Back to Journals » OncoTargets and Therapy » Volume 12
Estrogen Receptor Beta Inhibits The Proliferation, Migration, And Angiogenesis Of Gastric Cancer Cells Through Inhibiting Nuclear Factor-Kappa B Signaling
Authors Zhang Y, Wu Y, Zhou X, Yi B, Wang L
Received 5 May 2019
Accepted for publication 9 October 2019
Published 5 November 2019 Volume 2019:12 Pages 9153—9164
DOI https://doi.org/10.2147/OTT.S214529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
This paper has been retracted
Yiping Zhang, Yahua Wu, Xufeng Zhou, Benyi Yi, Lili Wang
Department of Biochemistry and Molecular Biology, Basic Medical College of Jiujiang University, Jiujiang City, Jiangxi Province 332000, People’s Republic of China
Correspondence: Yiping Zhang
Department of Biochemistry and Molecular Biology, Basic Medical College of Jiujiang University, No. 320, Xunyang East Road, Xunyang District, Jiujiang City, Jiangxi Province 332000, People’s Republic of China
Tel +86-13767275087
Email [email protected]
Purpose: This study aimed to investigate the regulatory roles of estrogen receptor beta (ERβ) on gastric cancer (GC) cells, and reveal the potential mechanisms relating to nuclear factor-kappa B (NF-κB) signaling.
Methods: GC cell lines SGC7901 and MKN45 were transfected with pEGFP-C1-ERβ to overexpress ERβ, and treated with PMA (a NF-κB activator) to activate NF-κB signaling. The cell proliferation and migration, as well as the formation of vessel-like structures in human venous endothelial cells (HUVECs) were detected. The expression of ERβ, NF-κB p65, p-NF-κB p65, Ki67 (a proliferation marker), vascular endothelial growth factor A (VEGF-A) and matrix metalloproteinase 2 (MMP-2), the DNA binding activity of NF-κB p65, the content of VEGF-A, and the activity of MMP-2 were detected in SGC7901 and MKN45 cells.
Results: The transfection of pEGFP-C1-ERβ significantly increased the expression of ERβ in SGC7901 and MKN45 cells (P < 0.05). Overexpression of ERβ in SGC7901 and MKN45 cells significantly decreased the cell activity, cell number in G2/M phase, cell migration, the expression of Ki67, VEGF-A and MMP-2, VEGF-A content, MMP-2 activity, as well as the number of vessel-like structures formed by HUVECs (P < 0.05). Overexpression of ERβ also significantly decreased the DNA binding activity and the expression of p-NF-κB p65 in SGC7901 and MKN45 cells (P < 0.05). The anti-tumor effect of ERβ overexpression on GC cells was reversed by the intervention of PMA (P < 0.05).
Conclusion: Overexpression of ERβ inhibited the proliferation, migration, and angiogenesis of GC cells through inhibiting NF-κB signaling.
Keywords: estrogen receptor beta, gastric cancer, nuclear factor-kappa B, angiogenesis, proliferation
Introduction
Gastric cancer (GC) is the fourth most common malignant tumor, and the second leading cause of cancer-related death in the world.1 As a fatal tumor that develops from the lining of the stomach, GC can be induced by diverse factors, such as diet, obesity, smoking, and chronic infection.2 In clinical practice, surgical resection remains the most effective therapeutic strategy against GC, and adjuvant chemotherapy and chemotherapy are also commonly used.3 However, the prognosis of GC patients remains poor, especially for those at advanced stages.4 The five-year survival rate is less than 20% for GC worldwide,5 and less than 10% for metastatic GC [6]. Researching of novel therapeutic targets for GC is urgently needed.
Estrogen receptor beta (ERβ) is a hormone-inducible transcription factor that downregulated in diverse cancers, such as colon cancer,6 breast cancer,7 ovarian cancer,8 and prostate cancer.9 A large number of previous studies have proved that ERβ plays a key regulatory role in the occurrence and development of cancers. For example, ERβ agonists significantly decrease the proliferation of OVCAR-3 and OAW-42 cells (ovarian cancer), and knockdown of ERβ increases the proliferation of OAW-42 cells about 1.9-fold.10 Overexpression of ERβ decreases the growth rate and motility of MCF-7 cells (breast cancer) in vitro, as well as the tumor volume in mice.11 Overexpression of ERβ inhibits the migration of HCT-116 cells (colon cancer),12 as well as the migration and invasion of MCF-7 cells.13 Noteworthily, ERβ is also downregulated in GC, and negatively associated with tumor stage, lymph node metastasis, poor overall survival, and recurrence of GC patients.14–16 However, the specific regulatory roles of ERβ on GC cells are not fully revealed.
Nuclear factor-kappa B (NF-κB) is an important transcription factor that involved in the regulation of diverse cellular processes in cancers, such as transformation, proliferation, migration, invasion, angiogenesis, chemoresistance, and radioresistance.17 The inhibition of NF-κB signaling has been considered as a therapeutic target for cancers.18 Diverse NF-κB-targeting agents have been identified to be effective in the treatment of GC, such as parthenolide,19 celastrol,20 propranolol,21 and toxicarioside A.22 However, whether the regulatory mechanisms of ERβ in GC cells are related with NF-κB signaling are still unclear.
In this study, ERβ was overexpressed in two GC cell lines, SGC7901 and MKN45 by the transfection of pEGFP-C1-ERβ. The effects of ERβ overexpression on the proliferation, migration and angiogenesis were evaluated. Based on the application of a NF-κB activator, PMA, the regulatory relationship between ERβ and NF-κB signaling was further analyzed. Our findings may provide a novel therapeutic target for GC, and open up new insights into the underlying mechanisms for the treatment of GC.
Materials And Methods
Cell Culture
Human gastric cancer cell lines SGC7901 and MKN45, and human venous endothelial cells (HUVECs) were purchased from Cell Bank of the Chinese Academy of Science (Shanghai, China). Cells were cultured in complete Roswell Park Memorial Institute (RPMI) 1640 medium (HyClon, Loga, UT, USA) containing 10% fetal bovine serum (FBS) and penicillin. Cells were maintained in an incubator at 37°C with 5% CO2, and passaged until 80% confluence. Logarithmic growth phase cells were used for further assays.
Cell Transfection And Treatments
The plasmids of pEGFP-C1-ERβ and pEGFP-C1 were purchased from Beijing Huada Gene Technology Co., Ltd. (Beijing, China). Cells were seeded in 6-well plates at a density of 6 × 105 cells/well, and cultured until 80% confluence. Then, cells were transfected with pEGFP-C1-ERβ (ERβ group), and pEGFP-C1 (ERβ-Control) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Positive transfected cells were identified by green fluorescence under an inverted fluorescence microscope (Olympus, Tokyo, Japan). After the transfection for 48h, pEGFP-C1-ERβ-transfected cells were treated with 10 μmmol/L PMA (a NF-κB activator, Beyotime, Beijing, China) for another 48 h (ERβ + PMA). Cells without treatment were considered as the control (Mock).
