Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 10
Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges
Authors Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P
Received 21 December 2016
Accepted for publication 16 May 2017
Published 13 September 2017 Volume 2017:10 Pages 375—383
DOI https://doi.org/10.2147/DMSO.S130810
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Elaine Chiquette,1 Elif A Oral,2 Abhimanyu Garg,3 David Araújo-Vilar,4 Praveen Dhankhar5
1Aegerion Pharmaceuticals, Cambridge, MA, USA; 2Brehm Center for Diabetes Research and Metabolism, Endocrinology and Diabetes Division, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, USA; 3Division of Nutrition and Metabolic Diseases, Department of Internal Medicine, Center for Human Nutrition, University of Texas Southwestern Medical Center, Dallas, TX, USA; 4Department of Medicine, UETeM, CIMUS School of Medicine, University of Santiago de Compostela, Santiago de Compostela, Spain; 5Complete HEOR Solutions (CHEORS), North Wales, PA, USA
Background: Lipodystrophy (LD; non-human immunodeficiency virus [HIV]-associated) syndromes are a rare body of disorders for which true prevalence is unknown. Prevalence estimates of rare diseases are important to increase awareness and financial resources. Current qualitative and quantitative estimates of LD prevalence range from ~0.1 to 90 cases/million. We demonstrate an approach to quantitatively estimate LD prevalence (all, generalized, and partial) through a search of 5 electronic medical record (EMR) databases and 4 literature searches.
Methods: EMR and literature searches were conducted from 2012 to 2014. For the EMR database searches (Quintiles, IMS LifeLink, General Electric Healthcare, and Humedica EMR), LD cases were identified by the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) code 272.6 (United Kingdom General Practice Research Database used other diagnostic codes to identify LD) plus additional LD-associated clinical characteristics (patients with HIV or documented HIV treatment were excluded). Expert adjudication of cases was used for the Quintiles database only. Literature searches (PubMed and EMBASE) were conducted for each of the 4 major LD subtypes. Prevalence estimates were determined by extrapolating the total number of cases identified for each search to the database population (EMR search) and European population (literature search).
Results: The prevalence range of all LD across all EMR databases was 1.3–4.7 cases/million. For the adjudicated Quintiles search, the estimated prevalence of diagnosed LD was 3.07 cases/million (95% confidence interval [CI], 2.30–4.02), 0.23 cases/million (95% CI, 0.06–0.59) and 2.84 cases/million (95% CI, 2.10–3.75) for generalized lipodystrophy (GL) and partial lipodystrophy (PL), respectively. For all literature searches, the prevalence of all LD in Europe was 2.63 cases/million (0.96 and 1.67 cases/million for GL and PL, respectively).
Conclusion: LD prevalence estimates are at the lower range of previously established numbers, confirming that LD is an ultra-rare disease. The establishment of diagnostic criteria and coding specific to the 4 major LD subtypes and future studies/patient registries are needed to further refine our estimates.
Keywords: adipose tissue, atypical diabetes, dyslipidemia, hypertriglyceridemia, insulin resistance, lipodystrophy, prevalence
Introduction
Physicians in daily practice are tasked with distinguishing an array of rare diseases from more common disorders in their differential diagnosis. Collectively, rare diseases are estimated to impact ~6–8% of individuals at some point in their lifetime, greatly reducing patient life expectancy and quality of life.1 Prevalence estimates of rare diseases are important to increase disease awareness and to marshal sufficient financial resources to improve detection and treatment. The European Union defines rare and ultra-rare diseases based on the prevalence rates of <5/10,000 individuals and <1/50,000 individuals, respectively.2,3 The US Food and Drug Administration has set a threshold of <200,000 people in the USA (or <7/10,000 people with a population of ~300 million) for an orphan drug that is intended to treat a rare disease or condition.4
Data for establishing a prevalence estimate may be derived from a variety of disparate information sources that are not standardized or are difficult to combine, including published case reports or systematic reviews, patient registries, expert opinion, and other anecdotal evidence.5 Analyses of claims databases can be very helpful in establishing a prevalence estimate; however, the lack of firmly established and specific diagnostic criteria or coding for a rare disease makes it challenging to know the true number of cases per population sample.1,6,7 Overestimation or underestimation of disease prevalence is likely, as the clinical findings and presentation of a patient with a rare disease may overlap with other common or rare diseases.8,9 Finally, some rare diseases may be more or less prevalent in a given geographic area than others.10,11
