Back to Journals » International Journal of Nanomedicine » Volume 11
Eupafolin nanoparticles protect HaCaT keratinocytes from particulate matter-induced inflammation and oxidative stress
Authors Lin Z, Lee C, Tsai M, Ko H, Fang J ![]() , Chiang Y
, Chiang Y ![]() , Liang C, Hsu LH, Hu SC, Yen F
, Liang C, Hsu LH, Hu SC, Yen F
Received 23 March 2016
Accepted for publication 5 June 2016
Published 11 August 2016 Volume 2016:11 Pages 3907—3926
DOI https://doi.org/10.2147/IJN.S109062
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Zih-Chan Lin,1,* Chiang-Wen Lee,2,3,* Ming-Horng Tsai,4 Horng-Huey Ko,5 Jia-You Fang,1,2 Yao-Chang Chiang,6,7 Chan-Jung Liang,8,9 Lee-Fen Hsu,10 Stephen Chu-Sung Hu,11,12 Feng-Lin Yen5,8,13
1Graduate Institute of BioMedical Sciences, Chang Gung University, 2Research Center for Industry of Human Ecology, Chang Gung University of Science and Technology, Kweishan, Taoyuan, 3Division of Basic Medical Sciences, Department of Nursing, Chang Gung Institute of Technology and Chronic Diseases and Health Promotion Research Center, Chiayi, 4Division of Neonatology and Pediatric Hematology/Oncology, Department of Pediatrics, Chang Gung Memorial Hospital, Yunlin, 5Department of Fragrance and Cosmetic Science, College of Pharmacy, Kaohsiung Medical University, Kaohsiung, 6Center for Drug Abuse and Addiction, China Medical University Hospital, 7Center for Drug Abuse and Addiction, China Medical University, Taichung, 8Center for Lipid and Glycomedicine Research, Kaohsiung Medical University, Kaohsiung, 9Center for Lipid Biosciences, Kaohsiung Medical University Hospital, 10Department of Respiratory Care, Chang Gung University of Science and Technology, Chiayi Campus, Chiayi, 11Department of Dermatology, College of Medicine, Kaohsiung Medical University, 12Department of Dermatology, Kaohsiung Medical University Hospital, 13Institute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung, Taiwan, Republic of China
*These authors contributed equally to this work
Abstract: Exposure to particulate matter (PM), a major form of air pollution, can induce oxidative stress and inflammation and may lead to many diseases in various organ systems including the skin. Eupafolin, a flavonoid compound derived from Phyla nodiflora, has been previously shown to exhibit various pharmacological activities, including antioxidant and anti-inflammatory effects. Unfortunately, eupafolin is characterized by poor water solubility and skin penetration, which limits its clinical applications. To address these issues, we successfully synthesized a eupafolin nanoparticle delivery system (ENDS). Our findings showed that ENDS could overcome the physicochemical drawbacks of raw eupafolin with respect to water solubility and skin penetration, through reduction of particle size and formation of an amorphous state with hydrogen bonding. Moreover, ENDS was superior to raw eupafolin in attenuating PM-induced oxidative stress and inflammation in HaCaT keratinocytes, by mediating the antioxidant pathway (decreased reactive oxygen species production and nicotinamide adenine dinucleotide phosphate oxidase activity) and anti-inflammation pathway (decreased cyclooxygenase-2 expression and prostaglandin E2 production through downregulation of mitogen-activated protein kinase and nuclear factor-κB signaling). In summary, ENDS shows better antioxidant and anti-inflammatory activities than raw eupafolin through improvement of water solubility and skin penetration. Therefore, ENDS may potentially be used as a medicinal drug and/or cosmeceutical product to prevent PM-induced skin inflammation.
Keywords: eupafolin, nanoparticles, particulate matter, oxidative stress, cyclooxygenase-2, keratinocytes
Introduction
Over the past decade, ambient air pollution (AAP), the contamination of the outdoor environment by airborne chemical, physical, or biological hazardous waste, has been linked to the destruction of the ecological environment.1,2 From the viewpoint of health, AAP is the world’s largest single environmental health risk, and long-term exposure to AAP is associated with various diseases, including acute lower respiratory tract disease, chronic obstructive pulmonary disease, ischemic heart disease, lung cancer, stroke, and neurodegenerative diseases.3–6 The World Health Organization reported that 3.7 million deaths were attributable to AAP worldwide in 2012. Of all these pollutants, particulate matter (PM) containing fine particles (particle size <2.5 μm, PM2.5) and coarse particles (particle size <10 μm, PM10) represent the real-time air quality index for monitoring the pollution level of a city. For example, the PM2.5 concentration of Shanghai city in the People’s Republic of China reached the “most severe” level.7 Previous reports have indicated that PM containing polyaromatic hydrocarbons can cause inflammatory reactions in the lung and may lead to systemic inflammatory reactions through activation of oxidative stress and mitogen-activated protein kinase (MAPK) signaling pathways.8–10 In particular, the skin constitutes the first line of defense of the human body to air pollutants. However, research investigating the effects of PM on the skin has been limited.
Recently, a number of studies have investigated the effects of PM on skin damage and its underlying molecular biological mechanisms. Air pollution containing PM had been found to increase the incidence of inflammatory skin diseases (including atopic dermatitis, acne, and psoriasis), extrinsic aging, androgenic alopecia, and skin cancer (including melanoma and squamous cell carcinoma). The underlying mechanisms include activation of aryl hydrocarbon receptors, generation of reactive oxygen species (ROS; such as superoxide and hydroxyl radicals), activation of MAPKs (ERK1/2, JNK, and p38), activation of transcription factors (nuclear factor-κB [NF-κB] and activator protein-1), production of inflammatory mediators (interleukin [IL]-1α, IL-6, IL-8, and tumor necrosis factor-α), activation of matrix metalloproteinases (MMPs; MMP-1, MMP-2, and MMP-9) with collagen degradation, and impairment of DNA repair system.11–17
Flavonoids (such as quercetin, kaempferol, myricetin, luteolin, and apigenin) are effective antioxidant and anti-inflammatory compounds and are abundantly found in fruits and plants.18 Izawa et al19 showed that quercetin may be protective against male reproductive toxicity induced by diesel exhaust particles. Tan et al20 also demonstrated that luteolin can inhibit cigarette smoke extract-induced oxidative damage and apoptosis through the Nrf2 pathway and may serve as a chemopreventive agent for lung cancer. Therefore, it is conceivable that flavonoids may be potentially useful for preventing PM-induced skin cell injury. Eupafolin is a flavonoid compound, which is purified from Phyla nodiflora, and previous studies have indicated that eupafolin exhibits anti-inflammatory, antioxidant, and anticancer effects.21–23 Moreover, the multiple pharmacological activities of flavonoids have been attributed to their structure–activity relationships, including the flavone backbone structure and the number of hydroxyl substitutions.24 However, the chemical structure of flavonoids, such as quercetin and naringenin, has also limited their administration and application in clinical medicine, due to their extremely low water solubility and poor bioavailability.25,26 Eupafolin has a similar flavone backbone structure and displays poor water solubility, resulting in poor cutaneous penetration following topical administration on skin. Therefore, many studies have attempted to develop nanoparticle delivery systems using nanoprecipitation and high pressure homogenization techniques to overcome the physicochemical drawbacks of the active compounds27,28 and thereby improve their solubility and skin absorption.29,30 However, there have been no previous reports about whether eupafolin nanoparticle delivery system (ENDS) shows improved solubility and skin penetration and whether it may attenuate PM-induced oxidative stress and inflammation in human HaCaT keratinocytes. In this study, polyvinyl alcohol (PVA) and Eudragit E100 were used as nanoparticle carriers to prepare ENDS, because of their nontoxicity and water solubility. PVA is a nontoxic polymer, which is frequently used as an emulsion stabilizer for nanoparticle formulations. Eudragit E100 is an acid-responsive cationic polymer, which is composed of butyl methacrylate, dimethylaminoethyl methacrylate, and methyl methacrylate. It is frequently used in oral and topical drug formulations since it is nontoxic and edible.31–34
The purpose of the present study is to investigate the solubility improvement of eupafolin through nanoprecipitation methods, by evaluation of particle size, crystalline to amorphous transformation, hydrogen bond formation, and in vitro skin penetration. In addition, we also investigated the molecular biological mechanisms underlying the antioxidant and anti-inflammatory effects of ENDS and raw eupafolin in PM-stimulated HaCaT keratinocytes, with regard to ROS production, cyclooxygenase-2 (COX-2) expression, prostaglandin E2 (PGE2) production, and MAPK phosphorylation.
