Back to Journals » Cancer Management and Research » Volume 11
Evaluating venetoclax and its potential in treatment-naïve acute myeloid leukemia
Authors Knight T, Edwards H, Taub JW, Ge Y ![]()
Received 10 January 2019
Accepted for publication 15 March 2019
Published 23 April 2019 Volume 2019:11 Pages 3197—3213
DOI https://doi.org/10.2147/CMAR.S180724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Beicheng Sun
Tristan Knight,1,2 Holly Edwards,3,4 Jeffrey W Taub,1–2,4 Yubin Ge2–4
1Division of Pediatric Hematology and Oncology, Department of Pediatrics, Children’s Hospital of Michigan, Detroit, MI, USA; 2Department of Pediatrics, Wayne State University School of Medicine, Detroit, MI, USA; 3Department of Oncology, Wayne State University School of Medicine, Detroit, MI, USA; 4Molecular Therapeutics Program, Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, MI, USA
Abstract: Venetoclax (ABT-199), a BH3-mimetic and selective BCL-2 inhibitor, was recently approved by the US Food and Drug Administration (FDA) for the treatment of acute myeloid leukemia (AML) in adult patients aged 75 years or older, or otherwise unable to tolerate intensive induction chemotherapy, in combination with either hypomethylating agents or low-dose cytarabine. In this review article, we discuss venetoclax’s mechanism of action, in relation to both the BCL-2 protein family in general and BH3-mimetic activity in particular. We then outline the pharmacological advances that preceded and facilitated its development, as well as providing an overview of key preclinical and clinical studies which lead to its use first in chronic lymphoid leukemia (CLL), then in small lymphocytic leukemia (SLL), and subsequently in AML. Finally, we seek to offer an overview of the challenges and opportunities encountered as venetoclax moves into more widespread use, including its use and activity against leukemia initiating cells and oxidative phosphorylation.
Keywords: ABT-199, acute myeloid leukemia, apoptosis, BCL-2 inhibitor, BH3-mimetic, venetoclax
Introduction
In late November 2018, the United States Food and Drug Administration (FDA) approved venetoclax plus either hypomethylating agents or low-dose cytarabine for first-line treatment of newly diagnosed acute myeloid leukemia (AML) among adult patients aged 75 years or older, or those with medical comorbidities which would otherwise preclude the usage of standard, intensive chemotherapy.
AML is characterized by the explosive clonal proliferation of malignant, immature myeloid elements, in conjunction with diminished apoptotic rate, relative to proliferation.1 The survival rates for both pediatric and adult patients remain frustratingly low, with 5-year survival rates approximating 65% and 25%, respectively.2 Two major contributors to these figures are chemotherapeutic resistance and a high rate of relapse—and at the time of relapse, the emergence of a unique, more resistant clonal population may be identified, molecularly distinct from that which was present at the time of the initial diagnosis–which hints that chemotherapeutically induced selection pressure may play a role in AML relapse.3 Standard induction therapy in AML has long been comprised of the 7+3 regimen, eg, 7 days of cytarabine plus 3 days of an anthracycline such as daunorubicin, followed by either a cytarabine-based consolidation regimen or allogeneic hematopoietic stem cell transplant (HSCT).4 The intensity and toxicity of this regimen is high, and in patients over the age of 60 (who constitute the bulk of those newly diagnosed with AML) and those patients with medical comorbidities, the 7+3 regimen may be poorly tolerated.4 New therapeutic options, which may additionally be safely used in more medically fragile patients, therefore, represent an ongoing and urgent need. Following a prolonged paucity in the development of efficacious new agents, the brief period from April 2017 to November 2018 was marked by the approval of multiple agents for the treatment of AML, including midostaurin (April 2017), liposomal daunorubicin/cytarabine (CPX-351) and enasidenib (both August 2017), gemtuzumab ozogamicin (September 2017), ivosidenib (July 2018), and glasdegib, gilteritinib, and venetoclax (all November 2018).5 During the preceding decades, improvements in AML outcomes were attributable to minor, incremental refinements in supportive care and therapeutic regimens6–evolutionary, rather than revolutionary changes, so to speak. Now, however, it would seem that AML management is poised upon the threshold of an era in which the promise of targeted small molecule inhibitors and biologic agents is finally being realized. A deepening understanding of the oncogenic driver mutations underlying AML pathogenesis has facilitated such progress, and it could reasonably be argued that the exponential growth in our understanding of its genetic heterogeneity and underpinnings make it one of the most genetically well characterized of all human diseases. With this explosive growth in knowledge comes the ability to supplement the previously lackluster therapeutic armamentarium available to frontline clinicians. In the vanguard of this new wave of pharmacological cavalry are agents such as venetoclax, which offer the ability to not only achieve improved outcomes for patients with AML without reliance on intensive existing regimens, but also a means to utilize the very mechanisms by which oncogenesis occurs against the resulting malignancy, and thereby achieve therapeutic success.
Providing a context for venetoclax
Venetoclax is a BH3 mimetic and selective inhibitor of the protein B-cell lymphoma 2 (BCL-2). Members of the BCL-2 protein family, named for the eponymous BCL-2 protein itself, are critical regulators of mitochondrial membrane potential (MMP) and therefore of the larger intrinsic apoptotic pathway as a whole, of which they are key constituents. The loss of MMP pushes the cell towards apoptosis as a result of the resultant release of previously sequestered proapoptotic proteins from the mitochondria, eg, cytochrome c, SMAC/DIABLO (second mitochondria-derived activator of caspases/direct IAP binding protein with low PI), and HTRA2/Omi (high temperature requirement).7 Once freed, these mediators facilitate the activation of caspases–a family of zymogen proteases which cleave their target substrate proteins using cysteine protease activity after specific aspartic acid residues, from which their name is derived: Cysteine-aspartic proteases, or Cysteine-dependent aspartate-directed proteases.8 It is the executioner caspases, particularly caspase 3, which drive apoptosis via the CAD-ICAD (caspase-activated DNAse/inhibitor of CAD) complex–an inactive heterodimer which, following activation, dimerizes and cleaves the DNA phosphodiester backbone to create a double-stranded DNA break.9
The BCL-2 family is comprised of over 20 individual proteins, subdivided into three sub-families according to their function (proapoptotic/antiapoptotic) and of the number of BCL-2 homology (BH) domains present.10 BCL-2, BCL-2A1, BCL-XL, BCL-W, and MCL-1 (myeloid cell leukemia 1) are members of the multi-domain BH1-4 antiapoptotic proteins and therefore contain all four BH domains.11 Members of this group facilitate cellular survival by antagonization of proapoptotic proteins. These are (1) the BH3-only proapoptotic proteins which, as their name suggests, possess only a single BH3 domain, and include (among others) BIM (BCL2L11; BCL2-interacting mediator of cell death), BAD (BCL2 antagonist of cell death), and BID (BH3-interacting domain death antagonist), and (2) the multi-domain proapoptotic proteins, sharing BH domains 1–3, and including BAX (BCL-2 associated X protein), and BAK (BCL-2 antagonist killer). Among members of the family which possess BH1, 2, and 3 domains, these make up a hydrophobic surface groove where the BH3-only proapoptotic proteins’ BH3 domain binds; subtle sequence variations exist, such that this binding is quite selective. When within the hydrophobic grove of the multi-domain proapoptotic proteins, the proapoptotic BH3-only family members facilitate homo-oligomerization and formation of a pore within the mitochondrial outer membrane.12 Pore size is reflective of the number of constituent monomers, with a minimum of four BAX monomers being required, for instance, to facilitate cytochrome c’s escape from the intermembrane space, and up to 20-monomers needed for the largest proapoptotic proteins to exit.12 Proapoptotic BCL-2 family members must be in relative excess to overcome inhibition by antiapoptotic family members and thereby induce apoptosis.12 This balance between proapoptotic and antiapoptotic proteins is a delicate one, and the relative degree to which a cell is “tilted” towards or away from apoptosis is referred to as “priming”–primed cells being more readily inducible towards apoptosis by virtue of a greater concentration of (and relative excess of) proapoptotic BCL-2 family proteins.13 Further increasing the concentration of BH3-only proteins, such as may occur via exposure to cellular stresses, may tip a primed cell towards apoptosis, while a malignant cell, by virtue of a greater concentration of antiapoptotic proteins (or a lower baseline expression of proapoptotic proteins) may evade apoptosis.10 The degree of a cell’s priming, and the BCL-2 family members responsible for that priming, may be measured via BH3 profiling, which uses synthetic BH3 peptides to assess the degree of resultant loss of mitochondrial membrane potential to assess these factors.13 This technique may also be useful in predicting resistance and chemotherapeutic response, as more “primed” patients demonstrate notably improved outcomes.14
