Back to Journals » OncoTargets and Therapy » Volume 13
ERK Activation-Mediated Autophagy Induction Resists Licochalcone A-Induced Anticancer Activities in Lung Cancer Cells in vitro
Authors Luo W, Sun R, Chen X, Li J, Jiang J, He Y, Shi S ![]() , Wen H
, Wen H
Received 2 September 2020
Accepted for publication 3 December 2020
Published 8 January 2021 Volume 2020:13 Pages 13437—13450
DOI https://doi.org/10.2147/OTT.S278268
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Wei Luo,1,* Ruifen Sun,2,* Xin Chen,3,* Ju Li,4 Jike Jiang,4 Yuxiao He,4 Shaoqing Shi,2 Heling Wen5
1Department of Respiratory and Critical Care Medicine, The People’s Hospital of Leshan, Leshan, Sichuan, People’s Republic of China; 2Center for Scientific Research, Yunnan University of Chinese Traditional Medicine, Kunming, Yunnan, People’s Republic of China; 3College of Chinese Medicine, Yunnan University of Chinese Medicine, Kunming, Yunnan, People’s Republic of China; 4College of Basic Medicine, Yunnan University of Chinese Medicine, Kunming, Yunnan, People’s Republic of China; 5Department of Cardiology, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, Chengdu, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Heling Wen
Department of Cardiology, Sichuan Academy of Medical Sciences and Sichuan Provincial People’s Hospital, No. 32th West Second Section First Ring Road, Qingyang District, Chengdu, Sichuan 610072, People’s Republic of China
Tel/Fax +86-28-87394174
Email [email protected]
Shaoqing Shi
Center for Scientific Research, Yunnan University of Chinese Traditional Medicine, No. 1076th Yuhua Road, Chenggong District, Kunming, Yunnan 650500, People’s Republic of China
Tel +86-871-65918050
Fax +86-871-65918049
Email [email protected]
Introduction: The incidence and mortality rates of lung cancer rank top in the different types of cancers in China. Licochalcone A (LA) is a flavonoid extracted from the roots of licorice with antitumor effects in various cancers in vitro and in vivo. However, the role of LA in non-small cell lung cancer (NSCLC) remains largely unclear.
Methods: The cell viability was measured by MTT assay, Edu staining and colony formation assay. Apoptosis was investigated using Annexin V/PI double-stained assays with flow cytometry. Real-time quantitative RT-PCR was carried out to investigate the expression of mRNA of related proteins. Western blotting was used to investigate the expression of related proteins.
Results: The results show that LA inhibits the proliferation of NSCLC cells in a dose-dependent manner and induces apoptotic cell death. Moreover, LA significantly suppresses the expression of c-IAP1, c-IAP2, XIAP, Survivin, c-FLIPL and RIP1 without influencing the level of mRNA. Cycloheximide chase assay demonstrates that LA greatly decreases the stability of Survivin, XIAP and RIP1. Mechanistic studies indicate that LA induces cytoprotective autophagy since block of autophagy with CQ greatly enhances LA-induced anticancer activities. Furthermore, LA rapidly induces ERK and p38 activation in a time-dependent manner in both A549 and H460 cells, but suppresses the activities of c-Jun N-terminal kinase (JNK); suppression of ERK not p38 with inhibitor attenuates LA-induced autophagy, while it remarkably enhances LA-induced cytotoxicity in lung cancer cells and further promotes the degradation of apoptosis-related proteins.
Discussion: The results of this study provide novel insights on the role of apoptosis-related proteins and the MAPKs pathway in the anticancer activities of LA.
Keywords: non-small cell lung cancer, NSCLC, licochalcone A, LA, inhibitors of apoptosis proteins, IAPs, RIP1, ERK, autophagy
Introduction
As one of the most common malignancies, lung cancer severely endangers the life and health of people worldwide. The incidence and mortality rates of this disease rank top of different types of tumors in China.1 Lung cancer mainly includes non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for about 85% of lung cancer diagnoses, the 5-year survival rate of which is only about 15%, which may be attributed to late stage of diagnosis and lack of effective therapies.2–4 Surgery, chemotherapy and radiotherapy are the main options for late-stage NSCLC patients, but the effects are still not satisfactory. Therefore, it is of great significance to explore novel anticancer agents for late-stage NSCLC treatment.
A flavonoid isolated from the Chinese medicinal herb Glycyrrhiza uralensis Fisch,5 licochalcone A (LA) possesses a wide spectrum of pharmacological effects, including antibacterial,6,7 anti-inflammatory,8,9 antioxidative,10,11 and anticancer properties. Recently its antitumor activities have attracted great interest of researchers. It has been demonstrated that LA inhibits the proliferation of different types of cancers, including gastric cancer BGC-823, AGS, SGC-7091 and MKN-45 cells,12,13 bladder cancer T24 and 5367,14 hepatocellular carcinoma HepG2 cells,15 breast cancer MDA-MB-231 cells,16 glioblastoma U87 cells,17 nasopharyngeal carcinoma HONE-1, NPC-39 and NPC-BM cells,18 cervical cancer SiHa and Hela cells,19 osteosarcoma HOS and MG-63 cells,20 lung cancer A549, H1299, H292 and H460,21–23 and so on. In vivo studies also reveal that LA is a potent anticancer agent in gastric cancer, cervical cancer and colon cancer. Notably, LA greatly attenuates cisplatin-induced kidney toxicity without influencing anticancer activities. Moreover, LA causes endoplasmic reticulum (ER) stress and apoptosis via upregulating miR-144-3p in human lung cancer cell line H292,22 it inhibits proliferation, causes G2/M cell cycle arrest and induces apoptosis in lung cancer A549 and H460 cells in a dose-dependent manner,23 and causes little cytotoxicity in Beas-2B and HELF cells, indicating LA selectively inhibits lung cancer cells. Additionally, LA induces autophagic induction in lung cancer, however, it seems that autophagy is not involved in LA-induced anticancer activities since block of autophagy with inhibitors or siRNA did not reverse LA-induced antitumor effects.21 LA suppresses the migration and invasion of lung cancer,24 hepatocellular carcinoma,25,26 oral cancer,27 and breast cancer cells,16,28 and also synergizes 5-fluorouracil29 and sorafenib25 to inhibit gastric cancer and hepatocellular cancer, respectively.
