Back to Journals » OncoTargets and Therapy » Volume 13
Epoxycytochalasin H: An Endophytic Phomopsis Compound Induces Apoptosis in A2780 Cells Through Mitochondrial Damage and Endoplasmic Reticulum Stress
Authors Wang J ![]() , Xu Z, Hu X
, Xu Z, Hu X ![]() , Yang Y, Su J, Liu Y
, Yang Y, Su J, Liu Y ![]() , Zhou L
, Zhou L ![]() , Qin J, Zhang D, Yu H
, Qin J, Zhang D, Yu H
Received 13 March 2020
Accepted for publication 13 May 2020
Published 3 June 2020 Volume 2020:13 Pages 4987—4997
DOI https://doi.org/10.2147/OTT.S253716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Nicola Silvestris
Jiabin Wang,1,2,* Zhonghang Xu,3,* Xiaoqing Hu,1 Yimeng Yang,1,2 Jing Su,1 Yanan Liu,1 Li Zhou,4 Jianchun Qin,5 Dawei Zhang,6 Huimei Yu1,6
1Key Laboratory of Pathobiology, Ministry of Education, Department of Pathophysiology, College of Basic Medical Sciences, Jilin University, Changchun 130021, People’s Republic of China; 2Department of Microbiology, College of Basic Medical Sciences, Jilin University, Changchun 130021, People’s Republic of China; 3Department of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital, Jilin University, Changchun 130033, People’s Republic of China; 4Department of Obstetrics & Gynecology, The First Hospital of Jilin University, Changchun 130021, People’s Republic of China; 5College of Plant Sciences, Jilin University, Changchun, Jilin 130062, People’s Republic of China; 6Center of Animal Experiment, College of Basic Medical Sciences, Jilin University, Changchun 130021, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dawei Zhang; Huimei Yu
College of Basic Medical Sciences, Jilin University, Changchun 130021, People’s Republic of China
Email [email protected]; [email protected]
Background: Natural compounds extracted from plants have been reported to have antitumor activity. A fungal metabolite from Phomopsis, identified as epoxycytochalasin H and isolated from the flowering plant Polygonatum sibiricum, was found to have significant antitumor activity. In this study, we report the antitumor effects and mechanism of action of epoxycytochalasin H in the ovarian cancer cell line A2780. Our data suggest that epoxycytochalasin H markedly reduces cell proliferation and induces apoptosis in ovarian cancer cells.
Materials and Methods: The viability, apoptosis and colony formation of A2780 cells, treated with epoxycytochalasin H, were detected by MTT assay, nuclear staining, flow cytometry, and clone formation assay. MitoROS and mitochondrial membrane potentials were detected by MitoSOX staining and flow cytometry. The expression of proteins associated with apoptosis, autophagy, and endoplasmic reticulum stress, in A2780 cells treated with epoxycytochalasin H, was detected by Western blot. Effects of mitophagy were detected in Parkin-overexpressing 293T cells.
Results: Our data suggested that epoxycytochalasin H could strongly reduce cell proliferation and induce apoptosis in ovarian cancer cell line A2780. Epoxycytochalasin H induced apoptosis through mitochondrial injury, mitophagy, and endoplasmic reticulum stress. Specifically, epoxycytochalasin H increased ROS level in cells, and in mitochondria, it decreased mitochondrial membrane potential, caused mitochondrial injury, activated macroautophagy and mitophagy, and subsequently induced apoptosis via the mitochondrial pathway. Additionally, it was discovered that epoxycytochalasin H could induce apoptosis more significantly in 293T cells overexpressing Parkin than in the parental cells. Thus, the mitophagy activated by epoxycytochalasin H could promote apoptosis. In addition, epoxycytochalasin H mediated endoplasmic reticulum stress-related apoptosis.
Conclusion: Epoxycytochalasin H could promote apoptosis of human ovarian cancer A2780 cells by activating mitochondrial and endoplasmic reticulum stress-related apoptotic pathways.
Keywords: natural compound, human ovarian cancer, apoptosis, mitochondrial damage, endoplasmic reticulum stress
Introduction
The potential of plant-derived natural compounds in cancer therapy has receiving increasing attention. Compared with traditional chemotherapeutic drugs, natural compounds derived from plants have significant advantages in their lower genotoxicity, while having antitumor activity.1 Much research has been done on the antitumor effects of natural compounds. For example, Fas ligand, a member of the tumor necrosis factor (TNF) family, can be induced by homoharringtonine, which is isolated from plants of the genus Cephalotaxus, to promote the apoptosis of HL-60 leukemia cells.2 Also, Rehmannia glutinosa oligosaccharide (LRPS) can increase the expression of the p53 gene in Lewis lung cancer cells and regulate their apoptosis.3 Therefore, the study of the mechanisms of plant-derived natural compounds may be helpful in developing new strategies for treating cancers.
Normal mitochondrial function is important for cancer cell survival.4 When mitochondria receive the stimulation signal for apoptosis, there is an increase in mitochondrial membrane permeability via the opening of the mitochondrial permeability transition pore (mPTP). Pro-apoptotic factors, such as cytochrome c (cyt c), are released into the cytoplasm, which initiates the caspase cascade reaction, induces PARP-1 activation, and leads to endogenous apoptosis.5,6 Therefore, evaluating the effects of drugs on mitochondrial function, in cancers and on mitochondrial apoptosis, is an important way to study changes to the apoptosis pathway, induced by natural compounds.
In addition, macrophage autophagy or mitophagy can be changed by direct stimulation by drugs or by impaired mitochondrial function.7 On one hand, in the case of cells with mitochondrial impairment, macrophage autophagy or mitophagy can remove damaged organelles or cytoplasmic proteins, and then play a protective role for tumor cells.8 On the other hand, the same process may also induce autophagic cell death or promote apoptosis. In view of the different potential roles of autophagy in cancers, this study considers how the level of autophagy in cancer cells can be influenced by drugs, and the mechanism of how autophagy affects cell survival may provide new therapeutic targets for natural compounds.