Quantitative Real-Time PCR (qrt-PCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen), and reverse-transcribed into cDNA using a cDNA Reverse Transcription Kit (Invitrogen) in accordance with manufacturers’ instructions. qRT-PCR was performed on ABI 7500 (Applied Biosystems, Foster City, CA, USA) using specific primers (Table 1). The PCR program included 95°C for 10 min, 40 cycles of 95°C for 10 s, 60°C for 20 s and 72°C for 34 s. β-actin was used as an internal control. The relative expression of target genes was calculated in accordance with the 2−ΔΔCt method.23
 |
Table 1 The Primer Sequences Used In Quantitative Real-Time PCR |
Western Blot
Cells were lysed in RIPA Lysis buffer (Invitrogen). Total proteins were quantified using a bicinchoninic acid assay kit (Invitrogen), separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidenefluoride membrane (Millipore, Billerica, MA, USA). The membrane was blocked with 5% skim milk in Tris-buffered saline Tween (TBST) for 2 h, and incubated with specific primary antibodies, including anti-ERβ, -NF-κB p65, -p-NF-κB p65, -vascular endothelial growth factor A (VEGF-A), -matrix metalloproteinase 2 (MMP-2), and -GAPDH (Rabbit anti-human, 1:1000, Abcam, Cambridge, MA, USA) overnight at 4°C. Then the membrane was washed with TBST for three times, and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-rabbit, 1:5000, Abcam) for 1 h at 25°C. The protein bands were visualized using HRP color development kit (Invitrogen).
Enzyme-Linked Immunosorbent Assay (ELISA) Assay
The DNA binding activity of NF-κB p65 was detected by using ELISA kit (Chemicon, Temecula, CA, USA) in accordance with the manufacturer’s instructions. Simply, the protein samples were incubated with anti-NF-κB p65 (1:100) for 1 h at 25°C. After washed with phosphate buffer saline (PBS) for 3 times, the samples were incubated with an HRP-conjugated secondary antibody (1:100) for 30 min at 25°C. Then, the samples were washed with PBS for 3 times and incubated with TMB substrate solution for 10 min at 30°C. TMB stop solution was added to stop the reaction. Optical density (OD) at 450 nm was detected by a microplate reader (Epoch 2, Biotek, Winooski, VT, USA). Similarly, the VEGF-A level was detected by using ELISA kit (Beyotime) in accordance with the manufacturer’s instructions.
MTT Assay
After the transfection for 24, 48, 72, and 96 h, MTT assay was performed to detect the cell activity in different groups. Simply, 200μL cells were seeded in 96-well plates at a density of 6 × 103/well, and incubated with 20 μL MTT (Sigma, St. Louis, MO, USA) for 4 h. Then, cells were incubated with 150 μL DMSO for 10 min. OD at 495 nm was detected by a Microplate Reader (Biotek).
Immunofluorescence
After the transfection for 48 h, immunofluorescence was performed to detect the expression of Ki67 (a proliferation marker) in cells of different groups. Simply, cells were fixed in 4% paraformaldehyde for 20 min at 4°C and permeated in 0.1% Triton X-100 for 5 min. After blocked with 5% BSA for 30 min, cells were incubated with anti-Ki67 (1:100, Abcam) overnight at 4°C. Then, cells were washed with PBS for 3 times and incubated with Alexa Fluor 488-conjugated secondary antibody (goat anti-rabbit, 1:500, Abcam) for 1h at 37°C. Followed by staining with DAPI (4,6-diamino-2-phenylindole), cells were observed under an inverted fluorescence microscope (Olympus).
Cell Migration Assay
After the transfection for 48 h, wound healing and transwell assay were performed to detect the migration ability of cells in different groups.
Wound Healing Assay
Cells were seeded in 6-well plates at a density of 5 × 105/well, and cultured until 90% confluence. A wound track was scored in each plate with a plastic scraper, and the cell debris was removed by washing with PBS. After 48 h of culture, the migration distance was measured under a microscope (Olympus), and the migration rate was calculated.
Transwell Assay
Transwell assay was performed by using trans-well chambers (Invitrogen), cells were seeded in the upper chamber at a density of 0.1 × 105/µL, and RPMI-1640 containing 10% FBS were added in the lower compartment. After 24 h of incubation at 37°C, cells on the upper chamber were removed with cotton swabs. Cells on the lower chamber were fixed in formaldehyde for 30 min and stained with 0.1% crystal violet for 20 min. Positive stained cells were counted under microscope (Olympus) in 10 randomly selected fields.
Flow Cytometry
The cell cycle of cells in different groups was detected by Flow cytometry. Simply, cells were washed with PBS, fixed in 70% pre-cooled ethanol, and stained with Muse Cell Cycle Reagent (Millipore, USA). After 30 min of incubation under darkness, the percentage of cells in different cell cycle phases was analyzed on Flow cytometry (Invitrogen).
Zymography
The MMP-2 activity of cells in different groups was detected by using MMP2/9 zymography kit (Xinfan, Shanghai, China) in accordance with the manufacturer’s instructions. Simply, the protein samples were separated by 10% gelatin-polyacrylamide gel electrophoresis, incubated with renaturation solution (Buffer A) for 24 h at 37°C. Then, the gel was incubated with incubation solution (Buffer B) for 20 h at 37°C, and stained with coomassie brilliant blue for 30 min. Negative stained bands were observed under Gel scanner (Invitrogen).
Angiogenesis Assay
The angiogenesis of HUVECs induced by cell supernatant was detected. Simply, cells were cultured for 3 days without medium refreshing. Cell supernatants were added into 24-well plates pre-coated with matrigel (matrigel and medium at a volume ratio of 1:9). Then, HUVECs were seeded in plates at a density of 5 × 105/well, and cultured for 72 h. Vessel-like structures were observed under an inverted fluorescence microscope (Olympus) and counted at five randomly selected fields.
Statistical Analyses
All data were expressed as mean ± standard deviation. Statistical analysis was performed by SPSS version 13.0 (SPSS Inc., Chicago, IL, USA). Comparison between different groups was determined by one-way ANOVA. A P-value of less than 0.05 represented significantly different.
