Back to Archived Journals » Hypoxia » Volume 4
Erythropoietin and a nonerythropoietic peptide analog promote aortic endothelial cell repair under hypoxic conditions: role of nitric oxide
Authors Heikal L ![]() , Ghezzi P, Mengozzi M, Stelmaszczuk B, Feelisch M, Ferns G
, Ghezzi P, Mengozzi M, Stelmaszczuk B, Feelisch M, Ferns G
Received 16 January 2016
Accepted for publication 23 March 2016
Published 16 August 2016 Volume 2016:4 Pages 121—133
DOI https://doi.org/10.2147/HP.S104377
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Video abstract presented by Lamia Heikal.
Views: 1104
Lamia Heikal,1 Pietro Ghezzi,1 Manuela Mengozzi,1 Blanka Stelmaszczuk,2 Martin Feelisch,2 Gordon AA Ferns1
1Brighton and Sussex Medical School, Falmer, Brighton, 2Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton General Hospital and Institute for Life Sciences, Southampton, UK
Abstract: The cytoprotective effects of erythropoietin (EPO) and an EPO-related nonerythropoietic analog, pyroglutamate helix B surface peptide (pHBSP), were investigated in an in vitro model of bovine aortic endothelial cell injury under normoxic (21% O2) and hypoxic (1% O2) conditions. The potential molecular mechanisms of these effects were also explored. Using a model of endothelial injury (the scratch assay), we found that, under hypoxic conditions, EPO and pHBSP enhanced scratch closure by promoting cell migration and proliferation, but did not show any effect under normoxic conditions. Furthermore, EPO protected bovine aortic endothelial cells from staurosporine-induced apoptosis under hypoxic conditions. The priming effect of hypoxia was associated with stabilization of hypoxia inducible factor-1α, EPO receptor upregulation, and decreased Ser-1177 phosphorylation of endothelial nitric oxide synthase (NOS); the effect of hypoxia on the latter was rescued by EPO. Hypoxia was associated with a reduction in nitric oxide (NO) production as assessed by its oxidation products, nitrite and nitrate, consistent with the oxygen requirement for endogenous production of NO by endothelial NOS. However, while EPO did not affect NO formation in normoxia, it markedly increased NO production, in a manner sensitive to NOS inhibition, under hypoxic conditions. These data are consistent with the notion that the tissue-protective actions of EPO-related cytokines in pathophysiological settings associated with poor oxygenation are mediated by NO. These findings may be particularly relevant to atherogenesis and postangioplasty restenosis.
Keywords: erythropoietin, pyroglutamate helix B surface peptide, scratch assay, proliferation, migration, apoptosis
Introduction
Inflammation and hypoxia are often associated with tissue injury and are involved in wound repair and atherogenesis.1,2 We have previously reported that the reparative effects of erythropoietin (EPO) on endothelial cells is more evident under 5% oxygen (O2) compared to atmospheric oxygen concentrations (21%).3 However, 5% O2 is close to physiological tissue oxygen concentrations, while persistent, more severe hypoxia (<1% O2), such as following ischemia, can be deleterious and is associated with endothelial injury due to effects on a variety of cellular processes.4 Hypoxia induces the expression of several cytokines such as vascular endothelial growth factor (VEGF) and EPO, which, in turn, help orchestrate the chronic adaptation to hypoxia. While the principal effect of EPO is to increase the number of red blood cells and thus improve oxygenation, it is also tissue protective and prevents ischemic injury of the vascular endothelium.5–7 These protective effects of EPO may be mediated in part by its stimulation of endothelial cell proliferation and migration (in cases of endothelial damage), inhibition of apoptosis and inflammation, and induction of angiogenesis.8–10 These effects are mediated by a tissue-protective receptor, which is distinct from the canonical homodimeric EPO receptor (EPOR) mediating its erythropoietic effects, and comprises a heterodimeric complex composed of EPOR and the common β-subunit of receptors for granulocyte-macrophage colony-stimulating factor, interleukin-3, and interleukin-5 (βCR, also known as CD131).
Nonerythropoietic analogs of EPO that are tissue protective but not erythropoietic may represent a potentially safer and more effective intervention for the treatment of vascular disease, as they lack the erythropoietic properties of EPO that may lead to some of its adverse vascular complications.5,11,12 EPO and its nonerythropoietic analogs have been shown to promote wound healing13,14 and confer protection in models of cardiovascular injury.15,16 We have previously shown that these nonerythropoietic analogs (carbamylated EPO and the synthetic peptide pyroglutamate helix B surface peptide [pHBSP]) exhibit tissue-protective effects that are similar to EPO in an in vitro vascular cell injury model at a low oxygen tension (5% O2) but not in normoxia.3 The aim of the present investigation was to study the potentially protective effects of EPO and its novel nonerythropoietic analog pHBSP under conditions in which the hypoxia was more pronounced (1% O2) and to determine the putative molecular mechanisms by which these two entities confer protection. For this purpose, we used an in vitro model of wound healing (the “scratch assay”) in bovine aortic endothelial cells (BAECs). The effects of EPO and pHBSP on cellular migration, proliferation, and apoptosis were studied and related to the formation of nitric oxide (NO) by measuring NO production and testing the effect of specific enzyme inhibitors and NO scavengers.
Materials and methods
All chemicals were from Sigma-Aldrich Co. (St Louis, MO, USA), unless otherwise stated. The peptide (pHBSP, or ARA290; pyroglu-EQLERALNSS) and the scrambled peptide (scr-pHBSP; pyroglu-LSEARNQSEL) used as a control peptide for our experiments were provided by Araim Pharmaceuticals (Tarrytown, NY, USA). Typically, 100× stock solutions were prepared fresh in phosphate-buffered saline (PBS) and sterile-filtered before addition to cells; 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide (PTIO) and staurosporine were kept frozen as 1,000× aliquots in dimethyl sulfoxide.
Cell culture
BAECs were obtained from European Collection of Authenticated Cell Cultures (ECACC) (Salisbury, UK) and used between passages 4 and 12. The cells were cultured in Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin/streptomycin (final concentration 100 IU/mL) and were cultured, prior to our experiments, at 37°C in a humidified atmosphere containing 5% CO2 and 21% O2 (corresponding to an effective O2 concentration of 18.6%).17 When indicated, hypoxic experiments were performed under 1% O2, 5% CO2, and 94% N2 in an acrylic chamber where O2 was maintained at such a low level using a CO2 and O2 controller (ProOx model c21, BioSpherix, Ltd, New York, USA).