Lipodystrophy (LD) syndromes are rarely inherited or acquired disorders characterized by near-total or partial loss of body fat.12,13 Excluding LD in human immunodeficiency virus (HIV)-infected patients, LD syndromes have been classified into 4 major subtypes: acquired generalized lipodystrophy (AGL) and congenital generalized lipodystrophy (CGL), referred to here as generalized lipodystrophy (GL), and acquired partial lipodystrophy (APL) and familial partial lipodystrophy (FPL), referred to as partial lipodystrophy (PL). Patients with GL syndromes have markedly low serum leptin levels, which reflects the widespread loss of adipose tissue and contributes to metabolic abnormalities (severe insulin resistance, lipoatrophic diabetes, hypertriglyceridemia, hepatic steatosis, and others) that increase patient morbidity and mortality. Although patients with PL may have variable loss of adipose tissue with corresponding variable levels of serum leptin, they also suffer from metabolic complications.13,14
Awareness of GL and PL is low due to its rarity, making an accurate estimate of disease prevalence very difficult. Patients with GL often have diabetes that is difficult to manage, and they may bear some dysmorphic features and coarse facies (the latter also a characteristic of acromegaly and gigantism).13,15,16 The overlap of clinical presentation with more common diseases may complicate the diagnosis. Similarly, patients with PL may also have diabetes that is difficult to manage and a physical appearance that may resemble an overweight or obese patient with metabolic syndrome or type 2 diabetes or a patient with Cushing’s syndrome.17,18 In patients with FPL type 1, a form of severe insulin resistance characterized by a lack of fat in the extremities, truncal obesity, and having components of the metabolic syndrome, there may be a polygenic contribution of up to 53 different loci that are also known to be associated with more common insulin-resistant states and obesity-related conditions.19 Including patients with FPL type 1 in a prevalence estimate, along with other well-defined monogenic forms of FPL that are more rare, could mistakenly inflate a prevalence estimate. Similarly, the lack of precise diagnostic criteria makes it hard to firmly establish the diagnosis of PL; patients may get missed, and this also complicates prevalence estimates. Finally, acquired forms of GL and PL have also been associated with autoimmune diseases, such as dermatomyositis, systemic lupus erythematosus, autoimmune hepatitis, and others.14,16,20
Not surprisingly, currently available worldwide and European prevalence estimates for the 4 major subtypes of LD combined are wide ranging and varied (~0.1–90 cases/million) depending on the information source (previously published reviews, expert opinion, or figures cited by the European Medicines Agency or Orphanet) and the methodology used for computation of the estimate (qualitative or quantitative).15,21–29 For a rare disease such as LD, documentation of disease prevalence is an important requirement mandated by many regulatory agencies for obtaining orphan drug designation.5,30 We aimed to demonstrate an approach to quantitatively estimate the prevalence of all LD, GL, and PL through a search of 5 electronic medical record (EMR) databases and 4 literature searches.
Methods
EMR database searches
A total of 5 EMR databases were individually searched from a period of 2012–2014—4 maintained in the USA (Quintiles, IMS LifeLink Health Claims, General Electric [GE] Healthcare, and Humedica EMR) and one maintained in the UK (United Kingdom General Practice Research Database [UK GPRD]) (Table 1). Collectively, the 5 EMR databases represent a total population of ~139 million de-identified patient records derived from a large number of medical institutions and providers. The level of patient detail was variable across each EMR database. At the time of the study, all databases captured basic demographic information and allowed for the search of International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) diagnostic codes (the UK GPRD used a system of searchable diagnostic coding that was unrelated to ICD-9-CM), but not all EMRs captured or linked patient records to laboratory data or physician notes that would lend additional support to review or confirm a patient’s diagnosis.
