Back to Journals » Drug Design, Development and Therapy » Volume 14
Evaluation of Bioequivalency and Pharmacokinetic Parameters for Two Formulations of Glimepiride 1-mg in Chinese Subjects
Authors Ju G ![]() , Yan K, Xu Y, Chen S
, Yan K, Xu Y, Chen S ![]() , Zheng Z, Qiu W
, Zheng Z, Qiu W
Received 12 February 2020
Accepted for publication 9 June 2020
Published 6 July 2020 Volume 2020:14 Pages 2637—2644
DOI https://doi.org/10.2147/DDDT.S249355
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Gehang Ju,1 Keyu Yan,1 Youwei Xu,2 Shilin Chen,3 Zhonghui Zheng,2 Wen Qiu4
1School of Pharmacy Lanzhou University, Lanzhou University, Lanzhou, People’s Republic of China; 2Research Institute, Shandong Xinhua Pharmaceutical Company Limited, Shandong, People’s Republic of China; 3The Department of Analysis, Chengdu Fanweixi Pharmaceutical Technology Company, Limited, Chengdu, People’s Republic of China; 4Phase I Clinical Unit, Lanzhou University Second Hospital, Lanzhou, People’s Republic of China
Correspondence: Wen Qiu Tel/ Fax +86-931-8487117
Email [email protected]
Purpose: Glimepiride, an FDA-approved oral hypoglycemic drug, is a long-acting sulfonylurea (SU), used for treating type 2 diabetes. The study aimed to evaluate the bioequivalence and safety profiles of two different formulations of glimepiride 1 mg from two different manufactures in healthy Chinese subjects in the fasting and fed state in order to acquire adequate pharmacokinetic evidence for registration approval of the test formulation.
Patients and Methods: This study is an open-label, two-period, two-sequence, randomized, two-way crossover pharmacokinetic study in healthy Chinese subjects in the fasting and fed state. Seventy-two subjects were randomly assigned to the fasting group and the fed group (n=36 each). We collected blood samples, 24-h post drug administration. The plasma concentration of glimepiride was assessed using HPLC coupled with mass spectrometry. The following parameters were evaluated: AUC0-inf, AUC0-last, Cmax, t1⁄ 2, Tmax, and λz. Safety was determined based on the occurrence of adverse events (AEs) and laboratory examinations (biochemistry, hematology, and urinalysis) throughout the entire study period.
Results: The geometric mean ratios (GMR) amongst the two glimepiride formulations for the primary pharmacokinetic parameters, ie, AUC0-inf, AUC0-last, and Cmax as well as the corresponding 90% CIs, were all within the range of 80.00– 125.00% in the fasting and fed state. The safety profile for both treatments was comparable.
Conclusion: PK analysis revealed that the test and reference formulations of glimepiride were bioequivalent and well tolerated in healthy Chinese subjects. Chinese Clinical Trials Registry identifier: CTR20171121.
Clinical Trial Registration Number: CTR20171121.
Keywords: glimepiride, bioequivalence, pharmacokinetics, HPLC-MS/MS, adverse event
Introduction
Glimepiride, a long-acting sulfonylurea (SU), is commonly used as an oral hypoglycemic drug. Its chemical name is 1-[[p-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido) ethyl] phenyl] sulfonyl]-3-(trans-4-methylcyclohexyl) urea. The recommended dosage of glimepiride is in the range of 1 to 4 mg daily for both adults and adolescents with type 2 diabetes mellitus.1 Other SU drugs include glibenclamide (glyburide), glipizide, gliclazide, etc.2 However, glimepiride, as a representative drug of SU, is clinically recommended for type 2 diabetes in several countries.3–5
Oral administration of a single dosage of glimepiride in healthy subjects revealed significant absorption from the gastrointestinal tract (G.I.) within 1 hour, with a Tmax of approximately 3 h. There was a dose-dependent increase in Cmax and AUC from 1 to 8 mg.6–8 The plasma protein binding and Vd for glimepiride were 99.4% and 8.8 L, respectively.9 It undergoes cytochrome P450 (CYP) 2C9 isozyme–mediated hydroxylation in vivo.10–12
There are previous reports on the pharmacokinetic (PK) evaluation of glimepiride in both Asian (Chinese) and non-Asian subjects.10,11,13–16 However, there are no reports on the comparison of bioequivalence of 1-mg glimepiride between test and reference tablets from two different manufactures in the fasting or fed state in healthy Chinese populations. Thus, this study aimed to lay the foundation for the future marketing of this generic drug formulation in China. (Clinical trial registration number CTR20171121)
Patients and Methods
Study Design
This study was an open-label, single-dose, randomized, Phase I trial with a 2 × 2 crossover bioequivalence design. We placed a 1-week washout period between the administration of doses to evaluate the bioequivalence of both glimepiride formulations (test and reference), which were prepared as 1-mg tablets by Shandong Xinhua Pharmaceutical Company Limited (Glimepirid; Test; Lot number, 1705039), with the brand name (Amaryl® 1-mg tablets, Sanofi-Aventis Deutschland GmbH; Reference; Lot number, B615). This study composed of two independent groups: the fasting group and the fed group.
