Back to Archived Journals » Biosimilars » Volume 5
Establishing a new marketplace for biologic therapy with biosimilar agents: importance of extrapolation of data
Authors Bressler B, Dingermann T
Received 12 December 2014
Accepted for publication 3 March 2015
Published 1 June 2015 Volume 2015:5 Pages 41—48
DOI https://doi.org/10.2147/BS.S78896
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ajay Singh
Brian Bressler,1 Theo Dingermann2
1St Paul’s Hospital, University of British Columbia, Vancouver, BC, Canada; 2Institute of Pharmaceutical Biology, Frankfurt, Germany
Abstract: Despite their enormous value for our health care system, biopharmaceuticals have become a serious threat to the system itself due to their high cost. Costs may be warranted if the medicine is new and innovative; however, it is no longer an innovation when its patent protection expires. As patents and exclusivities expire on biological drugs, biosimilar products defined as highly similar to reference biologics are being marketed. The goal of biosimilar development is to establish a high degree of biosimilarity, not to reestablish clinical efficacy and safety. Current sophisticated analytical methods allow the detection of even small changes in quality attributes and can therefore enable sensitive monitoring of the batch-to-batch consistency and variability of the manufacturing process. The European Medicines Agency (EMA), US Food and Drug Administration (FDA), and Health Canada have determined that a reduced number of nonclinical and clinical comparative studies can be sufficient for approval with clinical data from the most sensitive indication extrapolated to other indications. Extrapolation of data is a scientifically based principle, guided by specific criteria, and if approved by the EMA, FDA, and/or Health Canada is appropriate. Enablement of extrapolation of data is a core principle of biosimilar development, based on principles of comparability and necessary to fully realize cost savings for these drugs.
Keywords: biosimilars, Inflectra, infliximab, pharmacoeconomics, Canada, Europe
Introduction
Biologic drugs are highly complex molecules, which have added tremendous value to modern medicine. Although these drugs may be associated with adverse events, the risks of use are dramatically outweighed by the benefits achieved. In 2013, biologicals comprised an annual global market of US$170 billion, with recombinant insulin, human growth hormone, erythropoietins, and various monoclonal antibodies (mAbs) among the leading categories of products.1 The biopharmaceutical market is predicted to grow at greater than 20% annually as a result of a promising pipeline, approval for more common conditions, increased utilization, and expanded indications.2,3
Biologic drugs are structurally complex and may have several functional domains within a single molecule. For example, individual mAbs present a unique profile with respect to the characteristics of the antigen-binding region, the Fc cytotoxic effector function, and binding to Fc receptors.4 Since the introduction of biologic drugs, more sophisticated assays have been developed that allow for greater in-depth characterization of complex proteins, both on a physicochemical and a functional level. These modern assays permit greater precision for biologic drug development regarding potency and purity.4
Despite their enormous value for our health care system, biopharmaceuticals have become a serious threat to the system itself. Some of the treatments now available are extraordinarily expensive, costing over US$600,000 and more per patient.5 Because of the high cost many patients who qualify for treatment with a biologic drug may be left untreated. Costs may be warranted if the medicine is new and innovative; however, an innovation is finite, and it is no longer an innovation when its patent protection expires.
It is estimated that US$67 billion worth of patents on biologic agents will expire before 2020 (Table 1).6 With the expiry of patents and the development of these new assays, the door has been opened for the development of biosimilar versions of originator agents.
