Back to Journals » International Journal of Nanomedicine » Volume 11
Evaluation of β-cyclodextrin-modified gemini surfactant-based delivery systems in melanoma models
Authors Michel D, Mohammed-Saeid W, Getson H, Roy C, Poorghorban M, Chitanda JM, Verrall R, Badea I ![]()
Received 31 August 2016
Accepted for publication 15 October 2016
Published 12 December 2016 Volume 2016:11 Pages 6703—6712
DOI https://doi.org/10.2147/IJN.S121156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Deborah Michel,1 Waleed Mohammed-Saeid,1 Heather Getson,1 Caitlin Roy,1 Masoomeh Poorghorban,1 Jackson M Chitanda,2 Ronald Verrall,2 Ildiko Badea1
1Drug Design and Discovery Research Group, College of Pharmacy and Nutrition, 2Department of Chemistry, University of Saskatchewan, Saskatoon, SK, Canada
Abstract: Novel drug delivery systems are developed to improve the biological behavior of poorly soluble drugs and to improve therapeutic outcomes. In melanoma therapy, the goal is efficient drug delivery and mitigation of drug resistance. Melphalan (Mel), a currently used therapeutic agent for melanoma, requires solvent system for solubilization, leading to poor chemical stability. Moreover, drug resistance often renders the drug inefficient in clinical setting. A novel β-cyclodextrin-modified gemini surfactant (CDgemini) delivery system was developed to incorporate Mel in order to improve its physicochemical and biological behavior. Melphalan nanoparticles (Mel-NP) showed optimal particle size in the 200–250 nm range for endocytosis and induced significantly higher cell death compared with Mel (50% of inhibitory concentration [IC50] of 36 µM for the complexes vs 82 µM for Mel). The CDgemini delivery system did not alter the pathway of the cellular death triggered by Mel and caused no intrinsic toxicity to the cells. The Mel-NP complexes induced significant cell death in melanoma cells that were rendered resistant to Mel. These findings demonstrate in principle the applicability of the CDgemini delivery system as safe and efficient alternative to the current melanoma therapy, especially in chemoresistant cases.
Keywords: lipid nanoparticles, anticancer agent, drug resistance, apoptosis, spheroid, zeta potential, flow cytometry
Introduction
The advances in identifying the genetic, proteomic, and metabolomic contributions in cancer initiation and progression have led to a significant increase in the number of antineoplastic agents under investigation. However, most of the new chemical entities that show promising antitumor activity in the laboratory are discarded as a consequence of their inefficiency in preclinical or clinical studies. This inefficiency is usually associated with poor physicochemical properties of the molecule that negatively affect their pharmacologic and pharmacokinetic profiles. For instance, poor aqueous solubility is linked to low in vivo activity, nonspecific binding to plasma components, rapid elimination, and tumor resistance. Currently, there are efforts to address these deficiencies of both novel and commercially available cytotoxic agents by incorporating them in specifically designed drug delivery systems. Nanoparticle drug delivery systems have been used for the past two decades to improve drug solubility and pharmacokinetic profile of a series of chemotherapeutics.1 Their potential applicability is broad, given the successes achieved to date, such as Abraxane (nanoparticle albumin bound paclitaxel), Doxil® (liposomal doxorubicin), DaunoXome® (liposomal daunorubicin), and Marqibo® (liposomal vincristine).2,3 In addition, nanoparticle drug delivery systems can enhance cellular uptake of the drug; that is, overcome barriers such as the epithelial tight junctions and permeate protective barriers such as the stratum corneum of the skin.4 At the tissue level, they can improve the accumulation of the drug at the target site and achieve tumor targeting. For example, drugs encapsulated into nanoparticles preferentially accumulate in tumors due to the enhanced permeability and retention effect that permits the extravasation of the nanoparticles into the tumor through the leaky vasculature (passive targeting).5 Active tumor targeting can be achieved by modulating the chemical structures of the nanoparticles by attaching tumor-specific targeting moiety.6 At the cellular level, nanoparticles improve the effect of cytotoxic agents because they have the ability to accumulate the cytotoxic drug in the cells and prevent drug efflux from the diseased cell, thereby overcoming drug resistance.7,8 Hence, nanotechnology, such as polymer-based nanoparticles, dendrimers, liposomes, niosomes, cubosomes, and nanodiamonds, can contribute significant improvements to the efficiency of cancer therapy.9
Melanoma is a type of cancer that needs urgent attention and could benefit from nanotherapy. Malignant melanoma, in the USA, is the sixth most common cancer in males and the seventh in females, making this cancer one of the most lethal solid tumors. In 2016, it is estimated that there will be 76,380 new cases of melanoma of the skin, and an estimated 10,130 people will die due to this disease. It is becoming more prevalent in young adults.10 The treatment used for melanoma depends on its manifestation and stage, and options include surgery, radiotherapy, chemotherapy, isolated limb perfusion/infusion, immunotherapy, and biological therapy. Surgical excision of the tumor lesion is the primary treatment option for noninvasive and satellite metastatic melanoma with great prognosis for disease-free survival and low relapse.11 However, surgery is generally not feasible in advanced and metastatic melanoma as it can result in large, unsightly scars that might outweigh the risks of progression and death.12 In addition, surgery can be a complicated procedure in tumors that occur in cosmetically sensitive sites, such as lips and eyelids as in the case of lentigo maligna (a form of in situ melanoma).12 Therefore, there is a need to develop more efficient noninvasive therapies. Isolated limb perfusion (with tumor necrosis factor alpha and melphalan [Mel]) and systemic chemotherapy against in-transit melanoma have had limited success.13,14 Systemic therapy with cytotoxic agents is the first treatment option for most patients with metastatic melanoma, and dacarbazine, an alkylating agent, is the only US Food and Drug Administration approved (1975) chemotherapeutic for melanoma management.15 In addition, melanoma has limited options for chemotherapy because it is highly resistant to systemic anticancer agents.16 Thus, nanotechnology methodologies are needed to improve the efficiency of current therapeutic agents.