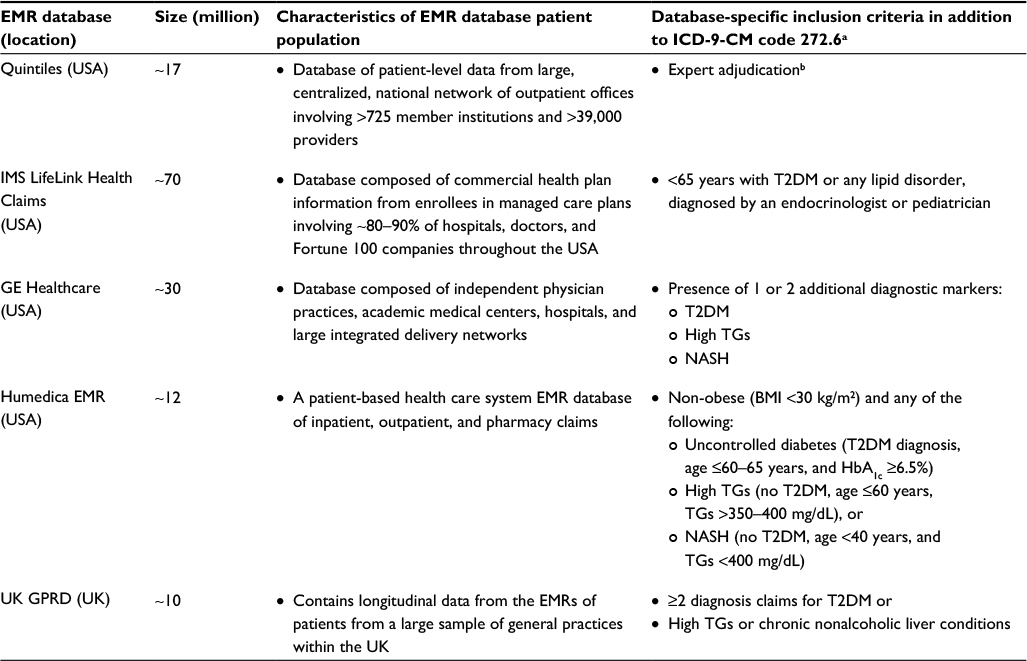 | Table 1 EMR database characteristics and search details Notes: aClinical characteristics or diagnostic criteria often associated with LD. EMR databases were queried in a 3-step process: 1) search for ICD-9-CM code 272.6 for LD (other diagnostic codes were used for UK GPRD); 2) removal of patients with documented HIV or HIV treatment; and 3) removal of patients who did not meet the mentioned above clinical characteristics or diagnostic criteria often associated with LD; bClinician experts reviewed physician notes for the phrases “lipodystrophy,” “lipoatrophy,” or “lipoatrophic”. Abbreviations: BMI, body mass index; EMR, electronic medical record; HbA1c, glycated hemoglobin; HIV, human immunodeficiency virus; ICD-9-CM, International Classification of Diseases, Ninth Revision, Clinical Modification; LD, lipodystrophy; NASH, nonalcoholic steatohepatitis; T2DM, type 2 diabetes mellitus; TGs, triglycerides; UK GPRD, United Kingdom General Practice Research Database. |
Because of variability in the data collected and disease terminology used across databases, search criteria were developed that were specific to each database; however, the overall search strategy used for each database was mostly similar. First, patient criteria that characterized LD were defined before the search of each database (Table 1). Second, the database was queried in the following sequence to generate a total number of patients with LD: 1) search for ICD-9-CM code 272.6 for LD (other diagnostic codes for UK GPRD); 2) removal of patients with documented HIV or HIV treatment (either can be associated with abnormal fat redistribution but not the rare disease of LD); and 3) removal of patients who did not meet the previously established clinical characteristics or diagnostic criteria often associated with LD, and this would minimize the likelihood of mistakenly including “typical” patients with diabetes or metabolic syndrome (Table 1). Owing to limited resources, simple searches were conducted for IMS LifeLink, GE Healthcare, Humedica, and UK GPRD using the 3-step sequence mentioned earlier. For the Quintiles database search only, positive cases identified from the 3-step sequence were further adjudicated by 2 clinician experts (AG and EAO). Expert clinicians manually reviewed de-identified physician notes to confirm or correct a diagnosis of LD and to further classify patients with LD by the 4 major LD subtypes. Cases with the ICD-9-CM diagnostic code 272.6 were removed if the visit was linked to a plastic surgery procedure, such as orbitoplasty or excess skin removal, as these visits uniformly contained the diagnostic code 272.6. Additionally, cases were excluded if the clinician experts did not agree that the case had LD after a review of the notes. Searches for undiagnosed cases of LD in the Quintiles EMR were not able to be performed, and the cases adjudicated to have the LD diagnosis had enough clinical detail in the medical records to feel confident that the cases were rare forms of LD. Where possible, both experts tried to refine the diagnosis further into the subtype of LD. The cases where both experts were not in agreement were reviewed jointly, and a consensus decision was reached where possible. If no consensus was possible, those cases were excluded from the count. All cases where the diagnosis of LD was possible, but not enough information was available to confirm or refute the diagnosis, were excluded from the count. Given the rigor of the review, the cases counted as LD were not ambiguous cases, and there was usually enough detail in the medical records to indicate that they were seen at reference centers such as the National Institutes of Health, University of Texas Southwestern, or University of Michigan for diagnosis, or that a consultation was arranged for verification. Although quite rigorous, this approach may have resulted in an underestimate of the prevalence.