BH3 mimetics then target such proteins as BCL-2 and bind within the hydrophobic groove, displacing sequestered proapoptotic BH3-only proteins and enabling them to activate BAK and BAX. Or, in less “primed” cells BH3-mimetics instead enter the hydrophobic groove and block the binding of subsequently expressed BH3-only proteins thereby reducing the concentration of these proteins needed to induce apoptosis.10 Effectively, BH3 mimetics increase the degree of priming and therefore the tendency towards apoptosis and sensitivity towards conventional chemotherapeutic agents.14
ABT-737 and navitoclax: precursors to venetoclax
Using a combination of nuclear magnetic resonance and fragment-based drug discovery, ABT-737 was developed by Oltersdorf et al in 2005.15 Briefly, two ligands (4ʹ-fluoro-biphenyl-4-carboxylic acid [Kd of 0.30±0.03 mM] and 5,6,7,8-tetrahydro-naphthalen-1-ol [Kd of 4.3±1.6 mM]) were identified with binding affinities to the P2 and P4 pockets within BCL-XL’s hydrophobic groove, respectively, and were subsequently combined into a single molecule with micromolar affinity. Multiple rounds of structural refinement followed, to both minimize unwanted protein binding and optimize P2 and P4 binding.15 Due to similarities in the hydrophobic groove between BCL-XL and its family members BCL-2 and BCL-w, ABT-737 displays a high affinity (eg, Ki <1 nmol/L) for these multi-domain BH1-4 antiapoptotic proteins, but a low affinity for BCL-2A1 and MCL-1.16 ABT-737 functionally mimics a BH3-only proapoptotic protein, and both competes with and displaces native BH3-only proteins from binding sites. By virtue of this mechanism, ABT-737 and its successors, including venetoclax, constitute the BH3-mimetic class of therapeutic agents. ABT-737 displayed excellent preclinical activity, but its poor oral bioavailability and limited solubility hindered its clinical utility. A successor, navitoclax (ABT-263) was therefore developed, demonstrating improved oral bioavailability, and pharmacokinetic and pharmacodynamics properties (including 20–50% oral bioavailability, and plasma half-life of approximately 9 hours).16,17 ABT-737 continues in widespread laboratory use, but is no longer in clinical use, and no clinical trials employing this agent exist. For its part, navitoclax is also a BH3-mimetic and, like its predecessor, acts similarly to the BH3-only protein BAD, with a high affinity (Ki of <1 nmol/L) for BCL-2, BCL-XL, and BCL-w, but low affinities for MCL-1 and BCL-2A1 (Ki 550 nmol/L and 354 nmol/L, respectively).16 Early studies of navitoclax showed reversal of apoptotic protection afforded by BCL-2 and BCL-XL overexpression models, and co-immunoprecipitation demonstrated decreased BIM:BCL-XL interaction following exposure to navitoclax, thereby demonstrating proof of its BH3-mimetic properties.16 In preclinical studies, in vivo use illustrated its efficacy in both small-cell lung carcinoma (SCLC) and acute lymphoblastic leukemia (ALL) models; it also demonstrated synergy with rituximab, bortezomib, and the rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone (R-CHOP) (cyclophosphamide, doxorubicin, vincristine, and prednisone) chemotherapeutic regimen in aggressive lymphoma models, prefacing its subsequent use in combination therapies.16 In the preclinical and clinical setting, however, navitoclax was found to cause thrombocytopenia via a now-understood mechanism, which precluded its use at higher doses. Platelet survival is governed by the intrinsic apoptotic pathway and dependent on adequate expression of BCL-XL to mitigate the activities of BAK.18 In their native state, platelets undergo a gradual degradation of BCL-XL, thereby priming aged platelets for apoptosis. The ratio of BCL-XL to BAK determines the rate at which this occurs.18 As such, in states where inhibition of BCL-XL occurs (such as during treatment with ABT-737 or navitoclax), platelet survival decreases. In this way, navitoclax exposure results in concentration-dependent, rapid, marked thrombocytopenia, which acts as a dose-limiting toxicity; indeed, this side effect may mean that it cannot be safely used in certain situations at doses necessary to exert a therapeutic effect, most particularly as monotherapy.19
The earliest clinical trial of navitoclax occurred in 2011 and established its efficacy in lymphoid malignancies, particularly in those patients with chronic lymphoid leukemia (CLL) and small lymphocytic leukemia (SLL)–diseases found to be especially sensitive to the drug.20 A subsequent Phase I study in relapsed/refractory CLL demonstrated a partial response rate of 35% (with 90% of patients seeing 50% or greater fall in leukocytosis);21 via combination with rituximab, further improvement in response rate (overall response rate of 70%) was seen.22 As noted, severe, dose-related thrombocytopenia was a common finding across all studies. At the time of writing, 10 active studies utilizing navitoclax were listed on the United States National Library of Medicine’s Clinicaltrials.gov website; all but two of these studies are examining its role as a component of combination therapy, in settings such as CLL, myelofibrosis, relapsed acute lymphoblastic leukemia/lymphoma, small-cell and non-small-cell lung cancer, and a range of relapsed/refractory solid tumors.
Increased precision and decreased collateral damage: preclinical and clinical development of venetoclax
Due to navitoclax’s dose-limiting, BCL-XL-mediated thrombocytopenia, it was subsequently redesigned via subtle alterations in drug structure to abrogate this particular side effect.19 Navitoclax was reverse engineered via removal of a thiophenyl unit and intercalation of an indole into the binding region, thereby allowing formation of a hydrogen bond between the novel agent and the Asp103 residue present on BCL-2, but not with the correspondent Glu96 residue present on BCL-XL.19 This redesign meant that venetoclax demonstrates subnanomolar affinity for BCL-2 (Ki <0.010 nM) but not towards other members of the BCL-2 family (Ki’s of 48 nM towards BCL-XL and 245 nM towards BCL-W; 5,000 times and 24,000 times lower than navitoclax, respectively), as well as negligible affinity towards MCL-1 (Ki >445 nM).19 Congruent with these affinities, venetoclax demonstrated efficacy during in vitro and in vivo testing against BCL-2-dependent malignancies, roughly corresponding to the degree to which BCL-2 was overexpressed; platelet toxicity was also minimal (EC50=5.5 vs 0.083 µM for navitoclax).19 Venetoclax is also orally available, but further improving upon navitoclax, demonstrates a threefold higher half-life in plasma; 26 hours, compared to navitoclax’s 9-hour half-life.23
Initial clinical trials examined venetoclax’s efficacy in CLL. Tumor lysis was a major concern during these early trials; the first Phase I study utilized a daily dose of 100–200 mg, with the first 3 patients all experiencing tumor lysis syndrome (TLS).24 Subsequent downward revision of the initial dose, to 50 mg daily, continued to be marked by clinically significant TLS (one fatal and one resulting in renal failure necessitating hemodialysis). An additional reduction in initial dosing, to 20 mg per day, followed by a gradual increase to 400 mg per day by week 5 of therapy, proved effective at avoiding early TLS, and no further cases were seen. Response rates were encouraging, 79% of patients showing a response, 20% showing a complete response, and 5% having negative minimal residual disease.24 Long-term follow-up of the initial cohort confirmed these findings–among patients receiving the full, 400 mg daily dose (after ramp-up), 81% overall response rate, 16% complete response rate, and 62% progression free survival were shown after 24 months on therapy.25 Subanalysis identified a 71% overall response rate among those with 17p deletion,24–a high risk cytogenetic marker–and a subsequent Phase II study was therefore conducted specifically of patients with relapsed/refractory CLL and a 17p deletion, achieving overall response rates and complete response rates of 79% and 8%, respectively, at median one year.26 In April 2016, venetoclax was therefore approved by the US FDA for treatment of patients with CLL and a 17p deletion, following at least one antecedent line of therapy. Subsequently, in June 2018, this approval was broadened to include those patients with CLL without a 17p deletion, as well as those with SLL, irrespective of 17p deletion status, who had already received one or more preceding line of treatment.