Scratch assay
The scratch assay was performed as previously described.3 The effects of EPO, its peptide analog (pHBSP), or a scrambled peptide (scr-pHBSP) as a control were investigated in either 21% or 1% O2. A reproducible scratch was produced in the endothelial monolayer as previously described, and thereafter, either EPO or pHBSP or scr-pHBSP was added to the cells and subsequently incubated in a 21% or 1% O2 for 24 hours. Optimal concentrations for each agonist were established in earlier experiments (Figure S1A).3 The defined area of the scratch was photographed under an inverted microscope (Olympus CKX41; Olympus Corporation, Tokyo, Japan) at 10× magnification using a Micropix 5 megapixel color complementary metal-oxide semiconductor digital camera (Olympus Cooperation). The position of the wound image was standardized each time against a horizontal line drawn on the base of the plate passing through the center of each well. The scratch area was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
In some experiments, either the nitric oxide synthase (NOS) inhibitor NG-nitro-l-arginine methyl ester (l-NAME; 300 μM) or l-NG-monomethyl-l-arginine (l-NMMA; 1 mM) was added 30 minutes prior to the addition of EPO. To test whether the effect observed was NO dependent regardless of its source, the NO scavenger PTIO (150 μM) was used and added 30 minutes prior to addition of EPO. l-NAME was freshly prepared for each experiment, while l-NMMA and PTIO were used from frozen stock solutions.
Cell viability assay
Cell viability was evaluated using the trypan blue exclusion test as previously described.3 Briefly, cells were seeded into 96-well plates at a density of 1×104 cells/mL (0.15 mL/well) in culture medium. After 24 hours, the supernatant was removed and replaced by 150 μL fresh medium containing EPO, pHBSP, or scr-pHBSP at a concentration of 1 ng/mL, and the cells were incubated at 21% or 1% O2. After 24 hours, trypan blue was added and the cells that were stained (dead) and unstained (live) were counted. Results were expressed as viable cell count per milliliter. To test whether the effects of EPO or its analog were mediated by NO, in some experiments, we used the NOS inhibitor l-NAME (300 μM).
Migration assay
A micro-Boyden chamber assay (NeuroProbe, Gaithersburg, MD, USA) was used to assess the effect of 1 ng/mL EPO, pHBSP, and scr-pHBSP on cell migration.18 Migrated cells were stained using Diff-Quick stain (Gamidor Technical Services Ltd, Didcot, UK) and counted under 40× magnification. To investigate whether the effects of EPO and its analog were mediated by endothelial NOS (eNOS). Some experiments were performed in the presence of 300 μM of the NOS inhibitor l-NAME.
Apoptosis
Caspase-3 activity measurement assay
Caspase-3/7 activity was measured using the Caspase-Glo 3/7 assay (Promega Corporation, Fitchburg, WI, USA). Cells were plated into 96-well plates at a seeding density of 5×104 cell/mL and cultured for 24 hours in 1% or 21% O2. Cells were then treated with either EPO (1 ng/mL) or vehicle (medium) for 3 hours, before apoptosis was induced by adding 500 nM staurosporine for 18 hours. Reconstituted Caspase-Glo 3/7 reagent was added to each well at a volume equal to that of the cell culture media (100 μL caspase-Glo reagent to 100 μL cell culture medium). Plates were mixed gently using a plate shaker at 300–500 rpm for 30 seconds and incubated at room temperature for 1 hour; 100 μL of each well was then transferred to a Corning Costar 96-well White Solid Plate (Thermo Fisher Scientific, Waltham, MA, USA) and luminescence was measured using a Synergy HTX plate reader (BioTek, Swindon, UK).
DeadEnd colorimetric TUNEL assay
Terminal deoxynucleotidyl transferase dUTP (2’-deoxyuridine 5’-triphosphate) nick end labeling (TUNEL) assay was also used to measure cell apoptosis. BAECs were cultured on poly l-ornithine coated cover slips until reaching confluence. Cells were then cultured under 21% or 1% O2 for 24 hours prior to treatment with EPO and staurosporine as described earlier. Thereafter, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 minutes at room temperature. Fixed cells were then washed with PBS and treated with 1% Triton X-100 in PBS (Sigma-Aldrich Co.) for 10 minutes at room temperature to permeabilize the plasma membrane. The proportion of apoptotic cells was then detected in the fixed permeabilized cells following a standard protocol (Promega Corporation).19 A light microscope (40× magnification) was used to quantify the staining.
Real-time quantitative polymerase chain reaction (qPCR)
Cells were seeded into 24-well plates and cultured until ~80% confluent and were then exposed to 1% or 21% O2 for 24 hours. The effect of 1 ng/mL EPO on gene expression was assessed at several time points (0, 0.5, 1, and 24 hours). Cells were then lysed using TRIzol (Thermo Fisher Scientific), and RNA was extracted and purified as described previously.20 RNA quality and concentration were determined using NanoDrop ND-1000 (NanoDrop Technologies, Thermo Fisher Scientific, Waltham, MA, USA).20 Reverse transcription and real-time qPCR for EPOR, βCR, VEGF, and β2-microglobulin (a housekeeping gene not affected by changes in O2 levels) were carried out on RNA samples using Taqman gene expression assays (Thermo Fisher Scientific) as previously reported.20,21 For gene expression quantification, the comparative threshold cycle (ΔΔCt) method was used following the guidelines of Thermo Fisher Scientific. Results were normalized to β2-microglobulin expression and expressed as arbitrary units using one of the normoxic samples as a calibrator as specified in the figure legend (Figure 4). VEGF, a hypoxia-induced gene, was measured as a positive control to validate the method and conditions used for the experiment.