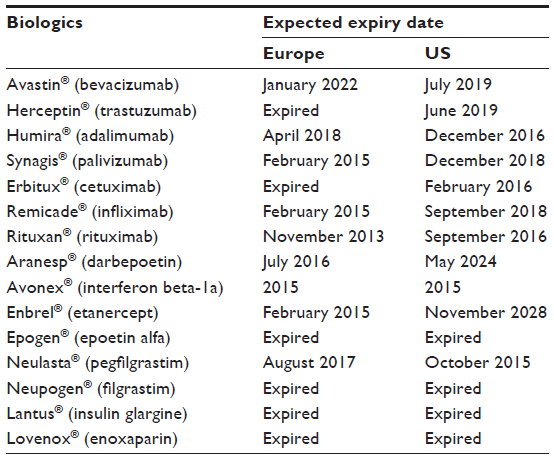 | Table 1 Expiry dates for major patents on best-selling biologicals |
In contrast to conventional, small-molecule drugs, it is impossible to manufacture identical copies of biologics, even from batch to batch within the same manufacturer. Hence, copies of biological agents are not categorized as generic versions of biologicals. They are rather termed “biosimilars” in Europe and the US or “subsequent entry biologics” in Canada.1 Several biosimilars have been approved in Europe and Canada. In the EU, the first patents on biopharmaceuticals expired in 2001, and the first biosimilar medicines were approved by the European Medicines Agency (EMA) in 2006 (Table 2).7 In Canada, the first biosimilar product was approved in 2009 (Omnitrope®, somatropin; Sandoz, Boucherville, QC, Canada).8 The introduction of biosimilars has resulted in substantial cost savings.1 Recently, with the approval of the first biosimilar mAb, the era of biosimilar drugs has entered a second phase.
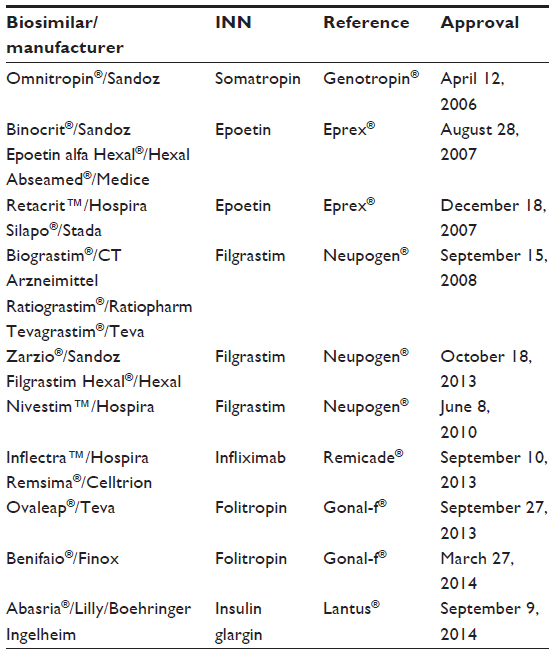 | Table 2 Biosimilar approval in Europe |
The purpose of this article is to provide a brief overview of the principles of biosimilar development and the associated pharmacoeconomic considerations, as well as to address concerns regarding the rationale and importance of extrapolation of data. This article describes the science and the regulatory reasoning for approval or disapproval of indications supported by data extrapolated from alternative disease states. Specifically, infliximab is the first biosimilar antibody discussed.
Regulatory mandate: highly similar (but not identical)
Copying a biotech medicine is much more complex than copying a chemically synthesized drug of low molecular weight.9 A certain degree of variability or “nonidenticality” is a normal principle in biotechnology. Even biomolecules isolated from a human source are heterogeneous to a certain degree on a molecular basis. A highly controlled biotech medicine manufacturing process is intrinsically important because control equals consistency, and consistency equals safety and efficacy. The “art” is to demonstrate that biosimilars are as close as possible or highly similar to their reference products in all relevant functional and structural aspects given the inherent variability.
Biologics are likely to be modified several times throughout their life cycles,10,11 thus even the original name-brand biologicals are not necessarily – after several changes to their original manufacturing process – identical to the original version that was approved by the regulatory body.11 When modifying biologics, the manufacturer must deliver a consistent product quality to guarantee a reproducible clinical performance. However, when the changes are beyond the accepted variances agreed to by the company and the health authority, studies are then required to ensure these changes are not clinically relevant. Current sophisticated analytical methods allow the detection of even small changes in quality attributes and can therefore enable sensitive monitoring of the batch-to-batch consistency and variability of the manufacturing process. Several different factors may account for changes in quality attributes. Although manufacturers try to prevent associated changes in quality attributes, such changes are not always avoidable.12 Evaluation of these changes follows a comparability exercise between the pre- and postchange product that is tightly regulated by the health authorities. The demonstration of comparability does not necessitate that the quality attributes of the prechange and postchange product are identical, but that they are highly similar and that the existing knowledge is sufficiently predictive to ensure similar quality.12 This comparability standard of “similar but not identical” applies to both changes of the original drug and evaluation of biosimilar products.