Venetoclax remains under intensive investigation in the CLL/SLL context; as of writing, 35 currently active clinical trials are listed under the United States National Library of Medicine’s Clinicaltrials.gov website, and an additional 11 are not yet recruiting. Many of these studies are examining its role in combination therapies—the results of the first such combination studies have recently become available, and optimism is justified. The earliest study combined rituximab combined rituximab with venetoclax among adult patients with relapsed/refractory CLL or SLL 86% of patients responded, with 60% of those patients having complete response (eg, a complete response rate in 51% of the total patients); 2-year progression free survival was approximately 82%.27 Although a maximum tolerated dose was not identified, 2 patients developed severe TLS after initiation of 50 mg daily venetoclax dosing. Subsequently, all patients initiated treatment at 20 mg daily, after which further cases of severe TLS were not observed.27 A goal dose of 400 mg per day of venetoclax was selected for further evaluation in subsequent studies.
Efforts at integrating venetoclax into CLL regimens followed, combining venetoclax with nitrogen mustard bendamustine and obinutuzumab or rituximab. Although limited by their recency, evidence suggests that these combinations show impressive efficacy. Phase I study data shows triple therapy with venetoclax, bendamustine, and either rituximab or obinutuzumab to be equally safe and tolerable, and with excellent overall response rates of 96% (relapsed/refractory CLL receiving venetoclax, bendamustine, and rituximab) to 100% (previously untreated patients receiving venetoclax, bendamustine, and rituximab and relapsed/refractory patients receiving venetoclax, bendamustine, and obinutuzumab).28 A Phase II trial of patients with untreated (54%) or relapsed/refractory (46%) CLL using the combination of bendamustine, obinutuzumab, and venetoclax achieved an overall response rate of 95% at end-induction assessment, including all treatment-naïve patients, and 87% of patients achieved a negative minimal residual disease (MRD).29
The results of a Phase III trial evaluating the combination of venetoclax and rituximab were recently published as well.30 In it, patients with relapsed or refractory CLL were randomized to either a venetoclax-rituximab group or bendamustine-rituximab group. Results were dramatic–venetoclax plus rituximab achieved an 84.9% progression-free survival rate at 2 years, vs the bendamustine plus rituximab arm, which achieved a 2-year progression-free survival rate of 36.3%.30 Sub-analysis of patients who had CLL and a 17p deletion (typically indicative of a poorer prognosis) likewise demonstrated a pronounced survival benefit with a 2-year progression-free survival rate of 81.5% on the venetoclax-rituximab arm vs 27.8% on the bendamustine-rituximab arm, respectively.30
Venetoclax in the treatment of AML
Clinical efficacy in AML
Although initial studies evaluated its use in CLL, venetoclax is presently undergoing evaluation in a range of clinical contexts, with recent data suggesting efficacy in an array of settings including ALL,31 non-Hodgkin's lymphoma (NHL),32 and multiple myeloma;33,34 and its role in these and other contexts (including diffuse large B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, marginal zone lymphoma, and Waldenström macroglobulinemia) has recently been reviewed.35 Case-report level examples of effective usage also exist in primary plasma cell leukemia36 and immunoglobulin light chain amyloidosis.37 As data continues to accrue, it seems likely that the therapeutic avenues opened will only continue to grow. Venetoclax’s recent FDA approval for newly diagnosed AML in those patients aged 75 years or older/those with comorbidities precluding intensive induction chemotherapy, plus either the hypomethylating agent azacitidine or decitabine, or low-dose cytarabine, was based upon two major studies.
The first38–40 paired venetoclax and low-dose cytarabine in a nonrandomized, open-label Phase I/II dose-escalation/expansion trial, among those with treatment-naïve AML aged 65 years or over and not eligible for intensive chemotherapy. Participants received subcutaneous cytarabine at a dose of 20 mg/m2 for days 1–10 of each 28-day cycle, as well as venetoclax at an eventual dose of 600 mg or 800 mg daily (following a 5-day ramp-up to target dose, starting from an initial dose of 50 mg/day) on days 2–28 of cycle 1, and days 1–28 of all subsequent 28-day cycles. Overall, the combination was well tolerated despite the relative frailty of the patient population, and no clinically significant episodes of tumor lysis syndrome were observed. Grade 3 or 4 adverse events were largely limited to cytopenias (neutropenia/thrombocytopenia/anemia) and febrile neutropenia; a more comprehensive discussion of adverse effects/toxicities is provided below. Overall response rate was 75%–5% partial response (PR), 70% complete response (CR)/complete