In addition to the mitochondrial apoptosis pathway and autophagy, endoplasmic reticulum (ER) stress is also involved in the regulation of apoptosis. Under various endogenous or exogenous stimuli, misfolded and unfolded proteins accumulate in the ER cavity, and activate the unfolded protein reaction (UPR).9 Excessive ER stress may promote the release of cyt c, initiate the caspase cascade reaction and activate endogenous apoptosis, simultaneously. Excessive ER stress can also activate caspase-4 and cause apoptosis by activation of caspase-3.10,11
In our study of the antitumor activity of natural compounds, it was found that epoxycytochalasin H, isolated from Polygonatum sibiricum (a flowering plant, commonly known as Solomon’s seal), could inhibit cell proliferation and promote apoptosis in ovarian cancer cells A2780. This study aimed to elucidate the mechanisms of apoptosis in human ovarian cancer cells induced by epoxycytochalasin H.
Materials and Methods
Culture and Isolation
The strain of Phomopsis sp. was isolated from the tissue of asymptomatic leaf of P. sibiricum. We identified the strain through the analysis of internal transcribed spacer (ITS) and morphological examination. Under 25°C condition, fungus was grown for 7 days on potato dextrose agar plates. After cultured, all mycelium was inoculated into an autoclaving sterilized solid medium including a formula of rice (60.0 g) and distilled water (80 mL) in Fernbach flasks (500 mL) for further fermentation at 25°C for another 40 days.
Epoxycytochalasin H Preparation
After fermented, the rice substrate was extracted with EtOAc (500 mL×3 times), and the organic solvent was dried under a vacuum to afford the crude extract. The original extract was fractionated on a silica gel column using petroleum ether-acetone (50:1–1:1) progressively to give ten fractions (fraction 1 to fraction 10). The fraction was further purified by semi-preparative HPLC (UV 210 nm, 55% MeOH in H2O) to yield Epoxycytochalasin H (9.0 mg, tR 15.2 min).
Cell Culture
A2780 ovarian cancer cells were purchased from American Tissue Culture Collection (Rockville, MD, USA) and were grown in RPMI‐1640 (Gibco Life Technologies, Carlsbad, CA) supplemented with 10% foetal bovine serum (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2.
Cell Viability Assay
A2780 cells were seeded in 96-well plates at a concentration of 8000 cells/well treated with Epoxycytochalasin H for 24 hours. Cellular viability was assessed using an MTT (Sigma‐Aldrich, USA) assay and absorbance values were measured at 490 nm using a Vmax Microplate Reader (Molecular Devices, USA).
Nuclear Staining by Hoechst 33342
40 μg/mL Epoxycytochalasin H treated cells for 24 hours, then fixed with 4% paraformaldehyde for 20 minutes, Hoechst 33342 (2μg/mL) staining for 30 minutes, washed three times with phosphate-buffer solution (PBS), and observed chromatin condensation with ZEISS (imager Z2) fluorescence microscope.
Colony Formation Assay
Cells were seeded at 500 cells per well in 24-well plates. With the treatments of Epoxycytochalasin H, cells continue to grow for a week. And then cells were washed three times with PBS and stained with 0.5% crystal violet. The stain was washed three times with ddH2O.
Cell Apoptosis Assay
Flow cytometry can detect early apoptosis and late apoptosis or necrosis of cells by Propidium iodide (PI) and Annexin V (Invitrogen, Carlsbad, CA) staining. Cells were seeded in six-well plates. When cells grew to about 80% confluence, they were treated with20, 40, 60μg/mL Epoxycytochalasin H for 24h. Then cells were collected and stained with propidium iodide (PI) and Annexin V to analyze apoptosis by guava easyCyte flow cytometry (Merck, USA).
Western Blot Analysis
Cultured A2780 cells were trypsinized, washed, and centrifuge at 4500 rpm for 5 min. Cells were lysed in RIPA buffer and boiled in loading buffer. Proteins were separated by SDS-PAGE and transferred to PVDF membrane. Membranes were blocked with skim 5% milk, followed by incubated with primary antibodies overnight at 4°C. The secondary antibodies (Proteintech group, Inc, USA) were incubated for 2h at room temperature the next day. The bound antibody complex was visualized using an ECL substrate kit (Thermo Fisher Scientific, USA) by Syngene Bio Imaging (Synoptics, Cambridge, UK).
Measurement of MitoROS Level
MitoROS levels were measured using MitoSOX Red mitochondrial superoxide indicator (Thermo Fisher, Scientific, USA). A2780 cells in 24-well plates were treated with Epoxycytochalasin H (20, 40, 60μg/mL) in RPMI‐1640 for 24h. After treatment, cells were incubated with 5μM MitoSOX working solution in the dark at 37°C for 10 min, washed three times with PBS, and micrographs were obtained using ZEISS (imager Z2) fluorescence microscope.
Mitochondrial Membrane Potential Assay
Mitochondrial membrane potential were measured using Mitochondrial membrane potential assay kit with JC-1 (Beyotime Biotech,China). Cells were seeded in six-well plates. When cells grew to about 80% confluence, they were treated with20, 40, 60μg/mL Epoxycytochalasin H for 24h. Then cells were collected and stained with JC-1 to analyze mitochondrial membrane potential by guava easyCyte flow cytometry (Merck, USA).
Results
Identification of Epoxycytochalasin H
H1-NMR (DMSO-d6, 300 MHz): 3.58 (1H, t, J = 7.0, H-3), 2.19 (1H,m, H-4), 1.58 (1H, m, H-5), 2.69 (1H, d, J = 5.5, H-7), 2.31 (1H, dd, J = 9.5, 5.5, H-8), 2.85 (2H, m, H-10), 0.76 (3H, d, J = 7.0, H-11), 1.16 (3H, s, H-12), 6.01 (1H, dd, J = 15.5, 9.5, H-13), 5.60 (1H, m, H-14), 2.06 (1H, m, H-15), 2.59 (1H, q, J = 11.0, H-15), 3.20 (1H, m, H-16), 3.15 (1H, br s, H-19), 3.51 (1H, br s, H-20), 5.61 (1H, br s, H-21), 1.18 (3H, d, J = 6.5, H-22), 1.51 (3H, s, H-23), 7.19 (2H, d, J = 7.5, H-2ʹ, 6ʹ), 7.32 (2H, t, J = 7.5, H-3ʹ, 5ʹ), 7.23 (1H, t, J = 7.5, H-4ʹ), 2.10 (3H, s, Actyl).