Mel, one of the therapeutic agents currently used to treat in transit melanoma, localized to limbs and limiting spread to lymph nodes, is a bifunctional DNA alkylator inhibitor of DNA and RNA synthesis in the cell, which ultimately leads to cell death.17 In clinical settings, Mel is a difficult drug to administer to patients by injection due to its low aqueous solubility, limited stability, and low biodistribution. Alkeran® for injection employs an initial dissolution of the Mel/povidone in sodium citrate, propylene glycol, ethanol, and sterile water for injection followed by dilution in saline. The stability of Mel, once diluted in saline, causes a loss of 1% of the label strength every 10 minutes due to hydrolysis. Also because of the presence of co-solvents, the intravascular injection causes local tissue damage if not administered slowly.18 Systemically, 60%–90% of Mel binds to plasma proteins and ~30% is covalently bound to the plasma proteins.19 One of the strategies used to combat such deficiencies is to incorporate Mel into two derivatives of β-cyclodextrin: HP-β-cyclodextrin and (SBE)7m-β-cyclodextrin. The encapsulation eliminates the need for organic co-solvents, the use of two vials for reconstitution, and improves long-term stability of the freeze-dried formulation. The shelf-life of the reconstituted Mel also increases from 1.69 to 5.01 hours.18
In this work, we report the results of an evaluation of the ability of a novel β-cyclodextrin-modified cationic gemini surfactant delivery system (CDgemini) [mono-6-O-(3(bis[3-(N-dodecyl-N, N-dimethylamino)propyl)carbamoyl]propanoyl)-beta-cyclodextrin]2+ to solubilize and deliver Mel and to improve its chemotherapeutic action by increased efficiency and by overcoming drug resistance. We hypothesized that CDgemini can encapsulate the partially insoluble Mel into the β-cyclodextrin ring, whereas the gemini surfactant moiety drives the formation of cationic nanoparticles, enhancing the cellular uptake of the drug by endocytosis.20 Thus, this nanoparticle delivery system has the compositional elements to improve solubilization of the drug, aid its cellular uptake, and prevent drug resistance.
Materials and methods
Materials
The [mono-6-O-(3(bis[3-(N-dodecyl-N, N-dimethylamino)propyl)carbamoyl]propanoyl)-beta-cyclodextrin]2+ surfactant (CDgemini) used in this study was synthesized as previously described.20 Mel was obtained from Sigma-Aldrich (Oakville, ON, Canada). The Dulbecco’s Modified Eagle’s Medium (DMEM) used for tissue culture was purchased from Fisher Scientific (Edmonton, AB, Canada). The fetal bovine serum (FBS) and antibiotic/antimycotic solution (containing penicillin, streptomycin, and amphotericin B) were obtained from Sigma-Aldrich. The MTT used for the cell viability assay was purchased from Invitrogen (Burlington, ON, Canada). The Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit was obtained from Biovision Inc (Milpitas, CA, USA). The Promega Caspase 3/7, 2, 8, and 9 kits were obtained from Fisher Scientific.
Nanoparticle formulation
Mel was dissolved in acidified ethanol (Commercial Alcohols Inc, Toronto, ON, Canada) at a concentration of 50 mM as per Sigma’s product specifications and used at appropriate concentrations for all experiments. To prepare the nanoparticles, the CDgemini was dissolved in pure water (Gibco) at a concentration of 10 mM. The Mel and delivery agent were mixed at a molar ratio of 1:2. This ratio was selected based on the previous studies of the encapsulation of poorly soluble compounds in CDgemini.20–22 The solvents were removed by speed vacuum, and the nanoparticles were re-suspended either in water or in a 0.5% methylcellulose (Medisca, Montreal, QC, Canada) gel.
Size measurements
Nanoparticles were prepared as described under “nanoparticle formulation” section. The size measurements were performed by using a Zetasizer Nano-Zs (Malvern Instruments, Malvern, UK) having Zetasizer Software version 7.01. The results are reported as the average of 3–5 measurements ± standard deviation.
Cell culture and spheroid development
A375, human amelanotic melanoma cells (American Type Culture Collection CRL-1619), were grown in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 25 ng/mL amphotericin B. Cells were cultured at 37°C in a humidified incubator under an atmosphere of 5% CO2 and 95% air. For all experiments, culturing conditions and passage numbers were kept constant.
For the spheroid development, A375 cells that were growing in a monolayer were harvested at second passage and seeded at a density of 1×105 cells per well in a 24-well tissue culture plate precoated with 1.33% agarose.23 Cells were incubated for 72 hours.24 Spheroid development was monitored by microscopy. Spheroids frozen in tissue freezing medium (Leica Biosystems, Concord, Ontario, Canada) were sectioned at 7 μm thickness and stained with eosine for light microscopy to verify stratification.