To arrive at a prevalence estimate of all LD for each EMR database, the total number of identified cases was divided by the total population of the respective EMR database (Table 1). For the Quintiles search with expert adjudication, it was possible to estimate the prevalence for diagnosed cases of GL (AGL and CGL) and PL (APL and FPL) in addition to all LD.
The use of the data from EMR databases did not require approval from an institutional review board. The EMR databases consisted of de-identified patient data.
Literature searches
PubMed and EMBASE literature searches were conducted through May 2012. Search terms and time frames for the literature searches are provided in Table 2. References identified by the searches were evaluated, and review articles or clinical case reports that summarized cases of LD in the European Union community population were retained. References that summarized cases of LD from countries outside of the European Union, repeated or updated presentations of the same patient, or general reviews of related syndromes or preclinical reviews were excluded.
 | Table 2 Literature search details Abbreviations: AGL, acquired generalized lipodystrophy; APL, acquired partial lipodystrophy; CGL, congenital generalized lipodystrophy; FPL, familial partial lipodystrophy; LD, lipodystrophy. |
A prevalence estimate was determined through a 3-step process. First, the total number of European cases identified for all LD (AGL + CGL + APL + FPL), GL (AGL + CGL), and PL (APL + FPL) was determined. Second, because it has been estimated that only 25% of all LD cases have been reported in the literature, the total number of cases identified was multiplied by 4.13 Third, the total number of cases for all LD, GL, and PL (after adjustment for underreporting) was divided by the total European Union community population as of 2012 figure (507,751,512 people).
Statistics
Prevalence estimates for each EMR database and literature search were summarized descriptively and presented as the number of cases per million. For the Quintiles EMR adjudicated search, 95% confidence intervals (CIs) were constructed using the Clopper–Pearson method based on the exact binomial distribution31 and then scaled to 1,000,000 patients. CIs were obtained using readily available software.32 A summary prevalence estimate combining the data from all 5 EMR databases was not possible because of differences in the search criteria used (each database used its own medical terminology), the lack of resources to perform an adjudicated search of each EMR, and the inability to minimize the double counting of patient cases across each database.
Results
The range of worldwide prevalence of all LD as determined by the simple EMR database searches was 1.3–4.7 cases/million (Figure 1). For the adjudicated Quintiles database search, the estimated prevalence of all diagnosed LD cases was 3.07 cases/million (95% CI, 2.30–4.02). When separated into diagnosed GL and PL, the estimated prevalence was 0.23 (95% CI, 0.06–0.59) and 2.84 (95% CI, 2.10–3.75) cases/million, respectively.
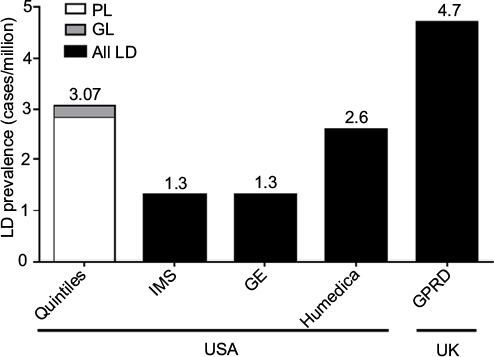 | Figure 1 LD prevalence by adjudicated Quintiles EMR search (diagnosed cases) and simple EMR database searches (IMS LifeLink, GE Healthcare, Humedica EMR, and UK GPRD). Note: Values above the bars report the prevalence of all LD for each database. Abbreviations: EMR, electronic medical record; GE, General Electric; GL, generalized lipodystrophy; LD, lipodystrophy; PL, partial lipodystrophy; UK GPRD, United Kingdom General Practice Research Database. |
Literature searches for AGL, CGL, APL, and FPL identified 7, 31, 11, and 40 publications, respectively, that met the search criteria (Figure 2). The total number of all LD, GL, and PL cases was 334, 122, and 212, respectively. When adjusted for underreporting and extrapolated to the European Union community population (507,751,512 people), the estimated prevalence of all LD was 2.63 cases/million (Figure 3). The estimated prevalence of GL and PL was 0.96 and 1.67 cases/million, respectively.