response with incomplete hematological recovery (Cri). Overall survival (OS) at 12 months was estimated at 74.7%, with a median time to best response of 30 days. In comparison, among patients likewise deemed unfit for intensive chemotherapy, prior examination of low-dose cytarabine monotherapy (at a dose of 20 mg twice daily for 10 days, administered in 4–6-week cycles) had demonstrated a CR of 18%, and 24% OS at 12 months.41 The subsequent recommended Phase II dose for venetoclax (when paired with low-dose cytarabine) was set at 600 mg daily. Follow-up reporting at one year40 (data upon which the actual FDA approval was granted) showed 64% achievement of CR/CRi/PR (including 26% CR and 2% PR), lasting a median of 14.9 months, with median OS of 11.4 months (and 18.4 months in those who achieved CR/Cri). Importantly, patients with NPM1, DNMT3A, FLT3-ITD, and SRSF2 mutations all achieved CR/CRi of over 75%;40 in contrast, for patients treated in the earlier study using low-dose cytarabine monotherapy, no patients with adverse cytogenetics responded to treatment.41
The second trial42 paired venetoclax with a hypomethylating agent (decitabine 20 mg/m2 on days 1–5 of each 28-day cycle, or azacitidine 75 mg/m2 on days 1–7 of each 28-day cycle) in a nonrandomized, open-label, dose-escalation Phase Ib study in patients with treatment-naïve AML aged 65 years or over and not eligible for intensive chemotherapy. Venetoclax administration was initiated on day 2 of cycle 1 and was escalated over 5 days to reach goal dose; 4 cohorts existed, with starting and target venetoclax doses of 20–100 mg and 400–1200 mg, respectively, and was continued daily for the full duration of each 28-day cycle. The combination was well tolerated, with thrombocytopenia, neutropenia, and febrile neutropenia being the most common grade 3/grade 4 adverse events; maximum tolerated doses were not reached, and no instances of tumor lysis syndrome were observed. Subsequent recommended Phase II dosing for venetoclax (paired with hypomethylating agents) was set at 400 mg daily (or 800 mg with interrupted dosing). Overall response rate was 68%; 61% achieved CR or CRi, with a median time to CR/CRi of 0.9–1.2 months, and median response duration of 11 months; OS at 12 months was estimated at 59%. Similar proportions of patients achieved a response whether venetoclax was paired with decitabine or azacitidine. Updated results43 following a median on-study time of 8.9 months show similar results, with 67% of patients achieving CR/CRi, median response duration of 11.3 months, and median OS of 17.5 months. A summary of the existing available results of studies of venetoclax in AML is provided in Table 1. In comparison, decitabine monotherapy (20 mg/m2 for 5 days, repeated every 28 days) had previously been shown to achieve an overall response rate of 25% and CR rate of 24%, with a median OS of 7.7 months.44 Direct assessment of azacitidine monotherapy is more difficult, but among a patient population with newly diagnosed AML with a low blast percentage (20–30%) and otherwise deemed ineligible for intensive chemotherapy, azacitidine (75 mg/m2 for 7 days, repeated every 28 days) achieved an 18% CR rate, with a median OS of 19.1 months; the authors note, however, that their study population (eg, those with 20–30% blasts) may demonstrate significant lead-time bias thereby limiting generalizability to the wider population of patients with AML, or even be more congruent with natural history of advanced myelodysplastic syndrome vs that of AML.45
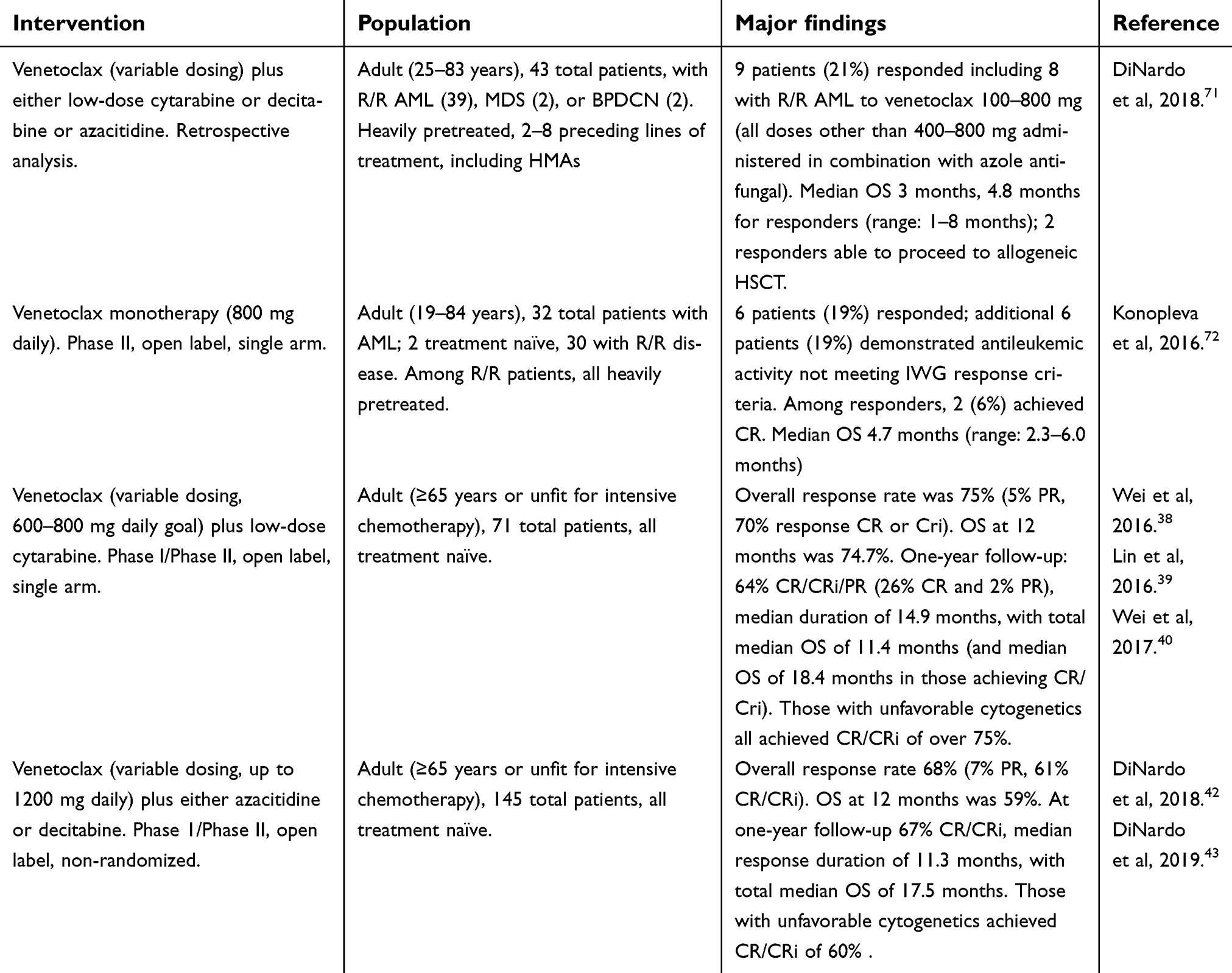 | Table 1 Completed studies of venetoclax in the treatment of AML |
Results of these preliminary studies of venetoclax’s efficacy are currently being validated in Phase III, randomized, double-blind, placebo-controlled trials in treatment-naïve patients ineligible for standard induction therapy: VIALE-A (NCT02993523) is examining azacitidine plus venetoclax vs azacitidine alone, and VIALE-C (NCT03069352) is assessing low-dose cytarabine plus venetoclax vs low-dose cytarabine alone. Both are actively recruiting, and no preliminary results were available as of writing. Venetoclax is also undergoing evaluation in multiple additional active clinical trials in the context of AML as of writing (Table 2). Other than the trial results outlined above, we were not able to identify additional outcomes or toxicity data via publically accessible databases.
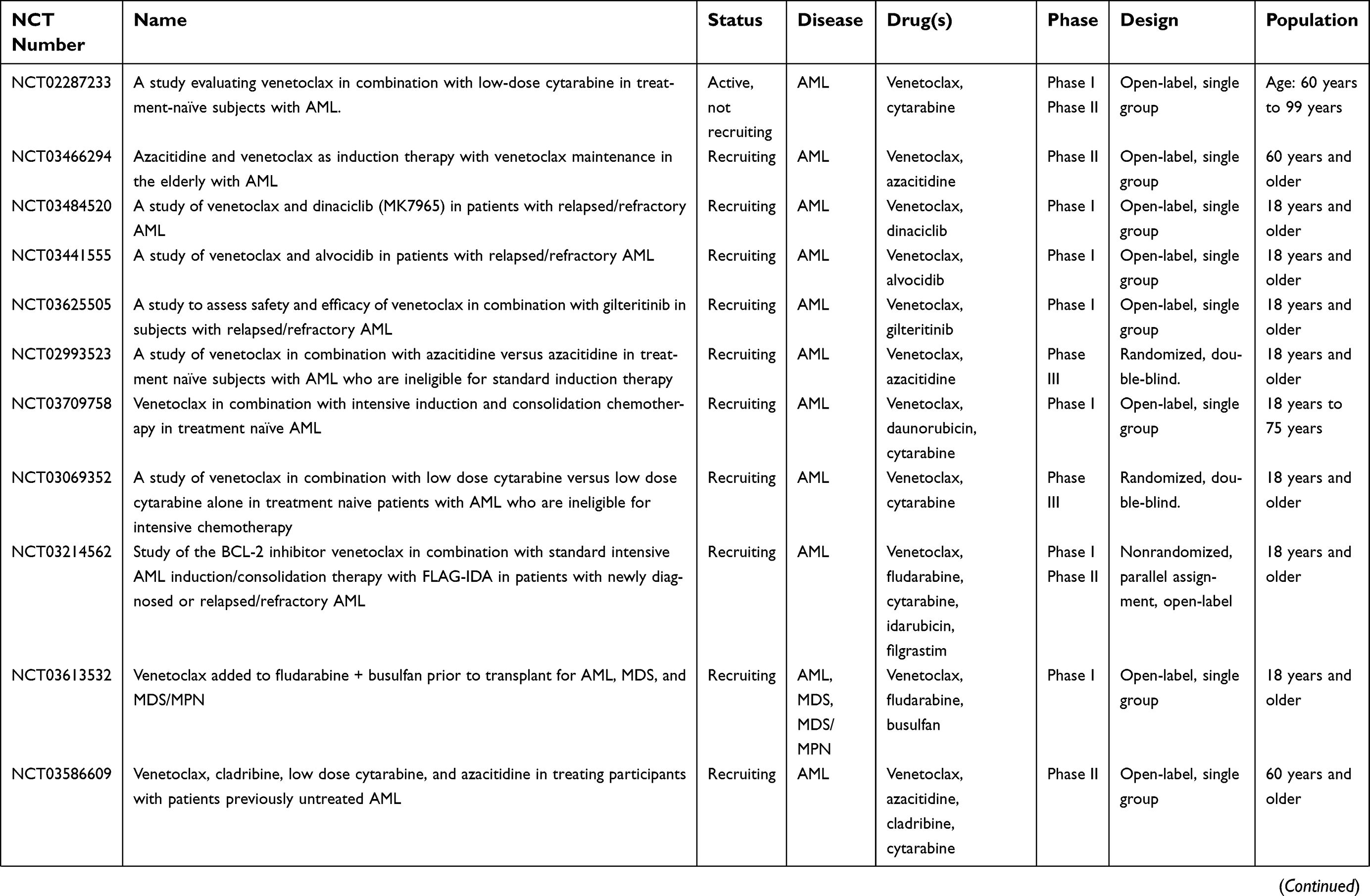 | Table 2 Active clinical trials of venetoclax in acute myeloid leukemia |
 | Table 2 (Continued). |
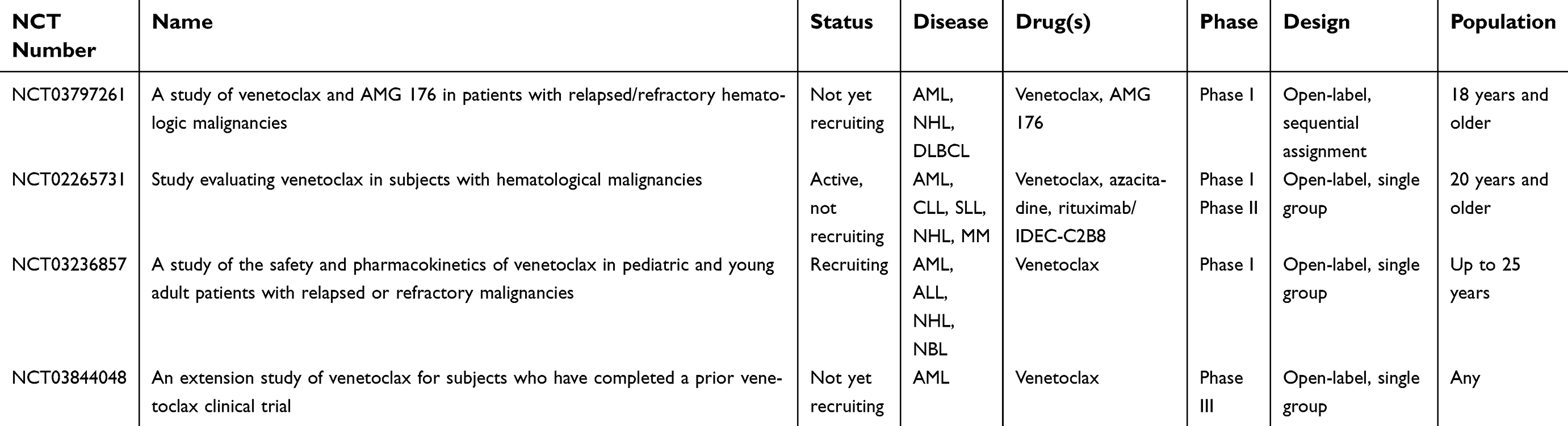 | Table 2 (Continued). |
Toxicities, side effects, dosing, and practical considerations46