Cell toxicity assay
The determination of cell viability was done using 3MTT as previously described.20 Briefly, cells were seeded at a density of 1×104 cells per well in 96-well plates. Cells were incubated for 24 hours. Mel (0.98–500 μM final concentrations) dissolved in 1% acidified ethanol or formulated in nanoparticles (Mel-NP) was added to each plate in triplicate wells. The formulations were prepared in aseptic conditions with sterile water. The ethanol concentration and drug/CDgemini ratio were kept constant in all wells. The spheroids were treated with either Mel or Mel-NP at their respective IC50 concentrations.
At 48 hours, fresh media containing 450 μg/mL of MTT were added to each well and incubated for 2 hours. The MTT solution was gently removed, and the plates were dried. Dimethyl sulfoxide was added to dissolve the generated formazan, and absorbance was read at 550 nm using a Synergy BioTek plate reader. The IC50 was determined by calculating the fraction of dead cells and plotting the data with a 4-parameter curve generated by GEN5 software from BioTek.
Flow cytometry – apoptosis and cell cycle analyses
Early-stage apoptosis was evaluated by measuring the externalization of the phosphotidylserine to the outer leaflet of the cell membrane using Annexin V-FITC Apoptosis Detection Kit (BioVision). Late stages of apoptosis were indicated by the loss of cell membrane integrity, permitting the permeation of propidium iodide (Sigma) into the cell. Cells were seeded in 6-well plates at a density of 5×105 cells per well and incubated for 24 hours before treatment. Treatments of Mel and Mel-NP at the IC50 concentration were applied, and the cells were incubated for 24 hours. After treatment, the cells were detached from the plates with trypsin and re-suspended in 500 μL Dulbecco’s phosphate-buffered saline (PBS; Sigma) containing calcium and magnesium. Cells were incubated with 5 μL FITC-conjugated Annexin V for 15 minutes in the dark followed by the addition of 5 μL propidium iodide. Cells were analyzed on an FACScalibur (Becton Dickinson, San Jose, CA, USA) with excitation from the 488 nm laser by simultaneously monitoring green fluorescence from the FITC-Annexin V in the 530/30 nm filter and propidium iodide red fluorescence in the 585/42 filter.
Cell cycle analysis was performed by seeding 5×105 cells per well in 6-well plates and incubated for 24 hours. After the treatment of cells with Mel and Mel-NP at the IC50 concentration, both adherent and floating cells were collected using trypsin. Approximately 1×106 cells were suspended in 300 μL PBS. Cold 100% ethanol was added drop by drop until a final concentration of 70% (v/v) was obtained. Cells were incubated for 24 hours at −20°C. The ethanol was removed by centrifugation at 480× g for 10 minutes at 4°C. The pellet was re-suspended in 1 mL PBS containing 0.3 mg/mL RNase. Propidium iodide at a final concentration of 20 μg/mL was added. The cells were analyzed on an FACScalibur with excitation at 488 nm and emission in the 585/42 filter. Cell cycle analysis was performed by using ModFit LT (Verity Software House).
Caspase-glo assays
The activities of Caspases 3/7, 2, 8, and 9 were performed in a one-step mixture, containing a proluminescent caspase substrate. In principle, in the presence of caspase, the substrate was cleaved to release aminoluciferin, a substrate for luciferase, for the generation of a luminescent signal. A375 cells, at the second passage, were seeded at a density of 5×103 cells per well in white-walled 96-well tissue culture-treated plates. After 24-hour incubation, the cells were treated with the Mel-NP at the IC50 concentration. All treatments were performed in triplicate wells per plate on three individual plates. The cells were incubated for 6, 12, 18, 24, 48, and 72 hours followed by the measurement of apoptosis pathways using the manufacturer’s protocols (Promega). Luminescence was measured by using a Synergy BioTek plate reader.
Development of Mel-resistant cells
A375 melanoma cells were cultured in 25 cm2 tissue culture flasks and treated with increasing concentrations of Mel from 100 nm to 20 μM over 7 weeks to induce drug resistance. The resistant cells were treated with 20 and 40 μM concentrations of Mel and with Mel-NP at the same concentrations, similar to the process described under the “Cell toxicity assay” section. The CDgemini alone was used at 80 μM concentration. MTT assay was used to evaluate the cell viability as described earlier.
Statistical analyses
Results are expressed as the average of n≥3±SD. Statistical analyses were performed using SPSS software v24.0. Independent t-test and one-way analysis of variance (Bonferroni’s post hoc tests) were used. Significant differences were considered at P<0.05 level.
Results
Particle size of formulation
For cellular uptake, the optimal particle size is in the range of 100–500 nm.25 The particle size of the Mel-NP formulated in water was 125±17 nm (Table 1). However, aggregation into large, 20 μm particles occurred within 10–15 minutes (results not shown). The addition of methylcellulose at a concentration of 0.5% (w/v) stabilized the system, and the particle size was maintained at 225±11 nm. To evaluate the behavior of the Mel-NP in the cell culture environment, further measurements, at the same methylcellulose concentration, were performed in the cell culturing medium DMEM. The size of 258±8 nm was consistent with the measurements in water.
 | Table 1 Particle size determined by dynamic light scattering |
Determination of the cellular toxicity of Mel in acidified ethanol and β-CDgemini nanoparticle in monolayer and spheroids
The IC50 of the Mel and Mel-NP was determined in a traditional monolayer cell culture system. In order to mimic melanoma in situ, tumor spheroids26 that show a three-dimensional stratification of the cells were also generated (Figure 1). In the monolayers, the IC50 was 82±3 μM for Mel and significantly lower (P<0.05) for Mel-NP at 36±1 μM (Table 2).