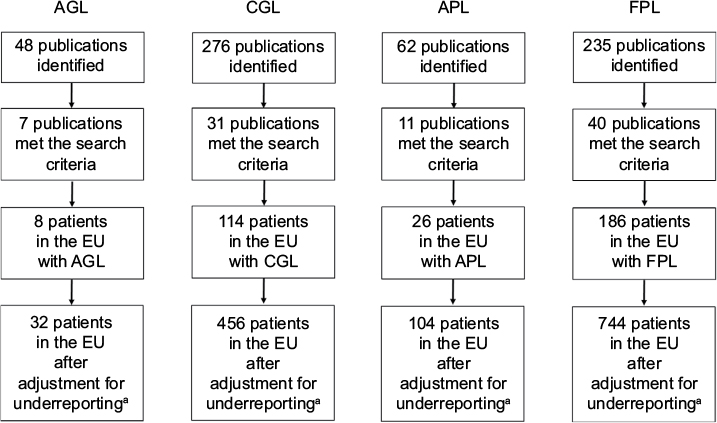 | Figure 2 Flow of literature search results. Note: aGarg13 has estimated that only 25% of all LD cases are reported. Abbreviations: AGL, acquired generalized lipodystrophy; APL, acquired partial lipodystrophy; CGL, congenital generalized lipodystrophy; EU, European Union; FPL, familial partial lipodystrophy; LD, lipodystrophy. |
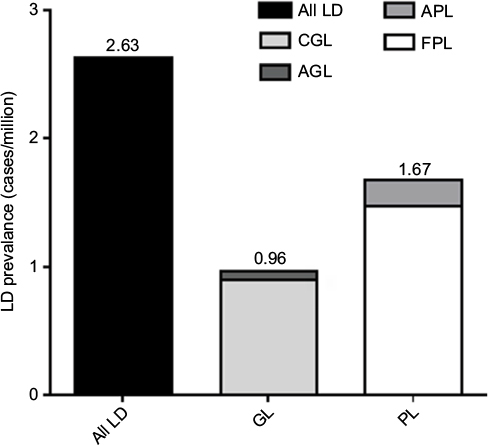 | Figure 3 LD prevalence by literature search. Note: Values above the bars report the prevalence. Abbreviations: AGL, acquired generalized lipodystrophy; APL, acquired partial lipodystrophy; CGL, congenital generalized lipodystrophy; FPL, familial partial lipodystrophy; GL, generalized lipodystrophy; LD, lipodystrophy; PL, partial lipodystrophy. |
Discussion
By evaluating the prevalence of LD from the 5 EMR database searches and 4 literature searches, we have demonstrated that the range of prevalence estimates for all LD (1.3–4.7 cases/million), GL (0.2–1.0 cases/million), and PL (1.7–2.8 cases/million) is smaller and narrower than the range of previously established estimates from multiple and disparate information resources (~0.1–90 cases/million).15,21–29 Our findings confirm that LD is an ultra-rare disease, as it is well below the thresholds established for an ultra-rare disease set forth by the European Union (<1/50,000 individuals).3 A recent report on the natural history of 33 patients with CGL from Turkey estimated a local prevalence of 0.5 cases/million for CGL.33 This estimate of CGL compares well with our estimate of 0.2–1.0 cases/million for GL, which includes both AGL and CGL.
Determining a prevalence estimate for LD was not an easy undertaking. One major limitation related to the EMR database searches was the use of the nonspecific LD ICD-9-CM diagnostic code (Table 3). Codes specific for the 4 major LD subtypes have not been established because the diagnostic criteria have not been finalized. Through the years, several expert opinions and reviews have been published suggesting diagnostic criteria for the 4 major subtypes of LD,12–15 but a unified consensus statement has been elusive to achieve. In the adjudicated Quintiles database search, it was observed that the ICD-9-CM code 272.6 was overused by dermatology and plastic surgery in the USA, causing many false-positive hits and illustrating the need for more accurate forms of verification of true cases. Localized LD has the same ICD-9-CM code and can confound prevalence estimates, especially in patients with diabetes mellitus receiving insulin therapy. It could also be argued that pairing the ICD-9-CM code 272.6 with a metabolic disorder or a diagnostic marker, as was done in the EMR database searches, may have excluded patients with LD who did not have active metabolic abnormalities or abnormalities that were presently controlled.