Materials and methods
Materials
Dulbecco’s Modified Eagle’s Medium:nutrient mixture F-12 (DMEM/F-12 medium) was obtained from Thermo Fisher Scientific (Waltham, MA, USA). Fetal bovine serum (FBS) was purchased from Hazelton Product (Denver, PA, USA). TRIZOL was obtained from Thermo Fisher Scientific. Western blotting enhanced chemiluminescence detection kit and Hyperfilms were purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). The bicinchoninic acid protein assay kit was obtained from Pierce (Rockford, IL, USA). The monoclonal antibodies COX-2, phospho-P65, and β-actin were obtained from Cell Signaling Technology (Danver, MA, USA). Phospho-ERK, p38, and JNK antibody kits were from Cell Signaling Technology. Phospho-c-Fos antibody was from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). PhosphoPlus p47phox antibody was purchased from Assay Biotechnology (Sunnyvale, CA, USA). Anti-GAPDH antibody was purchased from Biogenesis (Bournemouth, UK). N-acetylcysteine (NAC), apocynin (APO), and MAPK inhibitors (U0126, SB202190, and SP600125) were obtained from Biomol (Plymouth Meeting, PA, USA). PM was from the National Institute of Standards and Technology, Standard Reference Material® 1649b (Gaithersburg, MD, USA). Eudragit E100 was obtained from Röhm Pharma (Darmstadt, Germany), and PVA was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Eupafolin was extracted from P. nodiflora as described previously,35 and the purity of eupafolin was >95% as determined by high-performance liquid chromatography (HPLC). All other chemicals were of American Chemical Society reagent grade.
Preparation of ENDS
ENDS was prepared by nanoprecipitation with solvent evaporation method as described previously.36 Briefly, 100 mg of Eudragit E100 and 10 mg of eupafolin were, respectively, added into 5 mL of 95% alcohol and placed in an ultrasonic water bath until they dissolved (organic phase). Subsequently, 100 mg of PVA was added into 15 mL of pure water as aqueous phase. Then, the organic phase was immediately added into the aqueous phase and then homogenized at 22,000 rpm for 10 minutes. The mixed solution was processed by rotary vacuum evaporation to remove alcohol, and the remaining solution, consisting of ~10 mL as ENDS, was stored at 4°C until further use for biological assays.
Determination of particle size and morphology of ENDS
The particle size and polydispersity index (PI) of eupafolin and ENDS were determined by an N5 submicrometer particle size analyzer containing a 25 mW helium–neon laser lamp with a wavelength of 632.8 nm (Beckman Coulter, Brea, CA, USA). PI, the uniformity of the particle sizes, was determined by photon correlation spectroscopy. In addition, the morphology of the ENDS was photographed by a transmission electron microscope (JEOL JEM-2000 EXII TEM; JEOL, Tokyo, Japan). Briefly, ENDS was diluted 50-fold with pure water and then immediately stained with 0.5% (w/v) phosphotungstic acid (Sigma-Aldrich Co.) and fixed on a 200-mesh copper grid.
Determination of drug loading efficiency of ENDS
The drug loading efficiency of ENDS was determined by measuring the content of eupafolin encapsulated within the excipient. The eupafolin standard curve was obtained by HPLC analysis (Hitachi Ltd., Tokyo, Japan). The HPLC system consisted of an L-2130 pump, L-2200 autosampler, and L-2420 UV–vis detector. The analysis was carried out using the Mightysil RP-18 GP (250×4.6 mm2 internal diameter, 5 μm) column (Kanto Corporation, Portland, OR, USA). The mobile phase consisted of 10 mM potassium dihydrogen phosphate buffer (Scharlau, Barcelona, Spain) and acetonitrile (Honeywell, Morristown, NJ, USA; 65:35, v/v, pH adjusted to 2.8). Different eupafolin concentrations (from 1 μg mL−1 to 100 μg mL−1) were eluted at a flow rate of 1 mL min−1, and the retention times were detected by a UV-vis detector at a wavelength of 290 nm. The resulting calibration curve of eupafolin was linear (y=102,221x-42,045, r2=0.9996) and was used to determine the content of eupafolin in ENDS. The drug loading efficiency of ENDS was calculated by the method of Yen et al36 as follows:
|
|

Determination of crystalline to amorphous transformation
Powder X-ray diffractometry (XRD; Siemens D5000, Bruker, Germany) with Ni-filtered Cu-Kα radiation was used to determine the crystalline pattern of raw eupafolin and ENDS. XRD was performed at 40 kV and 25 mA, the scanned angle (2θ) setting was from 2° to 50°, and the scan rate was conducted at 1° min−1.
Determination of hydrogen bond formation
Proton nuclear magnetic resonance (1H NMR) analysis was used to determine the presence of hydrogen bond formation between eupafolin and excipient. A Varian Mercury Plus AS400 NMR System (Oxford Instrument Co., Abingdon, Oxfordshire, UK) was used to record the 1H NMR spectra of raw eupafolin and ENDS. In order to obtain the 1H NMR spectra, each sample (2 mg) was first dissolved in 0.8 mL of dimethyl sulfoxide (DMSO)-d6 (Sigma-Aldrich Co.) and then placed into the 1H NMR instrument.
Fourier transform infrared (FTIR) spectroscopy was also performed to evaluate the intermolecular interactions of the nanoparticle system, using a Perkin-Elmer 2000 spectrophotometer (PerkinElmer Inc., Waltham, MA, USA). Samples containing raw eupafolin or ENDS were mixed with potassium bromide (Sigma-Aldrich Co.), ground by an agate mortar, compressed into thin tablets, and then evaluated by infrared spectrophotometer. The scanning range for FTIR was 400–4,000 cm−1.
Solubility of ENDS and raw eupafolin
One milligram of eupafolin was added into a 1.5 mL tube containing 1 mL of pure water. ENDS corresponding to 1 mg of eupafolin was also added into 1 mL of pure water. The test samples were stirred with a shaker at 1,000 rpm for 10 minutes, followed by filtration through a 0.45 μm syringe filter. The content of eupafolin in the aqueous solution was determined by the HPLC method as described in the section “Determination of drug loading efficiency of ENDS”.
In vitro skin penetration
Fresh pig skin from the flank region was purchased from a local butcher and mounted on static Franz diffusion cells, in accordance with the COLIPA guidelines standard protocol.37 The dermal side of the skin disk was directed toward the receptor chamber and brought into contact with the receptor fluid (containing 0.14 M NaCl, 2 mM K2HPO4, 0.4 mM KH2PO4, 10% penicillin, pH 7.4). The corneum layer was exposed to air. The receptor fluid was stirred and maintained at a skin surface temperature of 32°C with in a thermostated water bath throughout the experiment. Initially, 200 μL of ENDS or raw eupafolin (n=6 for each formulation) was topically applied onto the skin surface. At the end of the experiment, the residual test samples were removed from the skin by a dropping pipette. The corneum layer was obtained by tape stripping for 15 times. The combined epidermis and dermis layers were cut into small pieces and extracted by immersing in methanol for 1 hour. Subsequently, the unknown drug concentrations in the corneum samples and the epidermis/dermis were, respectively, analyzed by HPLC using the corresponding standard curves as references.
Cell culture
The HaCaT keratinocyte cell line was kindly provided by Professor Jeff Yi-Fu Chen (Department of Biotechnology, Kaohsiung Medical University). Cells were cultured in DMEM/F-12 medium supplemented with 10% FBS and 1% penicillin–streptomycin and were incubated at 37°C in 5% CO2. HaCaT cells from passages 5 to 12 were used for experiments.
Primary native human epidermal keratinocytes were obtained from ScienCell Research Laboratories (San Diego, CA, USA), and were cultured in Keratinocyte Medium. Human dermal fibroblasts (Food Industry Research and Development Institute, Taiwan, Republic of China), human pulmonary alveolar epithelial cells (ScienCell Research Laboratories), and human fibroblast-like synoviocytes (Cell Applications, San Diego, CA, USA) were cultured in DMEM/F-12 medium with 10% FBS and 1% penicillin–streptomycin.
Preparation of PM suspension
Particulate matter particles, Standard Reference Material® 1649b, were added into phosphate-buffered saline (PBS), and then the PM suspension was sonicated for 30 minutes to avoid aggregation of particles. All experiments were conducted within 1 hour of PM suspension to avoid variability in PM components.
MTT cell viability assay
For 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay, HaCaT cells were seeded in 96-well plates. The cells were treated with ENDS for 24 hours, after which MTT (Sigma-Aldrich Co.) was added to the wells. After 3 hours of incubation, the plates were analyzed with a microplate reader using a wavelength of 550 nm.
Real-time cytotoxicity assay
The cytotoxic effect of ENDS on HaCaT cells was evaluated by the xCELLigence Real-Time Cell Analyzer (RTCA) (ACEA Biosciences, San Diego, CA, USA). This instrument monitors cell viability in real-time by determining the change in electrical impedance as the viable cells interact with the electronic microtiter plates. Cells were seeded at a density of 5×103 cells/well on electronic microtiter plates. After 24 hours of culture to allow cell adhesion and spreading, the culture medium was removed, and new medium containing 0 μM, 10 μM, 40 μM, and 100 μM ENDS was added. Each concentration of ENDS was performed in triplicate. Cell viability was measured from 0 hour to 72 hours. The cell index is a measure of cell viability and was normalized to the time point when ENDS was added. Analysis of data was carried out with the RTCA software.