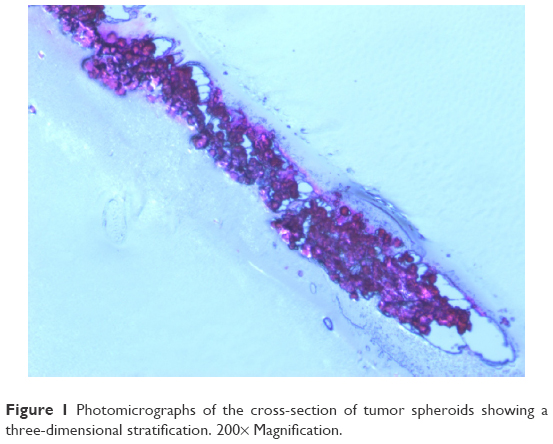 | Figure 1 Photomicrographs of the cross-section of tumor spheroids showing a three-dimensional stratification. 200× Magnification. |
 | Table 2 Cellular toxicity of melphalan and formulations in both monolayer and spheroid cultures |
The tumor spheroids, treated at the IC50 concentrations (82 μM for Mel and 36 μM for Mel-NPs), showed cellular death of 35% for Mel and 30% for Mel-NP, respectively. These values are slightly lower than the a priori expected 50% cell death in the monolayer at the IC50 concentrations.
Evaluation of cellular death
Early signs of apoptosis can be detected by measuring the translocation of phosphatidylserine from the inner leaflet to the cell surface. The cell populations of the untreated cells were predominately healthy (98.4% cell viability, Table 3). The combined early and late apoptotic cells represented 1.4% of the total population, and only 0.2% of the cells were in a necrotic state. Mel at a concentration of 36 μM, that is below the IC50 concentration, triggered apoptosis27 with 11.5% of the cells in early apoptotic state and 25.3% showing late apoptosis (Table 3). Unfortunately, flow cytometry could not be used to assess the cellular death in the cells treated with Mel-NP (Supplementary material, Figure S1A–D). We hypothesize that the lipid-like CDgemini coats the cell and prevents the Annexin V from binding to the exposed phosphatidylserine (Figure S1B and D);28 thus, caspases were measured to assess cell death for the Mel-NP.
 | Table 3 A375 cells were treated with melphalan and melphalan nanoparticles at 36 μM drug concentration and cell death determined by flow cytometry |
The cells treated with Mel expressed a significant amount of caspase 3 by 18 hours and reached a maximum by 24 hours (Figure 2A). It triggered both caspase 2 and caspase 9 at 18 hours and thereafter increased throughout the evaluation period. The treatment with the Mel-NP (Figure 2B) delayed the expression of caspases 3/7, 2, and 9 until 24 hours. The delivery system alone, CDgemini (Figure 2C), did not trigger a significant caspase cascade.
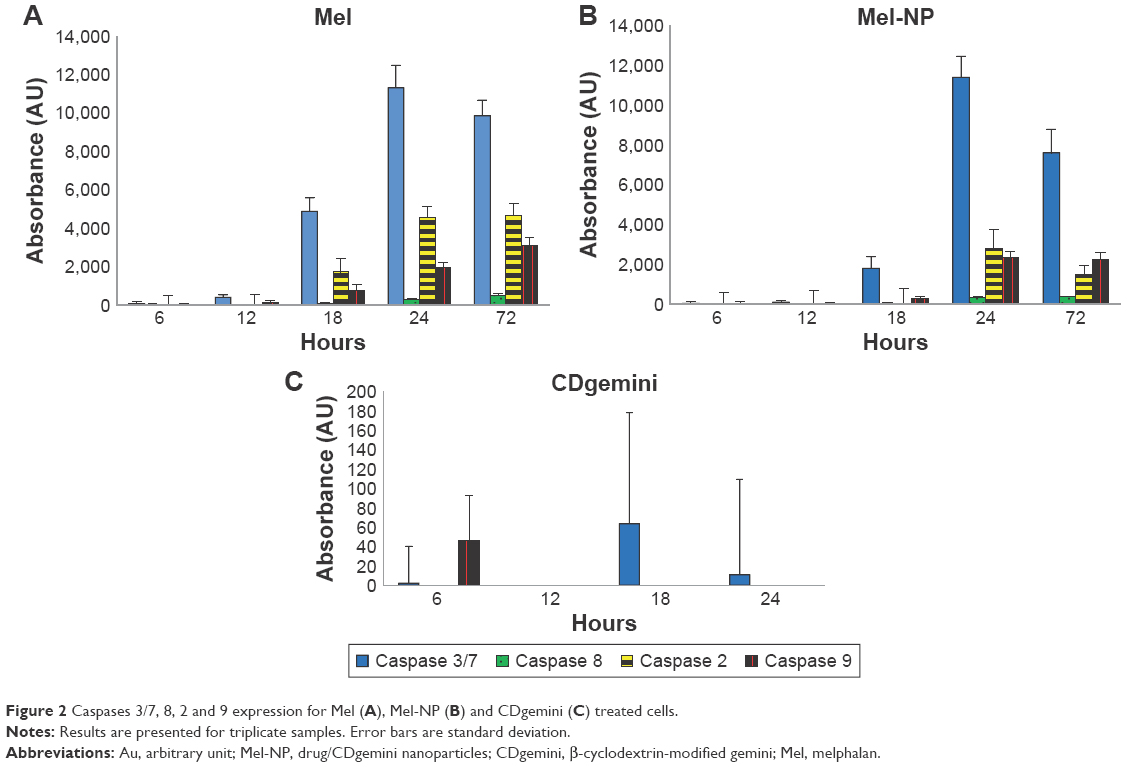 | Figure 2 Caspases 3/7, 8, 2 and 9 expression for Mel (A), Mel-NP (B) and CDgemini (C) treated cells. |
Determination of the effect of Mel/CDgemini nanoparticles on cell cycling
As shown in Table 4, the CDgemini-treated cells had only minimal effect on the cell proliferation, shifting the population only marginally to the G0/G1 phase. Mel triggered cellular arrest predominately in the S-phase shifting 64% of the population. Similarly, 65% of the Mel-NP-treated cells were arrested in the S-phase.