As noted, TLS is a prominent concern during the initiation of venetoclax therapy, and has prompted both numerous downward revisions of the recommended initial dose, and the development of escalating-dose treatment strategies. It is available in 10 mg, 50 mg, and 100 mg tablet formulations, with an initial recommended dose for patients with AML of 100 mg on day 1, 200 mg on day 2, 400 mg on day 3, and either 400 mg (patients receiving azacitidine or decitabine) or 600 mg (patients receiving low-dose cytarabine) on day 4 and beyond. To begin treatment in patients with AML, an initial white blood cell count of 25×109/L (25×103/µL) is recommended prior to venetoclax initiation; for those with higher levels, cytoreduction may be necessary to reduce the risk of TLS. In patients with CLL/SLL, for instance, in whom a high incidence of TLS was noted in early studies, dosing is initiated at 20 mg daily for 1 week, increasing to daily doses of 50 mg, 100 mg, 200 mg, and 400 mg every week, during weeks 2–5, respectively, with a goal dose of 400 mg daily. In both AML and CLL/SLL, strategies including hydration, close monitoring of blood chemistries and renal function, and use of antihyperuricemic agents may be warranted to reduce the risk of TLS, and patients taking venetoclax should be advised to maintain adequate hydration prior to and during venetoclax therapy.
Otherwise, toxicities are relatively mild across studies of venetoclax monotherapy, and include mild gastrointestinal side effects including diarrhea and nausea, as well as upper respiratory tract infections (at least one of which has been seen in approximately half of patients); hematological toxicities are also common, including neutropenia (~40–50% of patients), anemia (~20% of patients), and thrombocytopenia (~15% of patients).24,26,27 Studies involving patients with AML are somewhat more difficult to tease out, as azacitidine/decitabine/cytarabine were co-administered. However, side effects were not grossly disparate from those previously described. Commonly seen (in over 30% of patients) were nausea, vomiting, diarrhea, constipation, fatigue, neutropenia/febrile neutropenia, thrombocytopenia, and anemia; hematological toxicities were the most commonly noted severe side effects noted in all combinations. Dose adjustment and/or delay of subsequent cycles of chemotherapy may therefore be necessary in patients experiencing hematological toxicities, however, the impact of such alterations upon therapeutic efficacy and response to treatment are not known, and should be pursued with caution. One potential dose alteration was suggested in the original Phase I study of venetoclax plus hypomethylating agents, among patients with prolonged neutropenia.42 Among patients who have achieved CR/clearance of bone marrow leukemic blasts, granulocyte colony stimulating factor (GCSF) may be considered, potentially in conjunction with a reduced dosing schedule of venetoclax administered for 3 weeks continuously, followed by 1 week off, in 4-week cycles or, if necessary, in alternating cycles of 2 weeks on, 2 weeks off.42 In the setting of continued neutropenia, administration of GCSF and a 50% dose reduction for the paired hypomethylating agent may also be considered; again, however, the long-term outcomes of these changes is not known. Additionally, the possibility that delayed hematological recovery may be due to relapse, and not due to toxicity must be considered, prior to dose alteration.
Venetoclax is predominantly metabolized via hepatic CYP3A, and as such, concomitant use with strong CYP3A inhibitors (eg, antifungal agents itraconazole, voriconazole, and ketoconazole, macrolide antibiotics, and antivirals of the protease inhibitor class [eg, atazanavir, indinavir, ritonavir]) is contraindicated; the risk of TLS is increased due to reduced metabolism. Posaconazole 300 mg daily (eg, the recommended dose for prophylaxis against invasive aspergillus and candida infections) concurrently with venetoclax doses of 50–100 mg, appears to increase venetoclax’s Cmax by 7.1-fold and 24 hour area under the curve (AUC) by 8.8-fold.47 The authors of that study suggest posaconazole may be used for antifungal prophylaxis concurrent with venetoclax, provided the dose of the latter is reduced by 75%; the impact of this dose reduction on therapeutic efficacy is not known.47 Conversely, CYP3A inducers (eg, the rifamycin antibiotics, and anticonvulsants phenobarbital, phenytoin, and carbamazepine) decrease venetoclax plasma concentration. Venetoclax has also been shown to increase the concentrations of warfarin and digoxin, and concomitant usage should be avoided where possible. No data exists regarding its use in pregnancy or lactation in humans; murine data suggests a fetotoxic effect, without observed teratogenicity, and excretion has been documented in milk. As such, pregnancy testing prior to initiation of therapy and avoidance of subsequent pregnancy is recommended during treatment and for at least 30 days following cessation, as is abstention from breastfeeding.
Overcoming AML resistance to venetoclax
As promising as the initial early data appears, AML blasts and leukemia initiating cells (LICs) are nonetheless capable of either developing resistance to venetoclax, or of possessing intrinsic mechanisms of resistance present from the outset of therapy.