Results
The Transfection Of pEGFP-C1-ERβ Upregulated ERβ In GC Cells
In order to overexpress ERβ in GC cells, pEGFP-C1-ERβ was transfected into SGC7901 and MKN45 cells. As shown in Figure 1A, obvious green fluorescence was observed in SGC7901 and MKN45 cells transfected with pEGFP-C1 (ERβ-Control group) and pEGFP-C1-ERβ (ERβ group), but not in normal GC cells (Mock group) (Figure 1A). The transfection of pEGFP-C1-ERβ significantly increased the expression of ERβ in SGC7901 and MKN45 cells at the mRNA level (P < 0.05) (Figure 1B), and increased the protein level of exogenous ERβ (P < 0.05) (Figure 1C and D). The protein level of endogenous ERβ was not significantly influenced by the transfection of pEGFP-C1-ERβ in SGC7901 and MKN45 cells (Figure 1C and D).
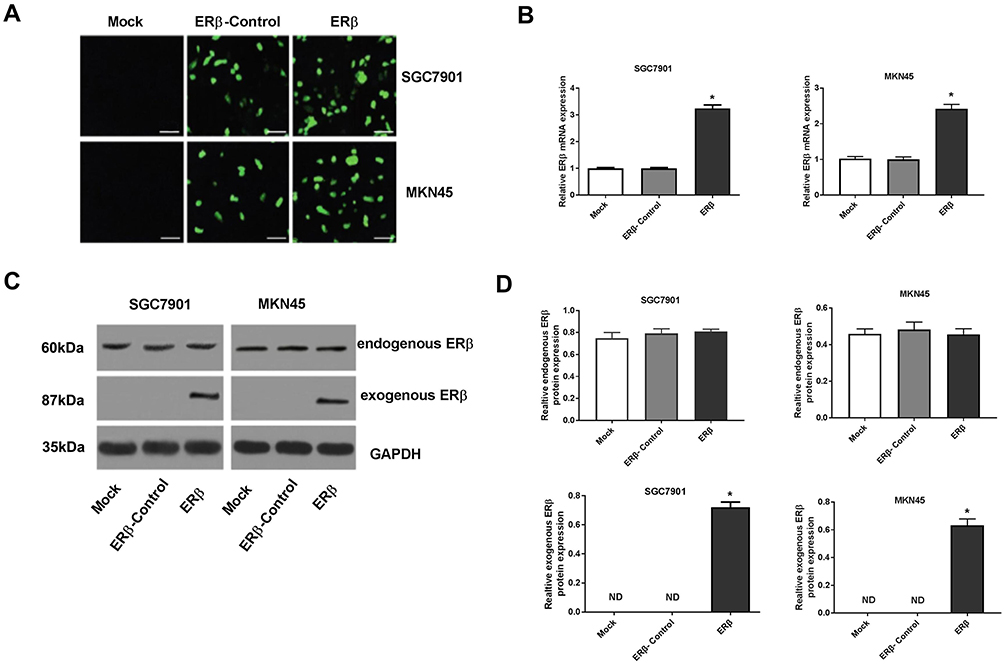 |
Figure 1 The expression of estrogen receptor beta (ERβ) in SGC7901 and MKN45 cells. (A) Green fluorescence under microscope (× 200, bar = 50 μm); (B) Relative ERβ expression detected by quantitative real-time PCR (mRNA level); (C) Protein bands of endogenous and exogenous ERβ detected by Western blot; (D) Relative endogenous and exogenous ERβ expression detected by Western blot (protein level). Mock, cells without transfection; ERβ-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ. *P < 0.05 vs Mock and ERβ-Control. |
Overexpression Of ERβ Downregulated p-NF-κB P65 In GC Cells
The regulatory relationship between ERβ and NF-κB p65 was evaluated in SGC7901 and MKN45 cells. As shown in Figure 2A, the DNA binding activity of NF-κB p65 (OD450 value) was significantly lower in the ERβ group than in ERβ-Control and Mock group (P < 0.05) (Figure 2A). In addition, the expression of p-NF-κB p65 was significantly lower in the ERβ group than in ERβ-Control and Mock group at the protein level (P < 0.05). However, the expression of NF-κB p65 in the ERβ group was not significantly different with ERβ-Control and Mock group (Figure 2B and C). No significant differences in the DNA binding activity and expression of p-NF-κB p65 were observed between ERβ-Control and Mock group (Figure 2A–C).
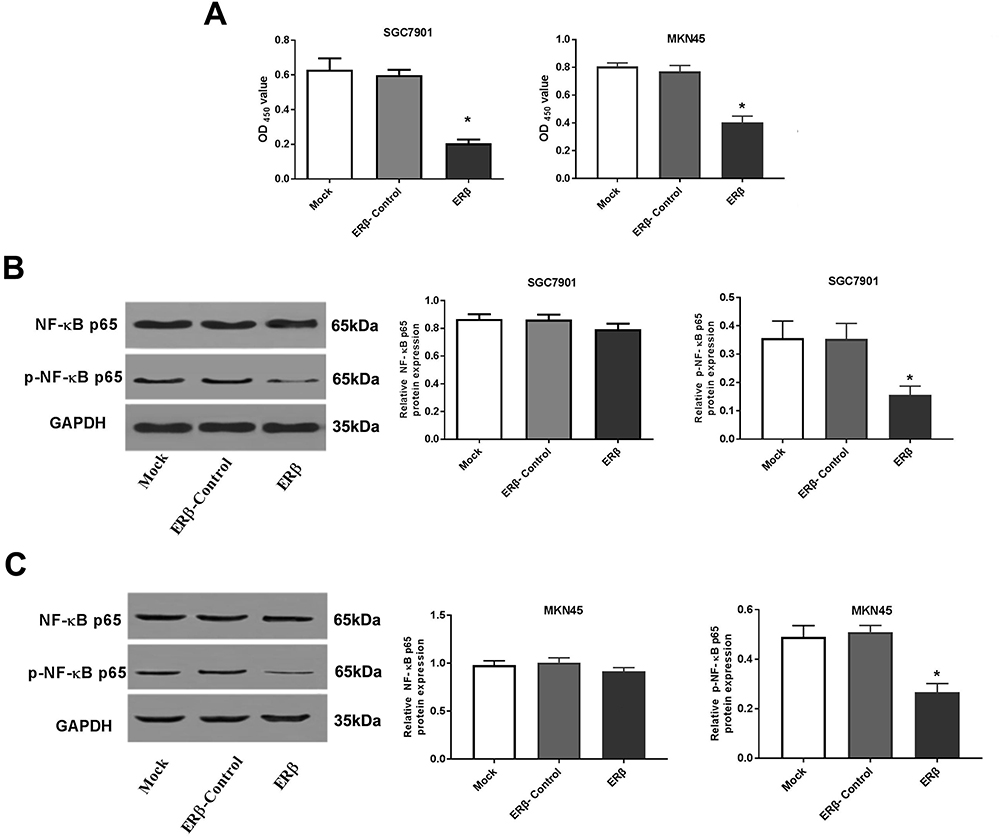 |
Figure 2 The DNA binding activity and nuclear factor-kappa B (NF-κB) p65 expression in SGC7901 and MKN45 cells. (A) DNA binding activity (OD450 value) detected by enzyme-linked immunosorbent assay; (B) Relative NF-κB p65 and p-NF-κB p65 expression detected by Western blot (protein level) in SGC7901 cells; (C) Relative NF-κB p65 and p-NF-κB p65 expression detected by Western blot (protein level) in MKN45 cells. Mock, cells without transfection; Estrogen receptor beta (ERβ)-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ. *P < 0.05 vs Mock and ERβ-Control. |
Overexpression Of ERβ Inhibited The Proliferation Of GC Cells