C13-NMR (DMSO-d6, 500 MHz): 175.5 (s, C-1), 54.5 (d, C-3), 51.2 (d, C-4), 36.7 (d, C-5), 57.3 (s, C-6), 62.4 (d, C-7), 44.8 (d, C-8), 50.8 (s, C-9), 45.7 (t, C-10), 12.4 (q, C-11), 19.6 (q, C-12), 131.5 (d, C-13), 131.1 (d, C-14), 37.6 (t, C-15), 41.8 (d, C-16), 216.3 (s, C-17), 76.4 (s, C-18), 59.9 (d, C-19), 52.9 (d, C-20), 72.7 (d, C-21), 19.1 (q, C-22), 21.9 (q, C-23), 136.8 (s, C-1ʹ), 129.0 (d, C-2ʹ, 6ʹ), 129.4 (d, C-3ʹ, 5ʹ), 127.4 (d, C-4ʹ), 20.8 (q, Actyl). (Figure 1).
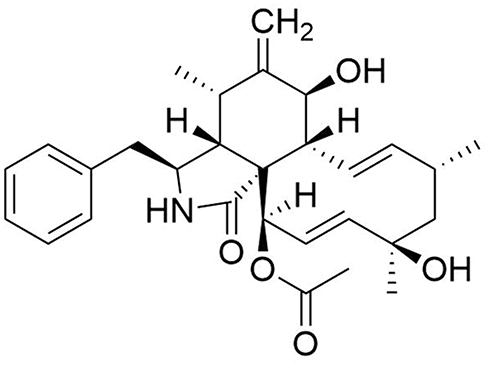 |
Figure 1 Chemical structure of Epoxycytochalasin H. |
Epoxycytochalasin H Inhibited Cell Viability and Induced Apoptosis of Ovarian Cancer Cells A2780
In order to analyze the effect of Epoxycytochalasin H on cell proliferation, A2780 cells were treated with different concentrations of Epoxycytochalasin H (6.25–100 μg/mL) for 24 h, and cell viability was measured by MTT assay. As shown in Figure 2A, Epoxycytochalasin H inhibited the viability of A2780 cells in a dose-dependent manner. In order to detect whether the decrease of cell viability was caused by the inhibition of cell proliferation or not, the clone formation assay was used.12 As shown in Figure 2B, Epoxycytochalasin H reduced the colony numbers of A2780 cells in a dose-dependent manner, suggesting that Epoxycytochalasin H could inhibit the proliferation of A2780 cells.
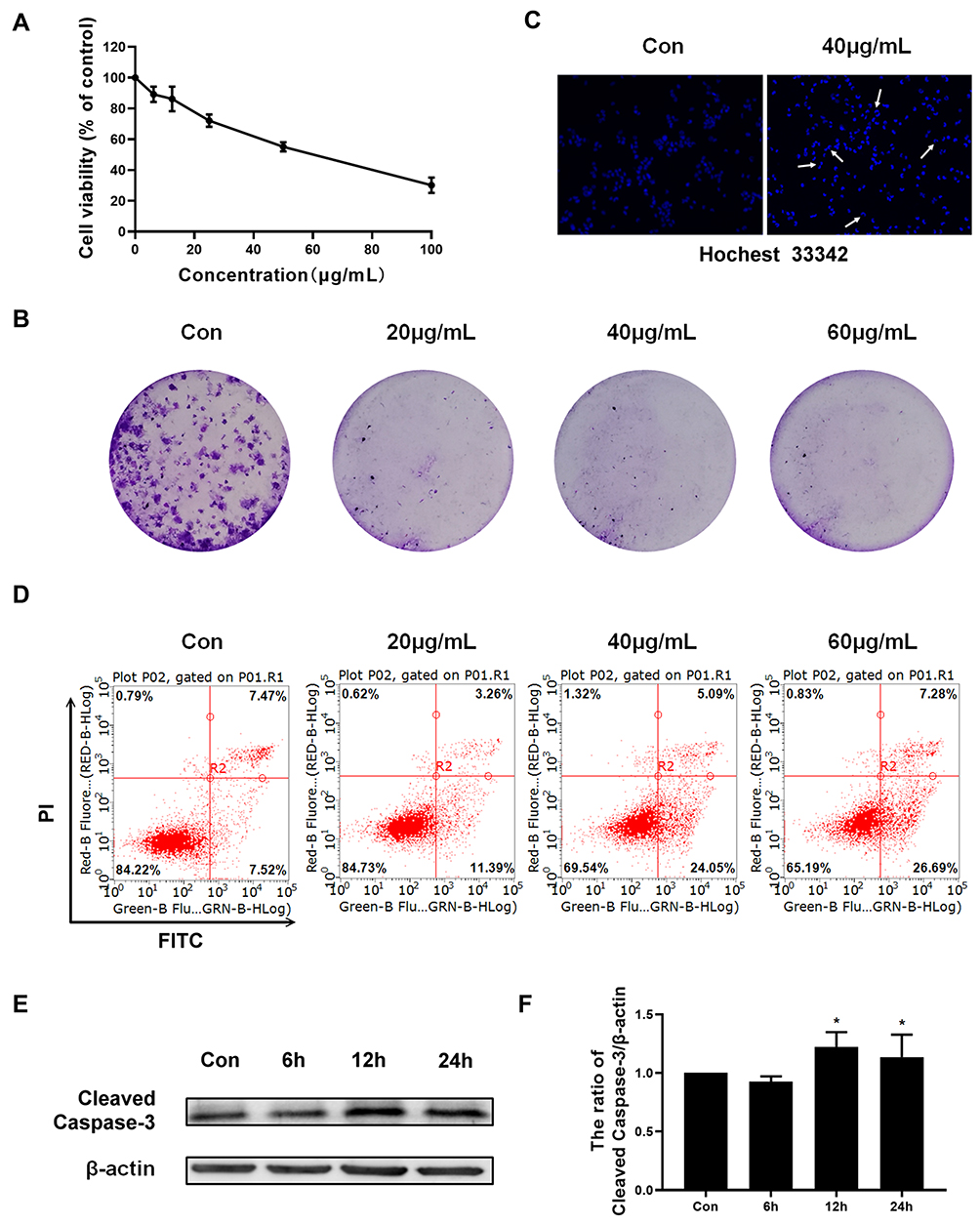 |
Figure 2 Epoxycytochalasin H inhibited the proliferation of A2780 cells and induced cell apoptosis. (A) After exposure to 6.25, 12.5, 25, 50, 100 μg/mL Epoxycytochalasin H for 24 h, cell viability was measured by MTT assay. The values were presented as mean ± SD, n =3. (B) The clone formation of A2780 cells treated with different concentrations of Epoxycytochalasin H for 7 days. (C) A2780 cells were treated with 60 μg/mL Epoxycytochalasin H for 24 h, and Hoechst 33342 was used to detect cell apoptotic chromatin condensation (arrows point to chromatin shrinkage showing apoptosis). Cell morphology was observed by laser microscope. (D) The effect of Epoxycytochalasin H induced A2780 cells apoptosis. A2780 cells were treated with different concentrations of Epoxycytochalasin H for 24 h, stained with PI and Annexin V-FITC, and the positive stained cells were counted using FACScan. Data were presented as a mean ± SD, n =3. (E) The expression of Cleaved Caspase-3 in A2780 cells treated with 60 μg/mL Epoxycytochalasin H for 6, 12 and 24 h was measured with Western blot. (F) The expression of cleaved Caspase-3 was measured with Western blot. Data were presented as a mean ± SD, n =3. *P< 0.05. |