Western blot
BAECs were seeded into 24-well plates at a seeding density of 1×105 cell/mL and cultured until 80% confluency, and were then exposed for a further 24 hours under 1% or 21% O2. Cells were then lysed and their protein quantified as previously described.3 Thirty micrograms of cellular proteins was separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (GE Healthcare UK Ltd, Little Chalfont, UK). After blocking with 5% skimmed milk (for EPOR and hypoxia inducible factor-1α [HIF-1α] detection) or 5% bovine serum albumin (for βCR, phosphorylated eNOS [p-eNOS], and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) detection) for 1 hour, the membranes were incubated with the appropriate primary antibody overnight, followed by horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. βCR, p-eNOS, and GAPDH (loading control) were detected using rabbit anti-βCR (N-20; Santa Cruz Biotechnology Inc., Dallas, TX, USA), rabbit anti-p-eNOS (9571S; New England Biolabs Ltd, UK), and rabbit anti-GAPDH (14C10; New England Biolabs Ltd, Hertfordshire, UK) at a dilution of 1:200, 1:2,000, and 1:1,000, respectively, and an antirabbit secondary antibody (A0545; Sigma-Aldrich Co.) at 1:5,000 dilution for βCR and 1:20,000 dilution for p-eNOS and GAPDH. EPOR was detected using goat anti-EPOR (W-20; Santa Cruz Biotechnology Inc.) at 1:200 dilution and an anti-goat secondary antibody (A8919, Sigma-Aldrich Co.) at 1:10,000 dilution. HIF-1α was detected using mouse anti-HIF-1α (NB 100-105, R&D systems, Abingdon, UK) at 1:500 dilution and an antimouse secondary antibody (ADI-SAB-100; Enzo Life Sciences Ltd, Exeter, UK) at 1:5,000 dilution. Protein bands were visualized by exposing the membranes developed with the Enhanced Chemiluminescence (ECL) reagent to chemiluminescence film (Hyperfilm ECL, GE Healthcare UK Ltd). Bands were quantified using ImageJ software (National Institutes of Health).
Measurement of NO
NO production in BAECs was quantitatively measured in the form of its stable oxidation products, nitrite (NO2−) and nitrate (NO3−), as previously described.22 Briefly, BAECs were seeded in six-well plates at a seeding density of 1×106 cells/mL. Cells were then either left untreated or treated with 1 ng/mL EPO, 1 mM l-NMMA, or a combination of EPO and l-NMMA and incubated at either 1% or 21% O2. After 24 hours, the cell culture medium was collected, snap frozen in liquid N2, and stored at −80°C for later analysis of extracellular nitrite and nitrate release, replace by; a measure of NOS activity. In addition, intracellular nitrite and nitrate levels were measured as follows. After removal of the supernatant, the treated BAECs were washed twice with PBS to minimize contamination with extracellular nitrite/nitrate and subjected to three successive freeze/thaw cycles in the presence of a limited volume (250 μL) of PBS. The fractured cells and cell lysates from each well were collected using a rubber policeman, quantitatively transferred to a prewashed Eppendorf vial, followed by methanol precipitation (1:1 v/v) of proteins and centrifugation at 11,000 ×g. The supernatant was transferred to a prewashed cryovial, snap frozen, and stored at −80°C for up to 1 week before analysis. Nitrite and nitrate were quantified simultaneously using a specific and sensitive high-performance liquid chromatography technique that employs ion chromatography with online reduction of nitrate to nitrite and subsequent postcolumn derivatization with Griess reagent (ENO-20; EiCom, Kyoto, Japan).22 Calibration curves for nitrite and nitrate standards (0.01–50 μM in PBS) were constructed daily, and an internal quality control was run after every ten samples; using a 20 μL injection loop, the detection limit was 25 nM for each anion. Cell culture medium without the cells being treated under otherwise identical conditions or PBS served as control. All values reported are corrected for the corresponding blanks.
Statistical analysis
All data were analyzed using GraphPad Prism 4 software (GraphPad Software, Inc., La Jolla, CA, USA). Differences in treatment (with or without EPO or its analogs) were tested for significance using one-way analysis of variance followed by a Bonferroni correction for multiple comparisons post hoc test. The t-test was used to compare the expression of EPOR or βCR under different O2 levels.
Results
Hypoxia stimulates reparative effects of EPO and its analogs
As shown in Figure 1A, EPO and pHBSP significantly enhanced scratch closure, 24 hours after treatment, in BAECs cultured under conditions of acute hypoxia (1% O2). However, EPO and its peptide analog did not significantly improve wound closure in cells maintained in 21% O2. No significant effect was observed after treatment with the scrambled peptide (scr-pHBSP) under either 21% or 1% O2.
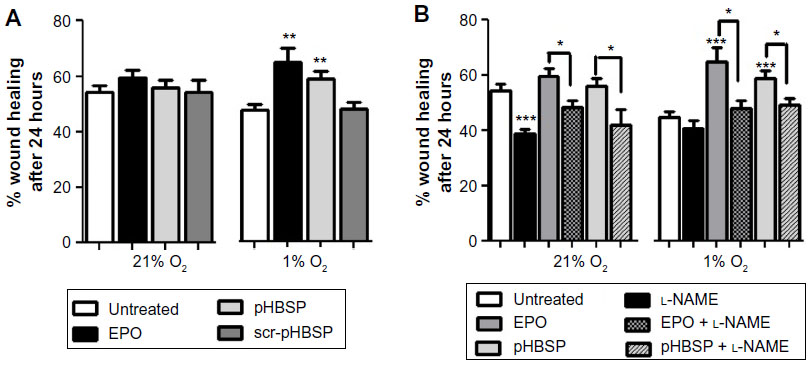 | Figure 1 EPO and pHBSP stimulate repair in a scratch assay model in BAEC under hypoxia. |
The eNOS inhibitor, l-NAME was used at the concentration giving optimum inhibition as verified by a concentration response curve on wound closure (300 μM) (Figure S1B). l-NAME inhibited wound closure significantly when added to endothelial cells kept at 21% O2 but showed no effect on untreated cells under 1% O2. l-NAME also inhibited wound closure in the presence of concomitant treatment with EPO or pHBSP at both 21% O2 and 1% O2 (Figure 1B).
EPO and pHBSP induce proliferation and migration of BAECs under hypoxic conditions
EPO and pHBSP had little effect on cell proliferation at 21% O2 but stimulated proliferation to a comparable extent in hypoxia (Figure 2A). Treatment with scr-pHBSP did not affect proliferation under either 21% or 1% O2. NOS inhibition by l-NAME had a significant inhibitory effect on proliferation of BAECs under 21% O2 (Figure 2B), but this effect was not observed under 1% O2. The EPO-induced enhancement of cell proliferation in hypoxia was completely inhibited by l-NAME (Figure 2B). Similar findings were observed on cell migration (Figure 2C and D).