Inhibitors of apoptosis proteins (IAPs, including c-IAP1, c-IAP2, XIAP, Survivin and livin) are the main inhibitors of cell proliferation and cell death.30–32 It has been reported that these proteins are highly expressed in many types of tumors, and predict poor diagnosis for several anticancer agents, including cisplatin.33,34 Previous studies have reported that LA reduces the mRNA level of Survivin in human hepatoma cells,35 and consistently decreases the expression of Survivin in oral squamous cell carcinoma36 and ovarian carcinoma cells.37 LA also downregulates the expression of Mcl-1 in oral squamous cell carcinoma36 and human osteosarcoma cells. Previous studies have demonstrated that chalcone and its derivatives are promising anticancer agents with little toxicity. We recently reported that chalcone derivative Chal-2438 synergizes cisplatin39 and TRAIL40-induced anticancer effects by decreasing the expression of IAPs and c-FLIPL in NSCLC cells. However, until now the role of IAPs and c-FLIPL in LA-induced anticancer effects remains undefined.
In the present study, we extend our interest to apoptosis-related proteins in LA-induced anticancer activities in lung cancer in vitro. We investigate the role of apoptosis-related proteins in LA-induced anticancer activities in NSCLC cells in vitro. We demonstrate that LA inhibits cell viability and induces apoptosis in NSCLC cells. LA significantly decreases the expression of apoptosis-related proteins without greatly influencing the expression of mRNA. Moreover, autophagy protects lung cancer cells from LA-induced cytotoxicity because suppression of autophagy with CQ significantly increased cell death. Furthermore, LA greatly induced ERK and p38 activation, but suppressed the activities of c-JunN-terminal kinase (JNK); block of ERK not p38 with inhibitor greatly attenuates LA-induced autophagic induction and remarkably enhances LA-induced cytotoxicity. The data from this study suggest ERK activation-mediated autophagy induction plays a protective part to resist LA-induced cell death in lung cancer.
Materials and Methods
Reagents and Antibodies
Anti-c-IAP1 (7065), c-IAP2 (3130), XIAP (2045), PARP (9542), RIP1 (3493), HSP90 (7065), phosphor-p38 MAPK (4511), LC3 (4108) and GAPDH (2118) were from Cell Signaling Technology (Danvers, MA, US). Antibody for JNK1 (201001), P44/42 MAPK (ERK1/2) (250222), p38 (220979), Survivin (71G4B7), β-tubulin (200608) was from Zen Bioscience (Chengdu, Sichuan, China). Anti-caspase 3 (ab32351) and c-FLIPL were from Abcam (Cambridge, UK). Anti-phospho-JNK (2679369) and phosphor-MAP kinase 1/2 (Erk1/2) were from Millipore. Licochalcone A, z-vad-fmk-1, U0126 and chloroquine (CQ) were from MCE MedChemExpress.
Cell Culture
Lung cancer cell line A549, H460, SPC-A1, H23 and H1299 were obtained from America Type Culture Collection (Manassas, VA, USA) and grown in RPIM 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U penicillin and 100 µg/mL streptomycin. All cells were cultured in standard incubator conditions at 37°C with 5% CO2. RPMI 1640 Medium without glucose and fetal bovine serum were purchased from Gibco (USA).
Cell Viability Assay
Cells were seeded in triplicate data density of 5×104/well and after staining with trypan blue were counted every day for 3 days. In some experiments, cells were seeded in triplicate at a density of 2.5×104/well. The next day and thereafter, cells were incubated with 20 mg/mL MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) for 3 h. MTT was then dissolved in DMSO and read at OD 570 nm. The results were expressed as relative cell number against the corresponding readings from the cells on day set to 100.
Colony Formation Assay
Lung cancer cells were seeded into 6-well plates at a density of 5×103 per well. After overnight growth, the cells were treated with LA (10 μM) for 7 days. Cells were fixed with methanol, then stained with 1% crystal violet dye. The visible colonies were photographed, and the colonies containing at least 50 cells were counted.
EdU Staining
DNA synthesis was analyzed using the Cell-Light EdU Apollo488 In Vitro Imaging Kit as per the manufacturer’s instructions (RiboBio, Guangzhou, China).
Annexin V-Fluorescein Isothiocyanate/PI Double-Stained Assays
Apoptosis was investigated using the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (BD Biosciences, San Jose, CA, USA). Lung cancer cells were seeded in 6-well plates and cultured with 0.1% DMSO or LA (20 μM) for 36 h. Afterward, the cells were collected and fixed and stained in binding buffer with 5 µL of PI solution and 5 µL of FITC-conjugated Annexin V for 15 min in the dark. After staining, FACS Calibur flow cytometry (BD FACSCalibur, Becton Dickinson Co.) was performed to detect apoptotic cells, and the data were analyzed with Flowjo software.
RNA Extraction
Total RNA was extracted from cells using Transzol (TaKaRa, Japan), chloroform (Sangon, Shanghai, China), isopropyl alcohol (Sangon, Shanghai, China), DEPC water (Sangon, Shanghai, China) and 75% ethanol (Sangon, China). The yield of RNA was determined using a NanoDrop 2000 spectrophotometer (Thermo Scientific, USA), and the integrity was evaluated using agarose gel electrophoresis stained with ethidium bromide.
Real-Time Quantitative RT-PCR
Quantification was performed with a two-step reaction process: reverse transcription (RT) and PCR. The PrimerScriptTM RT reagent Kit with gDNA Eraser (TaKaRa, Japan) was used for each RT reaction in a total volume of 10 μL. Reactions were performed in a GeneAmp® PCR System 9700 (Applied Biosystems, USA) for 15 min at 37°C, followed by heat inactivation of RT for 5 s at 85°C. The 10 μL RT reaction mix was then diluted × 10 in nuclease-free water and held at −20°C.
Real-time PCR was performed using ABI Prism 7900 Real-time PCR Instrument (Applied Biosystems, USA) with the TransStart® Top Green qPCR SuperMix (+Dye I/+Dye II)-qPCR kit (Transgen Biotech, China) in a 10 μL PCR reaction mixture. Reactions were incubated in a 96-well optical plate (Transgen Biotech, China) at 94°C for 30 min, followed by 40 cycles of 94°C for 50 s, 60°C for 30 s. Each sample was run in triplicate for analysis. At the end of the PCR cycles, melting curve analysis was performed to validate the specific generation of the expected PCR product. The primer sequences were designed in the laboratory and synthesized by Beijing TSINGKE, based on the mRNA sequences obtained from the NCBI database as follows: Primer sequences for real-time PCR are shown in Table 1.