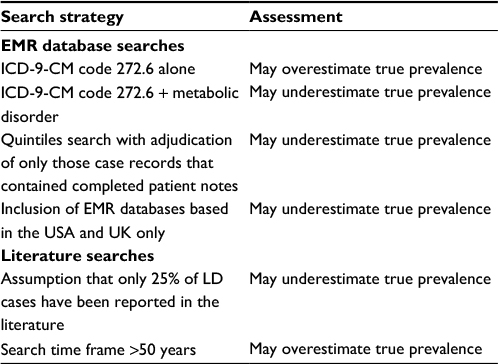 | Table 3 Factors impacting prevalence estimate Abbreviations: EMR, electronic medical record; ICD-9-CM, International Classification of Diseases, Ninth Revision, Clinical Modification; LD, lipodystrophy. |
A second major limitation in establishing a prevalence estimate for the EMR database searches was the variability in information sources. Humedica allowed for ranges of biochemical laboratory values to be searched in addition to the ICD-9-CM code for LD; thus, a more thorough search for LD cases was possible. Other EMR databases were more limited in their search capabilities due to the level of data captured within each database. The Quintiles database, for example, allowed individual patient records to be searched; however, not all patient records contained detailed physician notes, thus making expert adjudication more difficult. It is possible that our estimate from Quintiles underestimated the true prevalence due to only including patients with complete records for adjudication. A final point to variability was the lack of standardization of medical terminology between individual databases. The development of different search criteria for each database in our study could have introduced bias to the prevalence estimates. A third limitation to the EMR database searches was that databases were held and maintained in the USA and the UK. Samples from databases in other regions of the world where LD cases are known to occur would help to add to the range of prevalence estimates that have been presented here.14
One limitation concerning the literature searches is that despite efforts to avoid counting the same patient more than once, there is always the possibility that this occurred due to the lack of sufficient demographic details across publications. The number of cases derived from the literature search was based on the assumption that only 25% of cases are actually reported in the literature; however, this commonly used assumption has never been fully confirmed by a separate study because of the rarity of the disease. A second limitation is the use of a wide range of dates in the literature search (>50 years). It is possible that some patients who were included in the estimate may have been deceased. A third limitation was the decision to limit the literature search strategy to only the 4 major subtypes of LD. The exclusion of novel forms of LD that have not been fully characterized or that fit into the classification scheme of 4 major subtypes may have resulted in an underestimate of all LD prevalence.
It should also be stressed that the purpose of this paper was not to refine the prevalence of the genotypes for the genetic forms of LD, as these data are not readily available in the EMRs. Therefore, we did not specify the prevalence of the reported genotype from the specific papers in the medical literature when this information was sometimes available (but not uniformly). There are distinctive genetic causes for the different forms of LD,13 and more specific attention should be given to estimate the true prevalence of the different genotypes, which exceeds the scope of this paper. Finally, a general limitation in both the EMR database searches and the literature searches relates to the heterogeneity of LD syndromes (especially PL syndromes where patients with FPL type 1 may have been included) and the lack of specific diagnostic criteria to establish a firm diagnosis and, thus, a reliable prevalence estimate.
As investigations into rare diseases are inherently challenged because of a lack of patient cases to draw from, we consider the large number of information sources used in this study to be a strength. By casting a wide net with a search of 5 large claims databases held in the USA and the UK and 4 literature searches using PubMed and EMBASE, we have demonstrated that the range of possible prevalence was narrower and at the lower end of previously available estimates (~0.1–90 cases/million). A second strength of the study was the use of expert adjudication of LD cases in the Quintiles database search. The prevalence of all diagnosed cases of LD identified by Quintiles was 3.07 cases/million, which was approximately in the middle of estimates derived from the other 4 EMR databases using the simple search. To obtain a more precise prevalence estimate, it would have been ideal to have applied this additional layer of adjudication to the search of all 5 EMR databases and to include a search of undiagnosed cases of LD, but this was not possible due to the enormity of such an effort.
The finalization of diagnostic criteria and the establishment of diagnostic coding specific to the 4 major subtypes of LD will greatly help to refine future prevalence estimates derived from the database searches. Furthermore, there is a need to better differentiate FPL type 1 from the rest of FPL cases, as it appears to be distinctly different in etiology and creates confusion on the prevalence of the condition in general. The hope for more precise diagnostic codes assumes that endocrinologists and nonspecialist clinicians will be sufficiently informed and aware of LD to use the code and correctly distinguish from other conditions that may bear a similar clinical presentation to the LD syndromes or be able to differentiate between various LD subtypes. The establishment of registries is one recommended part of regional and national plans to increase the awareness of rare diseases and to provide the natural history of the disease.1 Data collected from a multicenter global registry initiative organized by major academic centers studying these conditions and the upcoming Metreleptin Effectiveness and Safety Registry (MEASuRE; NCT02325674) will help to improve the identification of new patients with LD and increase overall disease awareness. Prospective studies are also needed to validate algorithms independent of diagnostic codes to further define the prevalence of LD.
