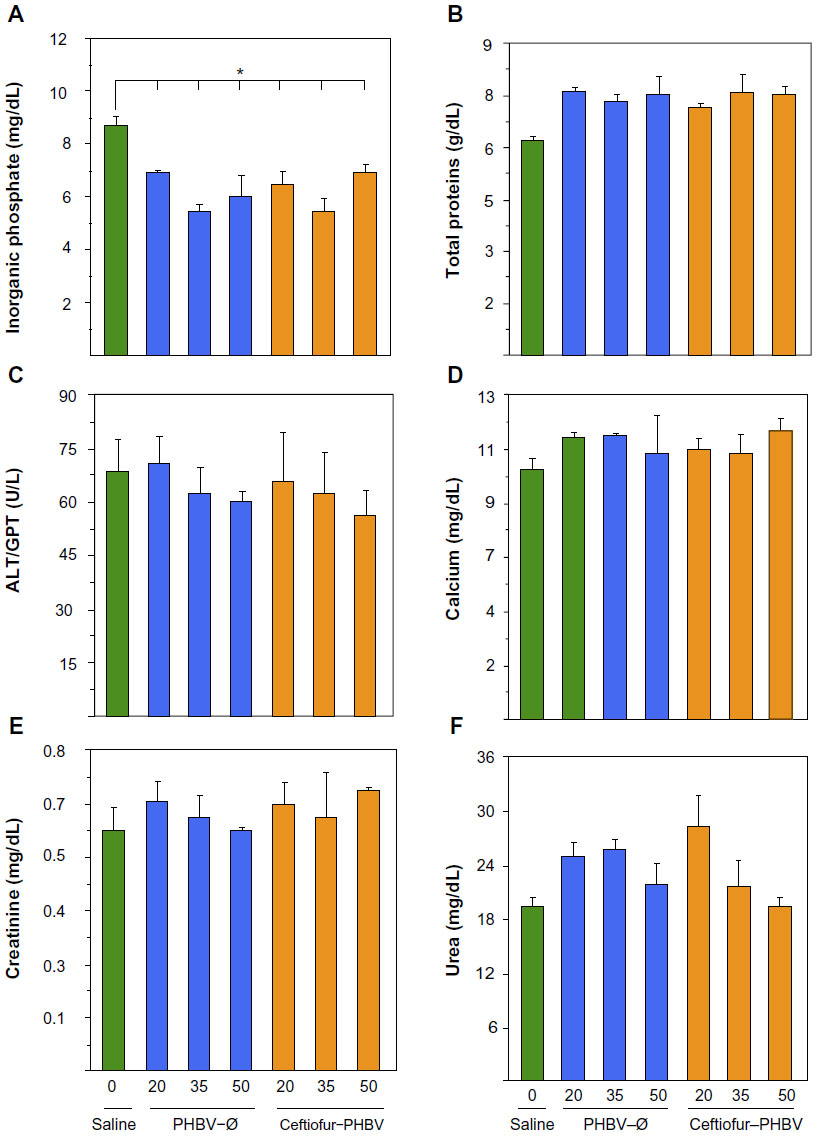 | Figure 6 Biochemical blood parameters of rats after 7 days of treatment with PHBV–Ø and ceftiofur–PHBV (doses of 20, 35, and 50 mg/kg) administrated intramuscularly (gastrocnemius). |
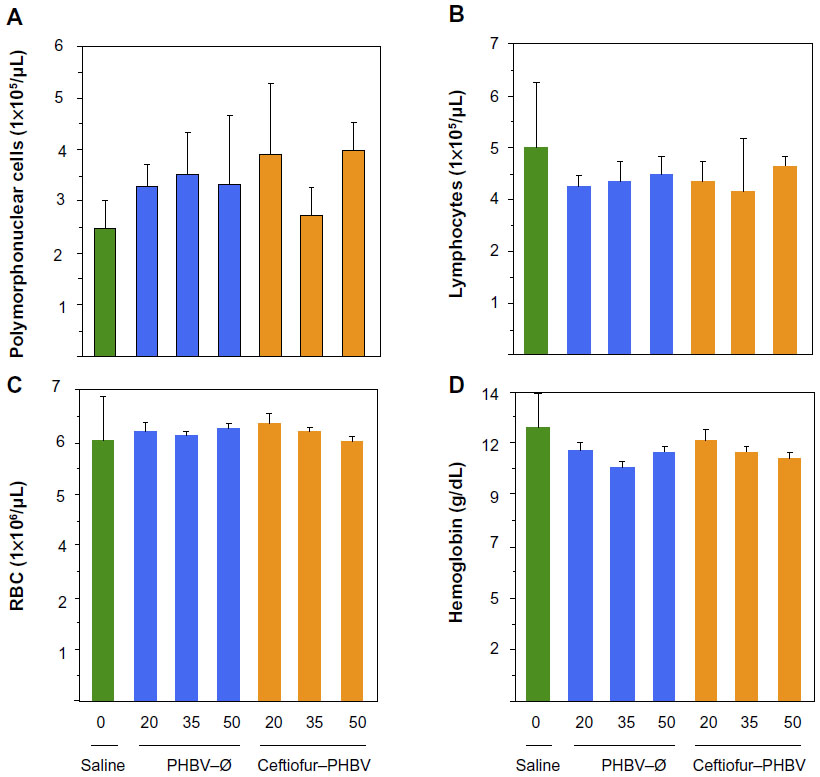 | Figure 7 Hematological blood parameters of rats after 7 days of treatment with PHBV–Ø and ceftiofur–PHBV (doses of 20, 35, and 50 mg/kg) administrated intramuscularly (gastrocnemius). |
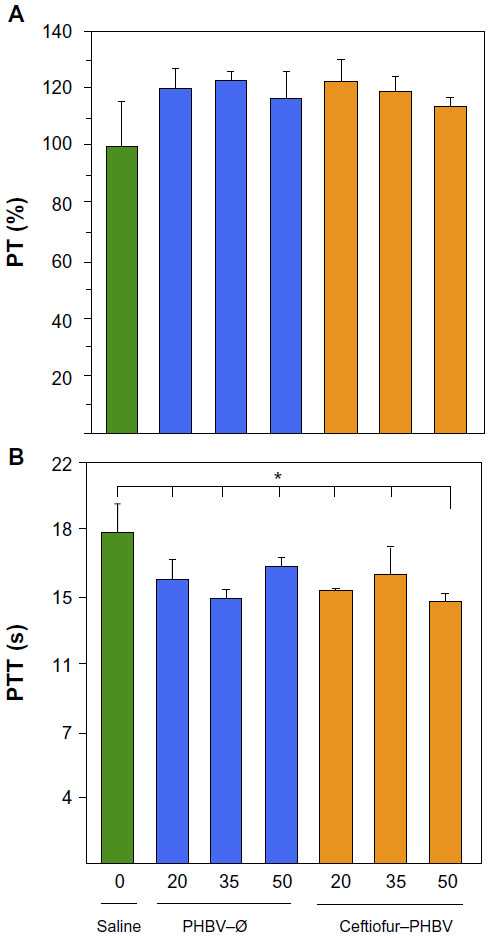 | Figure 8 Evaluation of coagulation parameters (A) prothrombin (PT), and (B) partial thromboplastin time (PTT) of rats after 7 days of treatment with PHBV–Ø and ceftiofur–PHBV (doses of 20, 35, and 50 mg/kg) administrated intramuscularly (gastrocnemius). |
The parameters associated with liver function (albumin, globulins, bilirubin and hepatic enzymes: alkaline phosphatase, gamma glutamyltransferase, lactate dehydrogenase, aspartate aminotransferase and alanine aminotransferase) did not exhibit significant changes in both toxicological experiments, demonstrating that the microparticles do not affect liver function at the doses tested. These results were confirmed by liver samples from the ceftiofur–PHBV group, processed for morphological analysis that showed signs of normal liver regeneration: cell proliferation, dyskaryostasis and formation of rows of liver cell trabeculae, without microparticle waste (Figure S3).
In addition, no changes were observed in creatinine and urea levels in all experimental groups, suggesting that microparticles and their degradation metabolites do not induce renal changes at the therapeutic and high doses administered. However, the levels of inorganic phosphate at high doses of microparticles showed a significant decrease in all groups. According to the literature, the hypophosphatemia could be generated by several mechanisms including internal redistribution, increased urinary excretion, decreased intestinal absorption, or a combination of all these abnormalities.37 The biochemical blood parameters measured and the healthy status of the animals during the experiment suggest that changes of inorganic phosphate metabolism do not involve changes in bone metabolism, liver damage, malnutrition or the presence of an infectious process. Moreover, animals did not receive medication such as diuretics, glucocorticoids or mineralocorticoids. With the data obtained in this study we cannot explain the changes observed in the metabolism of inorganic phosphate in animals that received high doses of microparticles.
The quantitative analysis of hematological parameters did not show changes, ruling out the presence of anemia or an adverse immune response generated by the microparticles or their degradation products. In addition, the observation of the peripheral blood smears exhibiting typical morphology of red and white blood cells of animals, confirmed these results.