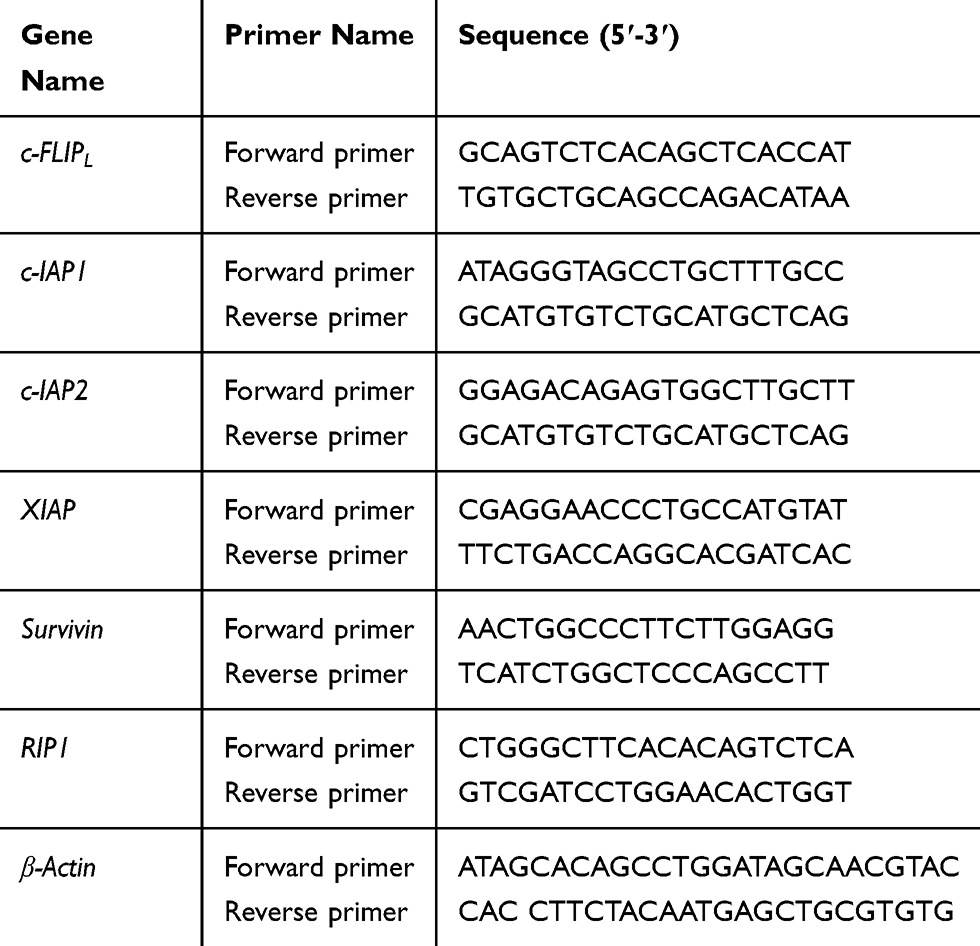 |
Table 1 Primer Sequences for Real-Time PCR |
Western Blotting
Cell lysates were prepared by suspending cells in M2 buffer (20 mM Tris-HCl pH 7.6, 0.5% NP40, 250 mM NaCl, 3 mM EDTA, 2 mM DTT, 0.5 mM phenylmethylsulfonylfluoride, 20 mM β-glycerophosphate, 1 mM sodium vanadate, and 1 µg/mL leupeptin). The samples were boiled for 5 minutes, equal amounts of proteins from each of the cell lysates were resolved by 8% or 12% SDS-PAGE and analyzed by Western blot. The proteins were visualized with enhanced chemiluminescence (Millipore) following the instructions of the manufacturer. Each experiment was repeated at least three times and representative results are shown.
Statistical Analysis
All data were expressed as mean ± SD and statistical significance was examined with one-way analysis of variance (ANOVA) pairwise comparison using SPSS 17.0 for Windows (SPSS Inc., Chicago, IL, USA). P < 0.05 was considered statistically significant.
Results
LA Promotes NSCLC Cells Apoptosis
To investigate the anticancer effects of LA, A549, H460, SPC-A1, H23 and H1299 cells were exposed to increasing concentrations of LA for 48 h, then cell viability was detected and qualified by MTT assay. As shown in Figure 1A, LA inhibited the proliferation of NSCLC cells in a dose-dependent manner. EdU staining and Colony formation assay also showed that LA suppressed the proliferation of lung cancer cells (Figure 1B and C). Consistent with previous studies, flow cytometry analysis showed that LA promoted apoptotic cell death in both A549 and H460 cells (Figure 2A), meanwhile LA remarkably induced caspase-3 activation and PARP cleavage (hallmarks of apoptosis) (Figure 2C), and pan-caspases inhibitor z-VAD-fmk blocked LA-induced cytotoxicity (Figure 2B), indicating LA induced apoptotic cytotoxicity in NSCLC cells in vitro. Together, these results indicate that LA mainly induces apoptotic cytotoxicity in lung cancer cells.
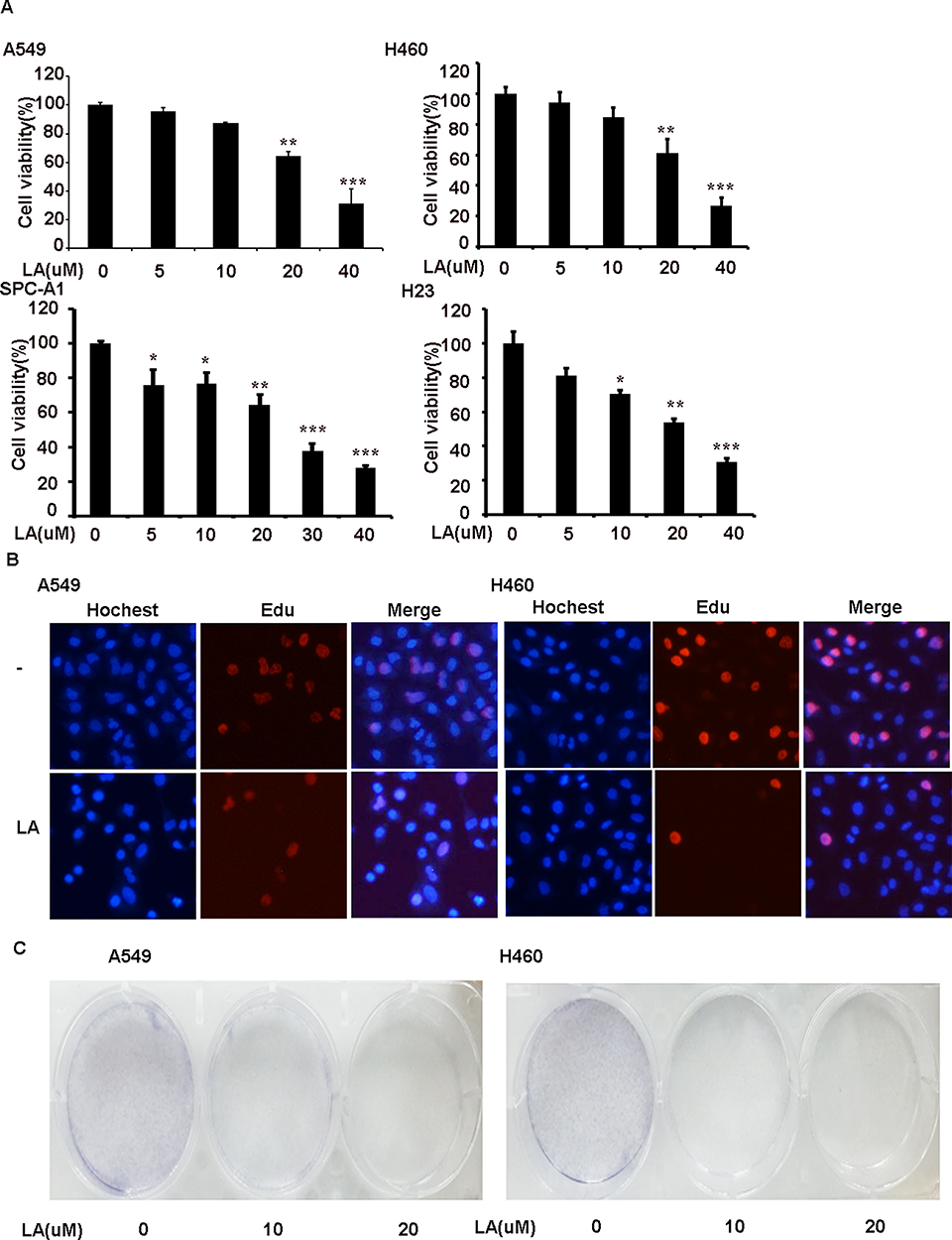 |
Figure 1 LA suppresses the viability of NSCLC cells in a dose-dependent manner. (A) A549 and H460 cell were exposed to indicated concentrations of LA for 48 h, cell viability was measured and quantified by MTT assay. (B) Lung cancer cells were exposed to LA for 8 h, then EdU staining was carried out. (C) 5×103 cells were seeded on a culture plate and treated with LA (10 µM) for 7 days. Cells were fixed with 1% formalin containing 1% crystal violet and colony formation inspected using an inverted microscope. N=3. *P<0.05, **P<0.0.1, ***P<0.001. |