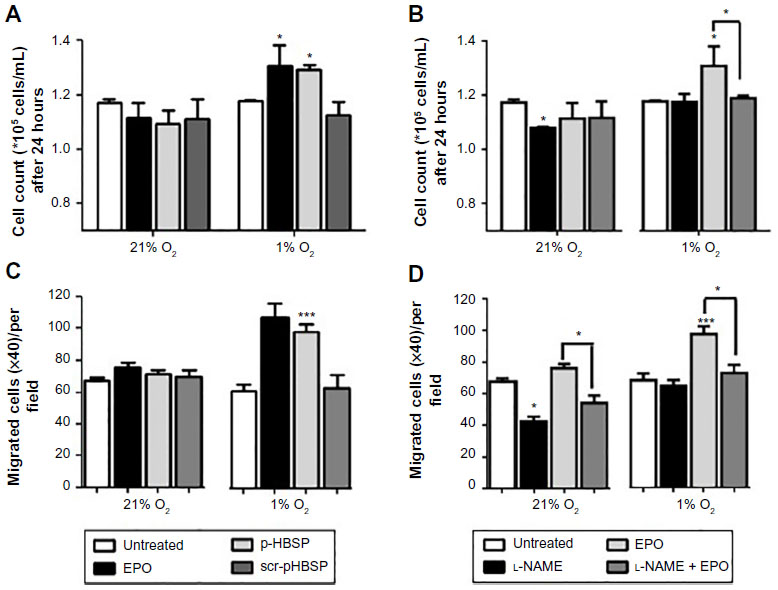 | Figure 2 EPO and pHBSP stimulate proliferation and migration of BAECs under hypoxia. BAECs were treated with EPO, pHBSP, and scr-pHBSP at 1 ng/mL and were then incubated under either 21% or 1% O2 for 24 hours. |
Antiapoptotic effect of EPO is stimulated under hypoxic conditions
To investigate the potential antiapoptotic effects of EPO and its nonerythropoietic analog, apoptosis was induced by exposing BAECs to 500 nM staurosporine for 18 hours. Both EPO and pHBSP had a significant antiapoptotic effect under 1% O2, but not under 21% O2. EPO inhibited staurosporine-induced apoptosis by approximately 50% on using either the TUNEL (Figure 3A and B) or caspase assay (Figure 3C).
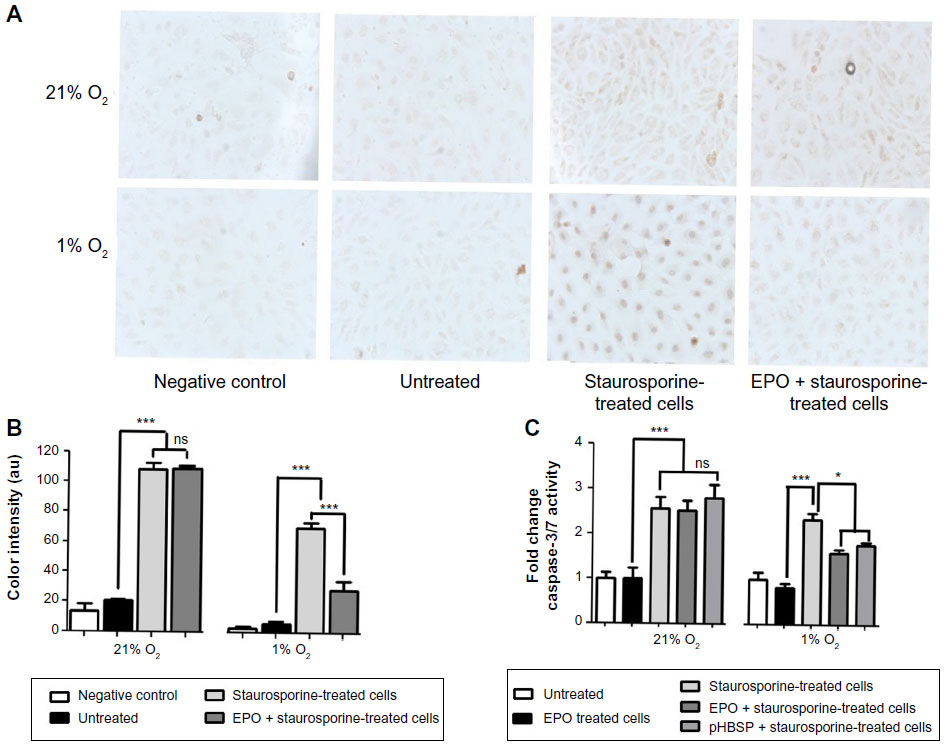 | Figure 3 Antiapoptotic effects of EPO and pHBSP under hypoxic, but not normoxic conditions. Apoptosis was induced in BAECs by treatment with 500 nM staurosporine for 18 hours after incubation with or without EPO or pHBSP for 3 hours. |
Hypoxia regulates different EPO-related downstream signaling pathways
qPCR and Western blot were used to study the effect of EPO on both the mRNA and protein expression, respectively, of selected genes and proteins. The effect of EPO on EPOR and βCR mRNA levels is shown in Figure 4; EPOR mRNA levels were significantly increased by nearly twofold in 1% O2. In contrast, the expression of βCR did not change significantly under the same conditions. Addition of EPO did not appear to affect either EPOR or βCR expression under hypoxic conditions at the time points investigated. VEGF was used as a positive control (representative hypoxia-inducible gene) and was found to increase fourfold to sixfold in 1% O2.