The study followed the principles of the Declaration of Helsinki as well as the amendments,17 the ICH-GCP,18 and the NMPA-GCP guidelines.19
Subjects
This study was conducted at the Lanzhou University Second Hospital’s Phase I Clinical Unit. Healthy Chinese volunteers, confirmed by performing a complete medical examination, from both sexes, were enrolled in this study. Exclusion criteria involved: smoking, frequent alcohol consumption within the last 6 months, used CYP enzymes inhibitor within the last 30 days, took any medication within the last 2 weeks, frequent use of Chinese traditional medicine, allergic to any medications, took part in clinical studies within the last 3 months, or medical history of clinically relevant disease.
The risk-benefit analysis of this study was explained in detail to all subjects. Additionally, written informed consent was collected from all participants before initiating the study, who were free to withdraw at any time.
Glimepiride Dosing
Healthy male and female subjects were administered two oral formulations of glimepiride at a dose of 1 mg in this two-period crossover design study. The SAS statistical software v9.13 was used to generate a random number table, and subjects were divided into Test/Reference (T/R) or Reference/Test (R/T) groups. A tablet of the test or reference drug with 240 mL of 20% glucose solution was given to the subjects in a standing posture in the fasting state. A standard high-fat (approximately 50%), high-calorie (800–1000 kcal) meal was given at 30 minutes before drug administration in the fed state. Among them, protein provides about 150 kcal, carbohydrates about 250 kcal, and fat about 500–600 kcal. The drugs were administered with 240 mL of warm water.
Blood Sampling
Blood samples (4 mL) were collected pre-dose (baseline) and post-dose at selected time points ie, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 6, 7, 8, 10, 12, 14, 16- and 24-h, followed by centrifugation for 5 min at 3000 rpm. The supernatant (plasma) was collected and frozen at −70±15°C within 2 h. Blood glucose content was determined periodically at 1.0 (±15min), 2.0 (±30min), 2.5 (±30min), 3.0 (±30min) and 4.0 (±30min) h after drug administration both in the fasting and fed state.
Analytical Determinations
A modified HPLC-MS/MS method was used to determine the plasma concentrations of glimepiride using a TEIPLE QUAN 5500 LC/MS/MS System (Applied Biosystems) in the positive ionization mode. A reversed-phase C18 column (50 × 2.0 mm; 5 μm) with a 4.0 × 3.0 mm, 5 μm C18 cartridge guard was used for chromatographic separation. The mobile phase consisted of 0.1% formic acid in acetonitrile at a flow rate of 0.4 mL/min. The mass-to-charge ratios (m/z) of 491.3→352.2 and 496.3→357.2 for glimepiride and d5-glimepiride and 507.2→352.2, respectively were assigned to monitor multiple reaction transitions. Analyst 1.6.3 and Watson LIMS 7.5sp were used for data acquisition and analysis.