Determination of cell viability using flow cytometry
Flow cytometric assessment of cell viability was performed using the FITC-annexin V/propidium iodide assay (Thermo Fisher Scientific). HaCaT cells were cultured in six-well plates and treated with ENDS for 24 hours, 48 hours, or 72 hours. Cells were detached by incubation with trypsin-EDTA and centrifuged at 1,500 rpm for 5 minutes. The cell pellet was collected and resuspended in 100 μL Annexin-Binding Buffer. FITC-annexin V and propidium iodide were then added to the cell suspension. After incubation at room temperature for 15 minutes, the stained cells were analyzed by flow cytometry (FACScan flow cytometer; BD, Franklin Lakes, NJ, USA), measuring the fluorescence emission at wavelengths of 530 nm and >575 nm.
Measurement of intracellular ROS concentration
Measurement of intracellular ROS concentration was performed using the fluorescent dichlorofluorescin diacetate assay (purchased from Sigma-Aldrich Co.). HaCaT cells were seeded on six-well plates and then incubated with the test compounds. Subsequently, cells were incubated with 10 μM dichlorofluorescin diacetate at 37°C for 30 minutes and then washed with PBS. Finally, the cell fluorescence level was measured using a Fluoroskan Ascent FL fluorescent plate reader (Thermo Fisher Scientific) with a 504 nm excitation filter and 524 nm emission filter.
Isolation of cell fractions
First, HaCaT cells were harvested and subjected to sonication for 10 seconds with a Qsonica Q700 sonicator (Delta Labo, Avignon, France). After centrifugation at 8,000 rpm at 4°C for 15 minutes, the pellet was collected and represents the nuclear fraction. Subsequently, the supernatant was centrifuged at 14,000 rpm for 1 hour at 4°C. The pellet was obtained and represents the cell membrane fraction.
Assessment of nicotinamide adenine dinucleotide phosphate oxidase activity
The cell membrane fraction was obtained from HaCaT cells using the method described in the section “Isolation of cell fractions”. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity was determined by the lucigenin chemiluminescence assay using 20 μM lucigenin (Sigma-Aldrich Co.) and 1 μM NADPH (Sigma-Aldrich Co.).
NADP+/NADPH ratio
Oxidative stress in HaCaT cells was also evaluated by determination of the NADP+/NADPH ratio using a colorimetric NADP/NADPH Assay Kit (Abcam, Cambridge, MA, USA) in accordance with the manufacturer’s instructions.
Flow cytometric assessment of cell surface NOX2 expression
For analysis of cell surface NOX2 expression by flow cytometry, HaCaT keratinocytes were cultured in six-well plates. Following treatment, cells were harvested and centrifuged at 1,000 rpm for 5 minutes. The cell pellet was collected and resuspended in PBS. Cells were stained at 4°C for 30 minutes with anti-NOX2/gp91phox antibody (Abcam), followed by incubation with Alexa Fluor 488-conjugated donkey anti-rabbit IgG secondary antibody (BioLegend, San Diego, CA, USA). After washing twice with PBS, stained cells were analyzed using a FACScan flow cytometer (BD).
Reverse transcription-polymerase chain reaction
Trizol reagent was used to isolate total RNA from HaCaT cells based on the manufacturer’s protocols. The Moloney murine leukemia virus reverse transcriptase (purchased from Thermo Fisher Scientific) was used to reverse transcribe cDNA from total RNA. Amplification of cDNA was performed by polymerase chain reaction (PCR). The sequences of the PCR primers were as follows: 5′-TGACGGGGTCACCCACACTGTGCCCATCTA-3′ (sense) and 5′-CTAGAAGCATTTGCGGTGGACGATG-3′ (anti-sense) for β-actin; 5′-CTCACACCACAGAAAGTTAAAAGAT-3′ (sense) and 5′-GCTACCACAGGCACATCACG-3′ (anti-sense) for COX-2. Subsequently, agar gel electrophoresis was performed, the PCR products were stained with ethidium bromide dye (Sigma-Aldrich Co.), and images were taken using a ChemiDoc XRS+ ultraviolet imaging system (Bio-Rad Laboratories Inc., Hercules, CA, USA).
COX-2 luciferase activity assay
A COX-2-luc plasmid was synthesized to measure COX-2 luciferase activity. A region extended from −459 bp to +9 bp of the COX-2 promoter gene was cloned into the pGL3-basic vector (purchased from Promega Corporation, Fitchburg, WI, USA). Subsequently, HaCaT cells were transfected with the COX-2-luc plasmid, and the COX-2 luciferase activity was determined using the luciferase assay (Promega Corporation). Following various treatments, 5 μL of the supernatant was added to 50 μL luciferase assay solution, and luminescence was measured using a Fluoroskan Ascent FL luminometer (Thermo Fisher Scientific). All experiments were performed at least three times.
The fragment sequence of the COX-2 promoter region was as follows: GACGTACAGACCAGACACGGCGGCGGCGGCGGGAGAGGGGATTCCCTGCGCCCCCGGACCTCAGGGCCGCTCAG
ATTCCTGGAGAGGAAGCCAAGTGTCCTTCTGCCCTCCCCCGGTATCCCATCCAAGGCGATCAGTCCAGAACTGG
CTCTCGGAAGCGCTCGGGCAAAGACTGCGAAGAAGAAAAGACATCTGGCGGAAACCTGTGCGCCTGGGGCGGTG
GAACTCGGGGAGGAGAGGGAGGGATCAGACAGGAGAGTGGGGACTACCCCCTCTGCTCCCAAATTGGGGCAGCT
TCCTGGGTTTCCGATTTTCTCATTTCCGTGGGTAAAAAACCCTGCCCCCACCGGGCTTACGCAATTTTTTTAAG
GGGAGAGGAGGGAAAAATTTGTGGGGGGTACGAAAAGGCGGAAAGAAACAGTCATTTCGTCACATGGGCTTGGT
TTTCAGTCTTATAAAAAGGAAGG.
Measurement of PGE2 production
HaCaT cells were seeded in 12-well plates. After treatment of cells with different test compounds, the culture supernatants were collected and stored at −80°C until further experiments. The concentration of PGE2 was determined using the PGE2 enzyme immunoassay kit (purchased from Cayman Chemical Company, Ann Arbor, MI, USA).
Western blot analysis
After treatment with different test compounds, HaCaT cells were harvested by scraping and incubated with lysis buffer. The bicinchoninic acid protein assay kit was used to measure the protein concentrations. Protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a 10% running gel. The proteins were then transferred to nitrocellulose membranes. The membranes were incubated with COX-2, phospho-ERK, ERK, phospho-p38, p38, phospho-JNK, JNK, phospho-c-Fos, and phospho-P65 primary antibodies overnight at 4°C. Subsequently, horseradish peroxidase-conjugated secondary antibodies were added for 1 hour at room temperature. Immunoreactive bands were detected by an enhanced chemiluminescence system and then developed by Hyperfilm.
Transient transfection with siRNAs
Human siRNAs for p47phox (SC-29422), NOX2 (gp91phox, SC35503), and scrambled siRNA (SC-37007) were obtained from Santa Cruz Biotechnology Inc. Transient transfection of HaCaT cells with siRNAs (100 nM) was performed using transfection reagent, according to the manufacturer’s instructions. The transfection efficiency in our experiments was ~60%, as evaluated by transfection with a negative control siRNA-EGFP (no homology in human, mouse, and rat mRNA database; Sigma-Aldrich Co.). The sequence of the negative control siRNA was GAUCAUACGUGCGAUCAGAdTdT (sense) and UCUGAUCGCACGUAUGAUCdTdT (anti-sense).
Statistical analysis of data
Data were processed using a GraphPad Prism Program (GraphPad Software, Inc., La Jolla, CA, USA) and expressed as the mean ± standard error of the mean. Statistical analysis was performed by one-way analysis of variance for multiple comparisons followed by Tukey’s post hoc test. All statistical tests were two sided. Statistical significance was defined at P<0.05.