Hemostasis is a physiological mechanism which causes bleeding to stop, which involves the participation of blood vessels, platelets, and the coagulation cascade. The coagulation cascade includes two pathways, intrinsic and extrinsic, and the cascade can be studied by the PT and PTT assays.38 The PT and PTT hemostasis assays exhibited normal clotting activity at a therapeutic dose, but at a high dose of PHBV–Ø and ceftiofur–PHBV the PTT showed a significant decrease compared to that of saline. PTT reflects the overall integrity of the intrinsic coagulation pathway and might be prolonged when there is a lack of factor XII, high molecular weight kininogen, prekallikrein, and factor deficiencies involving common pathways such as factor V, X, II and fibrinogen. However, a decrease in PTT could indicate procoagulant activity. Therefore, to determine the cause by which the high concentrations of microparticles reduce the PTT, further studies measuring the activity separate from factors and thrombin time (TT) will be required.
Therapeutic effectiveness of ceftiofur–PHBV microparticles in rat salmonellosis
The in vivo therapeutic effectiveness of ceftiofur–PHBV was analyzed in rats challenged with S. Typhimurium over 6 days. In the rats challenged with S. Typhimurium, the ceftiofur, ceftiofur–PHBV, PHBV–Ø, and saline were administrated 2 days before the S. Typhimurium inoculations. Based on the pharmacokinetic results, two days were necessary for ceftiofur and its metabolite to achieve MIC after the ceftiofur–PHBV injection.
The infected animals (controls) exhibited typical clinical manifestations of salmonellosis reported in rats.39 The analysis of body weight and food consumption displayed a significant decrease in all groups treated with S. Typhimurium compared to the uninfected group, suggesting that all of these animals experienced sickness, which was more or less severe depending on the treatment administered (data not shown). Specifically, the signs of disease in the group treated with ceftiofur such as presence of ruffled fur, reduced activity, persistent chills, increased frequency and depth of breathing, hunching, anorexia and a slightly aggressive behavior in response to manipulation, were quite similar to those of the infected control. In contrast, the ceftiofur–PHBV group showed only signs of mild disease, as presented in Table 2.
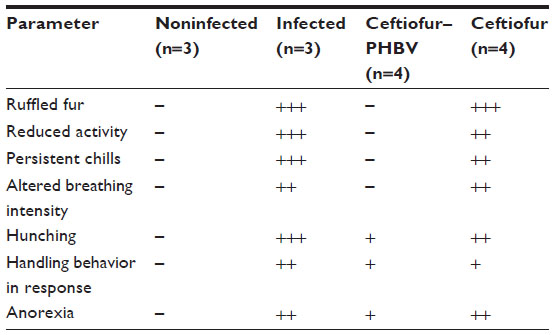 | Table 2 Evaluation of the general condition of the animals infected with Salmonella Typhimurium |
At the end of the experiment (6 days postinoculation of S. Typhimurium), animals were euthanized and examined. The evaluation of the internal organs of animals in the ceftiofur treatment group showed a remarkable presence of classical alterations previously reported in animal salmonellosis such as splenomegaly, intestinal exudate, and hepatomegaly with necrotic alterations in the lung and intestines (Table 3). Figure 9 demonstrates the appearance of internal organs in this group of animals. In addition, samples of liver, spleen, intestine, and colon were taken from all groups and cultured in SS agar to detect the presence of S. Typhimurium. The bacterial counts (CFU/mg of tissue) detailed in Table 4 showed that S. Typhimurium was found in all organs of the animals of the infected group, in the intestine and colon of animals treated with ceftiofur, and only in the colon of animals treated with ceftiofur–PHBV. The number of CFU/mg in animals treated with ceftiofur–PHBV was 70 and 100 fold less than those in the infected and ceftiofur groups respectively, which indicates that ceftiofur–PHBV treatment exerted a preventative effect upon Salmonella infection, reducing the systemic disease only to a localized infection of the colon.