Quality control samples with four times higher concentration than the upper limit of quantification (4 × uloq) were prepared, diluted (5x) with blank plasma, such that the concentration of the diluted sample lay within the linear range of the standard curve. Six parallel samples were prepared following a similar protocol. The dilution accuracy was described as mean ± SD of the recovery rate of six parallel samples with the same concentration. The average deviation of the accuracy of each concentration sample was between 85.00% and 115.00%, and the average CV% was ≤ 15.00%. The stability test items included the stability of drug-containing plasma after 24 h at room temperature, 24 h of treatment at 2–8°C, four times of freeze-thaw of drug-containing plasma at −30 to −10°C and −80 to −60°C, and 66 days of long-term storage of drug-containing plasma at −30 to −10°C and −80 to −60°C, the stability of glimepiride was acceptable. The linear range of glimepiride was from 0.5ng/mL to 150.0ng/mL. The deviation accuracy of the standard curve LLOQ was −7.98% to 10.46%, the deviation accuracy of other concentration samples except LLOQ was −12.00% to 12.88%, and R2 was 0.9932 to 0.9995.
PK Endpoints
All subjects who were randomly assigned to any group and who completed both Period 1 and Period 2 without any major protocol deviation were included in the PK analysis set (PKS). The primary endpoints were AUC0-inf (area under the curve, from t = 0 to infinity); AUC0-t (from t = 0 to the last quantifiable concentration); and Cmax (maximum serum concentration). The secondary endpoints were Tmax (time to Cmax), t1/2 (half-life), and λz (terminal elimination rate constant).
Pharmacokinetic and Statistical Analysis
Phoenix WinNonlin Software v7.0 was used to calculate the PK parameters using the noncompartmental method, followed by the construction of concentration-time profiles for individual plasma concentrations. Cmax and Tmax were derived from the acquired data. The linear trapezoidal method was used to calculate the AUC0-inf and the AUC0-t.
According to NMPA regulatory guideline,20 the logarithm (ln)-transformed PK parameters (AUC0-inf, AUC0-t, and Cmax) were analyzed using one-way analysis of variance (ANOVA), which included subject nested within the sequence as a random effect, and sequence, treatment, period as the fixed effects. Then, two-one-sided t-test was performed. The two drug formulations were considered bioequivalent, ie, if the difference between both parameters was statistically insignificant (P>0.05), and if 90% CIs for the geometric mean ratios (GMR) of Cmax, AUC0-t, and AUC0-inf were within 80% - 125%.
Tolerability and Safety
Changes in laboratory tests (biochemistry, hematology, and urinalysis), vital signs, and adverse events (AEs) were used to assess the safety of these formulations. Vital signs (systolic and diastolic blood pressure, body temperature, pulse rate) were recorded pre-dose and at 1, 2, 3, 4, and 24 h post-dose at each treatment visit. Meanwhile, the laboratory and physical examinations, along with ECG, were conducted at the beginning and the end of the study. The Medical Dictionary for Regulatory Activities v20.0 was used to code the AEs to the preferred term and system organ class. We summarized the incidence of serious adverse events (SAEs) based on the frequency and the number of subjects and the treatment-emergent adverse events (TEAEs) based on their severity and causality.
Results
Subjects
We screened one hundred and thirty-seven healthy Chinese subjects from September 24, 2017, to October 28, 2017 (Figure 1). Of these, the fasting group included 36 subjects (19 male and 17 female), and the fed group also included 36 subjects (20 male and 16 female). Table 1 lists the demographic characteristics of all the subjects.