Next, we investigated whether Epoxycytochalasin H could increase apoptosis of A2780 cell. The morphology of nuclei stained by Hoechst 33342 were observed with fluorescence microscope. As shown in Figure 2C, chromatin significantly condensed in the Epoxycytochalasin H treated cells compared to the control group. Then, the cell apoptosis was analyzed by flow cytometry. The results showed that Epoxycytochalasin H increased the apoptotic rates in a dose-dependent manner. (Figure 2D) Finally, we detected the expression of cleaved Caspase-3 in A2780 cells treated with Epoxycytochalasin H, and it found that the expression of cleaved Caspase-3 increased in a time-dependent manner. (Figure 2E, F)
The above results showed that Epoxycytochalasin H could inhibit the cell viability of A2780 cells, and its cytotoxicity was partly caused by inducing apoptosis.
Epoxycytochalasin H Damaged Mitochondrial Function in A2780 Cells
It was well known that impaired mitochondrial function could trigger the mitochondrial pathway apoptosis.13,14 Hence, we tried to analyze whether Epoxycytochalasin H could cause mitochondrial dysfunction and induce apoptosis of the mitochondrial pathway in A2780 cells. Impaired mitochondria could lead to increased production of ROS in mitochondria, damage mitochondrial DNA, and induce apoptosis.15 Therefore, we measured MitoROS levels to reflect mitochondrial function. A2780 cells were treated with different concentrations of Epoxycytochalasin H for 24 h and detected the level of MitoROS by MitoSOX staining. The results showed that red fluorescence gradually increased in a dose-dependent manner (Figure 3A). Next, JC-1 fluorescent probe was used to detect mitochondrial membrane potential by flow cytometry. As shown in Figure 3B, A2780 cells treated with Epoxycytochalasin H for 24h, the monomeric form of JC-1 increased and the aggregate form decreased, indicating that the mitochondrial membrane potential decreased in a dose-dependent manner. These results indicated that Epoxycytochalasin H could lead to impaired mitochondrial function in A2780 cells.
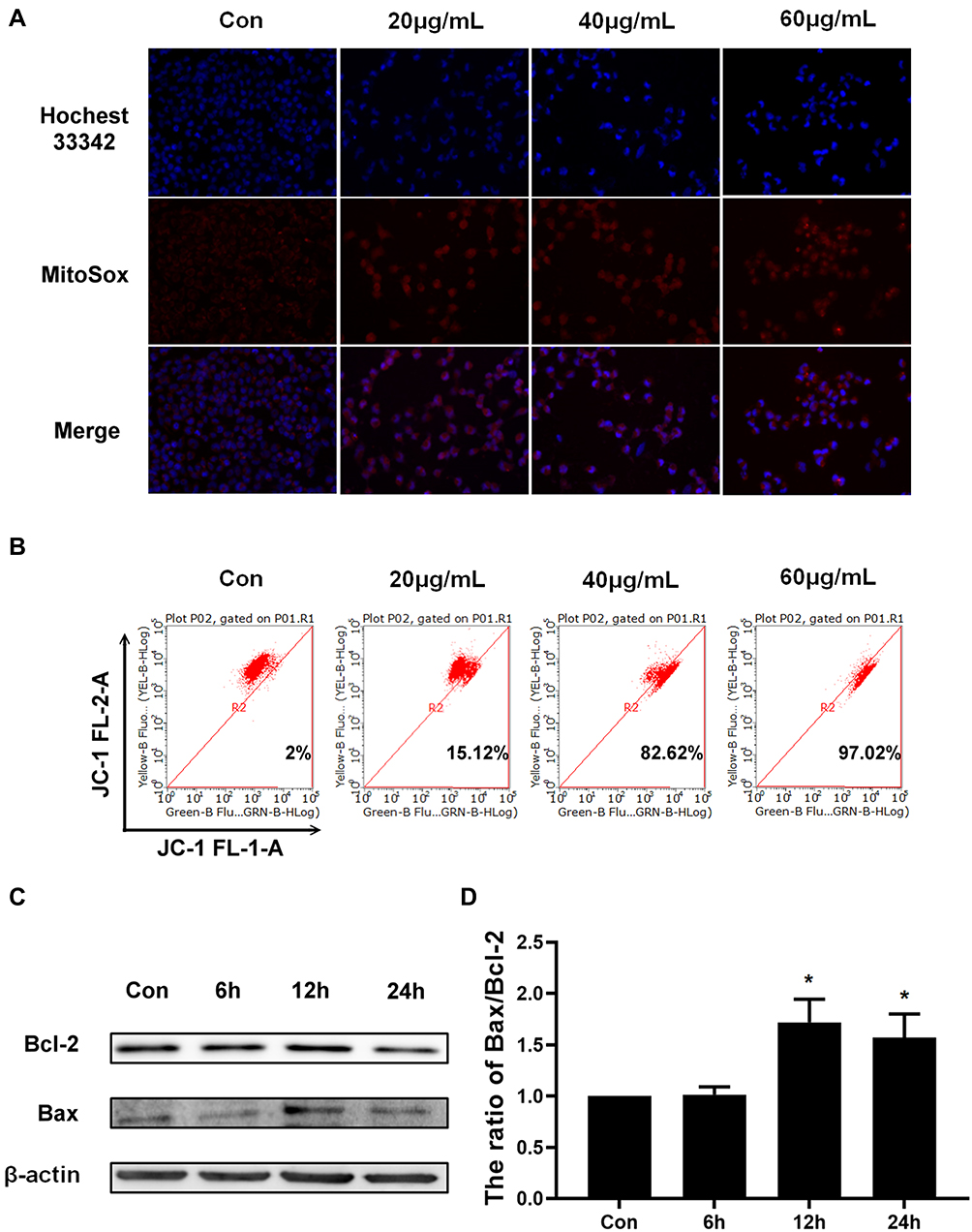 |
Figure 3 Epoxycytochalasin H damaged mitochondrial function and induced mitochondrial pathway apoptosis. (A) MitoSOX was used to detect MitoROS in A2780 cells treated with different concentrations of Epoxycytochalasin H for 24 h. (B) The effect of Epoxycytochalasin H induced A2780 cells mitochondrial membrane potential to decrease. A2780 cells were treated with different concentrations of Epoxycytochalasin H for 24 h, stained with JC-1, and the positive stained cells were counted using FACScan. Data were presented as a mean ± SD, n =3. (C) Western blot analysis for the expression of Bcl-2 and Bax in A2780 cells treated with 60 μg/mL Epoxycytochalasin H for 0, 6, 12 and 24 h. (D) The ratio of Bax/Bcl-2 protein level was measured with Western blot. Data were presented as a mean ± SD, n =3. *P< 0.05. |