Results
ENDS reduced the particle size of eupafolin and showed good drug loading and encapsulation efficiencies
First, we synthesized an ENDS through nanoprecipitation methods using the nanoparticle carriers Eudragit E100 and PVA. As shown in Table 1, the particle size of raw eupafolin was 4,782.9±1,021.1 nm and its PI was >1. ENDS, eupafolin after nanoparticle preparation, displayed a much lower particle size (70.7±5.2 nm) with a uniform particle size distribution (0.501±0.090). In addition, transmission electron microscopy analysis confirmed the nanoparticle morphology of ENDS (Figure 1) with small particle sizes (<100 nm), spherical shapes, and a uniform size distribution. Moreover, many publications have used drug loading and encapsulation efficiencies to determine the amount of active ingredients loaded in a nanoparticle formulation. As shown in Table 1, the drug loading and encapsulation efficiencies of ENDS were 36.5% and >99%, respectively. These results demonstrated that Eudragit E100 and PVA successfully reduced the particle size of eupafolin by nanoprecipitation method and also served as good excipients for encapsulating eupafolin into a nanoparticle formulation.
 | Table 1 Particle size distribution, drug loading, and encapsulation efficiency of eupafolin (Eup) and eupafolin nanoparticle delivery system (ENDS) |
 | Figure 1 Particle size distribution (A) and transmission electron microscopy photograph (B) of eupafolin nanoparticle delivery system (ENDS). |
Intermolecular interactions between eupafolin and excipients
The intermolecular interactions of the nanoparticle system were established by 1H NMR. The comparative 1H NMR spectra of raw eupafolin and ENDS are shown in Figure 2A. The spectrum of raw eupafolin showed protons on aromatic groups ranging from 6 ppm to 8 ppm and a strong intramolecular hydrogen bond at 13.01 ppm. In the case of ENDS, the result showed that the aromatic protons (H2′ and H8) of raw eupafolin were obviously shifted upfield and a hydroxyl group signal disappeared, indicating that an intermolecular hydrogen bond interaction had formed between raw eupafolin and Eudragit E100/PVA. Horisawa et al38 also observed similar results and concluded that the tertiary amine of Eudragit E100 formed an intermolecular hydrogen bond with an anti-inflammatory drug, which improved its water solubility.
 | Figure 2 Physicochemical properties of ENDS compared to raw eupafolin. |
In addition, the intermolecular interactions of ENDS were evaluated by FTIR spectroscopy. As shown in Figure 2B, the FTIR spectra of raw eupafolin showed an obvious characteristic absorption band of phenolic-OH group at 3,411 cm−1, a carbonyl group (C=O) stretching vibration at 1,661 cm−1, a C-O-H stretching band at 1,280 cm−1, and an aromatic C=C stretching band in the range of 1,612–1,386 cm−1. On the other hand, the FTIR spectra of ENDS exhibited a C=O ester stretch peak at 1,730 cm−1, a CH stretch peak at 2,948 cm−1 and a new broad peak of the hydroxyl group of Eudragit E100-PVA at 3,372 cm−1. These findings indicated that the phenolic-OH stretch of eupafolin formed a hydrogen bond with the aminoalkyl group of Eudragit E100 and became stably dispersed in PVA aqueous solution.
ENDS changed the structure of eupafolin from crystalline to amorphous form
It is well known that excipients can effectively change the crystalline structure of organic compounds into an amorphous form and thereby increase their aqueous solubility and bioavailability.39 The present study used XRD to determine the crystalline to amorphous transformation of eupafolin following the nanoengineering process. As shown in Figure 2C, diffraction peaks of eupafolin in the XRD pattern were recorded at 4.7°, 12.8°, 16.9°, 21.2°, 23.9°, 25.2°, and 28.4°, which indicated that eupafolin exhibits a crystalline structure. After nanoprecipitation preparation of eupafolin, the characteristic peaks of eupafolin in the XRD pattern were greatly diminished. These results indicated that Eudragit E100 and PVA had effectively encapsulated eupafolin, and the clustered crystalline structure had indeed been changed to an amorphous state in ENDS. A schematic diagram for the chemical structure of ENDS is presented in Figure 3.
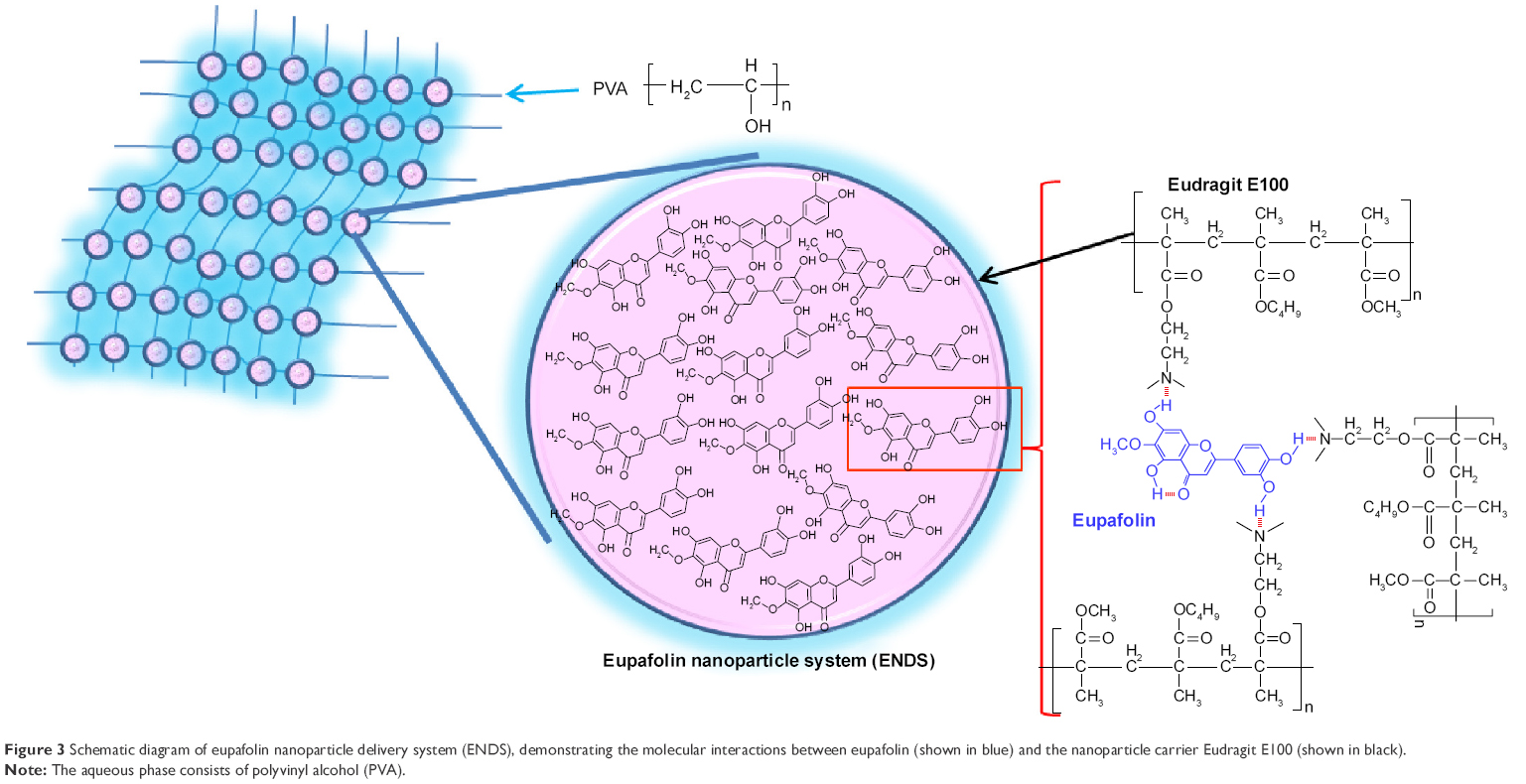 | Figure 3 Schematic diagram of eupafolin nanoparticle delivery system (ENDS), demonstrating the molecular interactions between eupafolin (shown in blue) and the nanoparticle carrier Eudragit E100 (shown in black). |
ENDS exhibited greater water solubility and in vitro percutaneous penetration compared to raw eupafolin
The solubility of eupafolin and ENDS in water is shown in Figure 4A. The aqueous solubility of raw eupafolin was 0.421±0.002 μg mL−1. In comparison, the aqueous solubility of ENDS was 14.757±0.222 μg mL−1, which was effectively increased to 35-fold compared to that of raw eupafolin. In addition, the comparison of percutaneous penetration of raw eupafolin and ENDS is important in the study of novel pharmaceutical delivery systems in order to verify their pharmacological effects. The in vitro percutaneous penetration of raw eupafolin and ENDS through porcine skin after topical treatment is presented in Figure 4B and C. Following topical administration of raw eupafolin in water, the amounts of drug retained in the corneum layer of porcine skin after 1 hour and 2 hours were 0.371 μg cm−2 and 0.293 μg cm−2, respectively. In comparison, topical treatment with ENDS for 1 hour and 2 hours resulted in corneum layer concentrations of 1.862 μg cm−2 and 1.351 μg cm−2, respectively. Furthermore, topical administration of raw eupafolin for 1 hour and 2 hours resulted in deposition of 1.621 μg cm−2 and 1.823 μg cm−2 of eupafolin in the epidermis and dermis, respectively. In comparison, topical treatment with ENDS led to increased skin penetration of eupafolin into the epidermis and dermis after 1 hour and 2 hours to 3.632 μg cm−2 and 5.032 μg cm−2, respectively. These results indicated that ENDS demonstrated better hydration effect within the stratum corneum and greater percutaneous penetration into the epidermis and dermis when compared to raw eupafolin. Therefore, ENDS can increase the skin penetration of eupafolin due to improvement in eupafolin’s aqueous solubility.