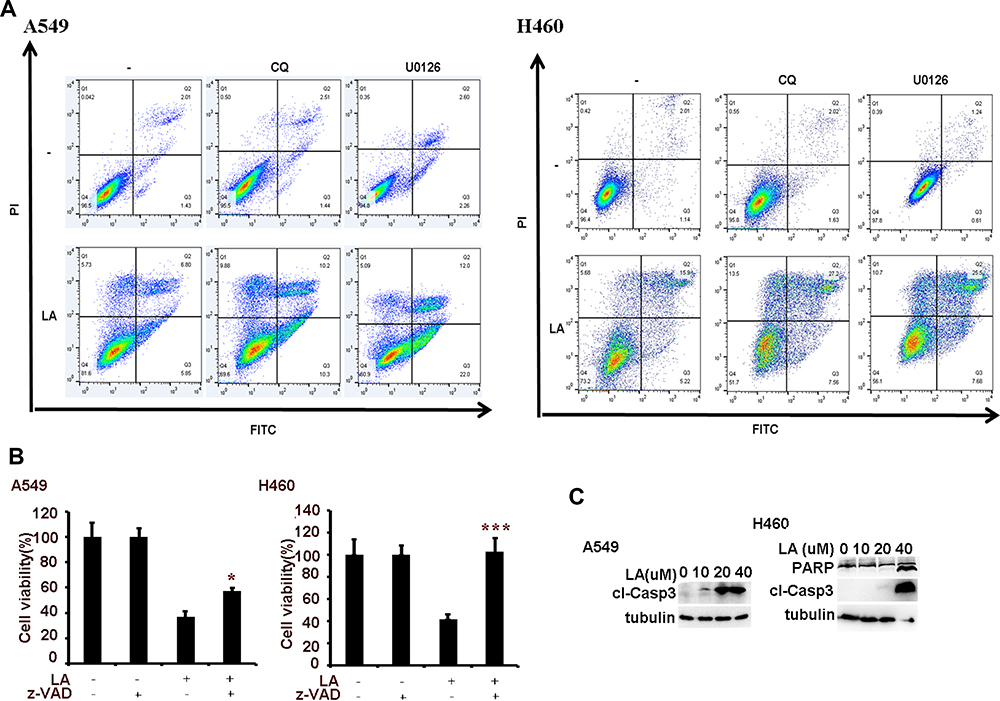 |
Figure 2 LA induces apoptotic cell death. (A) Lung cancer cells were pre-treated with CQ (20 µM) and U0126 (10 µM) for 1 h, then exposed to LA (20 µM) for 36 h, then flow cytometric analysis was carried out to investigate cell death. (B) Lung cancer cells were pre-treated with z-VAD-fmk (10 µM) for 1 h, then exposed to LA (20 µM) for 36 h, then cell viability was investigated by MTT assay. (C) Lung cancer cells were treated with indicated concentration of LA for 36 h, then the expression of indicated proteins was examined by Western blot. N = 3. *P<0.05, ***P<0.001. |
LA Suppresses the Expression of IAPs and c-FLIPL in NSCLC Cells
Accumulating evidence has demonstrated that IAPs and c-FLIPL are the main inhibitors of cell death, including apoptosis and necrosis, and previous studies have reported that LA suppresses the expression of Survivin in human hepatoma cells,36 oral squamous cell carcinoma36 and ovarian carcinoma cells.11 However, whether LA influences the expression of IAPs and c-FLIPL in NSCLC still remains elusive. As shown in Figure 3A, LA greatly decreased IAPs (c-IAP1, c-IAP2, XIAP, Survivin) and c-FLIPL expression in both A549 and H460 cells, however, LA did not greatly influence the expression of mRNA level of the above proteins (Figure 3B). To further elucidate the mechanism underlying LA-induced IAPs and c-FLIPL suppression, a cycloheximide chase assay was carried out to investigate the stability of IAPs and c-FLIPL. Lung cancer cells were pre-treated with LA (20 µM) or PBS for 4 h respectively, then exposed to cycloheximide (200 ug/mL) for indicated times, then Western blotting was carried out to investigate the expression of related proteins. As shown in Figure 3C, LA significantly accelerated the degradation of Survivin and XIAP in both A549 and H460 cells compared with the control group, but only marginally affected the expression of c-IAP1, c-IAP2 and c-FLIPL, suggesting that LA decreased the stability of Survivin and XIAP in lung cancer cells. Taken together, these results strongly suggest that LA kills lung cancer cells through degradation of IAPs and c-FLIPL in lung cancer cells.