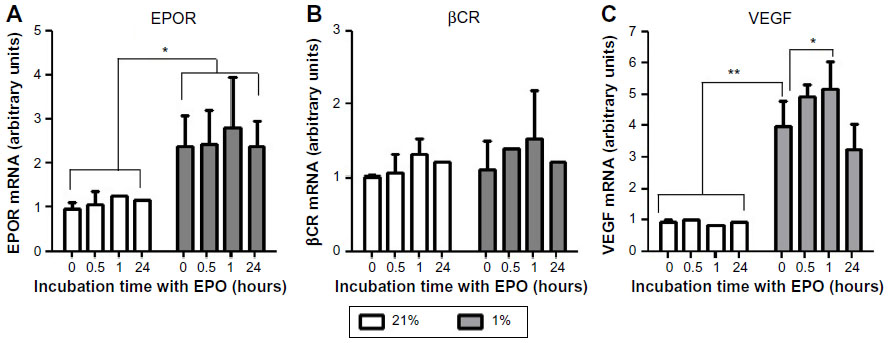 | Figure 4 Hypoxia increased gene expression of EPOR, but not the βCR in BAECs. Cells were cultured under 21% or 1% O2 for 24 hours and treated with EPO (1 ng/mL) for 0, 0.5, 1, and 24 hours. |
We also studied some of the EPO-related signaling pathways that might play a role in its reparative action. For this purpose, we measured the protein expression of EPOR, βCR, HIF-1α, and p-eNOS by Western blot. Consistent with the mRNA measurements by PCR, EPOR expression was increased in 1% O2 compared to 21% O2 while the expression of βCR was unaltered. Expression of p-eNOS, on the other hand, decreased significantly under hypoxic conditions, but this effect was offset by treatment with EPO (Figure 5). Moreover, hypoxia was found to increase inducible NOS (iNOS) expression and EPO to suppress this response, but unsurprisingly, overall levels of expression of this NOS isoform are extremely low when compared to eNOS (Figure S2). Taken together, both Western blot and PCR showed that hypoxia affects the regulation of several downstream pathways. Under our experimental conditions, this regulation did not appear to be affected by EPO, with the notable exception of eNOS phosphorylation at Ser-1177. The latter may be of relevance in determining NO output of endothelial cells as phosphorylation at Ser-1177 is known to be associated with enhanced NOS activity.23 Although likely to be of minor importance, we cannot exclude that iNOS may also contribute to NO production under hypoxic conditions.
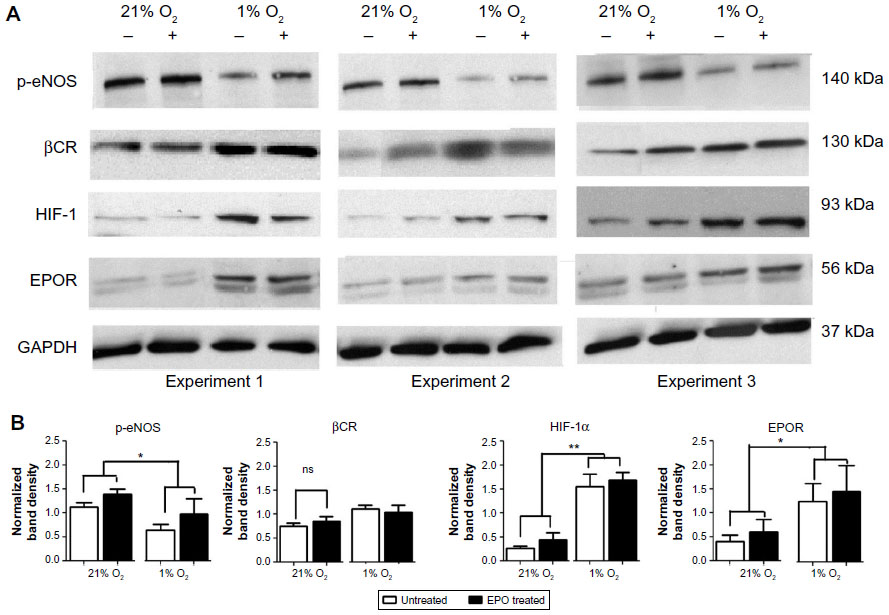 | Figure 5 EPOR but not βCR protein expression increases under hypoxic conditions. |
EPO stimulates NO production in hypoxia, but not in normoxia
NO production by BAECs was assessed by the formation of nitrate and nitrite, and both their extracellular and intracellular concentrations were determined. In general, endothelial NO production was significantly lower in cells incubated in 1% O2 compared to those incubated in 21% O2, as evidenced by decreases in both intracellular and extracellular nitrite and nitrate concentrations. In these experiments, l-NMMA was used as a NOS inhibitor instead of l-NAME because l-NAME metabolites were found to interfere with the determination of trace levels of nitrite by high-performance liquid chromatography and l-NMMA produced similar responses to l-NAME in the scratch assay (Figure S3). As shown in Figure 6A–D, EPO caused an increase in NO production in hypoxic cells compared to untreated cells. This effect was not observed in normoxia.
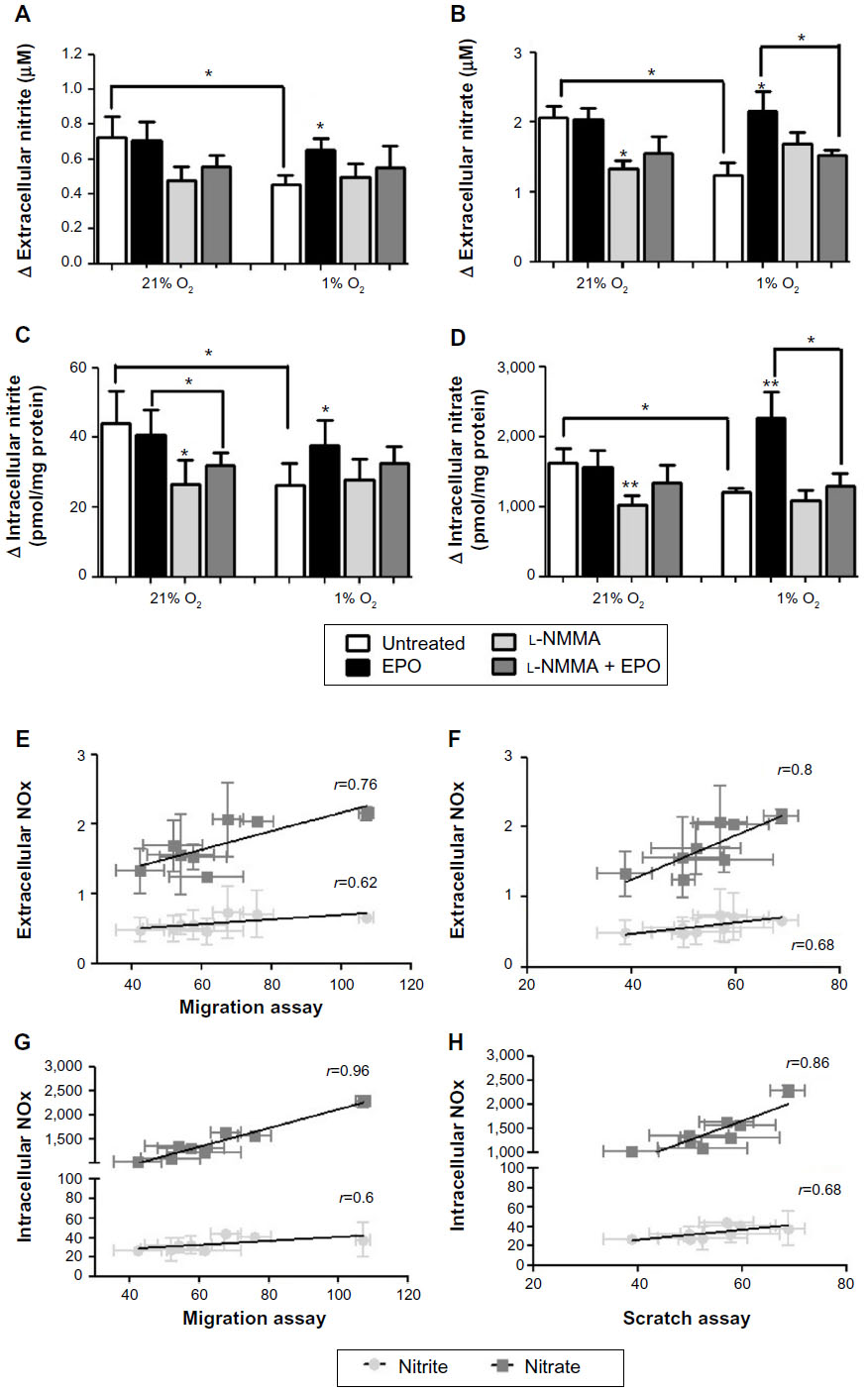 | Figure 6 Effect of EPO on NO production in 1% or 21% O2. |
l-NMMA not only decreased basal cellular NO production in normoxia but also partially counteracted the stimulatory effects of EPO on NO production in hypoxia (Figure 6A–D). We also tried to correlate the production of NO in the various experimental conditions reported in Figures 1, 2, and 6A–D. Association analyses reported in Figure 6E–H show that both cell migration and repair correlate with NO production, possibly suggesting that NO is important for the reparative effects of EPO in this model.