Clinical comparability: the product is the process
Because the registration of biosimilars leverages the clinical evidence of their originators, the clinical trials programs of biosimilars serve to confirm the totality of the comparability or biosimilarity evidence. This evidence is based on extensive physicochemical characterization of the reference and biosimilar molecules using advanced, state-of-the-art bioanalytical techniques to ensure the range of biosimilar molecule quality attributes are comparable to those of the originator.12
The EMA, US Food and Drug administration (FDA), and Health Canada have issued guidance on the development of biosimilars, including biosimilar mAbs, that provides specific direction with respect to the nonclinical and clinical requirements for biosimilar development. The emphasis of the use of step-wise approaches to the demonstration of biosimilarity is clear in the European guidance, as is the use of comparative studies in all aspects of development.13 These regulatory bodies require that both in vitro and in vivo assays should be sensitive enough to detect differences in bioactivity, pharmacology, and clinical safety and efficacy.
Developing the manufacturing process for biosimilars is challenging as the process must meet strict criteria for robustness. Compared with the manufacturing of organic compounds or traditional drugs, these agents require far greater stringency and documentation, including a greater number of batch records, more product quality tests, more critical process steps, and more process data entries. Fortunately, improvements over the last 20 years in the availability and sophistication of analytical techniques allow a thorough description of all chemical and physical aspects of the molecules and of possible contaminations and impurities. Although the degree of sophistication of current analytical tests has improved tremendously, the fact remains that the safety and efficacy of biosimilars need to be defined independently in formal clinical safety and efficacy trials.
Efficacy and safety
The comparative assessment of efficacy is a key component of the clinical assessment for biosimilars, including biosimilar mAbs. Relying on the totality of evidence learned from the reference product, extensive forethought is given to the setting in which clinical comparability is to be tested.13 This is essential with regard to extrapolation of data to other indications. Selecting a sensitive population with definable endpoints to compare similarity between the biosimilar drug and reference drug is critically important. A comparative trial designed to test for both efficacy and safety is ideal. Studies to compare the efficacy of mAbs should be randomized, double-blinded, and designed to test a hypothesis of equivalence. Equivalence margins should be predefined and supported by statistical estimation based on historical data available for the reference product and by a comparison of the current and prior study designs.13
The comparative assessment of safety also utilizes the totality of evidence approach for biosimilar drug development.13 A descriptive comparison of the types and severity of adverse events occurring after the initiation of treatment must be analyzed. The types and severity of adverse events of the biosimilar are compared to those observed throughout the reference product life cycle. Again, choosing a patient population that enhances the likelihood of detecting a difference is critical to the assessment of clinical differences. A homogeneous patient population may increase the ability to detect differences in safety by reducing contributing factors such as the use of prior therapies.13
Manufacturers of biosimilars must have adequate risk management plans and postmarketing surveillance mechanisms in place to differentiate between the adverse events associated with the proposed product and those associated with the reference product. This includes the identification of adverse events associated with the proposed product that have not been previously associated with the reference product. Authorities may also require a postmarketing study to evaluate certain safety risks. Since immunogenic responses may affect the safety, efficacy, and pharmacokinetics of biotherapeutics, it is essential that biosimilar studies assess the formation of antidrug antibodies in comparative clinical trials.13 The EMA’s Guideline on Immunogenicity Assessment of Monoclonal Antibodies Intended for In Vivo Clinical Use13,14 and the Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies – Non-Clinical and Clinical Issues13,15 provide recommendations on the types of assays that are useful for comparatively assessing the development of neutralizing and nonneutralizing antidrug antibodies.
Granting indication approval and necessity of extrapolation of data
Unlike traditional generics, biosimilars are not automatically granted approval for all indications of the reference product. Extrapolation of data is the leveraging of safety and efficacy data from clinical studies in the most sensitive indications to support the authorization of other less sensitive indications. The cornerstone of biosimilar development rests on “reversed engineering” and on a “reversed body of evidence.” Similarity must be analytically proven and then confirmed by relatively small clinical trials. The evidence originates from the lab and not from the clinic. Modern analytical tests are extremely sensitive, whereas clinical studies are imminently insensitive with regard to small differences in therapeutic outcome.