 | Figure 4 Water solubility and skin penetration of ENDS compared to raw eupafolin. |
Cell safety of ENDS
We next proceeded to evaluate the cell safety of ENDS in HaCaT keratinocytes using cell viability assays, and noncytotoxic concentrations of ENDS were applied in subsequent assays examining PM-induced inflammation in HaCaT keratinocytes. We first determined the effects on ENDS on cell viability using the MTT assay. As shown in Figure 5A, ENDS at concentrations ranging from 1 μM to 100 μM exhibited no cytotoxic effects on HaCaT cells after 24 hours treatment.
 | Figure 5 Effects of eupafolin nanoparticle delivery system (ENDS) on HaCaT keratinocyte cell viability. |
We also performed real-time cytotoxicity assay using the xCELLigence RTCA. As shown in Figure 5B, treatment of HaCaT keratinocytes with various concentrations of ENDS (10 μM, 40 μM, and 100 μM) for 0 hour to 72 hours had no significant effect on cell viability.
In addition, cell viability was assessed by flow cytometry following propidium iodide and annexin V staining. As shown in Figure 5C, treatment of HaCaT keratinocytes with various concentrations of ENDS (40 μM and 100 μM) for 24 hours, 48 hours, and 72 hours had no significant effect on cell viability.
ENDS suppressed PM-induced ROS production and NADPH oxidase activity
The overproduction of ROS by various cells and organs is known as oxidative stress. NADPH oxidase (such as NOX2/gp91phox) is a membrane-bound enzyme complex, which generates superoxide anions and other downstream ROS under various pathological conditions, including cardiovascular disease, and lung and skin inflammation.40 As show in Figure 6A, PM treatment significantly increased ROS production in HaCaT keratinocytes when compared to the control group (P<0.05). Pretreatment of cells with ENDS significantly reduced PM-induced ROS overproduction, and similar effects were seen with eupafolin in DMSO pretreatment. However, pretreatment with eupafolin in PBS did not significantly attenuate PM-induced ROS production. In addition, pretreatment with blank PVA-Eudragit E100 nanoparticle carriers (without eupafolin) had no significant effect on PM-induced ROS production. According to these data, ENDS displayed better antioxidant effect than raw eupafolin in PBS in PM-stimulated HaCaT cells.
 | Figure 6 Effects of ENDS on ROS production and NADPH oxidase activity in HaCaT keratinocytes. |
Moreover, Figure 6B shows that addition of PM induced an increase in NADPH oxidase activity (P<0.05) in HaCaT cells. Pretreatment of cells with ENDS and eupafolin in DMSO significantly decreased NADPH oxidase activity, but pretreatment with eupafolin in PBS had no significant effect. In addition, pretreatment with blank PVA-Eudragit E100 nanoparticle carriers (without eupafolin) had no significant effect on PM-induced NADPH oxidase activity.
Since NOX2 (NADPH oxidase-2) catalyzes the oxidation of NADPH to NADP+, the NADP+/NADPH ratio in cells is an indication of NOX2 activity and oxidative stress. The effects of PM and ENDS on NADP+/NADPH ratio in HaCaT cells were determined by colorimetric NADP/NADPH assay. We found that treatment of HaCaT cells with PM for 30 minutes increased NADP+/NADPH ratio, and this effect was significantly suppressed by pretreatment with ENDS (EUPA-N) but not raw eupafolin dissolved in PBS (EUPA-P; Figure 6C). Therefore, ENDS suppressed PM-induced increase in NADP+/NADPH ratio in HaCaT cells.
ENDS attenuated COX-2 gene and protein expression in PM-stimulated HaCaT keratinocytes
To compare the effects of ENDS and raw eupafolin on PM-induced COX-2 expression in keratinocytes, the COX-2 protein and gene expression patterns were determined by Western blotting and real-time reverse transcription PCR. As shown in Figure 7A, treatment of HaCaT cells with 50 μg cm−2 PM led to an increase in COX-2 protein expression. PM-induced COX-2 protein expression was significantly reduced by pretreatment with 0.1 μM, 1 μM, and 10 μM ENDS in a dose-dependent manner. On the other hand, pretreatment with raw eupafolin in PBS had no significant effects. Moreover, treatment of primary native human epidermal keratinocytes with PM alone induced increased levels of COX-2 protein expression, and these effects were suppressed by pretreatment with ENDS (Figures 7B). As seen in Figure 7C, PM-induced COX-2 mRNA expression was also suppressed by pretreatment with ENDS but not raw eupafolin in PBS. In addition, the present study also used the luciferase reporter assay to confirm the inhibitory effect of ENDS on COX-2 gene transcription. The COX-2-luciferase reporter construct was transfected into HaCaT keratinocytes. As shown in Figure 7D, addition of 50 μg cm−2 PM markedly stimulated COX-2 luciferase activity in HaCaT cells. Pretreatment of cells with ENDS and eupafolin in DMSO significantly inhibited PM-induced COX-2-luciferase activity (P<0.05); however, these effects were not observed with raw eupafolin in PBS. These results implied that ENDS exerts anti-inflammatory effects by reducing COX-2 gene and protein expression in PM-stimulated HaCaT keratinocytes.
 | Figure 7 Effects of ENDS on COX-2 expression and PGE2 generation in keratinocytes. |
Many publications have demonstrated that NOX2 and its regulatory subunit p47phox acts as an ROS producer to activate the COX-2-derived PGE2 inflammation signaling pathway.41–43 In order to confirm the involvement of NOX2 in PM-induced inflammatory responses, the present study used siRNA to inhibit NOX2 (gp91phox) and p47phox expression in HaCaT keratinocytes. As demonstrated by Western blotting in Figure 7E, transfection of cells with siRNA-p47phox and siRNA-gp91phox resulted in adequate inhibition of p47phox and gp91phox protein expression, respectively, while transfection of cells with scrambled siRNA had no significant effect. In particular, knockdown of NOX2 expression by siRNA-p47phox and siRNA-gp91phox suppressed PM-induced COX-2 protein expression in HaCaT cells, while transfection of cells with control scrambled siRNA had no significant effect. These findings indicate that NOX2 (including the p47phox and gp91phox subunits) plays a crucial role in PM-induced COX-2 protein expression in HaCaT keratinocytes.
In addition, transfection of HaCaT keratinocytes with siRNA was performed to inhibit NOX2/gp91phox expression in HaCaT keratinocytes. Cell surface NOX2 expression was measured by flow cytometry. As shown in Figure 7F, treatment of cells with PM alone induced an increase in NOX2 expression. Transfection of cells with siRNA-gp91phox suppressed PM-induced NOX2 expression, while transfection with scrambled siRNA had no significant effect. These findings indicate that PM-induced NOX2 expression is dependent on gp91phox.
During the activation process of NOX2, the cytosolic component p47phox becomes phosphorylated and translocates to the plasma membrane. The effects of PM and ENDS on phospho-p47 levels in HaCaT cell membrane fractions were determined using Western blotting. We found that treatment of HaCaT cells with PM for 30 minutes increased phospho-p47 levels in cell membrane fractions, and this effect was suppressed by pretreatment with ENDS (EUPA-N) but not with raw eupafolin dissolved in PBS (EUPA-P) or blank PVA-Eudragit E100 nanoparticle carrier (Figure 7G). Therefore, ENDS suppressed PM-induced increase in phospho-p47 levels in HaCaT cell membranes.
ENDS suppressed PGE2 production in PM-stimulated HaCaT keratinocytes
Figure 7H also shows that ENDS markedly decreased PM-induced PGE2 production in HaCaT keratinocytes in a similar manner to raw eupafolin in DMSO. On the other hand, raw eupafolin in PBS had no significant effect. In addition, silencing of NOX2 (gp91phox) and p47phox expression by siRNA suppressed PGE2 production in PM-stimulated HaCaT cells. Taken together, ENDS suppressed COX-2-derived PGE2 inflammation signaling by inhibiting NOX2/p47phox in PM-stimulated HaCaT cells, and its anti-inflammatory activities were superior to raw eupafolin in PBS.