Conclusion
LD syndromes are a rare body of diseases for which prevalence is currently estimated to be 1.3–4.7 cases/million. By evaluating multiple information sources, we have demonstrated that the range of possible prevalence is smaller and narrower than the range reported in previous estimates (~0.1–90 cases per million). Our methodology describes one approach to estimate the prevalence of a rare disease, which may be of use to other research groups investigating the prevalence of rare diseases. The true prevalence of LD is unknown, but future research will help to further refine and inform the estimates.
Acknowledgments
Robert Schupp, PharmD, CMPP, of inScience Communications, Springer Healthcare (Philadelphia, PA, USA), provided medical writing support funded by Aegerion Pharmaceuticals. Some of the data from this paper were presented in an abstract/poster form at the 2016 European Conference on Rare Diseases & Orphan Products in Edinburgh, Scotland; the 2015 American Diabetes Association Annual Meeting in Boston, MA; and the 2015 International Society for Pharmacoeconomics and Outcomes Research Annual Meeting in Philadelphia, PA. Funding of the study and development of the paper were supported by Aegerion Pharmaceuticals.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
EC is an employee and stockholder of Aegerion Pharmaceuticals. EAO has worked as a consultant/advisor to and has received grant/drug support from Amylin, Bristol-Myers Squibb, AstraZeneca, and Aegerion Pharmaceuticals (manufacturers of metreleptin through the years); Akcea Therapeutics; and Ionis Pharmaceuticals. Unrelated to the field of LD, EAO has received nonmaterial support from Boehringer Ingelheim and grant support from GI Dynamics. EAO is partially supported by R01 DK088114. AG has received research support from Aegerion Pharmaceuticals and Ionis Pharmaceuticals and has worked as a consultant for Aegerion Pharmaceuticals, Biomedical Insights, ClearView Healthcare Partners, Gerson Lehrman Group, and SmithSolve. DA-V has nothing to disclose. PD is an employee of Complete HEOR Solutions, which has done contract work for Aegerion Pharmaceuticals. The authors report no other conflicts of interest in this work.
References
EUROPLAN Partners. Recommendations for the Development of National Plans for Rare Diseases Guidance Document 20100601; 2010. Available from: http://www.europlanproject.eu/Resources/docs/2008-2011_2.EUROPLANRecommendations.pdf. Accessed August 23, 2016. | ||
Commission of the European Communities. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on Rare Diseases: Europe’s Challenges; 2008. Available from: http://ec.europa.eu/health/ph_threats/non_com/docs/rare_com_en.pdf. Accessed August 23, 2016. | ||
EUR-Lex. Regulation (EU) No 536/2014 of the European Parliament and of the Council of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC 2014. Available from: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32014R0536&qid=1421232837997&from=EN. Accessed August 23, 2016. | ||
United States Food and Drug Administration Department of Health and Human Services [webpage on the Internet]. 21CFR316.20 – Content and Format of a Request for Orphan-Drug Designation; 2015. Available from: http://www.ecfr.gov/cgi-bin/text-idx?SID=ad1729044e3018c44badef3eb3824efe&mc=true&node=se21.5.316_120&rgn=div8. Accessed August 23, 2016. | ||
European Medicines Agency. Points to Consider on the Calculation and Reporting of the Prevalence of a Condition for Orphan Designation; 2002. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Regulatory_and_procedural_guideline/2009/09/WC500003773.pdf. Accessed August 24, 2016. | ||
Harknett EC, Chang WY, Byrnes S, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM. 2011;104(11):971–979. | ||
Leadley RM, Lang S, Misso K, et al. A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases. Orphanet J Rare Dis. 2014;9:173. | ||
Hallal C, Kieling CO, Nunes DL, et al. Diagnosis, misdiagnosis, and associated diseases of achalasia in children and adolescents: a twelve-year single center experience. Pediatr Surg Int. 2012;28(12):1211–1217. | ||
Rother KI, Brown RJ. Novel forms of lipodystrophy: why should we care? Diabetes Care. 2013;36(8):2142–2145. | ||
Alpsoy E, Akman-Karakas A, Uzun S. Geographic variations in epidemiology of two autoimmune bullous diseases: pemphigus and bullous pemphigoid. Arch Dermatol Res. 2015;307(4):291–298. | ||
Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35(32):2146–2157. | ||