One readily understandable mechanism by which AML cells may resist venetoclax is via reliance upon alternative antiapoptotic protein; if BCL-2 overexpression is not the primary means by which a malignant cell has escaped apoptosis, or if a secondary means exists, no degree of isolated BCL-2 inhibition will be sufficient. MCL-1 is one such protein, playing a key part in many malignancies, including AML. In settings in which MCL-1 is overexpressed, it seems to play a crucial role in oncogenesis and in treatment resistance–failing to target MCL-1 in such contexts reduces therapeutic efficacy irrespective of whether there is adequate inhibition of the other antiapoptotic BCL-2 family members.48,49 Venetoclax, congruent with its activity as a BH3 mimetic, reduces association between BCL-2 and BIM, causing a compensatory increase in free BIM, but a subsequent increase in BIM/MCL-1 binding, particularly in venetoclax-resistant cell lines, which prevents BIM binding to BAX/BAK; this inhibits the loss of MMP and therefore reduces apoptotic potential.50 Concurrent inhibition of MCL-1, however, diminishes BIM/MCL-1 association and thereby abrogates venetoclax resistance occurring via this mechanism.49 Among the BCL-2 family, MCL-1 displays several relatively unique properties, which may be pharmacologically exploitable. MCL-1’s half-life is comparatively short at approximately 90 minutes, compared to BCL-2, BCL-XL, and BCL-W (half-lives all approximating 20 hours).51 Brief transcriptional downregulation, even for a brief period, may therefore be sufficient to induce apoptosis. MCL-1 also demonstrates a rapid response to proapoptotic and antiapoptotic signals, with marked resultant alterations in expression–cells are quite sensitive to MCL-1 fluctuations, and concentration may only need to be reduced for a short interval before apoptosis is irreversibly initiated.51 In vivo and in vitro efficacy of selective MCL-1 inhibitors such as A1210477 and S63845 has been demonstrated in AML cell lines which overexpress MCL-1 and are resistant to navitoclax and venetoclax; moreover, in cell lines with both MCL-1 and BCL-2 overexpression, combinations of venetoclax plus S63845 demonstrate marked efficacy.52,53 Although development of clinical agents capable of inhibiting MCL-1 has been somewhat laggardly, several compounds are now under evaluation in early trials, spurred by observations such as these. One of these will directly assess combined MCL-1 plus BCL-2 inhibition, via the use of S64315 and venetoclax in patients with relapsed/refractory AML (NCT03672695). As of writing however, there is a paucity of data regarding both efficacy and safety in humans. However, ex vivo BH3 profiling of primary patient samples appears to be able to accurately predict cellular response to selective MCL-1/BCL-2 inhibition, and demonstrates the clear benefit of combining these two approaches, lending credence to clinical trials involving combinations such as this.54 Indirect targeting of MCL-1 also appears to synergize with venetoclax, as well. Cyclin-dependent kinase 9 (CDK9) acts as a transcriptional regulator for MCL-1; inhibition therefore causes downregulation of its transcription and a reduction in concentration.55 This approach has been validated preclinically, and clinical evaluation of CDK9 inhibitors has begun in the AML context.56 Alvocidib (flavopiridol), in combination with standard AML therapy, demonstrates some efficacy but, due to concerns regarding toxicity, subsequent agents with improved selectivity have been developed, including voruciclib. Similarly, XPO1 is a nuclear exporter which is overexpressed in AML cells, the inhibition of which downregulates MCL-1. Combining the selective XPO1 inhibitor KPT-330 (selinexor) with venetoclax results in synergistic apoptotic induction in both AML cell lines and primary patient samples, due to concurrent downregulation of MCL-1 and BCL-2 inhibition.57 Exposure to agents with a directly toxic effect on DNA such as daunorubicin also reduces MCL-1 levels, which synergizes well with venetoclax’s anti-BCL-2 activity, and requires less pharmacological finesse, though at the cost of greater toxicity and side effects.58 Alterations within BCL-2 itself are also a means by which resistance may develop. BCL-2 phosphorylation, such that conformational change occurs within the hydrophobic binding groove, appears capable of altering affinity for BH3 mimetic compounds such as venetoclax, but does not alter affinity endogenous BH3-only proteins.59 Similarly, alterations impacting the BH3-binding domain prevent adequate pharmacological inhibition, as has been noted in situations where point missense mutations within BCL-2 induce a degree of venetoclax resistance.60,61 Downstream, loss-of-function mutations which alter BAX/BAK functionality or expression will also allow a degree of resistance to venetoclax, as the BH3-mimetic class as a whole relies upon the adequate and efficacious presence of at least one of these proteins to mediate apoptosis.60,61
Certain cell-surface receptors also appear to confer treatment resistance. FMS-like tyrosine kinase 3 (FLT3) internal tandem duplication (FLT3-ITD) causes BCL-XL and MCL-1 to be upregulated, which thereby confers venetoclax-resistance via the overexpression of other antiapoptotic proteins not inhibited by venetoclax.62 Fortunately however, the development of targeted, anti-FLT3 therapies has enabled the provision of agents which may abrogate this mechanism of resistance. The first generation FLT3 inhibitor midostaurin, and the second generation FLT3 inhibitor gilteritinib have both been recently approved by the FDA for use in FLT3-mutated AML (in April 2017 and November 2018, respectively). Preclinical data62 suggests that the combination FLT3 inhibitors with venetoclax is sufficient to overcome FLT3-induced resistance–and a clinical trial is currently evaluating gilteritinib plus venetoclax in patients with relapsed/refractory AML (NCT03625505).
The bone marrow microenvironment might also play a part in mediating venetoclax resistance, as well. MCL-1 expression in AML cells doubles when co-incubated with bone marrow derived mesenchymal stromal cells (BM-MSCs), resulting in demonstrable resistance to isolated BCL-2 inhibition–resistance which is overcome via concurrent pan-BCL-2 inhibition.63 Many unknowns remain in this realm however, and the role played by the bone marrow microenvironment remains to be fully elucidated.
Targeting leukemia initiating cells (LICs): a role for venetoclax?
Leukemia initiating cells (LICs), also referred to as leukemia stem cells (LSCs), are a subpopulation within the larger leukemic amalgam which appear to possess properties typically attributed to stem cells (hence their name) including self-renewal, engraftment and reconstitution, and initiation of leukemia.64 Moreover, their existence is a likely contributor to chemotherapeutic resistance65 and to AML’s unacceptably high relapse rate.66 LIC frequency has unequivocally been shown to increase in the setting of relapse, in terms of both absolute quantity and activity, and to demonstrate genetic and phenotypic alterations in comparison to LICs present at time of initial diagnosis.66 Specific LIC targeting is therefore an important consideration in the development of novel therapeutic agents for AML; a compound which eliminates circulating and bone marrow blasts, but leaves LICs untouched is not one which can be expected to produce a lasting remission or cure. LICs do appear to demonstrate characteristics which may make them uniquely vulnerable to venetoclax, including reactively low levels of reactive oxygen species (ROS-low), relative overreliance upon oxidative phosphorylation (OXPHOS), and overexpression of BCL-2 as means of apoptotic avoidance.67 Preclinical work on patient-derived AML samples demonstrated that inhibition of BCL-2 was effective at reducing OXPHOS and selectively eradicating quiescent LICs.67 These findings were subsequently validated via evaluation of LICs from patients initially treated on the Phase I clinical trial of venetoclax plus azacitidine.42 In comparing patients treated using the venetoclax/azacitidine combination with those treated via any other standard regimen, several striking differences emerged. Both AML blasts and phenotypically defined LICs (CD34+, CD38-, CD123+, Lin-) were rapidly eliminated (eg, within the first 96 hours of therapy), in comparison to standard chemotherapy, in which no significant reductions were seen.68 Pronounced downregulation of OXPHOS was seen in ROS-low cells (eg, LICs), with resultant reduction in adenosine triphosphate (ATP) levels, something again not seen in patients treated via conventional chemotherapy, or, critically, in patients receiving azacitidine monotherapy.68 Mechanistically speaking, disruption of OXPHOS appears to be mediated via altered tricarboxylic acid (TCA) cycle activity and disruption of the electron transport chain (ETC) complex II, which supplies the substrates upon which OXPHOS depends. Malate, fumarate, and α-ketoglutarate were all significantly decreased following exposure to venetoclax, while succinate levels were significantly elevated.68 Succinate levels, in particular, indicate a defect within the ETC complex II, succinate being this complex’s metabolic substrate and with increased levels therefore suggesting dysfunction. Reduced succinate dehydrogenase A glutathionylation, secondary to a reduction in available glutathione levels induced by the combination of venetoclax plus azacitidine, appears to be the means by which ETC complex II activity is reduced, resulting in the aforementioned TCA cycle perturbations.68 Importantly, pretreatment with cell-permeable glutathione resulted in an increase of intracellular glutathione, normalization of succinate levels, and rescued ETC complex II activity–similarly, this pretreatment was also able to rescue OXPHOS as determined by ATP levels in the presence of venetoclax/azacitidine.68 Finally, the authors noted that these activities and alterations appear to occur only in the presence of both venetoclax and azacitidine, but not with either agent in isolation.
Inhibition of OXPHOS appears to be an emerging area of cancer therapeutics. Although this mechanism of activity was not specifically predicted to be a means by which venetoclax functions, other agents have been developed with specific targeting of OXPHOS in mind. IACS-010759 is a novel small molecule inhibitor of agent which inhibits the ETC complex I, and has demonstrated promising in vitro and in vivo activity in AML models, with clear efficacy against LSCs.69 This agent is presently under clinical evaluation in two Phase I studies, one of which is being conducted among patients with relapsed/refractory AML (NCT02882321). If safety is demonstrated, addition of this agent to azacitidine/venetoclax is an intriguing possibility, as such a combination would theoretically inhibit the ETC at two separate sites (complex I and II, respectively), potentially resulting in synergistic LIC elimination.