ENDS suppressed MAPK and NF-κB signaling in PM-stimulated HaCaT keratinocytes
MAPKs, such as ERK, JNK, and p38, are phosphorylated in response to oxidative stress and inflammatory pathophysiological processes. The phosphorylation of MAPKs contributes to the activation of nuclear transcription factors including c-Fos and NF-κB in various cells exposed to different environmental stimuli.44,45 The present study investigated the effects of ENDS on PM-induced MAPKs phosphorylation, c-Fos phosphorylation, and NF-κB activation in HaCaT keratinocytes. Figure 8A–C shows that PM treatment significantly induced phosphorylation of MAPKs including ERK, p38, and JNK when compared to the control group (P<0.05). Pretreatment with ENDS significantly attenuated PM-induced ERK, p38, and JNK phosphorylation. However, pretreatment with eupafolin in PBS had no significant effects. In addition, our results also indicated that MAPK inhibitors, ROS inhibitor NAC, and NADPH oxidase inhibitor APO effectively suppressed the phosphorylation of ERK, p38, and JNK. Furthermore, PM treatment also markedly induced c-Fos phosphorylation and NF-κB activation (P65 phosphorylation) in HaCaT keratinocytes when compared to the control group (Figure 8D; P<0.05). Pretreatment with ENDS and eupafolin in DMSO markedly suppressed PM-induced c-Fos phosphorylation and NF-κB activation, whereas no similar effects were seen with eupafolin in PBS. These results indicated that ENDS downregulates MAPK phosphorylation and NOX activity and then inhibits c-Fos phosphorylation and NF-κB activation in PM-stimulated HaCaT keratinocytes.
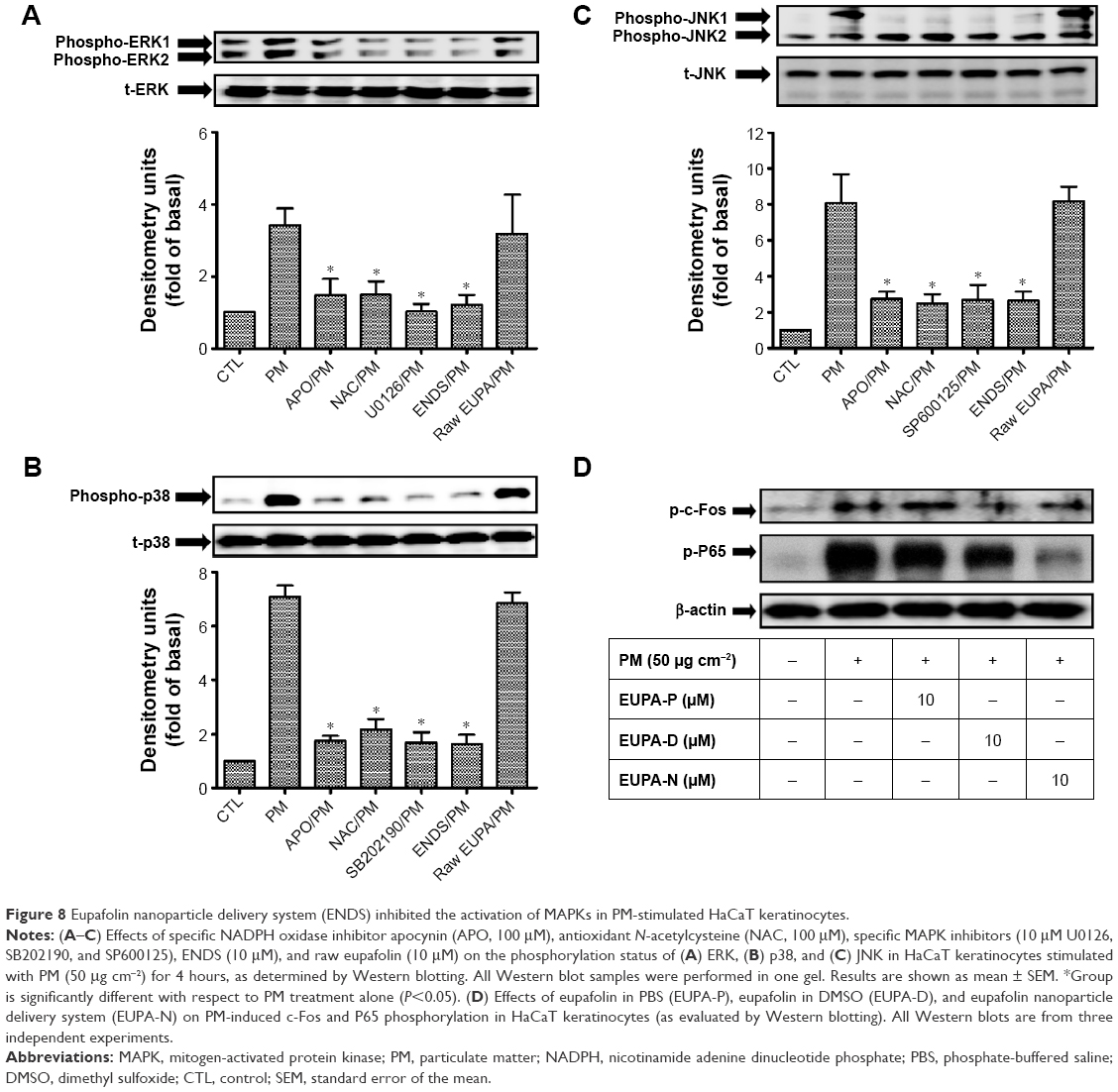 | Figure 8 Eupafolin nanoparticle delivery system (ENDS) inhibited the activation of MAPKs in PM-stimulated HaCaT keratinocytes. |
Discussion
The present study is the first to successfully synthesize an ENDS by a simple nanoprecipitation method, which effectively enhanced the aqueous solubility and percutaneous penetration of eupafolin by improving its physicochemical properties, including reduction of particle size, crystalline to amorphous transformation, and hydrogen bond formation with excipients. Additionally, our results also demonstrated for the first time that ENDS displayed better antioxidant and anti-inflammatory activities than raw eupafolin by downregulating MAPK signaling pathways to prevent PM-induced damage in HaCaT keratinocytes.
The skin is the largest organ of the human body and protects internal organs against toxic substances, pathogens, and organisms. Therefore, the skin acts as the largest interface between the human body and the external environment, as well as a permeability barrier. The permeability barrier properties of human skin, attributed mainly to the stratum corneum of the epidermis, play an important role in controlling the passage of substances in and out of the human body, including water, electrolytes, active biological substances, and toxic materials. The stratum corneum also impedes the skin penetration of topical drugs.46–48 A good topical delivery system can enhance the skin penetration of drugs and active ingredients. Paudel et al49 revealed that the output value of topical and transdermal drug delivery systems will reach 32 billion in the US market in 2015, including polymeric nanoparticles, solid lipid nanoparticles, liposomes, and nanoemulsions. In the past decade, nanoparticle formulations have been developed as novel topical drug delivery systems that can effectively improve the poor solubility of therapeutic and other active agents, enabling them to penetrate the stratum corneum via intercellular, intracellular, and transappendageal pathways for the treatment of skin diseases.50 Eupafolin, a flavonoid compound, is purified from P. nodiflora, and its flavone backbone structure displays poor water solubility and limits its skin absorption after topical administration. Our results demonstrated that ENDS can be successfully prepared by the nanoprecipitation method and can overcome the poor water solubility and skin penetration of raw eupafolin.
There are three viewpoints to explain the improvement in water solubility and skin penetration of eupafolin nanoparticles compared to raw eupafolin: 1) Many studies have demonstrated that reduction of particle size of raw materials can increase their surface area and enhance their skin absorption, such as quercetin and curcumin.25,29,30 Our results indicated that raw eupafolin may blend together with Eudragit E100 in the alcohol as hydrophobic phase, and then the hydrophilic end of PVA as an emulsion stabilizer can interpenetrate them during the nanoprecipitation process to finally form nanoparticles with reproducible size and uniform distribution; 2) The conversion of crystalline structure to amorphous phase may explain the increase in solubility and absorption of a drug. Our results demonstrated that raw eupafolin shows obvious characteristic Bragg’s peaks in its X-ray diffraction patterns, whereas no Bragg’s peaks were found in ENDS. The crystalline structure of eupafolin was therefore changed to an amorphous state; 3) The presence of hydrogen bond interactions between pure compound and polymer is another index for evaluating the enhancement of aqueous solubility and bioavailability.51 The results from 1H NMR and FTIR analyses revealed that the aromatic ring of eupafolin formed an intermolecular hydrogen bond with Eudragit E100 and PVA. As a consequence of the above factors, ENDS showed a 67.6-fold particle size reduction, which increased its water solubility. Thus, the skin penetration of ENDS into the stratum corneum and epidermis/dermis was increased by 4.6-fold and 2.8-fold compared to raw eupafolin, respectively. Müller et al52 proposed that nanoparticle formulations can reduce the particle size and increase the particle number of the active compound, which can then form a dense film on the skin surface. This phenomenon is called the occlusion effect and can increase the skin hydration effect of the nanoparticle system to enhance the skin penetration of the active compound. Our results supported the notion that ENDS exhibits an occlusion effect due to nanoparticle formation, which then increases its skin hydration effect and enables eupafolin to penetrate into the epidermis and dermis efficiently.