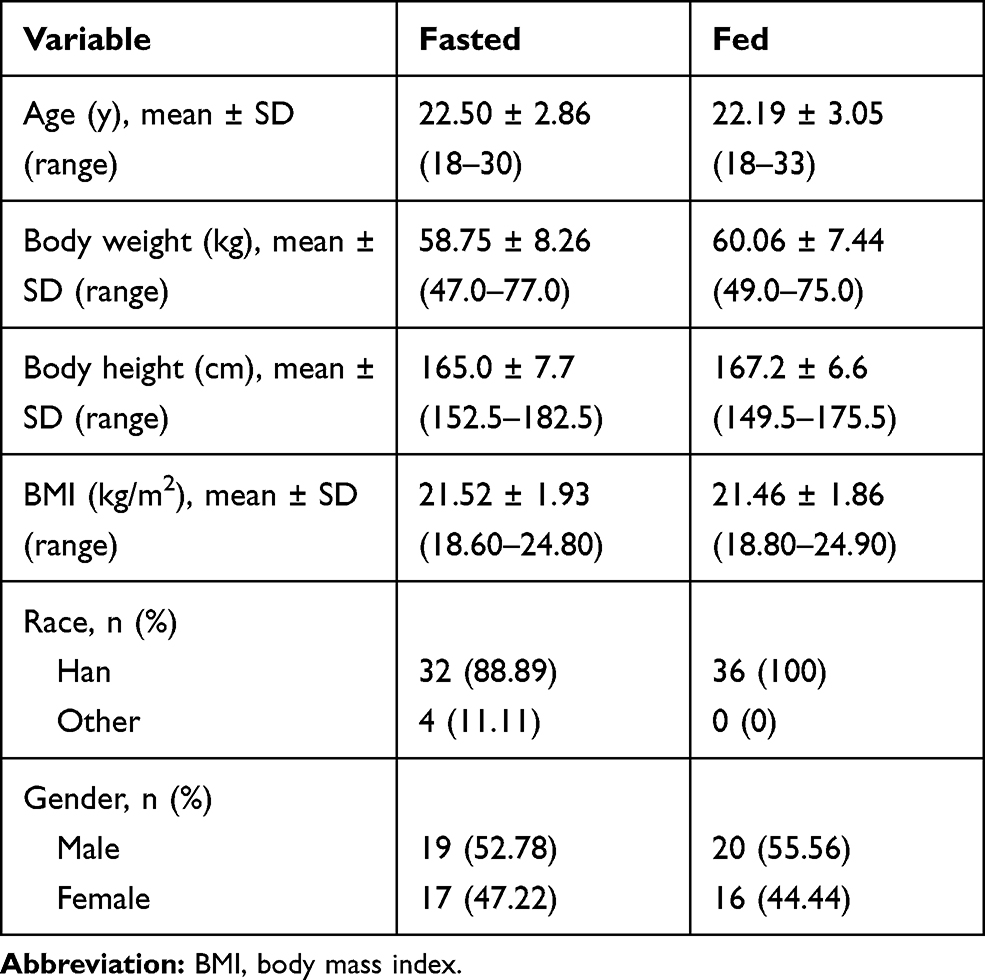 |
Table 1 Baseline Demographic of Subjects Under Fasted or Fed Conditions |
 |
Figure 1 Study design and disposition of subjects. |
PK Analysis
In the fasted study, 35 subjects completed both periods and were included in the pharmacokinetic analysis, 1 case in Group R/T was withdrawn due to personal reasons. 35 subjects completed both periods and were included in the pharmacokinetic analysis during the fed study, 1 case in Group R/T was withdrawn due to taking non-experimental drugs.
The mean plasma concentration-time profiles of glimepiride obtained after single oral administration of the test and the reference glimepiride products under fasted and fed conditions are shown in Figures 2 and 3. The primary PK parameters of glimepiride are summarized in Table 2 under fasted or fed conditions.
 |
Table 2 Pharmacokinetic Parameters of Glimepiride After Administration of Test and Reference Formulations Under Fasted or Fed Conditions |
 |
Figure 2 Mean plasma concentration-time profiles of test (n=35) and reference (n=36) formulations under fasted conditions. Note: Data represent the mean value, and error bars represent the SD. |
 |
Figure 3 Mean plasma concentration-time profiles of test (n=35) and reference (n=36) formulations under fed conditions. Note: Data represent the mean value, and error bars represent the SD. |
As for bioequivalence evaluation, in the fasted study, the statistical comparison of primary PK parameters between the test and the reference products showed that the geometric mean ratios (90% CI) for AUC0-inf, AUC0-last, and Cmax were 101.11% (98.45% to 103.84%), 102.68% (99.82% to 105.62%), and 94.27% (86.09% to 103.23%), respectively, with their corresponding 90% CIs within the pre-defined equivalence margin of 80.00% to 125.00%. In the fed study, the statistical comparison of primary PK parameters between the test and the reference products showed that the geometric mean ratios (90% CI) for AUC0-inf, AUC0-last, and Cmax were 103.38% (99.26% to 107.68%), 103.58% (99.53% to 107.80%), and 107.66% (97.88% to 118.42%), respectively, with their corresponding 90% CIs within the pre-defined equivalence margin of 80.00% to 125.00%. The analysis results of bioequivalence for plasma pharmacokinetic parameters of glimepiride under fasted or fed conditions are summarized in Table 3.
 |
Table 3 Analysis of Bioequivalence for Plasma Pharmacokinetic Parameters of Glimepiride Under Fasted or Fed Conditions |
In both the fasting and the fed group, data acquired from 35 subjects each, who completed both sequences, was used for the PK analysis. For the fasting group, one subject in the Group R/T withdrew due to personal reasons. In the fed group, one subject in the Group R/T withdrew due to the intake of non-experimental drugs.