Extrapolation of data is already an established scientific and regulatory principle that has been exercised for many years, as illustrated when major changes occur in the manufacturing process of originator biologicals.10 In these situations, it is customary for clinical data generated in one indication to be extrapolated to the other indications. There are no cases published where additional clinical studies with the changed product in other or even all approved indications have been considered necessary by regulators.10 Extrapolation into other disease categories is essential to keep the cost of biosimilars competitive. If all disease states must be studied, then the drug would be considered “new.” Without extrapolation of indications, time-consuming prior authorization would be necessary for certain disease states, possibly delaying treatment for the patient. Biosimilar manufacturers must display convincing and compelling data to the regulatory body before extrapolation is granted. Extrapolation guidelines exist in the EU, US, and Canada (Table 3).15–18
 | Table 3 Regulatory factors for biosimilar extrapolation of data |
The decision to extrapolate should be based primarily on the demonstration of similarity through extensive comparability studies that compare the physicochemical attributes and the biological activity between the biosimilar and reference product.13,19 More rigorous clinical testing is required for the indications that are considered the most sensitive, while considerably less clinical testing is required for the less sensitive indications. Analytics and bioanalytics are highly sensitive in detecting differences, whereas traditional clinical endpoints are not. Therefore a high degree of similarity based on analytics and bioanalytics is a very strong argument to rationalize extrapolation.
The EMA’s Committee for Medicinal Products for Human Use guideline on biosimilar mAbs14 states that extrapolation of clinical efficacy and safety data to other indications of the reference mAb, not specifically studied during the clinical development of the biosimilar mAb, is possible based on the overall evidence of comparability provided from the comparability exercise and with adequate justification.20
Biosimilars with extrapolation of data – EU experience
Concerns have been published in the medical literature about using biosimilars for indications that have been licensed on the basis of extrapolation of efficacy and safety data.13,21 Other publications have supported extrapolation of data and advocated for careful postauthorization pharmacovigilance of the biosimilar.10,22,23 Some of the published information regarding biosimilars suggest that clinicians must assume the responsibility of assessing properties or characteristics of these drugs for which they (clinicians) do not have the tools.21,24 It is the biosimilar manufacturer’s responsibility, with guidance from health authorities, to generate robust data and provide scientific justification for extrapolation.
Infliximab
Recently, the first biosimilar mAb, infliximab, was approved in the EU and provides an excellent example of extrapolation of data. The scientific literature suggests that the mechanism of action of infliximab is similar in rheumatologic indications and in psoriasis, through binding to soluble and membrane-bound tumor necrosis factor-alpha (TNFα). However, the Fc region of infliximab may be involved in other potential mechanisms (antibody-dependent cell-mediated cytotoxicity [ADCC] or complement-dependent cytotoxicity) that may play a role in inflammatory bowel disease (IBD).13 Therefore, extrapolation between these indications is not self-evident. The rationale for approval/nonapproval of indications for biosimilar infliximab through extrapolation is discussed.
Biosimilar infliximab (Inflectra™; Hospira, Lake Forest, IL, USA) was approved in the EU and included all indications of the originator product Remicade® (Janssen Pharmceutica, Beerse, Belgium), ie, rheumatoid arthritis (RA), ankylosing-spondylitis, psoriasis as well as IBDs (Crohn’s disease [CD] and ulcerative colitis), like in Korea and Japan.10 The EMA found convincing evidence that any difference detected in Inflectra compared to the reference product had no clinically relevant impact on the efficacy and safety, in particular in IBD.20 Additional in vitro data from human intestinal cells further supported extrapolation of the clinical data to IBD.20 Preliminary clinical data from a very small cohort of 23 patients with CD (15) or ulcerative colitis (eight) indicate similar response to Inflectra compared with historical data on Remicade.20,25 Enrollment of IBD patients in a postmarketing surveillance study and an additional comparative trial of Inflectra versus the reference infliximab (Remicade) in active CD is required by the EMA.20