The activity of SGC7901 and MKN45 cells in different groups was detected by MTT assay. As shown in Figure 3A, the activity of SGC7901 (OD495 value) was significantly increased in a time-dependent manner. The activity of SGC7901 cells was significantly lower in the ERβ group than in ERβ-Control and Mock group and was significantly higher in the PMA group than in ERβ-Control and Mock group starting from the 48 h (P < 0.05). The intervention of ERβ significantly decreased the activity of PMA-treated SGC7901 cells starting from the 48 h (P < 0.05) (Figure 3A). In addition, the expression of a proliferation marker, Ki67 was detected in SGC7901 and MKN45 cells by immunofluorescence. As shown in Figure 3B, the expression of Ki67 (fluorescence intensity) was significantly lower in the ERβ group and significantly higher in the PMA group than in ERβ-Control and Mock group at 72 h post-treatment (P < 0.05). The intervention of ERβ significantly decreased the expression of Ki67 in PMA-treated SGC7901 cells at 72 h post-treatment (P < 0.05) (Figure 3B). Flow cytometry further showed that the number of SGC7901 cells in G0/G1 phase was significantly higher in the ERβ group, and significantly lower in the PMA group than in ERβ-Control and Mock group at 72 h post-treatment (P < 0.05). On the contrary, the number of SGC7901 cells in the G2/M phase was significantly lower in the ERβ group and significantly higher in the PMA group than in ERβ-Control and Mock group at 72 h post-treatment (P < 0.05). The intervention of ERβ significantly reversed the effect of PMA on the cell cycle of SGC7901 cells (P < 0.05) (Figure 3C). No significant differences in the cell activity, Ki67 expression, and cell cycle were observed between ERβ-Control and Mock group. Consistent results with SGC7901 cells on cell proliferation were also observed in MKN45 cells (Figure 3A–C).
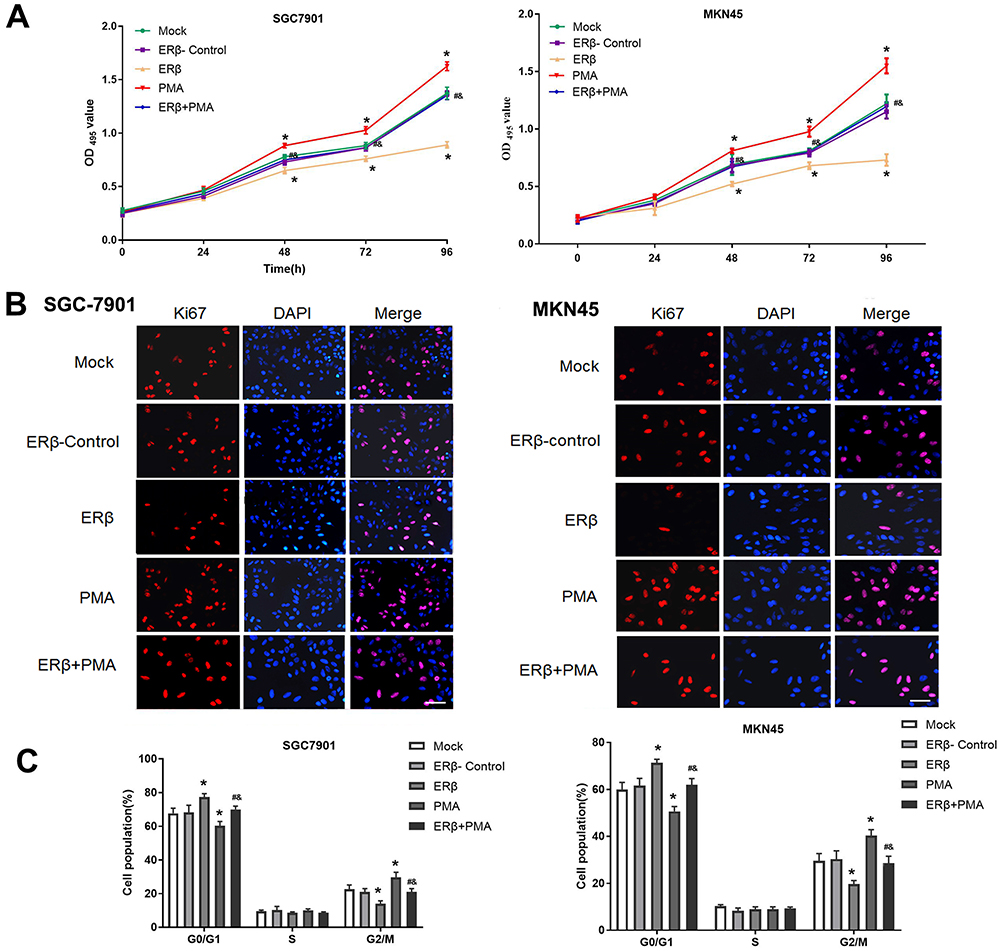 |
Figure 3 The proliferation of SGC7901 and MKN45 cells. (A) Cell activity (OD495 value) detected by MTT assay; (B) The expression of a proliferation marker, Ki67 (fluorescence intensity) detected by immunofluorescence (× 200, bar = 50 μm); (C) Cell cycle detected by Flow cytometry. Mock, cells without transfection; Estrogen receptor beta (ERβ)-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ; PMA, cells treated with PMA; ERβ + PMA, cells transfected with pEGFP-C1-ERβ and then treated with PMA. *P < 0.05 vs Mock and ERβ-Control; #, P < 0.05 vs ERβ; &, P < 0.05 vs PMA. |
Overexpression Of ERβ Inhibited The Migration Of GC Cells
The migration ability of SGC7901 and MKN45 cells was detected by wound healing and transwell assay. As shown in Figure 4A and B, the migration rate and the number of migration cells in SGC7901 cells were significantly lower in the ERβ group than in ERβ-Control and Mock group and were significantly higher in the PMA group than in ERβ-Control and Mock group (P < 0.05). The intervention of ERβ significantly decreased the migration rate and the number of migration cells in PMA-treated SGC7901 cells (P < 0.05). No significant difference in cell migration was observed between ERβ-Control and Mock group. Consistent results with SGC7901 cells on cell migration were also observed in MKN45 cells (Figure 4A and B).