Figures 2 and 3 show the mean plasma concentration-time profiles of glimepiride, post a single oral dosage of the test and the reference formulations in the fasting and fed state. Table 2 summarizes the primary PK parameters of glimepiride in the fasting or fed state.
For evaluating whether the formulations were bioequivalent, the statistical analysis of the primary PK parameters of the fasting group showed that the geometric mean ratios (90% CI) for AUC0-inf, AUC0-last, and Cmax were 101.11% (98.45% to 103.84%), 102.68% (99.82% to 105.62%), and 94.27% (86.09% to 103.23%), respectively, with their corresponding 90% CIs within 80.00% to 125.00%. For the fed group, the statistical comparison of primary PK parameters showed that the geometric mean ratios (90% CI) for AUC0-inf, AUC0-last, and Cmax were 103.38% (99.26% to 107.68%), 103.58% (99.53% to 107.80%), and 107.66% (97.88% to 118.42%), respectively, with their corresponding 90% CIs within 80.00% to 125.00%. Table 3 summarizes the results of bioequivalence analysis for plasma PK parameters of glimepiride in the fasting or fed state.
Tolerability and Safety
In the fasting group, data acquired from all 36 subjects were used for safety analysis. Of these, 21 AEs were recorded from 14 subjects. The most common adverse events include hypoglycemia, palpitation, fatigue, diarrhea, nausea, elevated bilirubin, hypotension, dizziness, hypertension, and sinus bradycardia; all were transient and labeled as mild or moderate. Similarly, in the fed group, data acquired from all 36 subjects were used for safety analysis, and 23 AEs were recorded in 14 subjects; all were transient and labeled as mild. No SAEs, including fatalities, were reported. Table 4 summarizes the incidence of AEs. We observed no clinically significant abnormalities in the laboratory and physical tests.
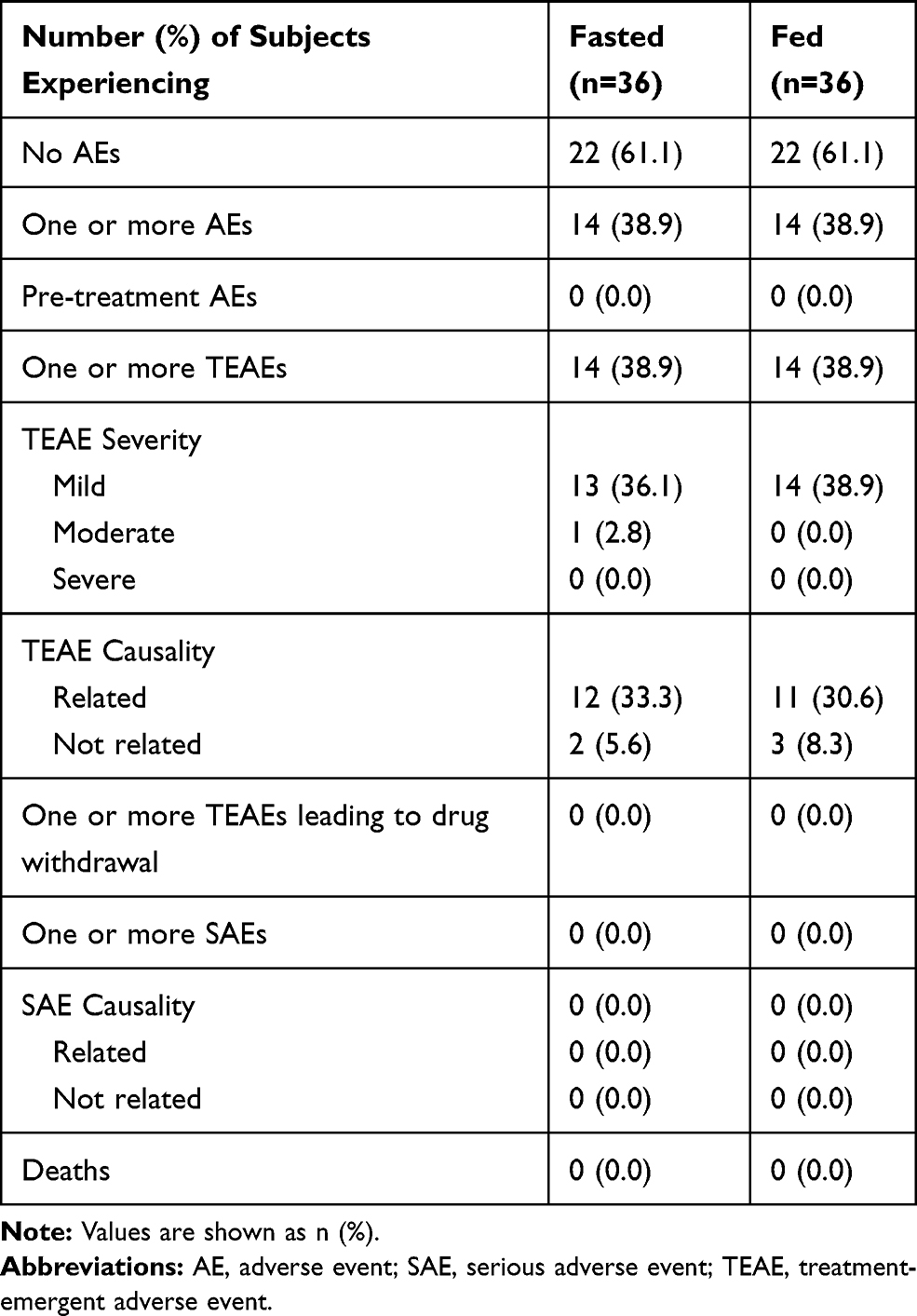 |
Table 4 Safety Profile (Safety Set) |
Discussion