 | Table 4 Cell cycle analysis in A375 cells |
Determination of the effect of the encapsulation of Mel into b-CDgemini nanoparticles on drug resistance
Compared to the nonresistant cells (Figure 3), the A375 cells rendered resistant to Mel showed little to no significant cellular death (P>0.05) when treated with Mel at 20 and 40 μM concentrations. However, the Mel-NP showed a significant decrease in cell viability compared with the naked Mel (P<0.05), from 109%±19% to 28%±8% at 20 μM concentration and from 91%±18% to 19%±3% at 40 μM concentration. Similar to the nonresistant A375, the delivery agent (CDgemini) alone had no detrimental effect on cells viability.
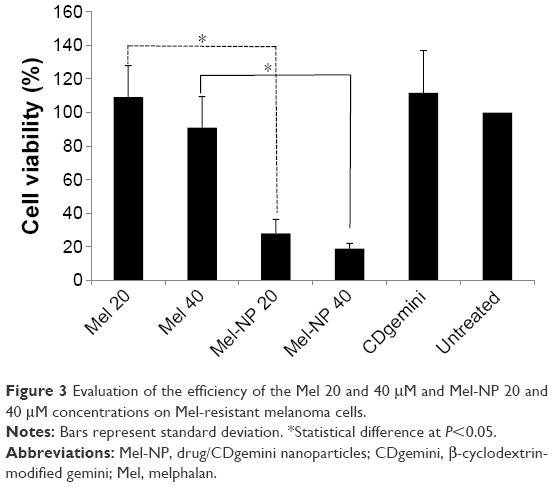 | Figure 3 Evaluation of the efficiency of the Mel 20 and 40 μM and Mel-NP 20 and 40 μM concentrations on Mel-resistant melanoma cells. |
Discussion
The results reported here show that Mel, a poorly water-soluble drug, was successfully included in the CDgemini nanoparticulate system. Inclusion complexes were created by freeze drying the components from an organic solvent and reconstituting in water. The complexes were completely soluble in water upon reconstitution having particle size around 125 nm (Table 1), suitable for promoting intracellular penetration.25,29 Unfortunately, pharmaceutical physical stability was very low, showing aggregation within 15 minutes. In the formulation development process, methylcellulose, a carbohydrate polymer, was used to prevent aggregation of the nanoparticles. Methylcellulose gel is a frequently used pharmaceutical excipient,30–32 which can form interpenetrating polymer network microspheres providing slow release of various drugs.31 While the particle size increased to 250 nm, it remained in the acceptable range for intracellular penetration and maintained its stability in the presence of cell culture medium. The assembly of the Mel-NPs resulted in similar sized nanoparticles to another poorly soluble compound, an 1,5-diaryl-3-oxo-1,4-pentadienyl curcumin analog.20 As our future goal is to develop a noninvasive treatment, methylcellulose could have potential benefit for topical absorption of the Mel-NP.
Comparing the cytotoxicity of the Mel-NP to the drug alone in a cell monolayer, it was found that the delivery agent enhanced the efficiency by 2.2-fold, without an intrinsic toxicity of the CDgemini (Table 2). This increase could be due to the fact that Mel is associated with the hydrophobic cavity of the CDgemini,22 thus protecting it from protein binding in the cell culture milieu33 and making it more available for cellular uptake. Alternatively, the nanoparticles could carry several molecules and be released, intracellularly, from the endocytosed nanoparticles. While there is no available information on the potential number of small molecules that the CDgemini nanoparticles can accommodate, Dong et al determined that similar sized gene delivery gemini nanoparticles can accommodate up to 35 rings of plasmid DNA per particle.34 Consequently, it could be assumed that each CDgemini nanoparticle can potentially carry hundreds of Mel molecules. Another factor leading to enhanced cytotoxicity is the ability of the delivery agent to bypass the cancer cells’ efflux pump for hydrophobic cytotoxic drugs.35 It has been documented that other forms of nanoparticles, such as dual-loading superparamagnetic nanoparticles,36 triblock copolymers,37 and peptide functionalized nanoparticles,38,39 can create a synergistic effect by evading the cancer cell’s efflux pump.
Interestingly, a slight decrease was observed in the efficiency between the two-dimensional and three-dimensional melanoma models. While the scaffold-free 3D cell models (Figure 1) show the characteristics of complexity of cell density and metabolic function of the tumor,36 they do not mimic entirely the in vivo tumors with regard to the presence of biomolecules, extracellular matrix, and angiogenesis.40 Nevertheless, we have demonstrated as a proof of principle (Table 2) that the cytotoxic drug can be delivered into 3D tumors.