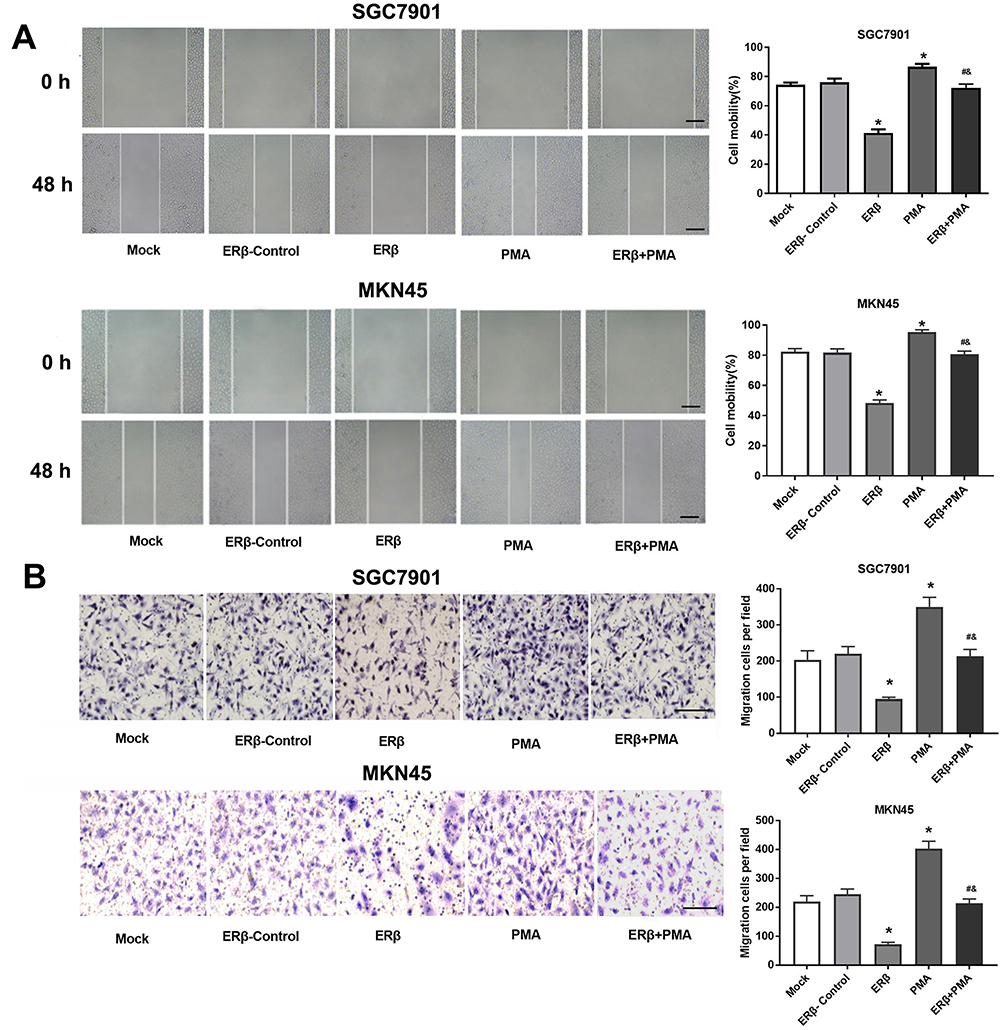 |
Figure 4 The migration of SGC7901 and MKN45 cells. (A) Cell mobility (%) detected by wound healing assay (× 50, bar = 200 μm); (B) Migration cells detected by transwell assay (× 100, bar = 100 μm). Mock, cells without transfection; Estrogen receptor beta (ERβ)-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ; PMA, cells treated with PMA; ERβ + PMA, cells transfected with pEGFP-C1-ERβ and then treated with PMA. *P < 0.05 vs Mock and ERβ-Control; #, P < 0.05 vs ERβ; &, P < 0.05 vs PMA. |
Overexpression Of ERβ Inhibited The Angiogenesis Of HUVECs
The regulatory role of ERβ on angiogenesis was further evaluated in HUVECs. As shown in Figure 5A, vessel-like structures were obviously formed by inducing with the supernatant of SGC7901 and MKN45 cells. The number of vessel-like structures was significantly lower in the ERβ group, and significantly higher in the PMA group than in ERβ-Control and Mock group (P < 0.05). The intervention of ERβ significantly decreased the number of vessel-like structures in PMA-treated SGC7901 and MKN45 cells (P < 0.05). No significant difference in the number of vessel-like structures was observed between ERβ-Control and Mock group (Figure 5B).