This study compared the PK properties and bioequivalency of the test and the reference glimepiride tablets in healthy Chinese volunteers in the fasting or fed state. This study found that the 90% CIs of the GMR, between the test and the reference glimepiride, for AUC0-inf, AUC0-last, and Cmax were within 80.00–125.00% and thus, were considered to be bioequivalent. Moreover, non-parametric analysis of Tmax indicated that the statistical difference between the test and the reference glimepiride formulations, both in the fasting and fed state, was insignificant.
On comparing the PK data between fasting and fed state, we observed that food influenced the PK parameters of the test and the reference glimepiride formulations. When glimepiride was given after a standard high-fat, high-calorie breakfast, a 50% increase in Cmax, approximately 74% increase in AUC0-t and AUC0-inf, approximately 20% decrease in Tmax was observed. Thus, food accelerated both the rate and extent of absorption of glimepiride.
Previous studies have reported that administration of glimepiride with meals resulted in 8% and 9%, respective reduction in the mean Cmax and AUC.7,8 Also, peak drug concentrations (Cmax) were found to be approximately 3 h post-dose, in studies where a single oral dosage of glimepiride was administered to healthy subjects and with multiple oral doses to patients having type 2 diabetes. However, our study showed an approximate increase of 50% and 74% in the mean Cmax and AUC and Tmax of approximately 4 h post-dose when glimepiride was given with a high-fat, high-calorie meal. This difference could be attributed to multiple factors, including inter-individual differences, ethnicity, environment, eating habits, CYP2C9 genotype (a major enzyme in the sulfonylurea drug metabolism).21 Its allele carriers show considerably lower apparent clearance of the sulfonylureas. However, it is difficult to infer specific influencing factors based on the current study accurately.