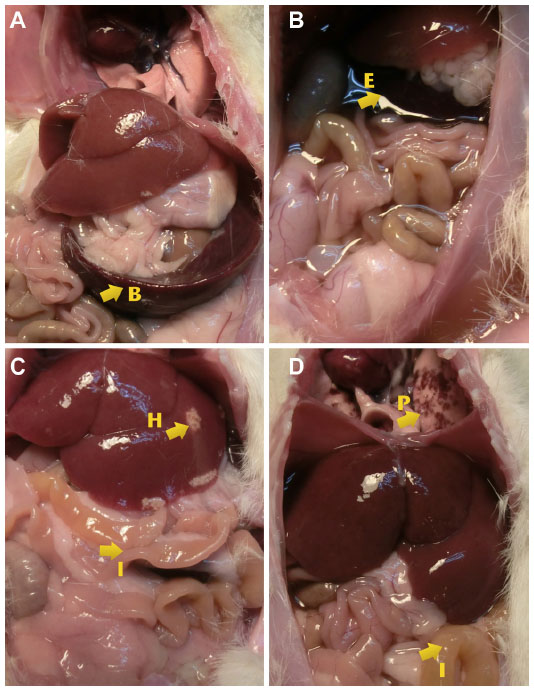 | Figure 9 Images showing the alterations found in the evaluation of internal organs. |
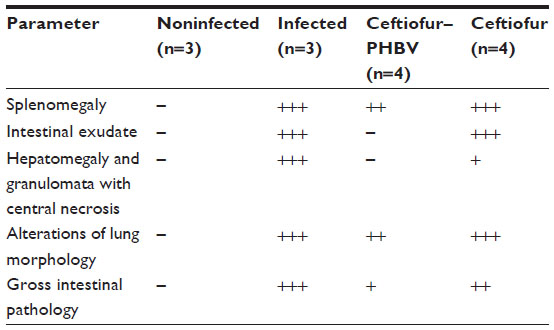 | Table 3 Evaluation of internal organs in animals infected with Salmonella Typhimurium |
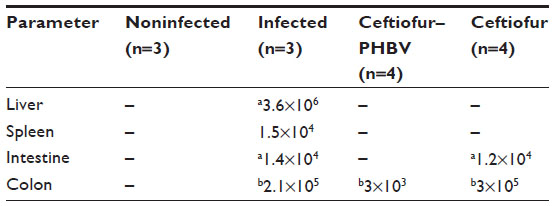 | Table 4 Detection of Salmonella Typhimurium in different organs removed at 6 days after infection |
On the other hand, the hematological parameters analyzed in blood samples at the end of the experiment displayed a significant increment of total leukocytes and polymorphonuclear cells in control infected and ceftiofur groups (>50%) compared with control noninfected and ceftiofur–PHBV, suggesting that S. Typhimurium infection was most noticeable in the infected and ceftiofur groups compared to the ceftiofur–PHBV group as shown in Table 5.
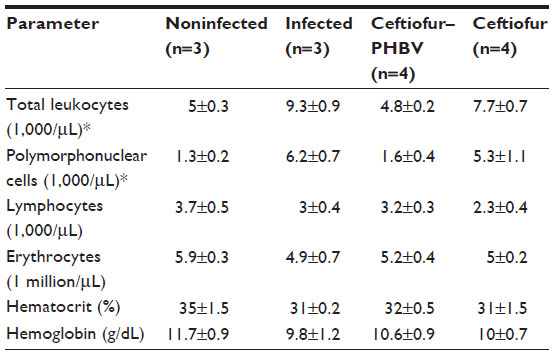 | Table 5 Hematological blood parameters evaluated in study of salmonellosis in rats |
During this study, two deaths were registered in the control infected, and one death in the group treated with ceftiofur, which demonstrates that the ceftiofur–PHBV had greater effectiveness than ceftiofur in rats infected with S. Typhimurium.
Conclusion
In this work, we analyzed the pharmacokinetics, toxicology, and the efficacy of ceftiofur-loaded PHBV microparticles in rats. In the pharmacokinetic study, the drug showed levels above the MIC for 17 days longer than that of nonencapsulated ceftiofur. In the toxicology study, no alterations in hepatic metabolism, renal, hematologic and coagulation measures were observed, indicating that the administration of microparticles at therapeutic doses can be used without changes in the toxicological parameters. In the rat infection model, the ceftiofur–PHBV treatment exposed remarkable therapeutic efficacy and preventative action against S. Typhimurium.
Ceftiofur–PHBV microparticles are a strong candidate for biotechnological applications in the veterinary industry due to the sustained release of ceftiofur at a therapeutic level, the low toxicity, and the high antimicrobial efficacy.
Acknowledgments
The authors acknowledge the financial support of CONICYT under BASAL Grant FB0807 and FONDECYT 1120952. We acknowledge Soung-Jae Bong for his contribution in editing and reviewing this manuscript. CV acknowledges the support of the Grant TPI06 and Becas-Chile fellowship CONICYT-Chile.
Disclosure
The authors report no conflicts of interest in this work.