 |
Figure 3 LA downregulates the expression of IAPs and c-FLIPL. (A) Lung cancer cells were treated with indicated concentration of LA for 36 h, then the expression of indicated proteins was examined by Western blot. (B) Lung cancer cells were exposed to LA (20 µM) for 8 h, then total RNA was extracted, and the mRNA level of indicated proteins was examined by q-RT-PCR, β-Actin was detected as an input control. (C) Lung cancer cells were pre-treated with LA (20 µM) or PBS for 4 h, then exposed to CHX (200 µg/mL) for indicated times, then the expression of indicated proteins was examined by Western blot. GAPDH or tubulin were detected as an input control. # P>0.05. |
LA Decreases the Expression of RIP1 in NSCLC Cells in a HSP90-Independent Way
It has been well documented that RIP1 plays a crucial role in cell survival and cell death. Our previous studies have shown that RIP1 maintains the expression of IAPs in lung cancer A549 and H460 cells,41,42 which leads us to investigate whether RIP1 is involved in LA-induced anticancer activities. Thereby, we continued to investigate whether LA influences the expression of RIP1 in lung cancer cells. As shown in Figure 4A, LA significantly suppressed the expression of RIP1 in both A549 and H460 cells. Considering the crucial role of HSP90 in the regulation of RIP1 expression in cancer cells, we explored whether LA influences the expression of HSP90. Unexpectedly, LA did not affect the expression of HSP90 in either A549 or H460 cells, suggesting that LA may decrease the expression of RIP1 in a HSP90-independent manner (Figure 4B). Surprisingly, LA slightly increased the level of mRNA of RIP1 (Figure 4C), which may be a compensation mechanism since RIP1 plays a key part in cell survival. To further explore the mechanism of LA-induced RIP1 downregulation, a cycloheximide chase assay was carried out to investigate whether LA affects the stability of RIP1. Lung cancer cells were pre-treated with LA (20 µM) or PBS for 4 h respectively, then exposed to cycloheximide (200 ug/mL) for indicated times. As shown in Figure 4D, LA significantly accelerated the degradation of RIP1 in both A549 and H460 cells compared with the control group, indicating that LA decreased the stability of RIP1 in NSCLC cells. Taken together, these results strongly suggest that LA induces apoptotic cytotoxicity in lung cancer cells by decreasing the stability of RIP1.
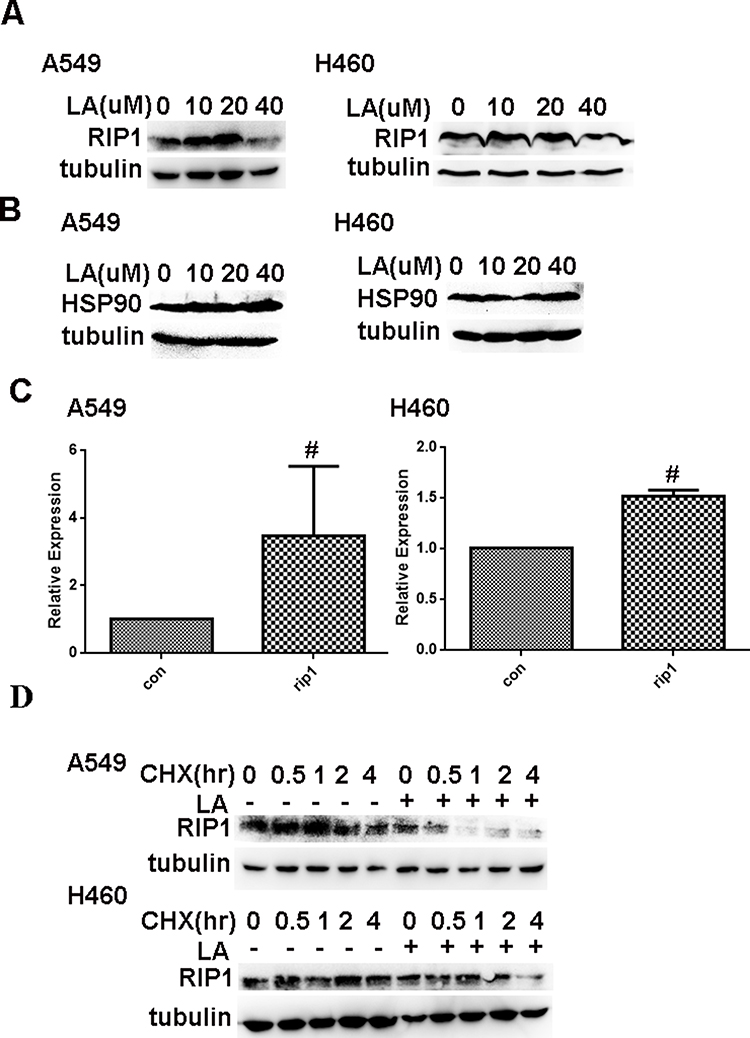 |
Figure 4 LA downregulates the expression of RIP1 in a HSP90-independent manner. (A) Lung cancer cells were treated with indicated concentration of LA for 36 h, then the expression of indicated proteins was examined by Western blot. (B) Lung cancer cells were treated with indicated concentration of LA for 36 h, then the expression of indicated proteins was examined by Western blot. (C) Lung cancer cells were exposed to LA (20 µM) for 8 h, then total RNA was extracted, and the mRNA level of indicated proteins was examined by q-RT-PCR, β-Actin was detected as an input control. (D) Lung cancer cells were pre-treated with LA (20 µM) or PBS for 4 h, then exposed to CHX (200 µg/mL) for indicated times, then the expression of indicated proteins was examined by Western blot. GAPDH or tubulin were detected as an input control. # P>0.05. |
LA Induces Protective Autophagy in Lung Cancer Cells
Recent studies suggest that LA induces autophagy in different types of cancers, however, the role of autophagy in LA-induced anticancer activities remains obscure. Our results showed that LA indeed increased autophagic activities in both A549 and H460 cells (Figure 5A), which was further validated by autophagic flux assay (Figure 5B). Intriguingly, inhibition of autophagy with CQ significantly enhanced LA-induced cytotoxicity in lung cancer cells (Figures 2A and 5C-D), meanwhile further downregulating the expression of apoptosis-related proteins (Figure 5E), indicating that autophagy plays a protective role in LA-induced cytotoxicity by maintaining the expression of apoptosis-related proteins in lung cancer.