Disruption of OXPHOS, while efficacious, underscores another potential mechanism of resistance to venetoclax therapy. Follow-up investigation of LICs from patients who experienced inferior responses demonstrated (a) increased reliance upon fatty acid metabolism, (b) increased movement of fatty acids into the TCA cycle, together resulting in (c) preservation of OXPHOS function and ATP generation.70 Elevated expression of carnitine palmitoyltransferase 1 (CPT1), a key enzyme in the beta-oxidation of long chain fatty acids, is a predictor of worsened OS in patients with AML in general, but also a predictor of on-therapy progression/poor response to venetoclax/azacitidine.70 Taken together, the authors inferred that increased reliance upon fatty acid metabolism (and therefore a concordant decrease in reliance upon the ETC) as mediated by increased CPT1 could be abrogated via concurrent inhibition of this enzyme plus venetoclax/azacitidine therapy. This was indeed the case: utilizing the CPT1 inhibitor etomoxir, LICs obtained from relapsed/refractory patients thereafter displayed reduced OXPHOS and were resensitized to venetoclax/azacitidine.70
Future directions, upcoming challenges, and conclusions
ABT-737’s discovery, subsequent refinement into the orally available navitoclax, and finally optimization into the selective BCL-2 inhibitor venetoclax has provisioned oncologists with a valuable weapon in the fight against AML. The first FDA-approved inhibitor of this family, venetoclax is the harbinger of a coming wave of targeted, high-precision agents directly targeting the BCL-2 family, and thereby reclaiming a key mechanism of clonal immortality. The two clinical trials which lead to its approval in treatment-naïve AML, in combination with either hypomethylating agents or low-dose cytarabine, are similarly at the vanguard of a large and ever-expanding number of clinical trials, exciting results from which may reasonably be expected. Despite this promise, however, challenges remain and there is much which remains unknown.
One area yet to be fully elucidated is how best to more broadly integrate venetoclax into the general therapeutic approach to AML. Patients with AML whom subsequently relapse or who’s disease is refractory to therapy are generally candidates for allogeneic HSCT. Venetoclax’s role in this regard is unclear; is it best used as one component of a multimodality approach to induce remission prior to transplant, as a maintenance therapy post-transplant, or some combination thereof? At present, the clearest benefit for patients receiving venetoclax appears to exist among those who would otherwise not be able to tolerate intensive chemotherapy. Theoretically, there is no reason to expect that venetoclax would not synergize well with traditional, intensive chemotherapeutic regimens such as 7+3 or FLAG-IDA (fludarabine, cytarabine, idarubicin, and GCSF); indeed studies evaluating this are currently in progress with results eagerly awaited. For the present however, venetoclax’s interaction with these regimens, and its benefit when combined with intensive chemotherapy remain unknown. Venetoclax may have some efficacy in relapsed/refractory AML–although the response seen is less than that observed in the treatment-naïve context. Among a heterogeneous, heavily pretreated population of patients with relapsed/refractory AML, who had received between two and eight antecedent lines of treatment, 21% of patients responded to venetoclax plus either low-dose cytarabine, decitabine, or azacitidine; median OS was 3 months among the entire cohort, and 4.8 months among responders, several of whom were able to proceed to allogeneic HSCT.71 The authors note that venetoclax plus either low-dose cytarabine or hypomethylating agents may therefore be a viable salvage option for patients with relapsed/refractory AML, particularly prior to progression to allogeneic HSCT.71 Venetoclax monotherapy appears to offer similar figures–among a similar patient population, a 19% overall response rate was achieved, and an additional 6 patients (19%) demonstrated antileukemic activity not meeting International Working Group response criteria.72 Among responders, 6% achieved CR, and median OS was 4.7 months (range: 2.3–6.0 months).72
Given the variant expression of members of the BCL-2 family, even among patients with the “same” disease, it is difficult to predict which patients might respond best to targeted inhibition and, just as importantly, what is the best means by which a physician might choose between various targeted inhibitors for an individual patient or group of patients sharing a similar mutation? As has been discussed, AML cells which are resistant to BCL-2 inhibition may be sensitive to MCL-1 inhibition, and vice versa. As availability continues to expand, comparative studies will be necessary. Complicating matters somewhat is the lack of available biomarkers–other than thrombocytopenia, as seen with inhibition of BCL-XL, they are notable for their absence. BH3 profiling may offer a partial answer to this quandary–it is not, however, widely available. Were this to change, however, and routine BH3 profiling widely integrated into standard care, selection of the optimal inhibitor or inhibitors (eg, in the case of combined reliance upon MCL-1 and BCL-2) could be done on this basis. Determination of treatment response and monitoring for relapse could also conceivably be performed via BH3 profiling, with assessment of altered protein expression potentially guiding treatment. With regard to LICs in particular, resistance to venetoclax has been seen to potentially be mediated via increased reliance upon fatty acid metabolism for the provision of adequate performance of OXPHOS.70 Along a similar line of thinking, it might therefore be prudent to consider use of techniques able to assess for altered metabolism prior to or shortly after initiation of therapy, so as to allow the addition of agents which might inhibit this activity and therefore preserve the anti-LIC component of venetoclax-based therapy.
Although large confirmatory Phase III trials are ongoing, the results of earlier studies justify a certain degree of excitement regarding venetoclax’s future as a member of the armamentarium of potent, selective agents newly becoming available for the treatment of AML. As current trials mature and further data accumulates, selective BCL-2 inhibition appears poised to become a revolutionary new therapeutic approach, with the potential for widespread integration into existent and future treatment protocols.
Acknowledgment
No funding was secured specifically for this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29(5):475–486. doi:10.1200/JCO.2010.30.2554
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA-Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
3. Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627–1635. doi:10.1182/blood-2016-10-696039
4. Short NJ, Rytting ME, Cortes JE. Acute myeloid leukaemia. Lancet. 2018;392(10147):593–606. doi:10.1016/S0140-6736(18)31041-9
5. Click ZR, Seddon AN, Bae YR, Fisher JD, Ogunniyi A. New food and drug administration-approved and emerging novel treatment options for acute myeloid leukemia. Pharmacotherapy. 2018;38(11):1143–1154. doi:10.1002/phar.2180
6. Rubnitz JE, Inaba H, Dahl G, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11(6):543–552. doi:10.1016/S1470-2045(10)70090-5
7. Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–163. doi:10.4161/cbt.4.2.1508
8. Kominami K, Nakabayashi J, Nagai T, et al. The molecular mechanism of apoptosis upon caspase-8 activation: quantitative experimental validation of a mathematical model. Biochim Biophys Acta. 2012;1823(10):1825–1840. doi:10.1016/j.bbamcr.2012.07.003
9. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
10. Knight T, Luedtke D, Edwards H, Taub JW, Ge Y. A delicate balance - The BCL-2 family and its role in apoptosis, oncogenesis, and cancer therapeutics. Biochem Pharmacol. 2019;162:250–261. doi:10.1016/j.bcp.2019.01.015
11. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–284. doi:10.1038/nrd.2016.253
12. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25(1):65–80. doi:10.1038/cdd.2017.186
13. Potter DS, Letai A. To prime, or not to prime: that is the question. Cold Spring Harb Symp Quant Biol. 2016;81:131–140. doi:10.1101/sqb.2016.81.030841
14. Ni Chonghaile T, Sarosiek KA, Vo TT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–1133. doi:10.1126/science.1206727
15. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi:10.1038/nature03579
16. Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi:10.1158/0008-5472.CAN-07-5836
17. Vogler M, Walter HS, Dyer MJS. Targeting anti-apoptotic BCL2 family proteins in haematological malignancies - from pathogenesis to treatment. Br J Haematol. 2017;178(3):364–379. doi:10.1111/bjh.14684
18. Mason KD, Carpinelli MR, Fletcher JI, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128(6):1173–1186. doi:10.1016/j.cell.2007.01.037
19. Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi:10.1038/nm.3048
20. Wilson WH, O’Connor OA, Czuczman MS, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–1159. doi:10.1016/S1470-2045(10)70261-8
21. Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488–496. doi:10.1200/JCO.2011.34.7898
22. Kipps TJ, Eradat H, Grosicki S, et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015;56(10):2826–2833. doi:10.3109/10428194.2015.1030638
23. Jones AK, Freise KJ, Agarwal SK, Humerickhouse RA, Wong SL, Salem AH. Clinical predictors of venetoclax pharmacokinetics in chronic lymphocytic leukemia and non-Hodgkin’s lymphoma patients: a pooled population pharmacokinetic analysis. AAPS J. 2016;18(5):1192–1202. doi:10.1208/s12248-016-9927-9
24. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322. doi:10.1056/NEJMoa1513257
25. Davids MS, Hallek M, Wierda W, et al. Comprehensive safety analysis of venetoclax monotherapy for patients with relapsed/refractory chronic lymphocytic leukemia. Clin Cancer Res. 2018;24(18):4371–4379. doi:10.1158/1078-0432.CCR-17-3761
26. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768–778. doi:10.1016/S1470-2045(16)30019-5
27. Seymour JF, Ma S, Brander DM, et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: a phase 1b study. Lancet Oncol. 2017;18(2):230–240. doi:10.1016/S1470-2045(17)30012-8
28. Stilgenbauer S, Morschhauser F, Wendtner CM, et al. Phase Ib Study (GO28440) of venetoclax with bendamustine/rituximab or bendamustine/obinutuzumab in patients with relapsed/refractory or previously untreated chronic lymphocytic leukemia. Blood. 2016;128:22. doi:10.1182/blood-2016-06-724161
29. Cramer P, von Tresckow J, Bahlo J, et al. Bendamustine followed by obinutuzumab and venetoclax in chronic lymphocytic leukaemia (CLL2-BAG): primary endpoint analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19(9):1215–1228. doi:10.1016/S1470-2045(18)30414-5
30. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. New Engl J Med. 2018;378(12):1107–1120. doi:10.1056/NEJMoa1713976
31. El-Cheikh J, Moukalled NM, El Darsa H, et al. Feasibility of the combination of venetoclax and asparaginase-based chemotherapy for adult patients with relapsed/refractory acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. 2018;18(10):e441–e444. doi:10.1016/j.clml.2018.07.289
32. de Vos S, Swinnen LJ, Wang D, et al. Venetoclax, bendamustine, and rituximab in patients with relapsed or refractory NHL: a phase Ib dose-finding study. Ann Oncol. 2018;29(9):1932–1938. doi:10.1093/annonc/mdy256
33. Rahbari KJ, Nosrati JD, Spektor TM, Berenson JR. Venetoclax in combination with bortezomib, dexamethasone, and daratumumab for multiple myeloma. Clin Lymphoma Myeloma Leuk. 2018;18(9):e339–e343. doi:10.1016/j.clml.2018.06.003
34. Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392–2400. doi:10.1182/blood-2017-06-788323
35. Mihalyova J, Jelinek T, Growkova K, Hrdinka M, Simicek M, Hajek R. Venetoclax: A new wave in hematooncology. Exp Hematol. 2018;61:10–25. doi:10.1016/j.exphem.2018.02.002
36. Jelinek T, Mihalyova J, Kascak M, et al. Single-agent venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am J Hematol. 2019;94(1):E35–E37. doi:10.1002/ajh.25331
37. Leung N, Thome SD, Dispenzieri A. Venetoclax induced a complete response in a patient with immunoglobulin light chain amyloidosis plateaued on cyclophosphamide, bortezomib and dexamethasone. Haematologica. 2018;103(3):e135–e137.