For the fasting state, we adopted a different drug delivery strategy than the FDA guidelines22 and previous studies16,23 for administrating glucose. The FDA guidelines for glimepiride bioequivalence studies recommend that subjects should be administered a test formulation or a reference formulation in 240 mL of 20% glucose solution and administered 60mL of 20% glucose solution every 15 min within 4 h to reduce the risk of hypoglycemia in the fasting condition. However, clinical research feedback showed that the Chinese subjects show poor tolerance to 20% glucose solution, and are prone to nausea and vomiting, which would interfere with the evaluation of adverse drug reactions. Therefore, in this study, we administered glimepiride with 20% glucose (240 mL) and administered 10% glucose (60 mL) every 15 minutes for the next 4 h and closely monitored blood glucose concentration for hypoglycemia. In the case of hypoglycemia, we administered 20% glucose (60 mL) each time. No serious adverse events were observed due to hypoglycemia, and only one subject had symptoms of hypoglycemia during the fasting study.
We observed no SAEs in the safety analysis. In the fasting group, a similar percentage of subjects experienced AEs in the test and the reference formulations (25.0% and 13.0% of subjects, respectively. p=0.3723). In the fed study, a similar percentage of subjects experienced AEs in the test and the reference formulations (19.4% and 19.4% of subjects, respectively. p=1.0000). Thus, both the test and the reference formulations of glimepiride were well tolerated in all subjects.
Conclusion
In this study, the evaluation of PK indicated that the 2 formulations were bioequivalent under fasted and fed conditions. The mainly pharmacokinetic parameters were within the acceptable range for bioequivalence (80.0% to 125.0%) as defined by the Pharmacopoeia of the People’s Republic of China guidelines. Two formulations of glimepiride were well tolerated. The bioequivalent form of the 1-mg oral tablet will offer an affordable, acceptable, and beneficial access to the medication in Chinese patients. However, the changes in pharmacokinetic parameters under fasted and fed condition were different from previous studies, which can be further discussed in later studies.
Here, the PK analysis showed that both the formulations were bioequivalent in the fasting and fed state. Based on the Pharmacopoeia of the People’s Republic of China guidelines, the primary PK parameters were within the acceptable range for bioequivalency (80.0% to 125.0%). Both formulations were well tolerated. Thus, the bioequivalent form of the 1-mg oral tablet will present an affordable, acceptable, and beneficial alternative to Chinese patients. However, the changes in PK parameters in the fasting and fed state were different from previous studies, which needs to be further evaluated.
Data Sharing Statement
All available data is displayed in the article. No study protocol, statistical analysis plan and other information would be provided.
Ethics Approval and Informed Consent
This study was approved by the Drug Clinical Trial Ethics Committee of the Lanzhou University Second Hospital and conducted in accordance with the Declaration of Helsinki and was approved by an ethics committee. All subjects provided written informed consent.
Acknowledgments
We thank all of the healthy subjects for their participation in this study and the sponsor, Shandong Xinhua Pharmaceutical Co, Ltd for their financial support.
Disclosure
Youwei Xu and Zhonghui Zheng are employees of Shandong Xinhua Pharmaceutical Company Limited. Shilin Chen is an employee of Chengdu Fanweixi Pharmaceutical Technology Company, Limited. The authors report no other conflicts of interest in this work.
References
1. Rosskamp R, Wernicke-Panten K, Draeger E. Clinical profile of the novel sulphonylurea glimepiride. Diabetes Res Clin Pract. 1996;31(Suppl):S33–S42. doi:10.1016/0168-8227(96)01228-4
2. Zhang H. ADRs of oral hypoglycemic drugs. Tianjin Pharm. 2017;29(2):75–82.
3. American Diabetes Association. Standards of Medical Care in Diabetes—2020 Abridgedfor Primary Care Providers. Clinical Diabetes. 2020;38(1):10–38. doi:10.2337/cd20-as01 https://clinical.diabetesjournals.org/content/diaclin/38/1/10.full.pdf.
4. The Japan Diabetes Society. Available from: Treatment guide for diabetes 2016–2017. http://www.fa.kyorin.co.jp/jds/uploads/Treatment_Guide_for_Diabetes_2016-2017.pdf.
5. Chinese Diabetes Society. Guidelines for the prevention and treatment of type 2 diabetes in China 2010. Available from: http://www.diab.net.cn/UploadFile/Ueditor/file/20160811/6360650768334000005174021.pdf.
6. Hoechst Roussel Pharmaceuticals Amaryl® (Glimepiride Tab- Lets) 1, 2, and 4 Mg. Kansas City: Hoechst-Roussel Pharmaceuticals; 2001.