To confirm this assumption, we performed additional experiments with the NO scavenger PTIO, which showed that, irrespective of its source of formation (ie, whether it is generated through a NOS-dependent or -independent pathway), NO is essential for the cellular wound healing process, regardless of the prevailing oxygen tension (Figure 7). PTIO also inhibited the stimulated reparative effects of EPO and pHBSP under hypoxic condition (Figure 7). This action of PTIO was not due to an effect on cell viability as confirmed using the trypan blue exclusion method. Cell viability in the presence and absence of PTIO was 89.8%±4.2% in untreated cells versus 87.1%±4.7% in PTIO-treated cells under normoxia (n=3) and 87.2%±3.4% in untreated cells versus 84.8%±9.1% in PTIO-treated cells under hypoxic condition (n=3).
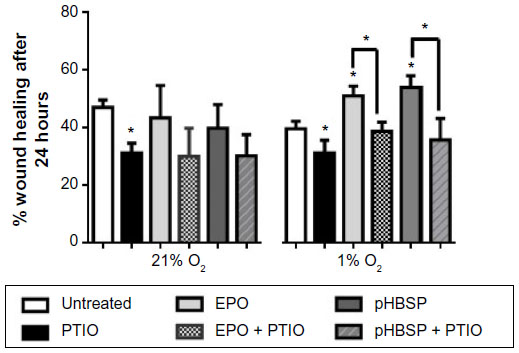 | Figure 7 PTIO (150 μM) inhibits repair in a scratch assay model in BAECs under both normoxia and hypoxia both in untreated cells (*P<0.05) and in EPO/pHBSP-treated cells (*P<0.05). |
Taken together, our findings demonstrate that hypoxia is an important determinant of the response to EPO and its analogs and that NO formation may be involved in the downstream signaling pathways.
Discussion
We have previously shown that EPO and its nonerythropoietic analogs enhance the regrowth of an endothelial monolayer following a scratch injury under low oxygen tension (5% O2), but not under normoxia (21% O2). The partial pressure of oxygen varies within the normal artery wall, even in health,24 and areas of profound hypoxia (<1% O2) may prevail in regions of diseased artery, for example, in macrophage-rich regions of atheroma.25 Furthermore, 5% O2 is not true hypoxia and is, in fact, a normal oxygen concentration for many tissues. In this present study, we demonstrated a priming effect of profound hypoxia on the reparative action of EPO and pHBSP. Hypoxia promotes a set of signaling pathways that directly affect vascular cell proliferation and survival, or act indirectly to influence the response to locally produced growth factors.26 These responses are largely mediated by the activation of the transcriptional factor, HIF-1, which controls the transcription of DNA to mRNA of more than a hundred target genes.27 There is increasing evidence that HIF-1 plays a critical role in mediating the protective effects of hypoxic episodes by inducing cytoprotective molecules such as EPO.28 In our experiments, we measured HIF-1α as a marker of cellular responses to hypoxia.
The expression of EPOR determines the responsiveness of cells to EPO.28 We have previously shown that at normal O2 tension (21%), BAECs express EPOR at low levels, leading to low EPO activity, and that at a lower O2 tension (5%), there is an increase in EPOR expression, priming endothelial cells to the reparative activity of EPO.29 This repair process appears to be due to the well-characterized ability of EPO and its analogs to promote cell proliferation and migration.26,30 We found in this study that these effects were also observed at 1% O2. As shown by others earlier, these effects may involve activation of PI3K/AKT phosphorylation, JAK2/STAT5, RAS/MAPK, and the antiapoptotic pathway involving Bcl-2 and Bcl-XL,28,31 but the proximal trigger for these changes has not been unequivocally identified.
The eNOS-mediated production of NO is a key regulator of vascular tone and blood flow.32 It is also involved in regulating endothelial permeability, platelet aggregation, leukocyte adhesion, and cell migration.32,33 Recent studies have suggested that eNOS has an essential role in the cytoprotective and tissue-protective effect of EPO.34,35 Wound repair in vivo is also modulated by NO, affecting several pathophysiological processes including inflammation, chemotaxis, antibacterial defenses, collagen production, and angiogenesis.36
Oxygen tension may influence NO signaling by affecting NOS-dependent NO production, a process that requires O2. At low O2 tension, eNOS expression and activity is thought to be reduced, and NO production by alternative, NOS-independent pathways, for example, the reduction of nitrite to NO, may occur.37,38 This notion is supported by the lack of l-NAME effect on the migration, proliferation, and wound closure of hypoxic cells compared to normoxic cells. In the present study, we found little evidence for the involvement of NOS-independent pathways in mediating the effects of EPO on BAEC proliferation, migration, and apoptosis under hypoxic conditions, since all of these reparative effects were reduced substantially by NOS inhibition. Therefore, under hypoxic conditions, it is possible that NO-mediated wound healing is decreased to a large degree by a combination of two effects, l-NAME inhibition of eNOS and the reduction of eNOS expression and activity by hypoxia. This does not rule out other, NOS-independent, pathways; however, our findings suggest that, at least in this experimental model, they may not play a major role.