 |
Figure 5 Autophagy plays a protective part in LA-induced anticancer effects. (A) Lung cancer cells were treated with indicated concentration of LA for 12 h, then the expression of indicated proteins was examined by Western blot. (B) Lung cancer cells were pre-treated with CQ (20 µM) for1 h, then exposed to LA (20 µM) for 12 h, then the expression of indicated proteins was examined by Western blot. (C–E) Lung cancer cells were pre-treated with CQ (20 µM) for 1 h, then exposed to LA (20 µM) for 36 h, then morphology was examined and cell viability was investigated by MTT assay, and the expression of indicated proteins was examined by Western blot. GAPDH or tubulin were detected as an input control. N = 3. ***P<0.001. |
ERK-Mediated Autophagy Activation Resists LA-Induced Cytotoxicity in NSCLC
Previous studies have reported that MAPKs (JNK, ERK and p38) contributes to LA-induced anticancer activities, however, the role of MAPKs is controversial in different types of cancers. Therefore, we continued to investigate the role of MAPKs in LA-mediated antitumor activities in lung cancer cells. LA indeed significantly induced ERK and p38 activation in a time-dependent manner in both A549 and H460 cells, strikingly, LA greatly suppressed the activity of JNK activities in lung cancer cells (Figure 6A). Then we continued to investigate whether ERK is involved in LA-induced autophagy induction. ERK inhibitor U0126 remarkably attenuated LA-induced LC3II accumulation in both A549 and H460 cells (Figure 6B), indicating that LA induced autophagy induction depending on ERK activation. To interrogate the biological role of MAPKs in LA-induced anticancer effects, U0126 and SB242235 were used to suppress the activities of ERK and p38 respectively. The results showed that U0126 but not SB242235 greatly increased LA-induced cytotoxicity in both A549 and H460 cells (Figure 2A and 6C-D), and suppression of ERK activities with U0126 further promoted LA-induced downregulation of IAPs, c-FLIPL and RIP1 in lung cancer cells (Figure 6E), suggesting ERK activation resisted LA-induced anticancer activities. Collectively, ERK-mediated autophagy activation plays a protective role in LA-induced cytotoxicity in NSCLC by maintaining the expression of IAPs, c-FLIPL and RIP1 in vitro.
 |
Figure 6 ERK-mediated autophagy resists LA-induced anticancer effects. (A) Lung cancer cells were treated with LA (20 µM) for indicated times, then the expression of indicated proteins was examined by Western blot. (B) Lung cancer cells were pre-treated with U0126 (10 µM) for 1 h, then exposed to LA (20 µM) for 12 h, then the expression of indicated proteins was examined by Western blot. (C–E) Lung cancer cells were pre-treated with U0126 (20 µM) for 1 h, then exposed to LA (20 µM) for 36 h, then morphology was examined and cell viability was investigated by MTT assay, and the expression of indicated proteins was examined by Western blot. GAPDH or tubulin were detected as an input control. N = 3. **P<0.0.1. |
Discussion
In the present study, we demonstrated that LA suppressed cell proliferation and induced apoptosis in NSCLC cells in a dose-dependent manner. LA significantly decreased the expression of apoptosis-related proteins, including IAPs, c-FLIPL and RIP1, and also greatly decreased the stability of Survivin, XIAP and RIP1. Mechanistic studies revealed that ERK-mediated autophagy activation played a cytoprotective role in LA-induced anticancer activities since suppression of ERK and autophagy remarkably enhanced LA-induced cytotoxicity and downregulation of apoptosis-related proteins (Figure 7). The results of this study extend the role of apoptosis-related proteins in LA-induced anticancer effects, furthering our knowledge of the underlying mechanisms of this agent.
 |
Figure 7 Model of LA-induced anticancer effects in lung cancer. LA induces ERK activation, leading to autophagy induction, autophagy maintains the expression of apoptosis-related proteins to resist LA-induced cytotoxicity. |
The IAPs proteins play an important role in cell survival. They are mediated by NK-kB activation and inhibit cell death.30–32 It has been well documented that IAPs proteins are overexpressed in many types of tumors, and predict poor prognosis for several anticancer agents, including cisplatin.33,34 Therefore, IAPs are promising targets for cancer therapy. IAPs are crucial factors of RIP1; RIP1 plays a key role in cell survival, apoptosis and necrosis and is essential for the formation of Ripoptosome complex. c-FLIPL inhibits the Ripoptosome-mediated cell death pathway. c-FLIP is often highly expressed in different types of cancers, conferring poor prognosis of patients.43,44 Accumulating evidence has demonstrated that downregulation of c-FLIP restores sensitivity to anticancer agents, including TRAIL, Fas and cisplatin.44,45 Our previous studies have reported that chalcone derivative Chal-24 alone or in combination of cisplatin39 and TRAIL40 promotes IAPs (c-IAP1, c-IAP2 and XIAP) and c-FLIPL degradation depending on JNK and ERK activation. Besides, our previous studies have demonstrated that RIP1 maintains IAPs expression to resist cisplatin-induced cytotoxicity in lung cancer cell.41,42 Our results show that LA decreases the expression of IAPs (c-IAP1, c-IAP2, XIAP, Survivin), c-FLIPL and RIP1. Moreover, LA significantly decreases the stability of Survivin, XIAP and RIP1, but not c-IAP1, c-IAP2 and c-FLIPL. Therefore, the underlying mechanisms of LA-induced c-IAP1, c-IAP2 and c-FLIPL downregulation warranted further investigation.
Autophagy may play a dual role in anticancer agents’ anticancer activities. Emerging evidence has suggested that LA induces autophagy in various cancer cells in vitro, however the role of autophagy in LA-induced anticancer activities is controversial. Autophagy may contribute to LA-induced anticancer effects in osteosarcoma cells.20 However, Jen-Pi Tsai and colleagues demonstrate that autophagy plays a protective role because autophagy inhibitors 3-methyladenine and bafilomycin A1 both increased LA-induced apoptosis.19 Additionally, Tang et al. reported that autophagy may be a bystander in LA-induced anticancer activities since block of autophagy with inhibitor or siRNA did not reverse LA-induced anticancer effects.21 In the present study, we indeed found that LA induced autophagic activation in both A549 and H460 cells, but autophagy plays a protective role in LA-induced apoptosis because the autophagy inhibitor CQ greatly enhanced LA-induced cytotoxicity and further promoted the degradation of apoptosis-related proteins. Our results also show that LA induces autophagy depending on ERK activation since block of ERK activity greatly decreased LA-induced autophagic activity.