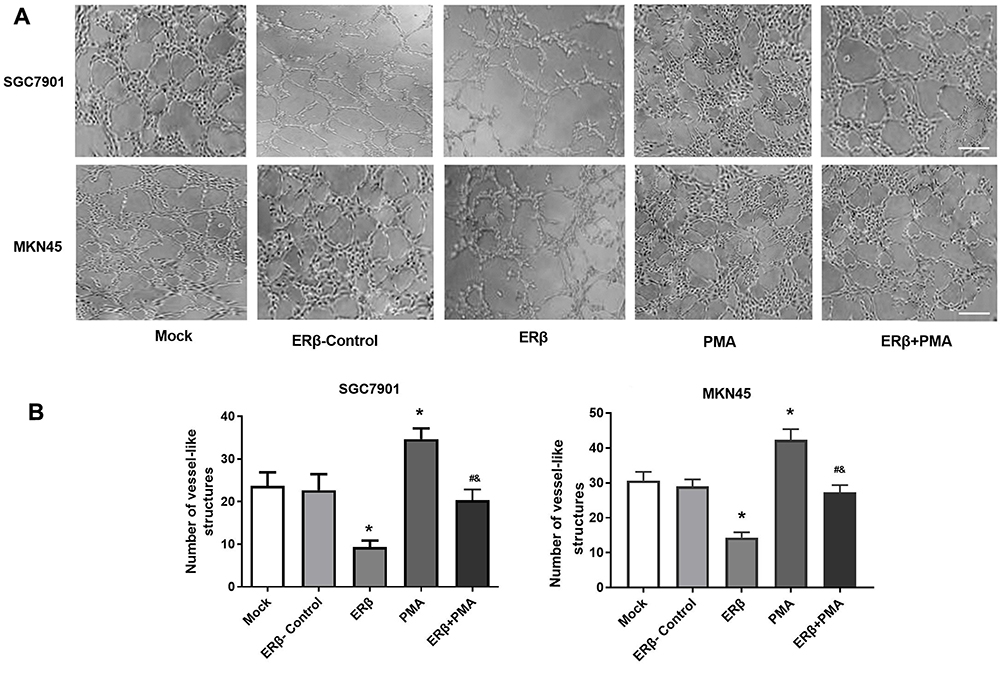 |
Figure 5 The angiogenesis of human umbilical vein endothelial cells (HUVECs) induced by the supernatants of SGC7901 and MKN45 cells. (A) Vessel-like structures under microscope (× 50, bar = 200 μm); (B) The number of vessel-like structures. Mock, cells without transfection; Estrogen receptor beta (ERβ)-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ; PMA, cells treated with PMA; ERβ + PMA, cells transfected with pEGFP-C1-ERβ and then treated with PMA. *P < 0.05 vs Mock and ERβ-Control; #, P < 0.05 vs ERβ; &, P < 0.05 vs PMA. |
Overexpression Of ERβ Decreased VEGF-A And MMP-2 Levels In GC Cells
The levels of VEGF-A and MMP-2 were detected in SGC7901 and MKN45 cells. As shown in Figure 6A and B, the expression of VEGF-A and MMP-2 was significantly lower in the ERβ group and significantly higher in the PMA group than in ERβ-Control and Mock group at both the mRNA and protein level (P < 0.05). The VEGF content and MMP2 activity were also significantly decreased, and increased by the intervention of ERβ, and PMA in SGC7901 and MKN45 cells, respectively (P < 0.05) (Figure 6C and D). The intervention of ERβ significantly downregulated VEGF-A and MMP-2 and decreased VEGF-A content and MMP2 activity in PMA-treated SGC7901 and MKN45 cells (P < 0.05) (Figure 6A–D). No significant differences in the levels of VEGF-A and MMP-2 were observed between ERβ-Control and Mock group (Figure 6A–D).
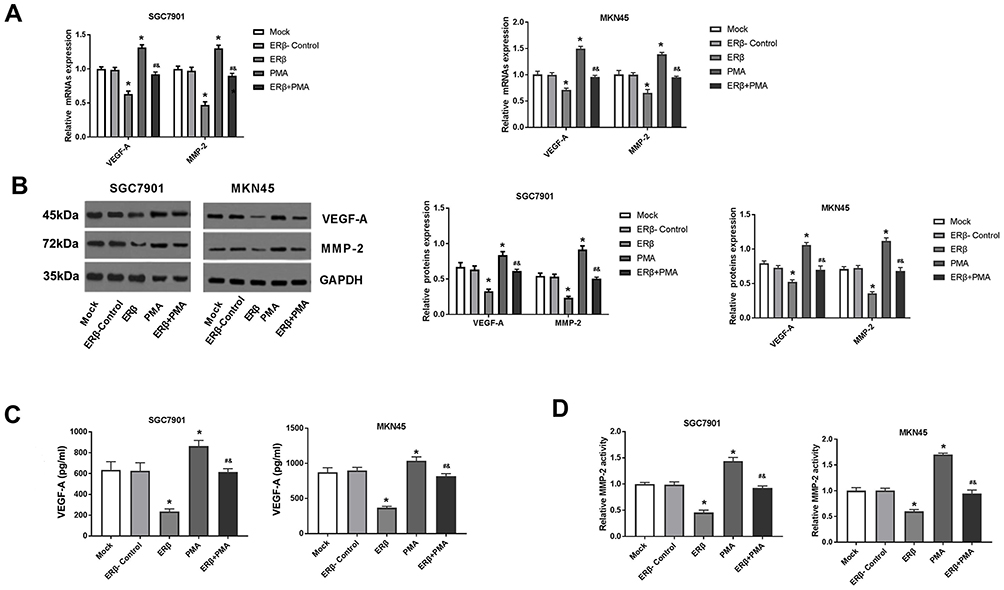 |
Figure 6 The levels of vascular endothelial growth factor A (VEGF-A), matrix metalloproteinase 2 (MMP-2) in SGC7901 and MKN45 cells. (A) Relative expression of VEGF-A and MMP-2 detected by quantitative real-time PCR (mRNA level); (B) Relative expression of VEGF-A and MMP-2 detected by Western blot (protein level); (C) The content of VEGF-A detected by enzyme-linked immunosorbent assay; (D) Relative MMP-2 activity detected by zymography. Mock, cells without transfection; Estrogen receptor beta (ERβ)-Control, cells transfected with pEGFP-C1; ERβ, cells transfected with pEGFP-C1-ERβ; PMA, cells treated with PMA; ERβ + PMA, cells transfected with pEGFP-C1-ERβ and then treated with PMA. *P < 0.05 vs Mock and ERβ-Control; #, P < 0.05 vs ERβ; &, P < 0.05 vs PMA. |
Discussion
ERβ is a specific estrogen receptor that acts as a tumor suppressor in diverse cancers, including colon cancer, breast cancer, ovarian cancer, and prostate cancer.6–9 A previous study has proved that the expression of ERβ is significantly lower in GC tissues than in normal tissues.14 ERβ may also exert great anti-tumor potential on GC, while related studies on the regulatory roles of ERβ on GC cells are still limited. In this study, ERβ was overexpressed in SGC7901 and MKN45 cells by the transfection of pEGFP-C1-ERβ. Then the specific effects of ERβ overexpression on GC cells were evaluated.
The anti-proliferation effects of ERβ on cancer cells have been identified by a large number of studies. For example, overexpression of ERβ results in a 38.7% decreased growth rate of MCF-7 cells.11 WAY200070 and ERB-041 (ERβ agonists) inhibit the growth of OAW-42 cells by 26.8%, and 24.4%, respectively.10 ERβ agonists significantly inhibit the growth of chemotherapy-resistant ovarian cancer cells (ES2 and SKOV-3TR), and sensitize them to apoptosis.24 In consistence with previous studies, we found that the activity of SGC7901 and MKN45 cells was significantly decreased by the transfection of pEGFP-C1-ERβ. Ki67 is not only known as a proliferation marker, but also a prognostic marker for GC.25,26 Previous studies have proved that Ki67 is upregulated in GC tissues, and positively correlated with the pathological T stage, poor overall survival and disease-free survival rate.27,28 In this study, Ki67 was significantly downregulated in SGC790 and MKN45 cells by the transfection of pEGFP-C1-ERβ. In addition, we also found that the transfection of pEGFP-C1-ERβ significantly increased the number of cells in the G0/G1 phase, and decreased the number of cells in the G2/M phase. These findings indicate that ERβ overexpression may inhibit the proliferation of GC cells through blocking cells in G0/G1.