To monitor whether the CDgemini had any effect on the bioactivity of the Mel, we measured the caspase activity affecting the DNA synthesis, mitochondrial activity, and/or surface characteristics of the melanoma cells. Mel is an alkylator41 and therefore binds to the DNA, arresting the proliferation of the cells and triggering apoptosis. When Mel binds to DNA and RNA, defective codes cannot be repaired, thus the cell triggers intrinsic apoptosis. Caspase 2 is activated, which in turn signals the mitochondria to decrease the membrane potential and trigger the release of cytochrome c. Cytochrome c activates caspase 9 causing the executioner caspases 3 and 7 to initiate programed cell death. During these events, cytochrome c plays a role in oxidizing and translocating the phosphatidylserine from the inner membrane to the outer leaflet of the cell.42,43 In this work, Mel alone caused necrosis in <1% of the cell populations and the cells passed through the apoptotic pathway (Table 3). As far as elucidating the caspase pathways in the treated cells, as predicted, the Mel alone triggered caspase 2, involved in the DNA degradation pathway and caspase 9 in the mitochondrial pathway of apoptosis (Figure 2A).27 Minimal cell death receptor caspase 8 expression was detected, and the cells went through apoptosis ultimately as caspase 3 was triggered.27 The cells treated with Mel-NP showed a similar profile to the Mel alone with lower levels of each caspase (Figure 2B) being observed. This may be due to a slow release of the Mel from the β-cyclodextrin ring to inside the cells. However, the difference in the caspase levels did not affect drug efficiency, as the IC50 of Mel-NP was 2-fold lower at 48 hours compared with Mel alone. The delivery agent, CDgemini, did not trigger any significant amount of caspase activity (Figure 2C), indicating that the CDgemini is safe to use as a pharmaceutical drug delivery system.
The most important benefit of the encapsulation of Mel in CDgemini nanoparticles is the potential benefit to overcome drug resistance. Several chemotherapeutic drugs are currently largely ineffective for many tumor types due to high levels of chemoresistance.44 Tumor resistance is a result of several mechanisms working synergistically.45 For example, melanoma expresses ABCB1 also known as MDR1 or P-glycoprotein, an ABC transporter that causes chemoresistance.46 Other recorded mechanisms for melanoma resistance include increased levels of phase II metabolism enzymes, and therefore increased drug inactivation; increased levels of antiapoptotic genes such as survivin and melanoma inhibitor of apoptosis, resulting in decreased cell death; and increased expression of interleukin-8, a signaling gene to activate survival pathways47 or upregulation of ATF3, CYR61, IER5, IL6, and PTGS2 genes.48 The heterogeneity of tumor cells makes bypassing tumor resistance at a molecular level difficult. Thus, incorporation of the chemotherapeutic agent in nanoparticulate systems that could overcome several mechanisms of drug inactivation can be a better therapeutic approach. Here, we have demonstrated that the Mel-resistant cells could be treated efficiently with Mel-NP, reverting drug resistance (Figure 3).
Conclusion
The CDgemini nanoparticulate system was shown to be effective in delivering a poorly soluble model drug, Mel. The cytotoxic agent was successfully incorporated into stable CDgemini nanoparticles that improved bioactivity significantly in cell monolayers. However, when applied to a spheroid tumor model, the delivery of the drug was not improved by the nanoparticles. As the CDgemini surfactant shows no intrinsic toxicity, Mel-NP administration could represent a clinically feasible alternative to the current practice of injecting a Mel solution. The nanoparticulate drug was able to overcome chemoresistance, expanding the potential use of the drug for patients who fail to improve by conventional Mel therapy.
Acknowledgments
This work was funded by the Natural Sciences and Engineering Research Council and the College of Pharmacy and Nutrition, University of Saskatchewan.