Further, we detected whether Epoxycytochalasin H could induce mitochondrial pathway apoptosis in A2780 cells. The expression of mitochondrial apoptosis-related proteins were detected by Western blot, and it was found that the ratio of Bax/Bcl-2 increased when cells were treated with Epoxycytochalasin H for 12h and 24h (Figure 3C, D).
Epoxycytochalasin H Activated the Autophagy and Mitophagy of A2780 Cells
In order to evaluate whether Epoxycytochalasin H could activate the autophagy of A2780 cells, we detected the expression of LC3 protein. The results showed that the expression of p62 was increased after treatment of Epoxycytochalasin H for 12h and 24h, the ratio of LC3-II/LC3-I increased gradually after treatment of Epoxycytochalasin H for 6h.(Figure 4A, B) Then, we tested whether the increased ratio of LC3-II/LC3-I was caused by autophagy activation or degradation inhibition, cells were treated with lysosomal inhibitor chloroquine (CQ) combined with Epoxycytochalasin H. The results showed that LC3-II/LC3-I was significantly increased in A2780 cells treated with Epoxycytochalasin H and CQ. (Figure 4C, D) This indicated that Epoxycytochalasin H could activate autophagy and increased autophagic flux of A2780 cells.
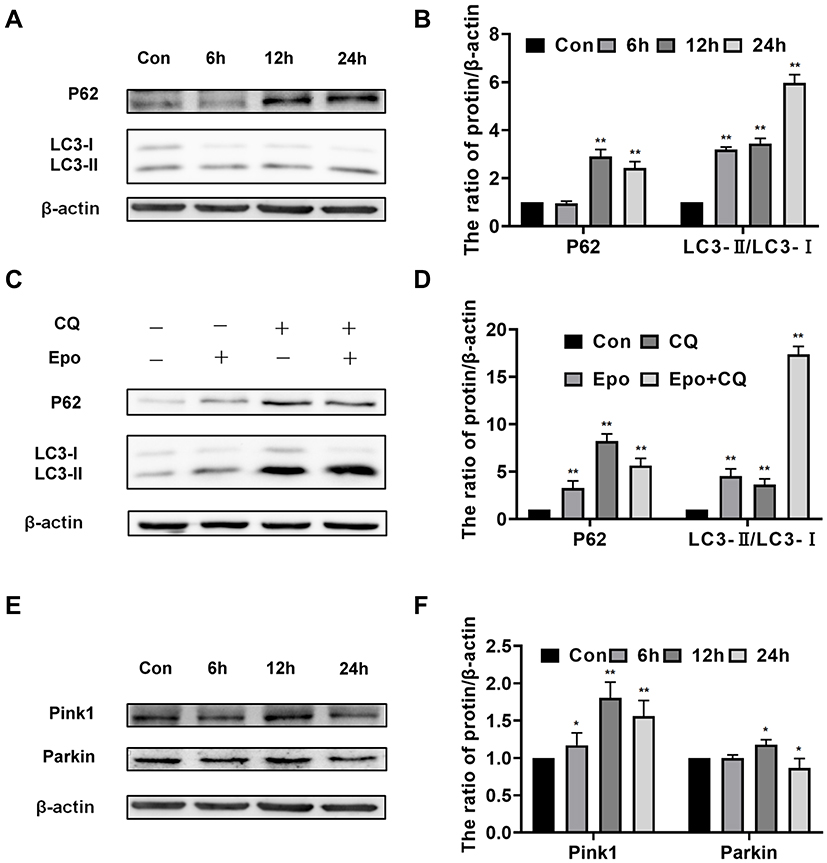 |
Figure 4 Epoxycytochalasin H activated autophagy and mitophagy of A2780 cells. (A) Western blot analysis for the expression of p62 and LC3 II/LC3 I proteins in A2780 cells treated with 60 μg/mL Epoxycytochalasin H for 0, 6, 12 and 24 h. (B) Quantitation of the expression of p62 and LC3 II/LC3 I proteins levels. Data are presented as mean ± SD, n =3, *P < 0.05, **P < 0.01. (C) Western blot analysis for the expression of p62, LC3 II/LC3 I proteins in A2780 cells treated with 60 μg/mL Epoxycytochalasin H (Epo) combined with 1μg/mL CQ for 6h. (D) Quantitation of the expression of p62and LC3 II/LC3 I proteins. Data are presented as mean ± SD, n = 3, *P< 0.05, **P< 0.01. (E) Western blot analysis for the expression of Parkin and Pink proteins in A2780 cells treated with 60 μg/mL Epoxycytochalasin H for 0, 6, 12 and 24 h. (F) Quantitation of the expression of Parkin and Pink proteins levels. Data are presented as mean ± SD, n =3, *P< 0.05, **P< 0.01. |
In addition, we evaluated whether mitochondrial induced by Epoxycytochalasin H could activate mitophagy. The expression of Pink1 and Parkin were used to evaluate mitophagy.16,17 The protein expression of Pink and Parkin were significantly increased when A2780 cells treated with Epoxycytochalasin H for 12 h (Figure 4E and F), indicating that the mitophagy of A2780 cells was activated.