Previous studies have reported that MAPKs play an important role in LA-induced anticancer activities in various cancers, however the role of MAPKs in LA-induced anticancer effects remains disputable. Chun-Yi Chuang and colleagues reported that LA increased JNK and p38 activation in human nasopharyngeal carcinoma cells, and block of JNK or p38 with JNK inhibitor (JNK-IN-8) or p38 inhibitor (SB203580) abolished LA-induced apoptosis.18 Hao et al. also found that LA induced the activation of JNK, ERK and p38 in human gastric cancer BGC-823 cells. Jae-Sung Kim and coworkers reported that LA rapidly induced ERK and p38 activation in oral cancer cells.46 However, Ke Yao and colleagues found that LA was a natural selective JNK1 inhibitor, in vivo and in vitro studies both demonstrate LA inhibited JNK1-mediated, but not JNK2-mediated c-Jun phosphorylation.47 Subsequent studies also confirmed that LA suppressed the activities of JNK in hepatocellular cell carcinoma.25,26 Huang et al. reported that LA significantly suppressed the activation of ERK in human glioma cells.48 Jian Wu and coworkers demonstrated that LA attenuated ERK activities in in gastric cancer.49 Xiangrong Chen and colleagues reported that LA suppressed the p38/JNK/ERK signaling pathway as well.50 Nevertheless, Tsai et al. demonstrated that LA only marginally affected the activities of MAPKs, including JNK, ERK and p38 in cervical cancer19 and lung cancer cells.24 The controversial role of MAPKs in various cancers may be attributed to different antibodies targets for JNK activation and time for LA exposure; MAPKs are usually activated rapidly, and long-term treatment may give misleading results. However, the role of MAPKs in LA-mediated anticancer effects in NSCLC remains unclear. The results from this study show that LA greatly caused ERK and p38 activation in a time-dependent manner in both A549 and H460 cells, however, LA remarkably inhibited the activity of JNK in lung cancer cells. Moreover, suppression of ERK not p38 kinase with inhibitor enhanced LA-induced cytotoxicity, while ERK inhibitor significantly enhanced LA-mediated downregulation of apoptosis-related proteins and attenuated LA-induced autophagic induction.
Conclusion
In summary, our results provide evidence that LA kills NSCLC cells by downregulating the expression of apoptosis-related proteins, and ERK-mediated autophagy activation resists LA-induced cytotoxicity by maintaining the expression of apoptosis-related proteins (Figure 7). Further in vivo and in vitro studies are warranted for exploring the specific role of apoptosis-related proteins in LA-induced anticancer activities.
Acknowledgments
This study was supported in part by National Natural Science Foundation of China (No. 81860412 to S. Shi, No. 81560429 to R. Sun), Yunnan Natural Science Foundation of China (2017FE468 [−151] to S. Shi, 2017FF116 (−023) and 2019FF002 (−011) to R. Sun), Yunnan Provincial Fund for High Level Reserve Talents in Health Science (H2018002 to S. Shi). And we would like to thank Mr. Zhuang Luo, Mr. Rui Zhang, Ms. Chunyan Liu and Xinyue Zhang for helping us to prepare this manuscript.
Disclosure
The authors report no conflicts of interest for this work and no financial dependence regarding this publication.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Zhang J, Gold KA, Lin HY, et al. Relationship between tumor size and survival in non-small-cell lung cancer (NSCLC): an analysis of the surveillance, epidemiology, and end results (SEER) registry. J Thor Oncol. 2015;10(4):682–690.
3. Neri S, Yoshida J, Ishii G, et al. Prognostic impact of microscopic vessel invasion and visceral pleural invasion in non-small cell lung cancer: a retrospective analysis of 2657 patients. Ann Surg. 2014;260(2):383–388.
4. Wanders R, Steevens J, Botterweck A, et al. Treatment with curative intent of stage III non-small cell lung cancer patients of 75 years: a prospective population-based study. Eur J Cancer. 2011;47(18):2691–2697.
5. Fu Y, Hsieh TC, Guo J, et al. Licochalcone-A, a novel flavonoid isolated from licorice root (Glycyrrhiza glabra), causes G2 and late-G1 arrests in androgen-independent PC-3 prostate cancer cells. Biochem Biophys Res Commun. 2004;322(1):263–270.
6. Messier C, Grenier D. Effect of licorice compounds licochalcone A, glabridin and glycyrrhizic acid on growth and virulence properties of Candida albicans. Mycoses. 2011;54(6):e801–6.
7. Ziegler HL, Hansen HS, Staerk D, Christensen SB, Hagerstrand H, Jaroszewski JW. The antiparasitic compound licochalcone a is a potent echinocytogenic agent that modifies the erythrocyte membrane in the concentration range where antiplasmodial activity is observed. Antimicrob Agents Chemother. 2004;48(10):4067–4071.
8. Kwon HS, Park JH, Kim DH, Kim YH, Shin HK, Kim JK. Licochalcone A isolated from licorice suppresses lipopolysaccharide-stimulated inflammatory reactions in RAW264.7 cells and endotoxin shock in mice. J Mol Med (Berl). 2008;86(11):1287–1295.
9. Feldman M, Grenier D. Cranberry proanthocyanidins act in synergy with licochalcone A to reduce Porphyromonas gingivalis growth and virulence properties, and to suppress cytokine secretion by macrophages. J Appl Microbiol. 2012;113(2):438–447.
10. Su X, Li T, Liu Z, et al. Licochalcone A activates Keap1-Nrf2 signaling to suppress arthritis via phosphorylation of p62 at serine 349. Free Radic Biol Med. 2018;115:471–483. doi:10.1016/j.freeradbiomed.2017.12.004
11. Liang M, Li X, Ouyang X, Xie H, Chen D. Antioxidant mechanisms of echinatin and Licochalcone A. Molecules. 2018;24(1):1. doi:10.3390/molecules24010003
12. Xiao XY, Hao M, Yang XY, et al. Licochalcone A inhibits growth of gastric cancer cells by arresting cell cycle progression and inducing apoptosis. Cancer Lett. 2011;302(1):69–75.
13. Hao W, Yuan X, Yu L, et al. Licochalcone A-induced human gastric cancer BGC-823 cells apoptosis by regulating ROS-mediated MAPKs and PI3K/AKT signaling pathways. Sci Rep. 2015;5:10336.
14. Zhou M, Qin B, Deng XS, et al. hNTCPexpressing primary pig hepatocytes are a valuable tool for investigating hepatitis B virus infection and antiviral drugs. Mol Med Rep. 2019;20(4):3820–3828.
15. Choi AY, Choi JH, Hwang KY, et al. Licochalcone A induces apoptosis through endoplasmic reticulum stress via a phospholipase Cgamma1-, Ca(2+)-, and reactive oxygen species-dependent pathway in HepG2 human hepatocellular carcinoma cells. Apoptosis. 2014;19(4):682–697.
16. Huang WC, Su HH, Fang LW, Wu SJ, Liou CJ. Licochalcone A inhibits cellular motility by suppressing E-cadherin and MAPK signaling in breast cancer. Cells. 2019;8:3.
17. Lu WJ, Wu GJ, Chen RJ, et al. Licochalcone A attenuates glioma cell growth in vitro and in vivo through cell cycle arrest. Food Funct. 2018;9(8):4500–4507.