References
Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. J Antibiot (Tokyo). 2009;62(1):5–16. | |
Sendil D, Gursel I, Wise DL, Hasirci V. Antibiotic release from biodegradable PHBV microparticles. Journal of Controlled Release: Official Journal of the Controlled Release Society. 1999;59(2):207–217. | |
Heslinga MJ, Mastria EM, Eniola-Adefeso O. Fabrication of biodegradable spheroidal microparticles for drug delivery applications. J Control Release. 2009;138(3):235–242. | |
Hewett GR. Sensitivity of A pleuropneumoniae and P multocida to ceftiofur. Vet Rec. 1994;134(8):200. | |
McDermott PF, Barry AL, Jones RN, et al. Standardization of broth microdilution and disk diffusion susceptibility tests for Actinobacillus pleuropneumoniae and Haemophilus somnus: quality control standards for ceftiofur, enrofloxacin, florfenicol, gentamicin, penicillin, tetracycline, tilmicosin, and trimethoprim-sulfamethoxazole. J Clin Microbiol. 2001;39(12):4283–4287. | |
Mann S, Siler JD, Jordan D, Warnick LD. Antimicrobial susceptibility of fecal escherichia coli isolates in dairy cows following systemic treatment with ceftiofur or penicillin. Foodborne Pathog Dis. 2011;8(8):861–867. | |
Simsek DG, Aycan Z, Ozen S, et al. Diabetes care, glycemic control, complications, and concomitant autoimmune diseases in children with type 1 diabetes in Turkey: a multicenter study. J Clin Res Pediatr Endocrinol. 2013;5(1):20–26. | |
Bossowski A, Moniuszko M, Dabrowska M, et al. Lower proportions of CD4+CD25(high) and CD4+FoxP3, but not CD4+CD25+CD127(low) FoxP3(+)T cell levels in children with autoimmune thyroid diseases. Autoimmunity. 2013;46(3):222–230. | |
Sainaghi PP, Bellan M, Nerviani A, et al. Superiority of a high loading dose of cholecalciferol to correct hypovitaminosis d in patients with inflammatory/autoimmune rheumatic diseases. J Rheumatol. 2013;40(2):166–172. | |
Kreuter A, Kryvosheyeva Y, Terras S, et al. Association of autoimmune diseases with lichen sclerosus in 532 male and female patients. Acta Derm Venereol. 2013;93(2):238–241. | |
Dang MN, Buzzetti R, Pozzilli P. Epigenetics in autoimmune diseases with focus on type 1 diabetes. Diabetes Metab Res Rev. 2013;29(1):8–18. | |