Overexpression of Parkin Promoted Apoptosis Induced by Epoxycytochalasin H
In the above study, we found that Epoxycytochalasin H could activate mitophagy. Therefore, we wanted to explore the effect on mitophagy in apoptosis induced by Epoxycytochalasin H. In stably transfected Parkin 293T cells, it was shown that the cell viability of the stably transfected Parkin 293T cells decreased significantly compared with the wild type 293T cells when treated with Epoxycytochalasin H. (Figure 5A) The expression of anti-apoptotic protein Bcl-2 decreased, and the ratio of Bax/Bcl-2 ratio increased (Figure 5B and C). It indicated that overexpression of Parkin could promote apoptosis induced by Epoxycytochalasin H.
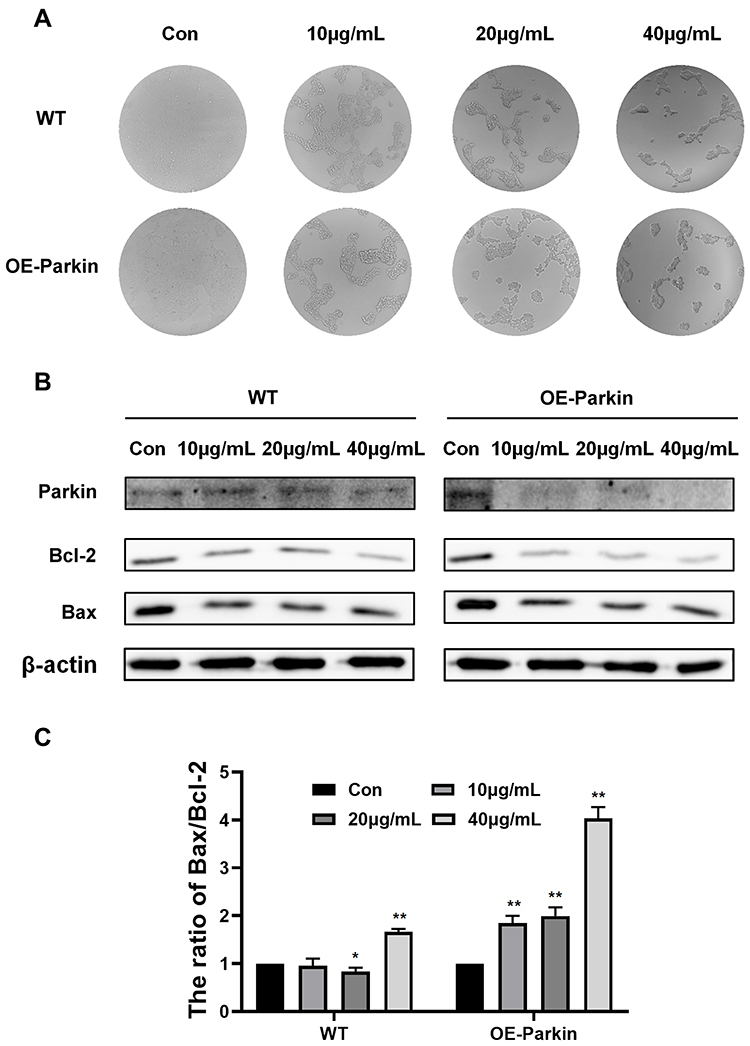 |
Figure 5 Overexpression of Parkin promoted apoptosis induced by Epoxycytochalasin H. (A) After exposure to 10μg/mL, 20μg/mL and40μg/mL Epoxycytochalasin H for 24 h, 293T and 293-Parkin cells were observed by ordinary microscope. (B) Western blot analysis for the expression of Bcl-2 and Bax in 293T and 293-Parkin cells treated with 10μg/mL, 20μg/mL and 40 μg/mL Epoxycytochalasin H for 24 h. (C) The ratio of Bax/Bcl-2 protein level. The ratio of Bax/Bcl-2 protein level was measured as described above, normalized to those of β-actin. Data were presented as a mean ± SD, n =3. *P< 0.05, **P < 0.01. |
Epoxycytochalasin H Induced Endoplasmic Reticulum Stress in A2780 Cells
In addition to mitochondrial apoptosis pathway, we wanted to detect whether endoplasmic reticulum apoptosis pathway was involved in Epoxycytochalasin H induced apoptosis of A2780 cells.18 GRP78, as an important endoplasmic reticulum chaperone protein, increased when endoplasmic reticulum stress activated.19,20 As the results shown in Figure 6A and B, the expression of GRP78 protein increased in a time-dependent manner after A2780 cells were treated by Epoxycytochalasin H for 6 to 24 h. It indicated that Epoxycytochalasin H activated endoplasmic reticulum stress in A2780 cells.