7. Amaryl (glimepiride tablets). Bridgewater, NJ: Sanofi-Aventis US LLC; 2018. Available from: http://products.sanofi.us/Amaryl/Amaryl.pdf.
8. Malerczyk V, Badian M, Korn A, Lehr KH, Waldhausl W. Dose linearity assessment of glimepiride (Amaryl) tablets in healthy volunteers. Drug Metabol Drug Interact. 1994;11(4):341–357. doi:10.1515/DMDI.1994.11.4.341
9. Rosenkranz B. Pharmacokinetic basis for the safety of glimepiride in risk groups of NIDDM patients. Horm Metab Res. 1996;28(9):434–439. doi:10.1055/s-2007-979833
10. Suzuki K, Yanagawa T, Shibasaki T, Kaniwa N, Hasegawa R, Tohkin M. Effect of CYP2C9 genetic polymorphisms on the efficacy and pharmacokinetics of glimepiride in subjects with type 2 diabetes. Diabetes Res Clin Pract. 2006;72(2):148–154. doi:10.1016/j.diabres.2005.09.019
11. Wang R, Chen K, Wen SY, Li J, Wang SQ. Pharmacokinetics of glimepiride and cytochrome P450 2C9 genetic polymorphisms. Clin Pharmacol Ther. 2005;78(1):90–92. doi:10.1016/j.clpt.2005.03.008
12. Badian M, Korn A, Lehr KH, Malerczyk V, Waldhausl W. Pharmacokinetics and pharmacodynamics of the hydroxymetabolite of glimepiride (Amaryl) after intravenous administration. Drug Metabol Drug Interact. 1996;13(1):69–85. doi:10.1515/DMDI.1996.13.1.69
13. Borges NC, Taveira Ydel A, Mazucheli JA, et al. Comparison study of two glimepiride formulations bioavailability in healthy volunteers of both sexes after a single dose administration. Arq Bras Endocrinol Metabol. 2007;51:950–955. doi:10.1590/S0004-27302007000600009
14. Matsuki M, Matsuda M, Kohara K, et al. Pharmacokinetics and pharmacodynamics of glimepiride in type 2 diabetic patients: compared effects of once-versus twice-daily dosing. Endocr J. 2007;54:571–576. doi:10.1507/endocrj.K06-052
15. Lei T, Yi-ling H, Lu H, et al. Pharmacokinetic studies of glimepiride and its hydroxyl-metabolite in healthy volunteers. Chin J Clin Pharmacol Ther. 2006;11:868–972.
16. Liu Y, Zhang MQ, Zhu JM, et al. Bioequivalence and pharmacokinetic evaluation of two formulations of glimepiride 2 mg: a single-dose, randomized-sequence, open-label, two-way crossover study in healthy Chinese male volunteers. Clin Ther. 2010;32(5):986–995. doi:10.1016/j.clinthera.2010.04.016
17. World Medica lAssociation (WMA) Declaration of Helsinki. Ethical principles for medical research involving human subjects. Available from: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/.
18. European Agency for the Evaluation of Medicinal Products (EMEA). ICH E6 (R2) guideline for good clinical practice; 2016. Available from: https://www.ema.europa.eu/en/ich-e6-r2-good-clinical-practice#current-version—revision-2-section.
19. National Medical Products Administration. Guideline for good clinical practice. Available from: http://www.nmpa.gov.cn/WS04/CL2101/329583.html.
20. National Medical Products Administration. Center for Drug Evaluation. Guideline for bioavailability and bioequivalence studies of generic drug products. Available from: http://www.nmpa.gov.cn/WS04/CL2093/331454.html.
21. Xu H, Murray M, McLachlan AJ. Influence of genetic polymorphisms on the pharmacokinetics and pharmacodynamics of sulfonylurea drugs. Curr Drug Metab. 2009;10(6):643–658. doi:10.2174/138920009789375388
22. Food and Drug Administration. Guidance on Glimepiride. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Glimepiride_tab_20496_RC11-04.pdf.
23. Jovanovic D, Stojsic D, Zlatkovic M, Jovic-Stosic J, Jovanovic M. Bioequivalence assessment of the two brands of glimepiride tablets. Vojnosanit Pregl. 2006;63(12):1015–1020. doi:10.2298/VSP0612015J
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.