38. Wei A, Strickland SA, Roboz GJ, et al. Safety and efficacy of venetoclax plus low-dose cytarabine in treatment-naive patients aged ≥ 65 years with acute myeloid leukemia. Blood. 2016;128:22. doi:10.1182/blood-2016-06-724161
39. Lin TL, Strickland SA, Fiedler W, et al. Phase Ib/2 study of venetoclax with low-dose cytarabine in treatment-naive patients age ≥ 65 with acute myelogenous leukemia. J Clin Oncol. 2016;34(15_suppl):7007. doi:10.1200/JCO.2016.34.15_suppl.7007
40. Wei A, Strickland SA, Roboz GJ, et al. Phase 1/2 study of venetoclax with low-dose cytarabine in treatment-naive, elderly patients with acute myeloid leukemia unfit for intensive chemotherapy: 1-year outcomes. Blood. 2017;130.
41. Burnett AK, Milligan D, Prentice AG, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109(6):1114–1124. doi:10.1002/cncr.22496
42. DiNardo CD, Pratz KW, Letai A, et al. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 2018;19(2):216–228. doi:10.1016/S1470-2045(18)30010-X
43. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752
44. Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, Phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol. 2010;28(4):556–561. doi:10.1200/JCO.2009.23.9178
45. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28(4):562–569. doi:10.1200/JCO.2009.23.8329
46. Venetoclax: Prescribing Information [Package Insert]. Chicago (IL): Abbvie Inc.; 2018.
47. Agarwal SK, DiNardo CD, Potluri J, et al. Management of venetoclax-posaconazole interaction in acute myeloid leukemia patients: evaluation of dose adjustments. Clin Ther. 2017;39(2):359–367. doi:10.1016/j.clinthera.2017.01.003
48. Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66(8):1326–1336. doi:10.1007/s00018-008-8637-6
49. Luedtke DA, Niu X, Pan Y, et al. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther. 2017;2:17012. doi:10.1038/sigtrans.2017.12
50. Niu X, Zhao J, Ma J, et al. Binding of released bim to Mcl-1 is a mechanism of intrinsic resistance to ABT-199 which can be overcome by combination with daunorubicin or cytarabine in AML cells. Clin Cancer Res. 2016;22(17):4440–4451. doi:10.1158/1078-0432.CCR-15-3057
51. Yang-Yen HF. Mcl-1: a highly regulated cell death and survival controller. J Biomed Sci. 2006;13(2):201–204. doi:10.1007/s11373-005-9064-4
52. Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):
53. Li Z, He S, Look AT. The MCL1-specific inhibitor S63845 acts synergistically with venetoclax/ABT-199 to induce apoptosis in T-cell acute lymphoblastic leukemia cells. Leukemia. 2019;33(1):262–266.
54. Ramsey HE, Fischer MA, Lee T, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018;8(12):1566–1581. doi:10.1158/2159-8290.CD-18-0140
55. Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15(4):519–527. doi:10.1080/15384101.2016.1138186
56. Boffo S, Damato A, Alfano L, Giordano A. CDK9 inhibitors in acute myeloid leukemia. J Exp Clin Cancer Res. 2018;37(1):36. doi:10.1186/s13046-018-0704-8
57. Luedtke DA, Su YW, Liu S, et al. Inhibition of XPO1 enhances cell death induced by ABT-199 in acute myeloid leukaemia via Mcl-1. J Cell Mol Med. 2018;22(12):6099–6111. doi:10.1111/jcmm.13886
58. Xie CZ, Edwards H, Caldwell JT, Wang G, Taub JW, Ge YB. Obatoclax potentiates the cytotoxic effect of cytarabine on acute myeloid leukemia cells by enhancing DNA damage. Mol Oncol. 2015;9(2):409–421. doi:10.1016/j.molonc.2014.09.008
59. Song T, Chai G, Liu Y, Yu X, Wang Z, Zhang Z. Bcl-2 phosphorylation confers resistance on chronic lymphocytic leukaemia cells to the BH3 mimetics ABT-737, ABT-263 and ABT-199 by impeding direct binding. Br J Pharmacol. 2016;173(3):471–483. doi:10.1111/bph.13370
60. Fresquet V, Rieger M, Carolis C, Garcia-Barchino MJ, Martinez-Climent JA. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood. 2014;123(26):4111–4119. doi:10.1182/blood-2014-03-560284
61. Tahir SK, Smith ML, Hessler P, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer. 2017;17(1):399. doi:10.1186/s12885-017-3383-5
62. Mali RS, Lasater EA, Doyle K, et al. FLT3-ITD activation mediates resistance to the BCL-2 selective antagonist, venetoclax, in FLT3-ITD mutant AML models. Blood. 2017;130.
63. Pan RQ, Ruvolo VR, Wei J, et al. Inhibition of Mcl-1 with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737 resistance in acute myeloid leukemia. Blood. 2015;126(3):363–372. doi:10.1182/blood-2014-10-604975
64. De Grandis M, Mancini SJ, Aurrand-Lions M. In quest for leukemia initiating cells in AML. Oncoscience. 2018;5(1–2):9–10. doi:10.18632/oncoscience.394
65. Doan PL, Chute JP. The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia. 2012;26(1):54–62. doi:10.1038/leu.2011.236
66. Ho TC, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128(13):1671–1678. doi:10.1182/blood-2016-02-695312
67. Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12(3):329–341. doi:10.1016/j.stem.2012.12.013
68. Pollyea DA, Stevens BM, Jones CL, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med. 2018;24(12):1859–1866. doi:10.1038/s41591-018-0233-1
69. Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–1046. doi:10.1038/s41591-018-0052-4
70. Pollyea DA, Jones CL, Stevens BM, et al. Relapsed acute myeloid leukemia is less sensitive to venetoclax + azacitidine due to leukemia stem cell resistance driven by fatty acid metabolism and can be overcome by pharmacologic inhibition of CPT1. Blood. 2018;132(Suppl 1):432.
71. Di Nardo CD, Rausch CR, Benton C, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol. 2018;93(3):401–407. doi:10.1002/ajh.25000
72. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi:10.1158/2159-8290.CD-16-0313
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.