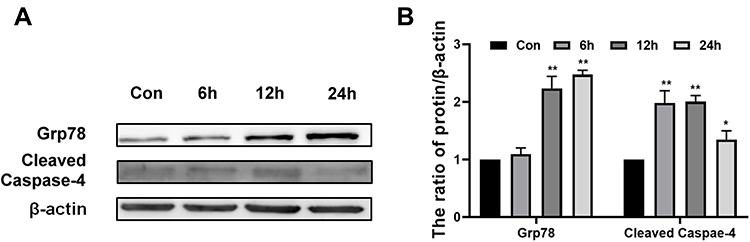 |
Figure 6 Epoxycytochalasin H induced endoplasmic reticulum stress in A2780 cells. (A) Western blot analysis for the expression of Grp78 and Cleaved Caspase-4 in A2780 cells treated with 60 μg/mL Epoxycytochalasin H for 0, 6, 12 and 24 h. (B) The ratio of Grp78 and Cleaved Caspase-4 proteins level. The ratios of Grp78 and Cleaved Caspase-4 proteins level were measured as described above, normalized to those of β-actin. Data were presented as a mean ± SD, n =3. *P< 0.05, **P< 0.01. |
Then, we wanted to test whether endoplasmic reticulum stress induced by Epoxycytochalasin H could activate endoplasmic reticulum stress related apoptosis pathway. In response to endoplasmic reticulum stress, Caspase-4, which is an ER-resident caspase, is required for apoptosis induced by endoplasmic reticulum stress.21 It was showed that the expression of cleaved Caspase-4 was increased specially when cells were treated with Epoxycytochalasin H for 6h, and peaked when treated for 12h. It was shown that Epoxycytochalasin H induced the endoplasmic reticulum apoptosis in the A2780 cells.
Discussion
Exploring the antitumor mechanisms of natural compounds may provide new strategies for the treatment of cancer and for overcoming drug resistance. In our study, we observed that epoxycytochalasin H could promote the mitochondrial pathway of apoptosis and ER stress pathways in ovarian cancer cell line A2780, as well as a change in the levels of macrophage autophagy and mitophagy.
Previous research has demonstrated that compounds from Phomopsis are able to induce apoptosis in cancer cells.22 In our study, we observed that epoxycytochalasin H could inhibit cell proliferation significantly and induce apoptosis in ovarian cancer cells. Although apoptosis is induced by endogenous and exogenous pathways, functional mitochondria are essential for the survival, growth, proliferation and metastasis of cancer cells.23,24 Therefore, the normal function of mitochondria in tumor cells and the mitochondria-related apoptotic pathway play important roles in the mechanisms of some antitumor drugs.25 According to our data, we found that epoxycytochalasin H could increase the ROS level in mitochondria and decrease mitochondrial membrane potential, which led to the impairment of mitochondrial function. Further, it was shown that epoxycytochalasin H could significantly elevate the ratio of protein Bax/Bcl-2 and the expression of cleaved caspase-3, suggesting that it may induce apoptosis of A2780 cells via the mitochondrial pathway.
In addition, we detected that other apoptotic pathways are involved in regulating apoptosis induced by epoxycytochalasin H. ER is not only the primary intracellular Ca2+ pool, but also the site of most protein synthesis, processing, and transport. Under the stimulation of drugs, ER homeostasis was destroyed. This induced the accumulation of misfolded and unfolded proteins in the ER lumen of cells and caused ER stress. The expression of chaperone proteins, such as GRP78, is increased in order to degrade misfolded proteins.26 However, when ER stress is excessive, damage to the cell cannot be alleviated, and apoptosis will be initiated. Therefore, in addition to endogenous and exogenous pathways, ER stress-related apoptosis is an important pathway to regulate apoptosis, overall.27 Therefore, we further investigated ER stress and apoptosis induced by the ER pathway, when cells were treated with epoxycytochalasin H. After treatment, the expression of GRP78 was increased significantly, indicating ER stress. After 6 h of epoxycytochalasin H treatment, the expression of caspase-4 increased, indicating the rapid induction of ER stress, and then ER stress-related apoptosis was observed, in A2780 cells.
In our study, we found that epoxycytochalasin H could impair mitochondrial function in A2780 cells, which may cause changes in autophagy. Furthermore, we observed autophagy of A2780 cells after epoxycytochalasin H treatment. According to the previous work, if autophagy is activated, LC3-I should be cut into LC3-II, and LC3-II should accumulate on the membrane.28 We measured the expression level of LC3-I and LC3-II. The results showed that the ratio of LC3-II/LC3-I was gradually increased after treatment with epoxycytochalasin H. The ratio of LC3-II/LC3-I was further increased when cells were treated with epoxycytochalasin H combined with lysosomal inhibitor CQ, indicating that epoxycytochalasin H could increase autophagic flux, in A2780 cells. At the same time, when the mitochondrial function is impaired, the mitochondrial membrane potential decreases, and PTEN-induced kinase 1 (Pink1) protein degrades slowly and aggregates in the mitochondrial outer membrane.29 Subsequently, Parkin, phosphorylated by Pink1, aggregated onto damaged mitochondria, with other proteins such as ubiquitinated VDAC1, and Mfn2, and became a signal to induce mitophagy.30 Here, the expressions of Pink1 and Parkin were detected, to indicate whether mitophagy was activated. When cells were treated with epoxycytochalasin H for 12 h, the expression of Pink1 and Parkin was increased, indicating that mitochondrial autophagy was activated. Meanwhile, it was noticed that the activation of mitophagy was later than that of macrophage autophagy.31 Moreover, after epoxycytochalasin H treatment for 12 h, the ratio of protein Bax/Bcl-2 and the expression of cleaved caspase-3 were significantly increased, leading to apoptosis, and indicating that epoxycytochalasin H induced mitochondrial autophagy and may promote apoptosis.
To further test whether mitophagy, induced by epoxycytochalasin H, can promote apoptosis, we used HEK293T cell lines, which overexpress Parkin, to detect the level of apoptosis. We found that the ratio of proteins Bax/Bcl-2 was higher in HEK293T cells overexpressing Parkin, suggesting that mitophagy induced by epoxycytochalasin H promoted the apoptosis of A2780 cells.