Earlier experiments have shown that EPO stimulates NO production in endothelial cells and that this effect is more marked in hypoxia.39 This effect of EPO could occur through an upregulation of eNOS phosphorylation at Ser-1177, which activates the enzyme, and our current experiments show that EPO restores eNOS phosphorylation under hypoxic conditions. The lower activity and expression of activated NOS in endothelial cells under hypoxic conditions could explain why wound healing is delayed in hypoxia. We further extend our earlier observations on the stimulation of NO production by EPO by demonstrating that the reparative effects of EPO and pHBSP are abrogated not only by NOS inhibition but also by the presence of an NO scavenger. Moreover, we showed for the first time that the extent of NO stimulation by EPO correlates with the magnitude of its biological effects under all experimental conditions, providing additional support for a crucial role of NO in mediating the action of EPO and its analogs.
We also studied the possible involvement of iNOS as a source of NO. As expected, in normoxic cells, we could only find a very faint band for iNOS by Western blot, but its expression was clearly increased under hypoxic conditions. This effect was partially inhibited when cells were treated with EPO. Due to the complexity of the system and possibility of multiple sources of NO production, the quantitative analysis of nitrite and nitrate was essential to determine the involvement of NO under different oxygen tensions.
Altogether, we found that hypoxia enhances the reparative effect of EPO (and its analogs), and this may explain the protective effects observed when these molecules are administered in several models of ischemic injury. Understanding the mechanisms responsible for these effects on the vascular endothelium could provide novel regenerative therapeutics in the treatment of cardiovascular disease, specifically in restenosis following angioplasty. Our findings strongly support the role of NO in mediating the reparative effects of EPO and its analogs under hypoxic conditions, corroborating and extending observations from other groups in different model systems.40
Conclusion
In this study, the reparative effects of EPO and its nonerythropoietic analog p-HBSP were assessed in cultured BAECs under hypoxic and normoxic conditions. We have shown that hypoxia primes the reparative cellular effects of EPO and p-HBSP and that these are largely mediated by eNOS activation and enhanced cellular NO production. Further work should be directed at disentangling the precise nature of this interaction and its possible relationship to cellular redox status and some of the signaling events downstream of the emerging EPO/EPOR/NO axis, underpinning its beneficial biological effects.
Disclosure
The authors report no conflicts of interest in this work.
References
Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: a comprehensive review of studies in mice. Cardiovasc Res. 2008;79(3):360–376. | |
Silvestre JS, Mallat Z, Tedgui A, Levy BI. Post-ischaemic neovascularization and inflammation. Cardiovasc Res. 2008;78(2):242–249. | |
Heikal L, Ghezzi P, Mengozzi M, Ferns G. Low oxygen tension primes aortic endothelial cells to the reparative effect of tissue-protective cytokines. Mol Med. 2015;21:709–716. | |
Ahluwalia A, Tarnawski AS. Critical role of hypoxia sensor – HIF-1 alpha in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr Med Chem. 2012;19(1):90–97. | |
Sanchis-Gomar F, Perez-Quilis C, Lippi G. Erythropoietin receptor (EpoR) agonism is used to treat a wide range of disease. Mol Med. 2013;19:62–64. | |
Martinez-Estrada OM, Rodriguez-Millan E, Gonzalez-de Vicente E, Reina M, Vilaro S, Fabre M. Erythropoietin protects the in vitro blood-brain barrier against VEGF-induced permeability. Eur J Neurosci. 2003;18(9):2538–2544. | |
Chong ZZ, Kang JQ, Maiese K. Angiogenesis and plasticity: role of erythropoietin in vascular systems. J Hematother Stem Cell Res. 2002;11(6):863–871. | |
Anagnostou A, Liu ZY, Steiner M, et al. Erythropoietin receptor messenger-RNA expression in human endothelial cells. Proc Natl Acad Sci U S A. 1994;91(9):3974–3978. | |
Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert Rev Mol Med. 2008;10:1–23. | |
Bahlmann FH, de Groot K, Spandau JM, et al. Erythropoietin regulates endothelial progenitor cells. Blood. 2004;103(3):921–926. | |
Coleman TR, Westenfelder C, Togel FE, et al. Cytoprotective doses of erythropoietin or carbamylated erythropoietin have markedly different procoagulant and vasoactive activities. Proc Natl Acad Sci U S A. 2006;103(15):5965–5970. | |
Bohr S, Patel SJ, Vasko R, et al. Modulation of cellular stress response via the erythropoietin/CD131 heteroreceptor complex in mouse mesenchymal-derived cells. J Mol Med (Berl). 2015;93(2):199–210. | |
Brines M, Patel NSA, Villa P, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A. 2008;105(31):10925–10930. | |