Metastasis is initiated when cancer cells migrated from the primary site to the surrounding tissues.29 The migration ability of cancer cells is considered as the rate-limiting step for metastasis.30 The anti-migration effects of ERβ have also been identified in cancer cells. It has been reported that ERβ ligands (raloxifene, tamoxifen, genistein and curcumin) significantly decrease the migration of DU145 and PC3 cells (prostate cancer).31 Overexpression of ERβ inhibits the migration of HCT-116 cells and MCF-7 cells.12,13 In this study, we found that the migration of SGC7901 and MKN45 cells was significantly inhibited by the transfection of pEGFP-C1-ERβ. Our findings are just consistent with previous studies, and further indicate that ERβ overexpression inhibits the migration of GC cells.
Angiogenesis, the recruitment of new blood vessels is essential for tumor growth and metastasis.32 In this study, we found that the number of vessel-like structures formed by HUVECs was significantly lower in the ERβ group than in ERβ-Control and Mock group. These findings are just consistent with previous studies that overexpression of ERβ decreases the number of blood microvessels in the tumor periphery,33 as well as the number of intratumoral blood vessels in T47D breast cancer xenografts.34 VEGF-A and MMP-2 are key angiogenesis factors that upregulated in GC tissues.35,36 It has been reported that resveratrol, an ERβ agonist significantly reduces the extracellular levels of VEGF in MDA-MB-231 cells (breast cancer).37 ERβ reduces the expression of MMP2 at the mRNA level and the enzymatic activity of MMP2 in ES2 cells.38 In this study, the transfection of pEGFP-C1-ERβ significantly downregulated VEGF-A and MMP-2, decreased VEGF-A content and MMP2 activity in SGC7901 and MKN45 cells. Our findings are just consistent with previous studies, and further indicate that ERβ overexpression inhibits the GC cells-induced angiogenesis through downregulating VEGF-A and MMP-2.
Since NF-κB signaling plays a key regulatory role in the pathogenesis of GC, it has become a therapeutic target for GC.2 It has been reported that parthenolide, an NF-κB inhibitor, inhibits the growth of GC cells (MKN-28, MKN-45 and MKN-74), and promotes the chemosensitivity to paclitaxel.19 Propranolol promotes the apoptosis and cell cycle arrest of SGC7901 cells via blocking NF-κB signaling.20 Toxicarioside A inhibits the proliferation, migration and invasion of SGC7901 cells via inhibiting NF-κB signaling.22 In this study, the DNA binding activity and expression of p-NF-κB p65 in SGC7901 and MKN45 cells were significantly decreased by the transfection of pEGFP-C1-ERβ. These results illustrate that overexpression of ERβ inhibits the NF-κB signaling in GC cells. ERβ overexpression may exert similar functions with parthenolide, propranolol, and toxicarioside A, and contribute to the inhibition of the proliferation, migration, and angiogenesis of GC cells. In addition, our further assays showed that the intervention of PMA, a NF-κB activator significantly increased the cell activity, migration, number of vessel-like structures, as well as VEGF-A and MMP-2 levels in pEGFP-C1-ERβ-transfected SGC7901 and MKN45 cells. These findings indicate that the activation of NF-κB signaling reversed the anti-tumor effect of ERβ overexpression on GC cells. A previous study has proved that ERβ acts as a gate-keeper of NF-κB p65 by inhibiting its expression and nuclear translocation.39 We suspect that ERβ overexpression may inhibit the proliferation, migration, and angiogenesis of GC cells through targeting NF-κB p65.
Conclusions
In conclusion, overexpression of ERβ blocked NF-κB signaling via targeting NF-κB p65, thereby contributing to the inhibition of the proliferation, migration, and angiogenesis of GC cells. Overexpression of ERβ may be a promising therapeutic target for GC. However, this study is still limited in the cellular level. The specific regulatory role of ERβ and related mechanisms on animal models is still needed to be studied.
Ethics Approval And Consent To Participate
This study was conducted after obtaining local ethical committee approval of Basic Medical College of Jiujiang University.
Acknowledgements
This study was funded by National Nature Science Foundation of China (No. 81360364).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global cancer incidence and mortality rates and trends—an update. Cancer Epidemiol Biomarkers Prev. 2016;25(1):16–27.
2. Naumann M, Sokolova O. NF-κB signaling in gastric cancer. Toxins. 2017;9(4):119.
3. Orditura M, Galizia G, Sforza V, et al. Treatment of gastric cancer. World J Gastroenterol. 2014;20(7):1635–1649.
4. Cutsem EV. The treatment of advanced gastric cancer: new findings on the activity of the taxanes. Oncologist. 2004;9 Suppl 2(suppl_2):9.
5. Ong HS, Smithers BM. Epidemiology of gastric cancer. Cancer Rev. 2012;02(1):1–7.
6. Wang Y, Xing T, Huang L, et al. Period 1 and estrogen receptor-beta are downregulated in Chinese colon cancers. Int J Clin Exp Pathol. 2015;8(7):8178.
7. Huang J, Woods P, Normolle D, et al. Downregulation of estrogen receptor and modulation of growth of breast cancer cell lines mediated by paracrine stromal cell signals. Breast Cancer Res Treat. 2016;161(2):229–243.
8. Bossard C, Busson M, Vindrieux D, et al. Potential role of estrogen receptor beta as a tumor suppressor of epithelial ovarian cancer. PLoS ONE. 2012;7(9):e44787.
9. Xiao L, Xiao M, Gao L, Xu W. Involvement of estrogen receptor β in androgen receptor-induced growth inhibition in prostate cancer PC-3 cells. Oncol Lett. 2017;14(3):2796–2802.
10. Schüler-Toprak S, Moehle C, Skrzypczak M, Ortmann O, Treeck O. Effect of estrogen receptor β agonists on proliferation and gene expression of ovarian cancer cells. BMC Cancer. 2017;17(1):319.
11. Hui L, Zhenzhen T, Lianxiao A, Zhiyu Q, Samuel A, Yueqing G. Inhibitory effects of ERβ on proliferation, invasion, and tumor formation of MCF-7 breast cancer cells–prognostication for the use of ERβ-selective therapy. Pharm Biol. 2012;50(7):839–849.