In conclusion, we evaluated the antitumor activity and mechanism of apoptosis, induced by epoxycytochalasin H, in ovarian cancer cell line A2780. Our results showed that epoxycytochalasin H could inhibit ovarian cancer cell viability, impair mitochondrial function, activate mitophagy and induce the mitochondrial apoptosis pathway. Also, epoxycytochalasin H caused ER stress, and further induced ER stress-related apoptosis. We investigated the mechanism of apoptosis induced by epoxycytochalasin H and found that this plant-derived compound exhibited effective antitumor activity in ovarian cancer cells, by inducing apoptosis of tumor cells through multiple pathways. This may provide a starting point for the development of new anticancer drugs.
Acknowledgment
This study was funded by the Natural Science Foundation of Jilin Province (no. 20180623021TC, 20200703010ZP), the Fundamental Research Funds for the Central Universities, JLU.
Disclosure
The authors declare that they have no conflict of interest.
References
1. Calcabrini C, Catanzaro E, Bishayee A, Turrini E, Fimognari C. Marine sponge natural products with anticancer potential: an updated review. Mar Drugs. 2017;15(10):310. doi:10.3390/md15100310
2. Wang ZY. Ham-Wasserman lecture: treatment of acute leukemia by inducing differentiation and apoptosis. Hematol-Am Soc Hematol Educ Program. 2003:1–13. doi:10.1182/asheducation-2003.1.1
3. Zhang R-X, Li M-X, Jia Z-P. Rehmannia glutinosa: review of botany, chemistry and pharmacology. J Ethnopharmacol. 2008;117(2):199–214. doi:10.1016/j.jep.2008.02.018
4. Zong W-X, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61(5):667–676. doi:10.1016/j.molcel.2016.02.011
5. Kim KY, Yu SN, Lee SY, et al. Salinomycin-induced apoptosis of human prostate cancer cells due to accumulated reactive oxygen species and mitochondrial membrane depolarization. Biochem Biophys Res Commun. 2011;413(1):80–86. doi:10.1016/j.bbrc.2011.08.054
6. Millan A, Huerta S. Apoptosis-inducing factor and colon cancer. J Surg Res. 2009;151(1):163–170. doi:10.1016/j.jss.2007.05.020
7. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. doi:10.1056/NEJMra1205406
8. Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. doi:10.1016/j.cell.2014.02.049
9. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168(4):692–706. doi:10.1016/j.cell.2016.12.004
10. Mohammad MK, Avila D, Zhang J, et al. Acrolein cytotoxicity in hepatocytes involves endoplasmic reticulum stress, mitochondrial dysfunction and oxidative stress. Toxicol Appl Pharmacol. 2012;265(1):73–82. doi:10.1016/j.taap.2012.09.021
11. Jiang CC, Chen LH, Gillespie S, et al. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007;67(20):9750–9761. doi:10.1158/0008-5472.CAN-07-2047
12. Xu C, Yang M, Tian J, Wang X, Li Z. MALAT-1: a long non-coding RNA and its important 3’ end functional motif in colorectal cancer metastasis.. Int J Oncol. 2011;39(1):169–175. doi:10.3892/ijo.2011.1007
13. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi:10.1007/s10495-007-0756-2
14. Manji H, Kato T, Di Prospero NA, et al. Impaired mitochondrial function in psychiatric disorders. Nat Rev Neurosci. 2012;13(5):293. doi:10.1038/nrn3229
15. Singh KK. Mitochondria damage checkpoint in apoptosis and genome stability. FEMS Yeast Res. 2004;5(2):127–132. doi:10.1016/j.femsyr.2004.04.008
16. Geisler S, Holmström KM, Treis A, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6(7):871–878. doi:10.4161/auto.6.7.13286
17. Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309. doi:10.1038/nature14893
18. Appierto V, Tiberio P, Villani MG, Cavadini E, Formelli F. PLAB induction in fenretinide-induced apoptosis of ovarian cancer cells occurs via a ROS-dependent mechanism involving ER stress and JNK activation. Carcinogenesis. 2009;30(5):824–831. doi:10.1093/carcin/bgp067
19. Liu G, Sun Y, Li Z, et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochem Biophys Res Commun. 2008;370(4):651–656. doi:10.1016/j.bbrc.2008.04.031
20. Yu Z, Luo H, Fu W, Mattson MP. The endoplasmic reticulum stress-responsive protein GRP78 protects neurons against excitotoxicity and apoptosis: suppression of oxidative stress and stabilization of calcium homeostasis. Exp Neurol. 1999;155(2):302–314. doi:10.1006/exnr.1998.7002
21. Chen J, Wei H, Xie B, Wang B, Cheng J, Cheng J. Endoplasmic reticulum stress contributes to arsenic trioxide-induced apoptosis in drug-sensitive and-resistant leukemia cells. Leuk Res. 2012;36(12):1526–1535. doi:10.1016/j.leukres.2012.08.018
22. Hazalin NAMN, Ramasamy K, Lim SM, Cole AL, Majeed ABA. Induction of apoptosis against cancer cell lines by four ascomycetes (endophytes) from Malaysian rainforest. Phytomedicine. 2012;19(7):609–617. doi:10.1016/j.phymed.2012.01.007
23. Townsend PA, Stephanou A, Packham G, Latchman DS. BAG-1: a multi-functional pro-survival molecule. Int J Biochem Cell Biol. 2005;37(2):251–259. doi:10.1016/j.biocel.2004.03.016
24. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–359. doi:10.1038/ncb3124
25. Florea A-M, Büsselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers. 2011;3(1):1351–1371. doi:10.3390/cancers3011351
26. Rao RV, Bredesen DE. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr Opin Cell Biol. 2004;16(6):653–662. doi:10.1016/j.ceb.2004.09.012
27. Guo Z, Li CS, Wang CM, Xie YJ, Wang AL. CSE/H2S system protects mesenchymal stem cells from hypoxia and serum deprivation‑induced apoptosis via mitochondrial injury, endoplasmic reticulum stress and PI3K/Akt activation pathways. Mol Med Rep. 2015;12(2):2128–2134. doi:10.3892/mmr.2015.3651
28. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503–2518. doi:10.1016/j.biocel.2004.05.009
29. Shiba-Fukushima K, Imai Y, Yoshida S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2(1):1002. doi:10.1038/srep01002
30. Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14(10):1929–1938. doi:10.1089/ars.2010.3799
31. Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164(5):896–910. doi:10.1016/j.cell.2015.12.057
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.