Hornish RE, Kotarski SF. Cephalosporins in veterinary medicine – ceftiofur use in food animals. Curr Top Med Chem. 2002;2(7):717–731. | |
Lisignoli G, Zini N, Remiddi G, et al. Basic fibroblast growth factor enhances in vitro mineralization of rat bone marrow stromal cells grown on non-woven hyaluronic acid based polymer scaffold. Biomaterials. 2001;22(15):2095–2105. | |
Tang S, Xiao J, Guo G, He J, Hao Z, Xiao X. Preparation of a newly formulated long-acting ceftiofur hydrochloride suspension and evaluation of its pharmacokinetics in pigs. J Vet Pharmacol Ther. 2010;33(3):238–245. | |
Beconi-Barker MG, Roof RD, Millerioux L, et al. Determination of ceftiofur and its desfuroylceftiofur-related metabolites in swine tissues by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1995;673(2):231–244. | |
Jaglan PS, Cox BL, Arnold TS, Kubicek MF, Stuart DJ, Gilbertson TJ. Liquid chromatographic determination of desfuroylceftiofur metabolite of ceftiofur as residue in cattle plasma. J Assoc Off Anal Chem. 1990;73(1):26–30. | |
Salmon SA, Watts JL, Yancey RJ Jr. In vitro activity of ceftiofur and its primary metabolite, desfuroylceftiofur, against organisms of veterinary importance. J Vet Diagn Invest. 1996;8(3):332–336. | |
Brown SA, Hanson BJ, Mignot A, et al. Comparison of plasma pharmacokinetics and bioavailability of ceftiofur sodium and ceftiofur hydrochloride in pigs after a single intramuscular injection. Journal of Veterinary Pharmacology and Therapeutics. 1999;22(1):35–40. | |
Mitomo H, Takahashi T, Ito H, Saito T. Biosynthesis and characterization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) produced by Burkholderia cepacia D1. Int J Biol Macromol. 1999;24(4):311–318. | |
Bokemeyer C, Bondarenko I, Makhson A, et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009;27(5):663–671. | |
Loo CY, Sudesh K. Biosynthesis and native granule characteristics of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) in Delftia acidovorans. Int J Biol Macromol. 2007;40(5):466–471. | |
Zhang C, Dong Y, Zhao L. Preparation and characterization of novel microparticles based on poly(3-hydroxybutyrate-co-3-hydroxyoctanoate). J Microencapsul. 2014;31(1):9–15. | |