After review, a biosimilar product may or may not be authorized for all routes of administration, doses, and indications for which the reference product is authorized.13 The EMA, Japan, and Korea granted the first approvals of two biosimilar versions of infliximab for identical indications to the reference product. However in Canada, not all indications were granted. It is postulated that the mechanism of action of infliximab is similar in the rheumatological indications and in psoriasis, ie, binding to soluble and membrane-bound TNFα. The Fc region of infliximab may be involved in other potential mechanisms (ADCC or complement-dependent cytotoxicity) that may influence IBD.13 Concerns based on slight differences in the fucose content of the glycostructure between Inflectra and the reference drug was expressed by Health Canada, that because of the multifaceted mechanisms of action of infliximab in IBD, the safety and efficacy determined from clinical studies in RA and AS where Fc-mediated functions do not appear to play a role may not be predictable.13 The EMA required additional data in order to clarify this concern. The initial differences noted on ADCC were only seen in vitro using target cells that were engineered to overexpress membrane TNF and using enriched natural killer cells from CD patients with the high affinity genotypes of the FcR. However, when ADCC activity was determined using more physiologic effector cells such as whole blood or isolated peripheral blood mononuclear cells, the difference in fucosylation for Inflectra and the reference drug did not impact ADCC.20 This is in line with the opinion of some experts who argue against a major role of the Fc region of infliximab in IBD because IBD patients respond to treatment with Cimzia® (certolizumab pegol), a pegylated anti-TNAα antibody derivative without an Fc region.26
Postmarket monitoring
Regulatory bodies require a comprehensive risk management plan (pharmacovigilance) to be put in place for all biosimilars upon approval. These plans monitor and detect the known and potentially unknown safety signals associated with the use of the biologic agents. Detailed information is sought particularly with respect to the evaluation of immunogenicity of the biosimilar mAb.
First reports show similar immunogenicity between biosimilar infliximab and reference biosimilar. In a Phase III study performed in patients with active RA, antibodies to infliximab were detected in 25.4% (n=69) and 25.8% (n=70) of patients for biosimilar infliximab and reference infliximab at week 14 and 48.4% (n=122) and 48.2% (n=122) of patients for biosimilar infliximab and reference infliximab, respectively, at week 30.27 No significant difference in response was found between reference infliximab and the biosimilar mAb. A Phase I study in patients with active ankylosing spondylitis detected antibodies to infliximab in 9.1% (n=11) and 11.0% (n=13) of patients for biosimilar infliximab and reference infliximab at week 14 and 27.4% (n=32) and 22.5% (n=25) of patients for biosimilar infliximab and reference infliximab, respectively, at week 30.28 No significant differences were found between the infliximab biosimilar and reference infliximab. Although these data are encouraging, postmarketing surveillance of these products, and additional clinical studies, are needed to obtain a good overview on immunogenicity of biosimilar mAb.29 In the above two studies, antibodies against infliximab biosimilar or reference infliximab were measured in a paired manner using an electrochemiluminescent immunoassay method utilizing the Meso Scale Discovery platform (MSD, Rockville, Maryland, USA), and thus were found highly similar to each other. Assay results are highly dependent on the individual sensitivity and specificity of the assay. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medication, and underlying disease. For these reasons, comparison of the incidence of antibodies to infliximab with the incidence of antibodies to infliximab in other studies or to other products may be misleading.30
Pharmacoeconomic considerations
Cost containment is vitally important to the global health care system. This is especially true as the population ages. Globally, many countries are experiencing significant economic pressures. The advent of biosimilars is a crucial factor for establishing a fare market trade with biologic drugs. Without biosimilar development leading to competition, there would be little hope of price curtailment. The cost savings attributed to treatment of patients with biosimilars can be redistributed, possibly improving access to these or newer treatments and reducing cost saving pressures elsewhere.31,32
Health care budget cuts and the mounting evidence on the relative safety of approved biosimilars are contributing to more prescribing of biologics including biosimilars. Since biologics were first introduced, their use has increased greatly, and indeed more patients are receiving biologics than ever before. In 2010, biosimilars’ overall market share in Europe was 15% and is now projected to be over 20%.1 It is estimated that between 2007 and 2020 the use of biosimilars will result in an overall savings of between US$15.5 billion and US $43.5 billion, with most of the cost reductions focused in France, Germany, and the UK.1
Specifically for infliximab,33 a prevalence-based model constructed for budget impact analysis predicted a net savings of US$20 million to US$27 million in six EU countries over 3 years. If budget savings were spent on reimbursement of additional biosimilar infliximab treatment, approximately 1,200 to 1,800 more patients could be treated in the six countries within 3 years.