Eupafolin is a good antioxidant and anti-inflammatory compound that can protect the body against oxidative stress;21,22 however, pharmaceutical preparations of eupafolin may influence its biological activities. For these reasons, we compared the antioxidant and anti-inflammatory activities of raw eupafolin and ENDS in PM-stimulated human keratinocytes. PM is a major form of air pollution and poses a significant risk to human health.53 Andreau et al54 indicated that PM consists of a complex mixture of solid particles and liquid droplets commonly referred to as fine particles (PM2.5) and coarse particles (PM10). The contents of PM include hydrocarbons, organic compounds, pesticides, metals, and minerals. The skin functions as the largest interface between the human body and the surrounding environment and acts as the primary site of contact for PM. Choi et al55 revealed that prolonged exposure to airborne dust particles originating from seasonal Asian dust storms activates the aryl hydrocarbon receptors in human epidermal keratinocytes and then induces cellular oxidative stress and overproduction of proinflammatory cytokines, which may be one of the reasons for increased incidence of dermatitis. Krutmann et al56 reviewed many studies and suggested that long-term contact with PM may increase the amounts of organic compounds bound to PM in the skin and induce oxidative stress and inflammatory reaction, resulting in disruption of skin integrity.
It is well known that COX2-derived PGE2 overexpression plays an important role in skin inflammation pathways after exposure to different stimuli, including UVB and lipopolysaccharide.57,58 Our results revealed that ENDS and eupafolin in DMSO can effectively suppress PM-induced COX-2 mRNA and protein expression and PGE2 production in HaCaT keratinocytes, but similar effects were not found for eupafolin in PBS. In addition, NADPH oxidase (NOX)-mediated ROS overproduction is involved in mediating inflammatory reactions in different cells. NOX2 is a member of the NOX family, which is composed of cytosolic regulatory subunits (including p47phox, p40phox, p67phox), membrane catalytic subunits (including gp91phox and p22phox), and the small GTPase Rac. Sareila et al59 indicated that following phosphorylation, the cytosolic component p47phox is translocated together with the p67phox subunit to the plasma membrane, which then activates the membrane catalytic NOX2 enzyme complex (gp91phox). It has been shown that p47phox and NOX2 enzyme complex may play an important role in acute and chronic skin inflammation.59 Our present results indicated that siRNA-mediated inhibition of NOX2 (gp91phox) and p47phox expression effectively inhibited PM-induced PGE2 production. Similar results were seen following treatment with ENDS and eupafolin in DMSO but not for eupafolin in PBS. Therefore, ENDS treatment shows greater anti-inflammatory activity than eupafolin in PBS treatment and suppresses PM-induced PGE2 overexpression through the inhibition of NOX2/p47phox/ROS pathway in HaCaT keratinocytes.
MAPK signaling pathways, including ERK, p38, and JNK, can regulate cellular responses to proliferation, apoptosis, differentiation, and inflammation in humans. MAPKs also play an important role in phosphorylating transcription factors, such as NF-κB (P65) and activator protein-1, and subsequently modulate the ROS-mediated COX-2 inflammation pathway.44,45 Previously, Jeong et al60 reported that cigarette smoke induced MAPK activation and tumor necrosis factor-α expression to initiate the development of inflammation in HaCaT keratinocytes. Cheng et al61 also indicated that cigarette smoke particle-induced ROS generation is mediated through a c-Src/NADPH oxidase/MAPK pathway in human tracheal smooth muscle cells. Therefore, we speculated that MAPK signaling pathways may be involved in the PM-induced ROS-mediated COX-2 inflammation pathway in HaCaT keratinocytes. Our present results demonstrated that PM markedly induced the phosphorylation of ERK, p38, and JNK. ENDS and eupafolin in DMSO effectively inhibited the phosphorylation of MAPKs after PM exposure, but eupafolin in PBS showed no obvious effect. We also found that pretreatment of cells with U0126, SB202190, and SP600125 suppressed PM-induced phosphorylation of ERK, p38, and JNK, respectively. In addition, pretreatment with ROS inhibitor NAC and NADPH oxidase inhibitor APO inhibited the phosphorylation of all three MAPKs after PM exposure. Furthermore, PM acts as an inflammation inducer to increase the phosphorylation of P65 and c-Fos in HaCaT keratinocytes. ENDS and eupafolin in DMSO reduced the phosphorylation of P65 and c-Fos after PM exposure, but in contrast eupafolin in PBS showed no obvious effect. According to these results, ENDS shows greater inhibitory effects than eupafolin in PBS on MAPK phosphorylation in PM-induced HaCaT keratinocytes, and this is mediated by inhibition of the MAPKs/P65/c-Fos-dependent pathway.
Conclusion
Our results showed that ENDS can effectively overcome the physicochemical drawbacks of raw eupafolin with respect to water solubility and skin penetration, through reduction of particle size and formation of an amorphous state with hydrogen bonding. The present study also found that ENDS can suppress PM-induced ROS production, COX-2 expression, and PGE2 content through downregulation of MAPKs-mediated NF-κB and c-Fos signaling pathways in HaCaT keratinocytes, which is consistent with the antioxidant and anti-inflammatory activities of eupafolin. In contrast, raw eupafolin in PBS did not display significant antioxidant and anti-inflammatory activity, which is likely due to its poor water solubility. A schematic diagram summarizing the findings of this study is presented in Figure 9. In conclusion, we suggest that ENDS may potentially be used as a medicinal drug and/or cosmeceutical product to prevent PM-induced skin oxidative stress and inflammation in the future.
 | Figure 9 Schematic diagram showing the molecular mechanisms through which eupafolin nanoparticle delivery system (ENDS) inhibited PM-induced oxidative stress and inflammation in the skin. |
Acknowledgments
This work was supported by grants from the National Science Council, Taipei, Taiwan, Republic of China (NSC 99-2313-B-255-001-MY3), Chang Gung Medical Research Program Foundation (CMRPF6A0082 and CMRPF6A0083), China Medical University Hospital (DMR-105-028, DMR-103-081, and DMR-104-074), and Kaohsiung Medical University Hospital (KMUH104-4T01, KMUH104-4R49).
Disclosure
The authors report no conflicts of interest in this work.
References
Sofianopoulou E, Rushton SP, Diggle PJ, Pless-Mulloli T. Association between respiratory prescribing, air pollution and deprivation, in primary health care. J Public Health (Oxf). 2013;35(4):502–509. | ||
Richardson EA, Pearce J, Tunstall H, Mitchell R, Shortt NK. Particulate air pollution and health inequalities: a Europe-wide ecological analysis. Int J Health Geogr. 2013;12:34. | ||
Mehta S, Shin H, Burnett R, North T, Cohen AJ. Ambient particulate air pollution and acute lower respiratory infections: a systematic review and implications for estimating the global burden of disease. Air Qual Atmos Health. 2013;6(1):69–83. | ||
Ko FW, Hui DS. Air pollution and chronic obstructive pulmonary disease. Respirology. 2012;17(3):395–401. | ||
Mannucci PM, Harari S, Martinelli I, Franchini M. Effects on health of air pollution: a narrative review. Intern Emerg Med. 2015;10(6):657–662. | ||
Tzivian L, Winkler A, Dlugaj M, et al. Effect of long-term outdoor air pollution and noise on cognitive and psychological functions in adults. Int J Hyg Environ Health. 2015;218(1):1–11. | ||
World Health Organization [webpage on the Internet]. Burden of Disease from Ambient Air Pollution for 2012; 2014. Available from: http://www.who.int/phe/health_topics/outdoorair/databases/AAP_BoD_results_March2014.pdf. Accessed March 1, 2015. | ||
Kelly FJ, Fussell JC. Linking ambient particulate matter pollution effects with oxidative biology and immune responses. Ann N Y Acad Sci. 2015;1340:84–94. | ||
Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health. 2013;10(9):3886–3907. | ||
Rohr AC. The health significance of gas- and particle-phase terpene oxidation products: a review. Environ Int. 2013;60:145–162. | ||