Stangel M, Baumann U, Borte M, et al. Treatment of neurological autoimmune diseases with immunoglobulins: first insights from the prospective SIGNS registry. J Clin Immunol. 2013;33 (Suppl 1):S67–S71. | |
Motlekar N. Optimization of experimental parameters for the production of LMWH-loaded polymeric microspheres. Drug design, development and therapy. 2009;2:39–47. | |
Vilos C, Constandil L, Herrera N, Solar P, Escobar-Fica J, Velasquez LA. Ceftiofur-loaded PHBV microparticles: A potential formulation for a long-acting antibiotic to treat animal infections. Electron J Biotechnol. 2012;15(4):1–13. | |
Guide for the Care and Use of Laboratory Animals. The National Academies Press; 1996. | |
Jaglan PS, Kubicek MF, Arnold TS, et al. Metabolism of ceftiofur. Nature of urinary and plasma metabolites in rats and cattle. J Agric. Food Chem. 1989;37(4):1112–1118. | |
Havelaar AH, Garssen J, Takumi K, et al. A rat model for dose-response relationships of Salmonella Enteritidis infection. J Appl Microbiol. 2001;91(3):442–452. | |
Thygesen P, Martinsen C, Hougen HP, Hattori R, Stenvang JP, Rygaard J. Histologic, cytologic, and bacteriologic examinations of experimentally induced Salmonella typhimurium infection in Lewis rats. Comp Med. 2000;50(2):124–132. | |
Rodenburg W, Keijer J, Kramer E, et al. Salmonella induces prominent gene expression in the rat colon. BMC Microbiol. 2007;7:84. | |
Goudah A. Pharmacokinetics of ceftiofur after single intravenous and intramuscular administration in camels (Camelus dromedarius). J Vet Pharmacol Ther. 2007;30(4):371–374. | |
Drew ML, Johnson L, Pugh D, Navarre CB, Taylor IT, Craigmill AL. Pharmacokinetics of ceftiofur in llamas and alpacas. Journal of Veterinary Pharmacology and Therapeutics. 2004;27(1):13–20. | |
Liu S, Guo D, Guo Y, Zhou W. Preparation and pharmacokinetics of ceftiofur sodium liposomes in cows. J Vet Pharmacol Ther. 2011;34(1):35–41. | |
Collard WT, Cox SR, Lesman SP, et al. Pharmacokinetics of ceftiofur crystalline-free acid sterile suspension in the equine. J Vet Pharmacol Ther. 2011;34(5):476–481. | |
Duran N, Alvarenga MA, Da Silva EC, Melo PS, Marcato PD. Microencapsulation of antibiotic rifampicin in poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Archives of Pharmacal Research. 2008;31(11):1509–1516. | |