Disclosure
The authors report no conflicts of interest in this work.
References
Friberg S, Nyström AM. Nanotechnology in the war against cancer: new arms against an old enemy – a clinical view. Future Oncol. 2015;11(13):1961–1975. | ||
Barenholz YC. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. | ||
Loo Y, Grigsby CL, Yamanaka YJ, et al. Comparative study of nanoparticle-mediated transfection in different GI epithelium co-culture models. J Control Release. 2012;160(1):48–56. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Sa LT, Albernaz Mde S, Patricio BF, et al. Biodistribution of nanoparticles: initial considerations. J Pharm Biomed Anal. 2012;70:602–604. | ||
Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab. 2009;10(8):836–841. | ||
Buse J, El-Aneed A. Properties, engineering and applications of lipid-based nanoparticle drug-delivery systems: current research and advances. Nanomed (Lond). 2010;5(8):1237–1260. | ||
Bei D, Meng J, Youan BB. Engineering nanomedicines for improved melanoma therapy: progress and promises. Nanomed (Lond). 2010;5(9):1385–1399. | ||
National Cancer Institute, SEER stat fact sheets: melanoma of the skin; 2016. Available from: http://seer.cancer.gov/statfacts/html/melan.html. Accessed August 12, 2016. | ||
Weide B, Faller C, Büttner P, et al. Prognostic factors of melanoma patients with satellite or in-transit metastasis at the time of stage III diagnosis. PLoS One. 2013;8(4):e63137. | ||
Bowen GM, Bowen AR, Florell SR. Lentigo maligna: one size does not fit all. Arch Dermatol. 2011;147(10):1211–1213. | ||
Klaase JM, Kroon BB, van Geel AN, Eggermont AM, Franklin HR. Systemic leakage during isolated limb perfusion for melanoma. Br J Surg. 1993;80(9):1124–1126. | ||
Di Giacomo AM, Danielli R, Calabro L, et al. Ipilimumab experience in heavily pretreated patients with melanoma in an expanded access program at the University Hospital of Siena (Italy). Cancer Immunol Immunother. 2011;60(4):467–477. | ||
Eggermont AM, Kirkwood JM. Re-evaluating the role of dacarbazine in metastatic melanoma: what have we learned in 30 years? Eur J Cancer. 2004;40(12):1825–1836. | ||
Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22(20):3138–3151. | ||
Fernberg JO, Lewensohn R, Skog S. Cell cycle arrest and DNA damage after melphalan treatment of the human myeloma cell line RPMI 8226. Eur J Haematol. 1991;47(3):161–167. | ||
Ma DQ, Rajewski RA, Stella VJ. New injectable melphalan formulations utilizing (SBE)(7m)-β-CD or HP-β-CD. Int J Pharm. 1999;189(2):227–234. | ||
Gera S, Musch E, Osterheld H, Loos U. Relevance of the hydrolysis and protein binding of melphalan to the treatment of multiple myeloma. Cancer Chemother Pharmacol. 1989;23(2):76–80. | ||
Michel D, Chitanda JM, Balogh R, et al. Design and evaluation of cyclodextrin-based delivery systems to incorporate poorly soluble curcumin analogs for the treatment of melanoma. Eur J Pharm Biopharm. 2012;81(3):548–556. | ||
Poorghorban M, Das U, Alaidi O, et al. Characterization of the host-guest complex of a curcumin analog with beta-cyclodextrin and beta-cyclodextrin-gemini surfactant and evaluation of its anticancer activity. Int J Nanomed. 2015;10:503–515. | ||
Poorghorban M, Karoyo AH, Grochulski P, Verrall RE, Wilson LD, Badea I. A 1H NMR study of host/guest supramolecular complexes of a curcumin analogue with beta-cyclodextrin and a beta-cyclodextrin-conjugated gemini surfactant. Mol Pharm. 2015;12(8):2993–3006. | ||
Odot J, Albert P, Carlier A, Tarpin M, Devy J, Madoulet C. In vitro and in vivo anti-tumoral effect of curcumin against melanoma cells. Int J Cancer. 2004;111(3):381–387. | ||
McGary EC, Heimberger A, Mills L, et al. A fully human antimelanoma cellular adhesion molecule/MUC18 antibody inhibits spontaneous pulmonary metastasis of osteosarcoma cells in vivo. Clin Cancer Res. 2003;9(17):6560–6566. | ||
Gratton SE, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Nat Acad Sci U S A. 2008;105(33):11613–11618. | ||