Erbayraktar Z, Erbayraktar S, Yilmaz O, Cerami A, Coleman T, Brines M. Nonerythropoietic tissue protective compounds are highly effective facilitators of wound healing. Mol Med. 2009;15(7–8):235–241. | |
Fiordaliso F, Chimenti S, Staszewsky L, et al. A nonerythropoietic derivative of erythropoietin protects the myocardium from ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2005;102(6):2046–2051. | |
Ueba H, Shiomi M, Brines M, et al. Suppression of coronary atherosclerosis by helix B surface peptide, a nonerythropoietic, tissue-protective compound derived from erythropoietin. Mol Med. 2013;19:195–202. | |
Wenger R, Kurtcuoglu V, Scholz C, Marti H, Hoogewijs D. Frequently asked questions in hypoxia research. Hypoxia. 2015;3:35–43. | |
Zwezdaryk KJ, Coffelt SB, Figuero YG, et al. Erythropoietin, a hypoxia-regulated factor, elicits a pro-angiogenic program in human mesenchymal stem cells. Exp Hematol. 2007;35(4):640–652. | |
Chen Z-Y, Wang L, Asavaritkrai P, Noguchi CT. Up-regulation of erythropoietin receptor by nitric oxide mediates hypoxia preconditioning. J Neurosci Res. 2010;88(14):3180–3188. | |
Mengozzi M, Cervellini I, Villa P, et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci U S A. 2012;109(24):9617–9622. | |
Cervellini I, Annenkov A, Brenton T, Chernajovsky Y, Ghezzi P, Mengozzi M. Erythropoietin (EPO) increases myelin gene expression in CG4 oligodendrocyte cells through the classical EPO receptor. Mol Med. 2013;19:223–229. | |
Rassaf T, Bryan NS, Kelm M, Feelisch M. Concomitant presence of N-nitroso and S-nitroso proteins in human plasma. Free Radic Biol Med. 2002;33(11):1590–1596. | |
Chen C-A, Druhan LJ, Varadharaj S, Chen Y-R, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283(40):27038–27047. | |
Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999;19(4):870–876. | |
Buscombe JR. Exploring the nature of atheroma and cardiovascular inflammation in vivo using positron emission tomography (PET). Br J Radiol. 2015;88(1053):20140648. | |
Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling (vol 16, pg 771, 2002). FASEB J. 2006;20(9):1573–1573. | |
Marsch E, Sluimer JC, Daemen MJAP. Hypoxia in atherosclerosis and inflammation. Curr Opin Lipidol. 2013;24(5):393–400. | |
Marzo F, Lavorgna A, Coluzzi G, et al. Erythropoietin in heart and vessels: focus on transcription and signalling pathways. J Thromb Thrombolysis. 2008;26(3):183–187. | |
Beleslin-Cokic BB, Cokic VP, Yu XB, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood. 2004;104(7):2073–2080. | |
Trincavelli ML, Da Pozzo E, Ciampi O, et al. Regulation of erythropoietin receptor activity in endothelial cells by different erythropoietin (EPO) derivatives: an in vitro study. Int J Mol Sci. 2013;14(2):2258–2281. | |
Lester RD, Jo M, Campana WM, Gonias SL. Erythropoietin promotes MCF-7 breast cancer cell migration by an ERK/mitogen-activated protein kinase-dependent pathway and is primarily responsible for the increase in migration observed in hypoxia. J Biol Chem. 2005;280(47):39273–39277. | |
Atochin DN, Huang PL. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch. 2010;460(6):965–974. | |
Murohara T, Witzenbichler B, Spyridopoulos I, et al. Role of endothelial nitric oxide synthase in endothelial cell migration. Arterioscler Thromb Vasc Biol. 1999;19(5):1156–1161. | |
Burger D, Lei M, Geoghegan-Morphet N, Lu X, Xenocostas A, Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovasc Res. 2006;72(1):51–59. | |
d’Uscio LV, Smith LA, Santhanam AV, Richardson D, Nath KA, Katusic ZS. Essential role of endothelial nitric oxide synthase in vascular effects of erythropoietin. Hypertension. 2007;49(5):1142–1148. | |
Madigan MC, McEnaney RM, Shukla AJ, et al. Xanthine oxidoreductase function contributes to normal wound healing. Mol Med. 2015;21:313–322. | |
Hickok JR, Vasudevan D, Jablonski K, Thomas DD. Oxygen dependence of nitric oxide-mediated signaling. Redox Biol. 2013;1(1):203–209. | |
Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7(2):156–167. | |
Cokic BBB, Cokic VP, Suresh S, Wirt S, Noguchi CT. Nitric oxide and hypoxia stimulate erythropoietin receptor via MAPK kinase in endothelial cells. Microvasc Res. 2014;92:34–40. | |
Zhang Y, Wang L, Dey S, et al. Erythropoietin action in stress response, tissue maintenance and metabolism. Int J Mol Sci. 2014;15(6):10296–10333. |
Supplementary materials
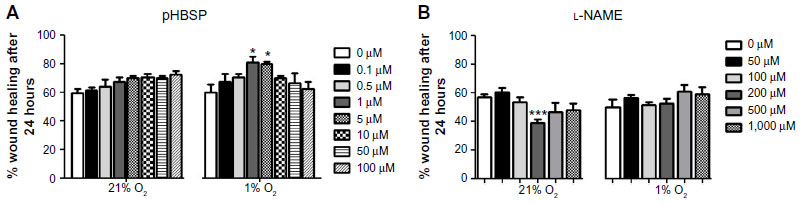 | Figure S1 Figure S1 Effect of different concentrations of (A) pHBSP (0–100 μM) and (B) l-NAME (0–1,000 μM) on wound closure in BAECs under 21% or 1% O2. |
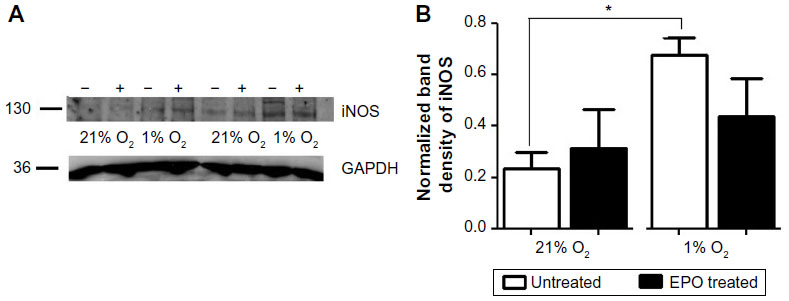 | Figure S2 Figure S2 iNOS expression increased under hypoxic conditions |
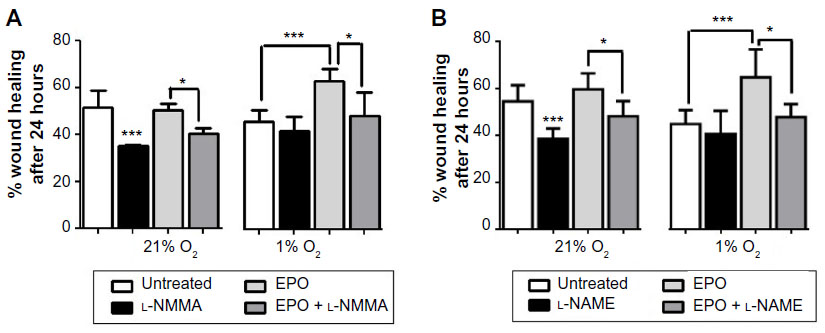 | Figure S3 Both l-NAME (300 μM) and l-NMMA (1 mM) decreased wound closure for untreated cells under 21% O2, but showed no significant effect under 1% O2. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.