Li H, Chang J. Preparation, characterization and in vitro release of gentamicin from PHBV/wollastonite composite microspheres. Journal of Controlled Release: Official Journal of the Controlled Release Society. 2005;107(3):463–473. | |
McPherson R, Matthews P. Henry’s Clinical Diagnosis and Management by Laboratory Methods, 22nd ed. 2011. | |
Tanaka K, Davie EW, Ikeda Y, SpringerLink (Online service). Recent Advances in Thrombosis and Hemostasis 2008. Tokyo: Springer; 2008: http://dx.doi.org/10.1007/978-4-431-78847-8. Accessed December 3, 2013. | |
Langer R. Selected advances in drug delivery and tissue engineering. J Control Release. 1999;62(1–2):7–11. |
Supplementary material
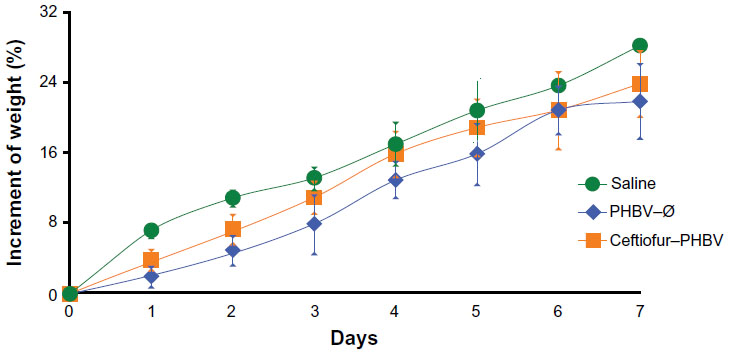 | Figure S1 Increment of weight of rats obtained in the toxicological evaluation at the therapeutic dose. |
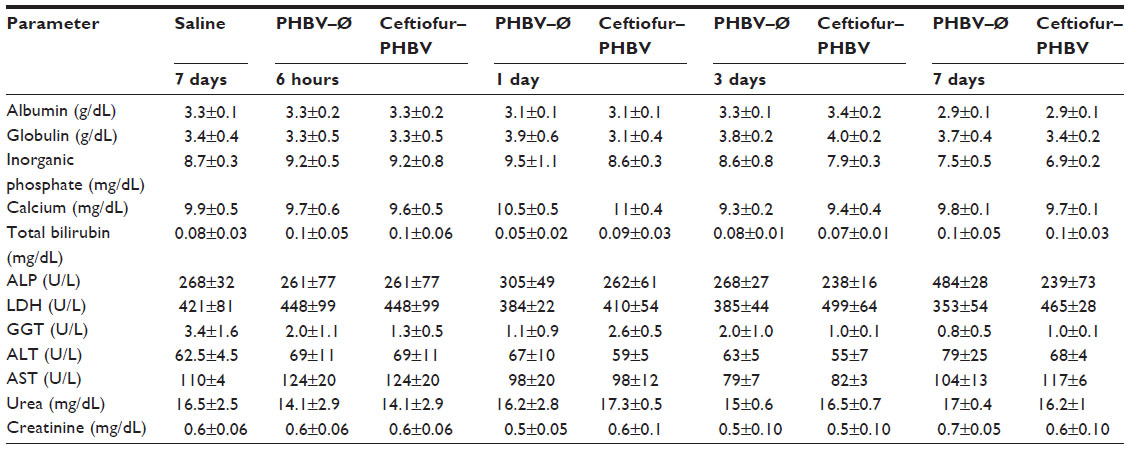 | Table S1 Plasma biochemical parameters obtained in the toxicological evaluation at the therapeutic dose |
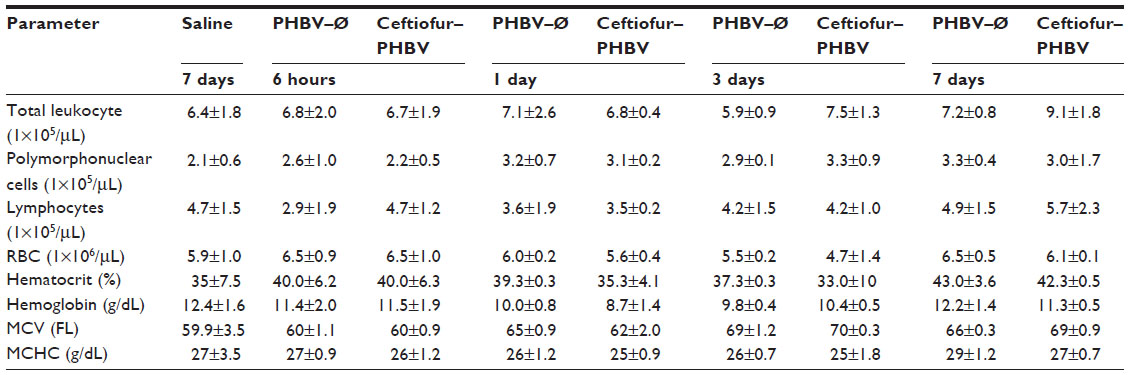 | Table S2 Plasma hematological parameters of rats obtained in the toxicological evaluation at the therapeutic dose |
 | Table S3 Evaluation of plasma coagulation parameters of rats obtained in the toxicological evaluation at the therapeutic dose |
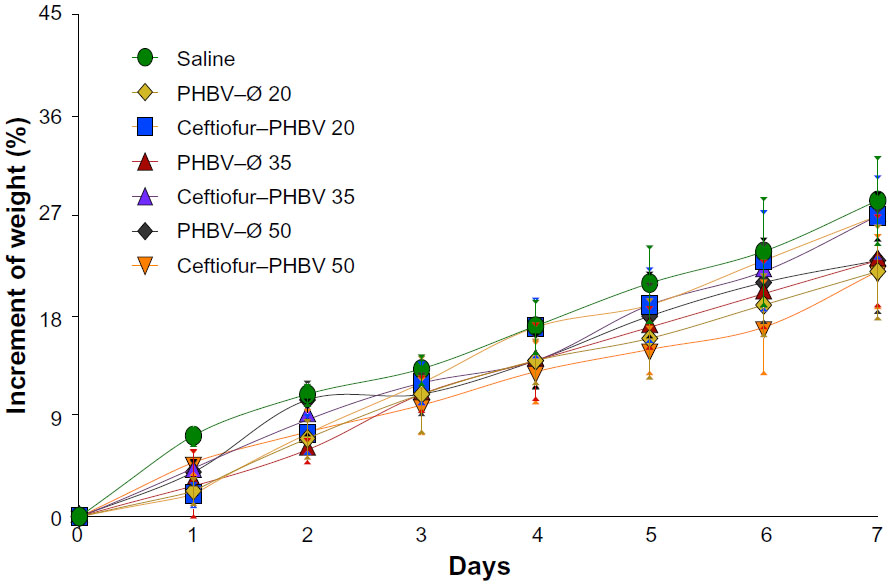 | Figure S2 Increment of weight of rats obtained in the toxicological evaluation at high doses of microparticles. |
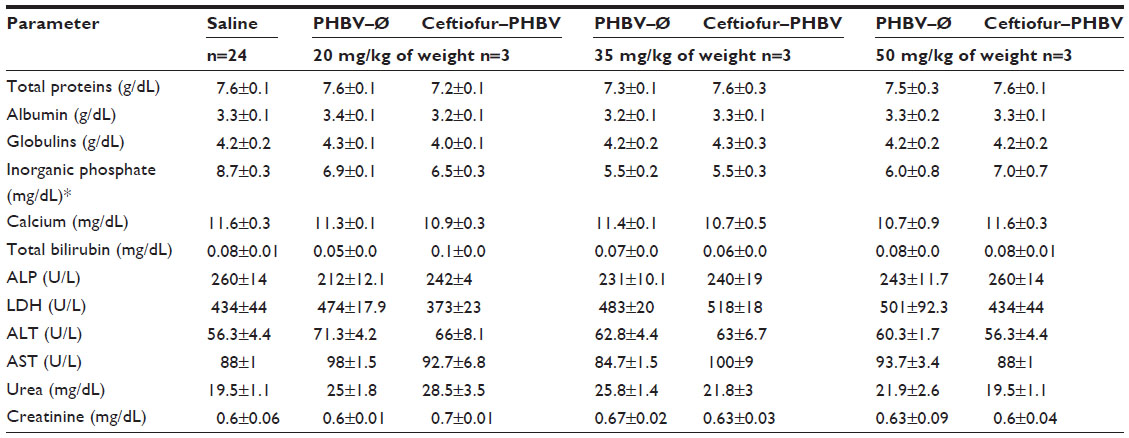 | Table S4 Plasma biochemical parameters of rats obtained in the toxicological evaluation at high doses of microparticles |
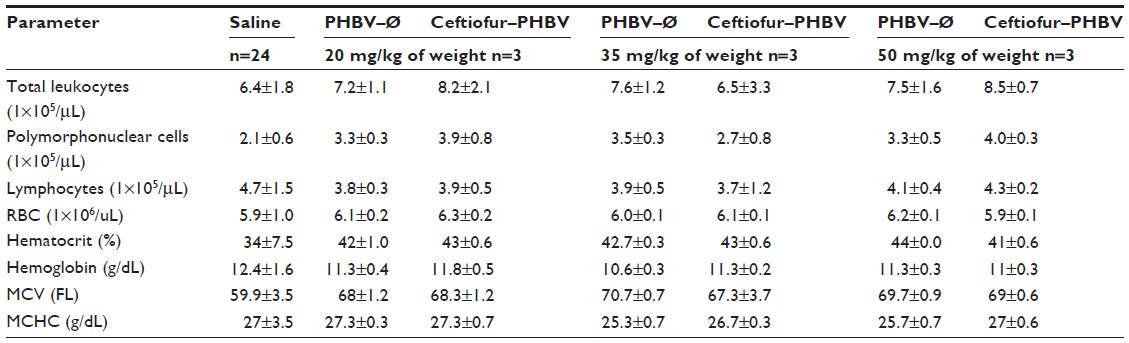 | Table S5 Plasma hematological parameters of rats obtained in the toxicological evaluation at high doses of microparticles |
 | Table S6 Evaluation of plasma coagulation parameters of rats obtained in the toxicological evaluation at high doses of microparticles |
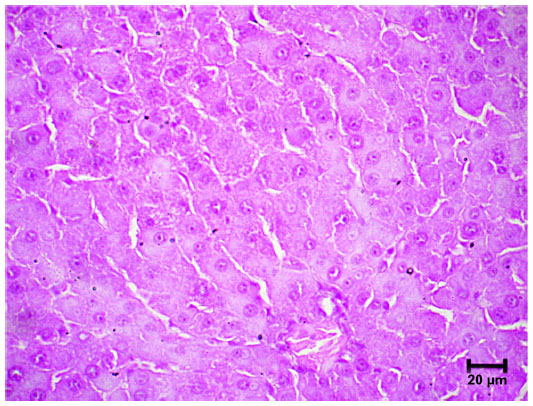 | Figure S3 Image showing morphological signs of liver regeneration, without microparticle waste in the ceftiofur–PHBV group. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.