18. Chuang CY, Tang CM, Ho HY, et al. Licochalcone A induces apoptotic cell death via JNK/p38 activation in human nasopharyngeal carcinoma cells. Environ Toxicol. 2019;34(7):853–860.
19. Tsai JP, Lee CH, Ying TH, et al. Licochalcone A induces autophagy through PI3K/Akt/mTOR inactivation and autophagy suppression enhances Licochalcone A-induced apoptosis of human cervical cancer cells. Oncotarget. 2015;6(30):28851–28866.
20. Shen TS, Hsu YK, Huang YF, Chen HY, Hsieh CP, Chen CL. Licochalcone A suppresses the proliferation of osteosarcoma cells through autophagy and ATM-Chk2 activation. Molecules. 2019;24:13.
21. Tang ZH, Chen X, Wang ZY, et al. Induction of C/EBP homologous protein-mediated apoptosis and autophagy by licochalcone A in non-small cell lung cancer cells. Sci Rep. 2016;6:26241.
22. Chen G, Ma Y, Jiang Z, et al. Lico A Causes ER stress and apoptosis via up-regulating miR-144-3p in human lung cancer cell line H292. Front Pharmacol. 2018;9:837.
23. Qiu C, Zhang T, Zhang W, et al. Licochalcone A inhibits the proliferation of human lung cancer cell lines A549 and H460 by Inducing G2/M cell cycle arrest and ER stress. Int J Mol Sci. 2017;18:8.
24. Huang HC, Tsai LL, Tsai JP, et al. Licochalcone A inhibits the migration and invasion of human lung cancer cells via inactivation of the Akt signaling pathway with downregulation of MMP-1/-3 expression. Tumour Biol. 2014;35(12):12139–12149.
25. Wu MH, Chiu YF, Wu WJ, et al. Synergistic antimetastatic effect of cotreatment with licochalcone A and sorafenib on human hepatocellular carcinoma cells through the inactivation of MKK4/JNK and uPA expression. Environ Toxicol. 2018;33(12):1237–1244.
26. Tsai JP, Hsiao PC, Yang SF, et al. Licochalcone A suppresses migration and invasion of human hepatocellular carcinoma cells through downregulation of MKK4/JNK via NF-kappaB mediated urokinase plasminogen activator expression. PLoS One. 2014;9(1):e86537.
27. Shen H, Zeng G, Tang G, et al. Antimetastatic effects of licochalcone A on oral cancer via regulating metastasis-associated proteases. Tumour Biol. 2014;35(8):7467–7474.
28. Komoto TT, Bernardes TM, Mesquita TB, et al. Chalcones Repressed the AURKA and MDR Proteins Involved in Metastasis and Multiple Drug Resistance in Breast Cancer Cell Lines. Molecules. 2018;23:8.
29. Lin X, Tian L, Wang L, Li W, Xu Q, Xiao X. Antitumor effects and the underlying mechanism of licochalcone A combined with 5-fluorouracil in gastric cancer cells. Oncol Lett. 2017;13(3):1695–1701.
30. Bertrand MJ, Milutinovic S, Dickson KM, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30(6):689–700.
31. LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene. 2008;27(48):6252–6275.
32. Straub CS. Targeting IAPs as an approach to anti-cancer therapy. Curr Top Med Chem. 2011;11(3):291–316.
33. Silke J, Vucic D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014;545:35–65.
34. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nat Rev Drug Discov. 2012;11(2):109–124.
35. Wang J, Zhang YS, Thakur K, et al. Licochalcone A from licorice root, an inhibitor of human hepatoma cell growth via induction of cell apoptosis and cell cycle arrest. Food Chem Toxicol. 2018;120:407–417.
36. Cho JJ, Chae JI, Yoon G, et al. Licochalcone A, a natural chalconoid isolated from Glycyrrhiza inflata root, induces apoptosis via Sp1 and Sp1 regulatory proteins in oral squamous cell carcinoma. Int J Oncol. 2014;45(2):667–674.
37. Lee CS, Kwak SW, Kim YJ, et al. Guanylate cyclase activator YC-1 potentiates apoptotic effect of licochalcone A on human epithelial ovarian carcinoma cells via activation of death receptor and mitochondrial pathways. Eur J Pharmacol. 2012;683(1–3):54–62.
38. Mitchell AJ, Green A, Jeffs DA, Roberson PK. Physiologic effects of retinopathy of prematurity screening examinations. Advan Neonatal Care. 2011;11(4):291–297.
39. Shi S, Wang Q, Xu J, et al. Synergistic anticancer effect of cisplatin and Chal-24 combination through IAP and c-FLIPL degradation, Ripoptosome formation and autophagy-mediated apoptosis. Oncotarget. 2015;6(3):1640–1651.
40. Xu J, Xu X, Shi S, et al. Autophagy-mediated degradation of IAPs and c-FLIP(L) potentiates apoptosis induced by Combination of TRAIL and Chal-24. J Cell Biochem. 2016;117(5):1136–1144.
41. Wang Q, Shi S, He W, et al. Retaining MKP1 expression and attenuating JNK-mediated apoptosis by RIP1 for cisplatin resistance through miR-940 inhibition. Oncotarget. 2014;5(5):1304–1314.
42. Wang Q, Chen W, Bai L, et al. Receptor-interacting protein 1 increases chemoresistance by maintaining inhibitor of apoptosis protein levels and reducing reactive oxygen species through a microRNA-146a-mediated catalase pathway. J Biol Chem. 2014;289(9):5654–5663.
43. Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332(2):141–150.
44. Subramaniam K, Hirpara JL, Tucker-Kellogg L, Tucker-Kellogg G, Pervaiz S. FLIP: a flop for execution signals. Cancer Lett. 2013;332(2):151–155.
45. He W, Wang Q, Xu J, et al. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy. 2012;8(12):1811–1821.
46. Kim JS, Park MR, Lee SY, et al. Licochalcone A induces apoptosis in KB human oral cancer cells via a caspase-dependent FasL signaling pathway. Oncol Rep. 2014;31(2):755–762.
47. Yao K, Chen H, Lee MH, et al. Licochalcone A, a natural inhibitor of c-Jun N-terminal kinase 1. Cancer Prev Res (Phila). 2014;7(1):139–149.
48. Huang CF, Yang SF, Chiou HL, et al. Licochalcone A inhibits the invasive potential of human glioma cells by targeting the MEK/ERK and ADAM9 signaling pathways. Food Function. 2018;9(12):6196–6204.
49. Wu J, Zhang X, Wang Y, et al. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. Oncol Rep. 2018;39(3):1181–1190.
50. Chen X, Liu Z, Meng R, Shi C, Guo N. Antioxidative and anticancer properties of Licochalcone A from licorice. J Ethnopharmacol. 2017;198:331–337.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.