Patel NR, Aryasomayajula B, Abouzeid AH, Torchilin VP. Cancer cell spheroids for screening of chemotherapeutics and drug-delivery systems. Ther Deliv. 2015;6(4):509–520. | ||
Lee CK, Wang S, Huang X, Ryder J, Liu B. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 2010;296(2):233–240. | ||
Bose S, Tuunainen I, Parry M, Medina OP, Mancini G, Kinnunen PK. Binding of cationic liposomes to apoptotic cells. Anal Biochem. 2004;331(2):385–394. | ||
Mailänder V, Landfester K. Interaction of nanoparticles with cells. Biomacromolecules. 2009;10(9):2379–2400. | ||
Duddu SP, Vakilynejad M, Jamali F, Grant DJ. Stereoselective dissolution of propranolol hydrochloride from hydroxypropyl methylcellulose matrices. Pharm Res. 1993;10(11):1648–1653. | ||
Dewan M, Bhowmick B, Sarkar G, et al. Effect of methyl cellulose on gelation behavior and drug release from poloxamer based ophthalmic formulations. Int J Biol Macromol. 2015;72:706–710. | ||
Muankaew C, Jansook P, Stefánsson E, Loftsson T. Effect of γ-cyclodextrin on solubilization and complexation of irbesartan: influence of pH and excipients. Int J Pharm. 2014;474(1–2):80–90. | ||
Kuznetsova NR, Sevrin C, Lespineux D, et al. Hemocompatibility of liposomes loaded with lipophilic prodrugs of methotrexate and melphalan in the lipid bilayer. J Control Release. 2012;160(2):394–400. | ||
Dong C, Badea I, Poorghorban M, Verrall R, Foldvari M. Impact of phospholipids on plasmid packaging and toxicity of gemini nanoparticles. J Mater Chem B Mater Biol Med. 2015;3(45):8806–8822. | ||
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219–234. | ||
Dilnawaz F, Sahoo SK. Enhanced accumulation of curcumin and temozolomide loaded magnetic nanoparticles executes profound cytotoxic effect in glioblastoma spheroid model. Eur J Pharm Biopharm. 2013;85(3 Pt A):452–462. | ||
Scarano W, de Souza P, Stenzel MH. Dual-drug delivery of curcumin and platinum drugs in polymeric micelles enhances the synergistic effects: a double act for the treatment of multidrug-resistant cancer. Biomater Sci. 2015;3(1):163–174. | ||
Valetti S, Maione F, Mura S, et al. Peptide-functionalized nanoparticles for selective targeting of pancreatic tumor. J Control Release. 2014;192:29–39. | ||
Wang H, Zhao Y, Gong J, et al. Low-molecular-weight protamine-modified PLGA nanoparticles for overcoming drug-resistant breast cancer. J Control Release. 2014;192:47–56. | ||
Carvalho MR, Lima D, Reis RL, Correlo VM, Oliveira JM. Evaluating biomaterial- and microfluidic-based 3D tumor models. Trends Biotechnol. 2015;33(11):667–678. | ||
Loeber R, Michaelson E, Fang Q, Campbell C, Pegg AE, Tretyakova N. Cross-linking of the DNA repair protein Omicron6-alkylguanine DNA alkyltransferase to DNA in the presence of antitumor nitrogen mustards. Chem Res Toxicol. 2008;21(4):787–795. | ||
Jangi SM, Díaz-Pérez JL, Ochoa-Lizarralde B, et al. H1 histamine receptor antagonists induce genotoxic and caspase-2-dependent apoptosis in human melanoma cells. Carcinogenesis. 2006;27(9):1787–1796. | ||
Galluzzi L, Vitale I, Vacchelli E, Kroemer G. Cell death signaling and anticancer therapy. Frontiers Oncol. 2011;1:5. | ||
Gao Z, Zhang L, Sun Y. Nanotechnology applied to overcome tumor drug resistance. J Control Release. 2012;162(1):45–55. | ||
Hu X, Zhang Z. Understanding the genetic mechanisms of cancer drug resistance using genomic approaches. Trends Genet. 2016;32(2):127–137. | ||
Saha S, Adhikary A, Bhattacharyya P, Das T, Sa G. Death by design: where curcumin sensitizes drug-resistant tumours. Anticancer Res. 2012;32(7):2567–2584. | ||
Chen KG, Valencia JC, Gillet JP, Hearing VJ, Gottesman MM. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigm Cell Melanoma Res. 2009;22(6):740–749. | ||
Wouters J, Stas M, Govaere O, Van den Eynde K, Vankelecom H, van den Oord JJ. Gene expression changes in melanoma metastases in response to high-dose chemotherapy during isolated limb perfusion. Pigm Cell Melanoma Res. 2012;25(4):454–465. |
Supplementary material
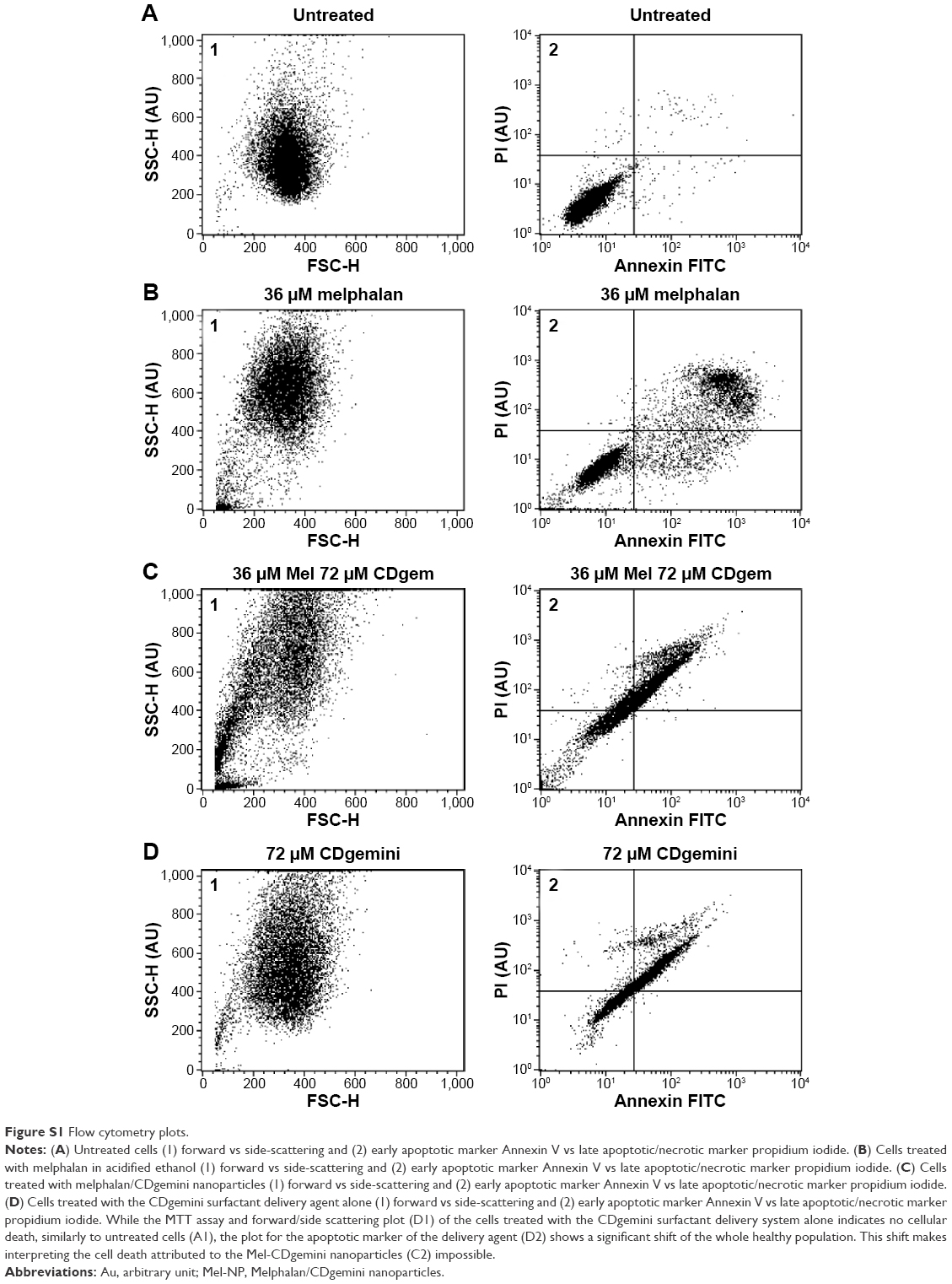 | Figure S1 Flow cytometry plots. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.