Conclusion
For the affordability of our health care system and to improve care for patients, a reduction of the costs of biologics should be a priority. Competition after patent expiry is one reasonable tool. By eliminating the monopoly on biologic drugs, manufacturers will be encouraged to concentrate on new innovations. Many unmet medical needs are waiting for safe and efficacious intervention options. Also, potential exists for improving the characteristics of specific molecules towards second or third generation drugs with the newer, sophisticated analytics.
Biosimilars have been available in Europe, with more than 100 million patient days of treatment with biosimilars without unexpected adverse events.34 Extrapolation of data is a scientifically based principle, guided by specific criteria, and if approved by the EMA, FDA, and/or Health Canada is appropriate. Enablement of extrapolation of data is a core principle of biosimilar development based on principles of comparability and necessary to fully realize cost savings for these drugs.
Prescribers, payers, and patients should feel confident in the safety and efficacy of biosimilars with indications and uses granted by their regulatory body. It is believed that the greater the clinicians’ understanding of biosimilars, the more likely they are to grasp the extrapolation concept and why clinical trials are not necessary for all indications. Biosimilar products are now a reality, at least in Europe, and have been marketed without unintended effects, demonstrating similar safety and efficacy to reference products as long as appropriate regulatory supervision is in place.
Acknowledgments
Editorial assistance was provided by contract medical writer Anne Gentry, PharmD (Gentry Medical Communications, LLC [Dublin, OH, USA]) supported by Hospira.
Author contributions
Theo Dingermann and Brian Bressler were equally and primarily responsible for the content of the manuscript, including development of the first draft, writing or approving all subsequent revisions, and/or critically reviewing it for intellectual content.
Disclosure
Brian Bressler is an advisor/speaker for Shire, Ferring, Janssen, AbbVie, and Takeda; an advisor for Pendopharm, Genentech, and Celltrion; and a speaker for Actavis. He has been involved in clinical trials for GlaxoSmithKline, Inc., Bristol-Myers Squibb, Amgen, AbbVie, Janssen, Genentech, Merck, and Boehringer Ingelheim.
Theo Dingermann has received honoraria for oral presentation from the following companies and associations: Hospira, Mundipharma, Teva, Progenerika, and the European Generic Medicines Association. Not relevant to the article, but in the interests of full disclosure, the author declares that he has also received in the past honoraria for oral presentation from the following Biotech companies: Amgen, AbbVie, Genzyme, Merck-Serono, Novo-Nordisk, Roche, Sanofi, Stada, and Pfizer.
References
Cohen JP, Felix AE, Riggs K, Gupta A. Barriers to market uptake of biosimilars in the US. GaBi J. 2014;3(3):107–114. | |
Simoens S. Biosimilar medicines and cost-effectiveness. Clinicoecon Outcomes Res. 2011;3:29–36. | |
Simon F. Market access for biopharmaceuticals: new challenges. Health Aff (Millwood). 2006;25(5):1363–1370. | |
Committee for Medicinal Products for Human Use. Guideline on Similar Biological Medicinal Products containing Monoclonal Antibodies. London: European Medicines Agency; 2010. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/11/WC500099361.pdf. Accessed December 2, 2014. | |
Herper M. The world’s most expensive drugs [webpage on the Internet]. Forbes.com; 2010. Available from: http://www.forbes.com/2010/02/19/expensive-drugs-cost-business-healthcare-rare-diseases.html. Accessed December 3, 2014. | |
US$67 billion worth of biosimilar patents expiring before 2020 [webpage on the Internet]. GaBi Online; 2012 [updated January 20, 2014]. Available from: http://www.gabionline.net/Biosimilars/General/US-67- billion-worth-of-biosimilar-patents-expiring-before-2020. Accessed December 2, 2014. | |
European public assessment reports [webpage on the Internet]. London: European Medicines Agency; 2014. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&mid=WC0b01ac058001d124&searchTab=searchByAuthType&alreadyLoaded=true&isNewQuery=true&status=Authorised&keyword=Enter+keywords&searchType=name&taxonomyPath=&treeNumber=&searchGenericType=biosimilars&genericsKeywordSearch=Submit. Accessed December 3, 2014. | |