12. Tu Z, Ma Y, Tian J, et al. Estrogen receptor β potentiates the antiproliferative effect of raloxifene and affects the cell migration and invasion in HCT-116 colon cancer cells. J Cancer Res Clin Oncol. 2012;138(7):1091–1103.
13. Yan Z, Jia M, Yan X, Yi Z, Jun J. ERβ1 inhibits the migration and invasion of breast cancer cells through upregulation of E-cadherin in a Id1-dependent manner. Biochem Biophys Res Commun. 2015;457(2):141–147.
14. Xu CY, Guo JL, Jiang ZN, et al. Prognostic role of estrogen receptor alpha and estrogen receptor beta in gastric cancer. Ann Surg Oncol. 2010;17(9):2503–2509. doi:10.1245/s10434-010-1031-2
15. Ryu WS, Kim JH, Jang YJ, et al. Expression of estrogen receptors in gastric cancer and their clinical significance. J Surg Oncol. 2012;106(4):456–461. doi:10.1002/jso.23097
16. Ge H, Yan Y, Tian F, Wu D, Huang Y. Prognostic value of estrogen receptor α and estrogen receptor β in gastric cancer based on a meta-analysis and The Cancer Genome Atlas (TCGA) datasets. Int J Surg. 2018;53:24–31. doi:10.1016/j.ijsu.2018.03.027
17. Aggarwal BB, Sung B. NF-κB in cancer: a matter of life and death. Cancer Discov. 2011;1(6):469–471. doi:10.1158/2159-8290.CD-11-0260
18. Orlowski RZ, Baldwin AS. NF-κB as a therapeutic target in cancer. Trends Mol Med. 2002;8(8):385–389.
19. Sohma I, Fujiwara Y, Sugita Y, et al. Parthenolide, an NF-κB inhibitor, suppresses tumor growth and enhances response to chemotherapy in gastric cancer. Cancer Genomics Proteomics. 2011;8(1):39.
20. Min S, Jun Y, Li-Xin Z, Zheng-Yun L, Ya-Bao C, Jun-Xing H. Celastrol induces apoptosis of gastric cancer cells by miR-21 inhibiting PI3K/Akt-NF-κB signaling pathway. Pharmacology. 2014;93(2):39–46. doi:10.1159/000357683
21. Xinhua L, Xiangming C, Wei Z, Danjie Z, Tieqiang B, Guanghui W. The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor κB signaling. Oncol Rep. 2010;24(6):1669–1676. doi:10.3892/or_00001032
22. Guo JL. Toxicarioside A inhibits SGC-7901 proliferation,migration and invasion via NF-κB/bFGF signaling. World J Gastroenterol. 2012;18(14):1602–1609. doi:10.3748/wjg.v18.i14.1602
23. Livak KJST. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T))method. Methods. 2001;25(4):402–408. doi:10.1006/meth.2001.1262
24. Sareddy GR, Chavez JE, Salazar H, et al. Abstract 606: therapeutic efficacy of ERβ agonists on ovarian cancer. Cancer Res. 2014;74(19 Supplement):606.
25. Lee WS, Ko GH, Go SI, Lee JH, Ha WS. Abstract 4930: ki-67 serves as a prognostic marker in early gastric cancer, but not in advanced gastric cancer. Cancer Res. 2016;76(14 Supplement):4930.
26. Tzanakis NE, Peros G, Karakitsos P, et al. Prognostic significance of p53 and Ki67 proteins expression in Greek gastric cancer patients. Acta Chir Belg. 2009;109(5):606–611. doi:10.1080/00015458.2009.11680496
27. Yongli Z, Yandong L, Jianyun Z, Kaige L, Hongmei Z. Detecting of gastric cancer by Bcl-2 and Ki67. Int J Clin Exp Pathol. 2015;8(6):7287–7290.
28. Badary DM, Abdel-Wanis ME, Hafez MZ, Aboulhagag NA. Immunohistochemical analysis of PTEN, HER2/neu, and ki67 expression in patients with gastric cancer and their association with survival. Pathophysiology. 2017;24(2):99. doi:10.1016/j.pathophys.2017.02.006
29. Ryu S, Park KM, Lee SH. Gleditsia sinensis thorn attenuates the collagen-based migration of PC3 prostate cancer cells through the suppression of α2β1 integrin expression. Int J Mol Sci. 2016;17(3):328. doi:10.3390/ijms17030328
30. Wang J, Si W, Yun L, et al. Overexpression of NEDD9 in renal cell carcinoma is associated with tumor migration and invasion. Oncol Lett. 2017;14(6):8021–8027. doi:10.3892/ol.2017.7231
31. Piccolella M, Crippa V, Messi E, Tetel MJ, Poletti A. Modulators of estrogen receptor inhibit proliferation and migration of prostate cancer cells. Pharmacol Res. 2014;79(1):13–20. doi:10.1016/j.phrs.2013.10.002
32. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–1370. doi:10.1038/nm.2537
33. Hartman J, Lindberg K, Inzunza J, Wan J, Ström A, Gustafsson J. Estrogen receptor β represses breast tumor growth and angiogenesis in vivo. J Clin Oncol. 2006;(18Suppl):10101.
34. Johan H, Karolina L, Andrea M, José I, Anders SM, Jan-Ake G. Estrogen receptor beta inhibits angiogenesis and growth of T47D breast cancer xenografts. Cancer Res. 2006;66(23):11207–11213. doi:10.1158/0008-5472.CAN-06-0017
35. Wang G, Liu H, Rong MA, Jiao H, Hospital BC. Expression and clinical significance of Vav3 and MMP-2 in human gastric cancer. Chin Clin Oncol. 2016.
36. Yurong O, Min K, Lei Z, Zenong C, Sulan T, Donghong Y. [Infection with L-form of Helicobacter pylori and expressions of MIF, MMP9 and VEGF in gastric carcinoma]. J South Med Univ. 2014;34(2):180–187.
37. Stina G, Karin O, Charlotta D. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006;231(1):113–122. doi:10.1016/j.canlet.2005.01.031
38. Jing Z, Keqin H, Hong S, Yinhua Y, Hongyan J, Youji F. Re-expression of estrogen receptor β inhibits the proliferation and migration of ovarian clear cell adenocarcinoma cells. Oncol Rep. 2011;26(6):1497. doi:10.3892/or.2011.1266
39. Paul M, Jiarong L, Sanjoy S, Mercurio AM. ERβ regulation of NF-kB activation in prostate cancer is mediated by HIF-1. Oncotarget. 2015;6(37):40247–40254. doi:10.18632/oncotarget.5377
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.