Kim KE, Cho D, Park HJ. Air pollution and skin diseases: adverse effects of airborne particulate matter on various skin diseases. Life Sci. 2016;152:126–134. | ||
Magnani ND, Muresan XM, Belmonte G, et al. Skin damage mechanisms related to airborne particulate matter exposure. Toxicol Sci. 2016;149(1):227–236. | ||
Lefebvre MA, Pham DM, Boussouira B, et al. Consequences of urban pollution upon skin status. A controlled study in Shanghai area. Int J Cosmet Sci. 2016;38(3):217–223. | ||
Xiao W, Sarsour EH, Wagner BA, et al. Succinate dehydrogenase activity regulates PCB3-quinone-induced metabolic oxidative stress and toxicity in HaCaT human keratinocytes. Arch Toxicol. 2016;90(2):319–332. | ||
Wang IJ, Tung TH, Tang CS, Zhao ZH. Allergens, air pollutants, and childhood allergic diseases. Int J Hyg Environ Health. 2016;219(1):66–71. | ||
Li M, Vierkötter A, Schikowski T, et al. Epidemiological evidence that indoor air pollution from cooking with solid fuels accelerates skin aging in Chinese women. J Dermatol Sci. 2015;79(2):148–154. | ||
Mancebo SE, Wang SQ. Recognizing the impact of ambient air pollution on skin health. J Eur Acad Dermatol Venereol. 2015;29(12):2326–2332. | ||
Rubió L, Motilva MJ, Romero MP. Recent advances in biologically active compounds in herbs and spices: a review of the most effective antioxidant and anti-inflammatory active principles. Crit Rev Food Sci Nutr. 2013;53(9):943–953. | ||
Izawa H, Kohara M, Aizawa K, et al. Alleviative effects of quercetin and onion on male reproductive toxicity induced by diesel exhaust particles. Biosci Biotechnol Biochem. 2008;72(5):1235–1241. | ||
Tan X, Jin P, Feng L, et al. Protective effect of luteolin on cigarette smoke extract-induced cellular toxicity and apoptosis in normal human bronchial epithelial cells via the Nrf2 pathway. Oncol Rep. 2014;31(4):1855–1862. | ||
Lin FJ, Yen FL, Chen PC, et al. HPLC-fingerprints and antioxidant constituents of Phyla nodiflora. ScientificWorldJournal. 2014;2014:528653. | ||
Tsai MH, Lin ZC, Liang CJ, Yen FL, Chiang YC, Lee CW. Eupafolin inhibits PGE2 production and COX2 expression in LPS-stimulated human dermal fibroblasts by blocking JNK/AP-1 and Nox2/p47(phox) pathway. Toxicol Appl Pharmacol. 2014;279(2):240–251. | ||
Chung KS, Choi JH, Back NI, et al. Eupafolin, a flavonoid isolated from Artemisia princeps, induced apoptosis in human cervical adenocarcinoma HeLa cells. Mol Nutr Food Res. 2010;54(9):1318–1328. | ||
Lago JH, Toledo-Arruda AC, Mernak M, et al. Structure-activity association of flavonoids in lung diseases. Molecules. 2014;19(3):3570–3595. | ||
Men K, Duan X, Wei XW, et al. Nanoparticle-delivered quercetin for cancer therapy. Anticancer Agents Med Chem. 2014;14(6):826–832. | ||
Khan AW, Kotta S, Ansari SH, Sharma RK, Ali J. Self-nanoemulsifying drug delivery system (SNEDDS) of the poorly water-soluble grapefruit flavonoid Naringenin: design, characterization, in vitro and in vivo evaluation. Drug Deliv. 2015;22(4):552–561. | ||
Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1–2):113–142. | ||
Kathe N, Henriksen B, Chauhan H. Physicochemical characterization techniques for solid lipid nanoparticles: principles and limitations. Drug Dev Ind Pharm. 2014;40(12):1565–1575. | ||
Caddeo C, Díez-Sales O, Pons R, Fernàndez-Busquets X, Fadda AM, Manconi M. Topical anti-inflammatory potential of quercetin in lipid-based nanosystems: in vivo and in vitro evaluation. Pharm Res. 2014;31(4):959–968. | ||
Esposito E, Ravani L, Mariani P, et al. Effect of nanostructured lipid vehicles on percutaneous absorption of curcumin. Eur J Pharm Biopharm. 2014;86(2):121–132. | ||
Tzeng CW, Yen FL, Wu TH, et al. Enhancement of dissolution and antioxidant activity of kaempferol using a nanoparticle engineering process. J Agric Food Chem. 2011;59(9):5073–5080. | ||
Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–114. | ||
Rowe RC, Sheskey PJ, Cook WG, Fenton ME. Handbook of Pharmaceutical Excipients. 7th ed. London: Pharmaceutical Press; 2012. | ||
Huang PH, Hu SC, Lee CW, Yeh AC, Tseng CH, Yen FL. Design of acid-responsive polymeric nanoparticles for 7,3′,4′-trihydroxyisoflavone topical administration. Int J Nanomedicine. 2016;11:1615–1627. | ||
Ko HH, Chiang YC, Tsai MH, et al. Eupafolin, a skin whitening flavonoid isolated from Phyla nodiflora, downregulated melanogenesis: role of MAPK and Akt pathways. J Ethnopharmacol. 2014;151(1):386–393. | ||
Yen FL, Wu TH, Lin LT, Cham TM, Lin CC. Naringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl(4)-induced acute liver failure. Pharm Res. 2009;26(4):893–902. | ||
Cosmetics Europe [webpage on the Internet]. COLIPA Guidelines: Guidelines for Percutaneous Absorption/Penetration, 1997, Cosmetics Europe. Available from: http://www.jacvam.jp/files/doc/05_01/05_01_Z3.pdf. Accessed February 1, 2015. | ||
Horisawa E, Danjo K, Haruna M. Physical properties of solid dispersion of a nonsteroidal anti-inflammatory drug (M-5011) with Eudragit E. Drug Dev Ind Pharm. 2000;26(12):1271–1278. | ||
Brough C, Williams RO 3rd. Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int J Pharm. 2013;453(1):157–166. | ||
Brandes RP, Weissmann N, Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–226. | ||
Murakami A, Ohigashi H. Targeting NOX, INOS and COX-2 in inflammatory cells: chemoprevention using food phytochemicals. Int J Cancer. 2007;121(11):2357–2363. | ||
Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. 2012;84(5):581–590. | ||
Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol. 2013;64(4):409–421. | ||
Arthur JS. MSK activation and physiological roles. Front Biosci. 2008;13:5866–5879. | ||
Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol. 2006;38(10):1654–1661. | ||
Richmond JM, Harris JE. Immunology and skin in health and disease. Cold Spring Harb Perspect Med. 2014;4(12):a015339. | ||
Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev. 2015;67(2):259–279. | ||
Blickenstaff NR, Coman G, Blattner CM, Andersen R, Maibach HI. Biology of percutaneous penetration. Rev Environ Health. 2014;29(3):145–155. | ||
Paudel KS, Milewski M, Swadley CL, Brogden NK, Ghosh P, Stinchcomb AL. Challenges and opportunities in dermal/transdermal delivery. Ther Deliv. 2010;1(1):109–131. | ||
Raphael AP, Garrastazu G, Sonvico F, Prow TW. Formulation design for topical drug and nanoparticle treatment of skin disease. Ther Deliv. 2015;6(2):197–216. | ||
Loftsson T, Brewster ME. Physicochemical properties of water and its effect on drug delivery. A commentary. Int J Pharm. 2008;354(1–2):248–254. | ||
Müller RH, Petersen RD, Hommoss A, Pardeike J. Nanostructured lipid carriers (NLC) in cosmetic dermal products. Adv Drug Deliv Rev. 2007;59(6):522–530. | ||
Lu F, Xu D, Cheng Y, et al. Systematic review and meta-analysis of the adverse health effects of ambient PM2.5 and PM10 pollution in the Chinese population. Environ Res. 2015;136:196–204. | ||
Andreau K, Leroux M, Bouharrour A. Health and cellular impacts of air pollutants: from cytoprotection to cytotoxicity. Biochem Res Int. 2012;2012:493894. | ||
Choi H, Shin DW, Kim W, Doh SJ, Lee SH, Noh M. Asian dust storm particles induce a broad toxicological transcriptional program in human epidermal keratinocytes. Toxicol Lett. 2011;200(1–2):92–99. | ||
Krutmann J, Liu W, Li L, et al. Pollution and skin: from epidemiological and mechanistic studies to clinical implications. J Dermatol Sci. 2014;76(3):163–168. | ||
Chun KS, Langenbach R. A proposed COX-2 and PGE(2) receptor interaction in UV-exposed mouse skin. Mol Carcinog. 2007;46(8):699–704. | ||
Wyns H, Plessers E, De Backer P, Meyer E, Croubels S. In vivo porcine lipopolysaccharide inflammation models to study immunomodulation of drugs. Vet Immunol Immunopathol. 2015;166(3–4):58–69. | ||
Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R. NOX2 complex-derived ROS as immune regulators. Antioxid Redox Signal. 2011;15(8):2197–2208. | ||
Jeong SH, Park JH, Kim JN, et al. Up-regulation of TNF-alpha secretion by cigarette smoke is mediated by Egr-1 in HaCaT human keratinocytes. Exp Dermatol. 2010;19(8):e206–e212. | ||
Cheng SE, Lee IT, Lin CC, Kou YR, Yang CM. Cigarette smoke particle-phase extract induces HO-1 expression in human tracheal smooth muscle cells: role of the c-Src/NADPH oxidase/MAPK/Nrf2 signaling pathway. Free Radic Biol Med. 2010;48(10):1410–1422. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.