Canada’s biosimilar portal [webpage on the Internet]. Toronto: Gowlings; 2014. Available from: http://www.biosimilars.ca/. Accessed December 2, 2014. | |
Crommelin D, Bermejo T, Bissig M, et al. Pharmaceutical evaluation of biosimilars: important differences from generic low-molecular-weight pharmaceuticals. Eur J Hosp Pharm Sci. 2005;11(1):11–17. | |
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196. | |
Schneider CK. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72(3):315–318. | |
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–312. | |
Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: A Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation. J Clin Pharmacol. 2015;55(Suppl 3):S123–S132. | |
Committee for Medicinal Products for Human Use. Guideline on Immunogenicity Assessment of Monoclonal Antibodies Intended for In Vivo Clinical Use. London: European Medicines Agency; 2010 [updated May 24, 2012]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128688.pdf. Accessed December 2, 2014. | |
Committee for Medicinal Products for Human Use. Guideline on Similar Biological Medicinal Products Containing Monoclonal Antibodies – Non-Clinical and Clinical Issues. London: European Medicines Agency; 2010 [updated May 30, 2012]. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed December 2, 2014. | |
Feagan BG, Choquette D, Ghosh S, et al. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42(4):177–183. | |
Health Products and Food Branch, Health Canada. Guidance for sponsors: information and submission requirements for subsequent entry biologics (SEBs) [webpage on the Internet]. Ottawa: Health Canada; 2010. Available from: http://www.hc-sc.gc.ca/dhp-mps/brgtherap/applic-demande/guides/seb-pbu/seb-pbu_2010-eng.php. Accessed December 2, 2014. | |
US Food and Drug Administration. Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (Draft Guidance). Rockville, MD; US Food and Drug Administration; 2012. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed December 2, 2014. | |
Klein AV, Wang J, Bedford P. Subsequent entry biologics (biosimilars) in Canada: approaches to interchangeability and the extrapolation of indications and uses. GaBi J. 2014;3(3):150–154. | |
Committee for Medicinal Products for Human Use. Inflectra Assessment Report. London: European Medicines Agency; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002778/WC500151490.pdf. Accessed December 2, 2014. | |
Danese S, Gomollon F; Governing Board and Operational Board of ECCO. ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis. 2013;7(7):586–589. | |
Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111–5117. | |
Ebbers HC. Biosimilars: in support of extrapolation of indications. J Crohns Colitis. 2014;8(5):431–435. | |
Griffith N, McBride A, Stevenson JG, Green L. Formulary selection criteria for biosimilars: considerations for US health-system pharmacists. Hosp Pharm. 2014;49(9):813–825. | |
Kang YS, Moon HH, Lee SE, Lim YJ, Kang HW. Clinical experience of the use of CT-P13, a biosimilar to infliximab in patients with inflammatory bowel disease: a case series. Dig Dis Sci. Epub October 14, 2014. | |
Mozaffari S, Nikfar S, Abdolghaffari AH, Abdollahi M. New biologic therapeutics for ulcerative colitis and Crohn’s disease. Expert Opin Biol Ther. 2014;14(5):583–600. | |
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–1620. | |
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–1612. | |
Brinks V. Immunogenicity of biosimilar monoclonal antibodies. GaBI J. 2013;2(4):188–193. | |
Remicade® Highlights of Prescribing Information [prescribing information]. Horsham, PA: Janssen Biotech, Inc.; 2013 [revised November 2013]. Available from: http://www.remicade.com/shared/product/remicade/prescribing-information.pdf. Accessed December 2, 2014. | |
Gascón P, Tesch H, Verpoort K, et al. Clinical experience with Zarzio® in Europe: what have we learned? Support Care Cancer. 2013;21(10):2925–2932. | |
Olry de Labry A, Giménez E, Lindner L, García L, Espín J, Rovira J. Biosimilars in the European market. GaBI J. 2013;2(1):30–35. | |
Brodszky V, Baji P, Balogh O, Péntek M. Budget impact analysis of biosimilar infliximab (CT-P13) for the treatment of rheumatoid arthritis in six Central and Eastern European countries. Eur J Health Econ. 2014;15(Suppl 1):S65–S71. | |
Sandoz: Pioneer and global leader in biosimilars [webpage on the Internet]. Sandoz Biopharmaceuticals. Available from: http://www.sandoz-biosimilars.com/aboutus/biosimilars_at_sandoz.shtml. Accessed December 2, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.