Brown RJ, Araujo-Vilar D, Cheung PT, et al. The diagnosis and management of lipodystrophy syndromes: a multi-society practice guideline. J Clin Endocrinol Metab. 2016;101(12):4500–4511. | ||
Garg A. Clinical review: lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313–3325. | ||
Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine. 2004;83(1):18–34. | ||
Garg A. Lipodystrophies. Am J Med. 2000;108(2):143–152. | ||
Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore). 2003;82(2):129–146. | ||
Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 1999;84(1):170–174. | ||
Handelsman Y, Oral EA, Bloomgarden ZT, et al. The clinical approach to the detection of lipodystrophy – an AACE consensus statement. Endocr Pract. 2013;19(1):107–116. | ||
Lotta LA, Gulati P, Day FR, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. | ||
Savage DB, Semple RK, Clatworthy MR, et al. Complement abnormalities in acquired lipodystrophy revisited. J Clin Endocrinol Metab. 2009;94(1):10–16. | ||
European Medicines Agency. Public Summary of Opinion on Orphan Designation: Metreleptin for the Treatment of Berardinelli-Seip Syndrome; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/08/WC500131635.pdf. Accessed August 30, 2016. | ||
European Medicines Agency. Public Summary of Opinion on Orphan Designation: Metreleptin for the Treatment of Lawrence Syndrome; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/08/WC500131630.pdf. Accessed August 30, 2016. | ||
European Medicines Agency. Public Summary of Opinion on Orphan Designation: Metreleptin for the Treatment of Barraquer-Simons Syndrome; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/08/WC500131630.pdf. Accessed August 30, 2016. | ||
European Medicines Agency. Public Summary of Opinion on Orphan Designation: Metreleptin for the Treatment of Familial Partial Lipodystrophy; 2015. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Orphan_designation/2012/08/WC500131629.pdf. Accessed August 30, 2016. | ||
Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220–1234. | ||
van Maldergem L [database on the Internet]. Berardinelli-Seip Congenital Lipodystrophy; 2009. Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=1007&Disease_Disease_Search_diseaseGroup=congenital-generalized-lipodystrophy&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Berardinelli-Seip-congenital-lipodystrophy&title=Berardinelli-Seip-congenital-lipodystrophy&search=Disease_Search_Simple. Accessed August 30, 2016. | ||
Vantyghem M-C [database on the Internet]. Partial Acquired Lipodystrophy; 2006. Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11131&Disease_Disease_Search_diseaseGroup=79087&Disease_Disease_Search_diseaseType=ORPHA&Disease(s)/group%20of%20diseases=Partial-acquired-lipodystrophy&title=Partial-acquired-lipodystrophy&search=Disease_Search_Simple. Accessed August 30, 2016. | ||
Vantyghem M-C [database on the Internet]. Acquired Generalized Lipodystrophy; 2009. Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11130&Disease_Disease_Search_diseaseGroup=acquired-generalized-lipodystrophy&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Acquired-generalized-lipodystrophy&title=Acquired-generalized-lipodystrophy&search=Disease_Search_Simple. Accessed August 30, 2016. | ||
Vigouroux C [database on the Internet]. Familial Partial Lipodystrophy; 2015. Available from: http://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=13323&Disease_Disease_Search_diseaseGroup=familial-partial-lipodystrophy&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Familial-partial-lipodystrophy&title=Familial-partial-lipodystrophy&search=Disease_Search_Simple. Accessed August 30, 2016. | ||
United States Food and Drug Administration Department of Health and Human Services [webpage on the Internet]. FAQ Document 2015; 2015. Available from: https://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/ucm240819.htm. Accessed March 1, 2017. | ||
Brown LD, Cat TT, DasGupta A. Interval estimation for a proportion. Stat Sci. 2001;16:101–133. | ||
Sergeant ESG [webpage on the Internet]. Epitools Epidemiological Calculators; 2017. Available from: http://epitools.ausvet.com.au/content.php?page=CIProportion. Accessed May 4, 2017. | ||
Akinci B, Onay H, Demir T, et al. Natural history of congenital generalized lipodystrophy: a nationwide study from Turkey. J Clin Endocrinol Metab. 2016;101(7):2759–2767. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.