Back to Journals » OncoTargets and Therapy » Volume 10
Epithelial-to-mesenchymal transition correlates with gefitinib resistance in NSCLC cells and the liver X receptor ligand GW3965 reverses gefitinib resistance through inhibition of vimentin
Authors Hu Y, Zang J, Qin X, Yan D, Cao H, Zhou L, Ni J, Yu S, Wu J, Feng JF ![]()
Received 15 October 2016
Accepted for publication 11 March 2017
Published 28 April 2017 Volume 2017:10 Pages 2341—2348
DOI https://doi.org/10.2147/OTT.S124757
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yong Hu,1,* Jialan Zang,2,* Xiaobing Qin,1 Dali Yan,1 Haixia Cao,1 Leilei Zhou,1 Jie Ni,1 Shaorong Yu,1 Jianzhong Wu,1 Ji-Feng Feng1
1Research Center for Clinical Oncology, Affiliated Cancer Hospital of Nanjing Medical University, Jiangsu Cancer Hospital and Jiangsu Institute of Cancer Research, Nanjing, Jiangsu, 2Department of Oncology, The First Hospital of Harbin City, Harbin, Heilongjiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: The role of epithelial-to-mesenchymal transition in cancer drug resistance is increasingly acknowledged. We examined whether epithelial-to-mesenchymal transition affects gefitinib resistance in non-small cell lung cancer (NSCLC) cells. Cell viability was detected by CCK-8 assay, VIM expression levels were determined by quantitative real-time polymerase chain reaction. Western blot and immunocytochemistry were performed to determine the protein expression level of vimentin. We observed morphologic differences between gefitinib-sensitive and -insensitive cells. Compared with the sensitive parental cell line, HCC827, vimentin expression levels were increased in HCC827 cells with acquired gefitinib resistance. Vimentin expression was also markedly upregulated in cells with intrinsic gefitinib resistance, and upregulated vimentin expression was correlated with gefitinib sensitivity. Our previous study demonstrated that coadministration of gefitinib and GW3965 resulted in decreased cell proliferation and induced apoptosis. Therefore, we investigated the relationship among GW3965, vimentin, and gefitinib resistance in NSCLC cells by analysis of the expression of vimentin in cells treated with a combination of gefitinib and GW3965. Gefitinib treatment led to increased levels of intracellular vimentin, while combined treatment with gefitinib and GW3965 resulted in decreased vimentin expression levels through reduction of gefitinib drug resistance in NSCLC cells. Overall, these findings suggest that vimentin expression is associated with sensitivity to gefitinib, and our study highlights the potential usefulness of the drug, GW3965, for reversal of gefitinib resistance through inhibition of vimentin expression.
Keywords: gefitinib resistance, LXR, EMT, GW3965, vimentin
Introduction
Lung cancer is the leading cause of cancer-related mortality,1 and tobacco use is the main risk factor for lung cancer development.2 The majority of patients with non-small cell lung cancer (NSCLC) are diagnosed at an advanced stage because the disease can be symptomless in its early stages.
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) consist of a large number of different small-molecule EGFR antagonists which have been used for the treatment of advanced NSCLC. EGFR-TKIs are well tolerated and have effective antitumor activity. Gefitinib is an orally administered selective EGFR-TKI that can inhibit proliferation of NSCLC cells. EGFR-mutated lung cancer responds well to gefitinib; however, all patients eventually develop resistance to the drug.3 Therefore, exploring the mechanisms of acquired gefitinib resistance could lead to changes in lung cancer therapy and potential clinical benefits. Recent research has revealed that smoking abolishes the therapeutic effects of EGFR-TKIs in NSCLC patients with EGFR mutations.4 Current smokers showed lower disease control rates and objective response rates and shorter progression-free and overall survival, compared with former smokers and never smokers when they received EGFR-TKI therapy.5 The stimulation of epithelial–mesenchymal transition (EMT) by cigarette smoking has been observed in lung cancer.6 For some patients, acquired resistance to EGFR inhibitors is associated with the phenomenon of EMT.7
EGFR has an important role in lung cancer tumor progression and metastasis, a key indicator of poor prognosis. Furthermore, the EMT process is receiving increasing attention due to its role in acquired drug resistance. This study focused on the link between EMT and drug resistance.8 EMT is a complex process, which is primarily characterized by downregulation of markers commonly expressed in epithelial cells, such as E-cadherin, and increased expression of mesenchymal markers, such as N-cadherin and vimentin.9 EMT has an important role in tumor invasion and metastasis. A recent in vitro study suggested a molecular and phenotypic association between EMT and chemoresistance.10 The acquisition of EMT features by tumors resistant to EGFR inhibition has been described previously, both in preclinical studies and in investigations using clinical samples.11 E-cadherin expression is significantly elevated following treatment of EGFR-TKI-resistant cell lines with TKIs. A previous study indicated that restoration of E-cadherin expression increased sensitivity to EGFR-TKIs.12 EMT progression is characterized by the loss of proteins involved in cell junctions, such as E-cadherin,13 and the gain of mesenchymal markers, such as vimentin.9 Genes affected by EMT include those associated with adherens junctions, such as E-cadherin (CDH1, CDH2, and CDH12), β-catenin (CTNNB1), mesenchymal markers such as VIM, and genes that regulate EMT gene expression, such as ZEB1, ZEB2, and SNAI1, SNAI3.13 Upregulation of mesenchymal-associated proteins, such as vimentin, is a signature of gefitinib resistance.13
Vimentin is a member of the intermediate filament protein family. Together with microtubules and actin filaments, vimentin is a component of the cytoskeleton. Vimentin expression is typically involved in cell adhesion, migration, apoptosis, and cell signal transduction. The research of Xu et al indicated that vimentin can modulate and control cellular processes, such as migration, and possibly also proliferation.14 High expression of vimentin is significantly associated with cell invasiveness. Furthermore, vimentin is expressed at significantly higher levels in drug-resistant cancer cells, including paclitaxel-resistant prostate cancer, GR malignant glioma,15 and tamoxifen-resistant breast cancer.16
Liver X receptor (LXR) belongs to the nuclear receptor family of ligand-dependent transcription factors, and ligands of LXR, such as TO901317 or GW3965, regulate cholesterol, glucose, and fatty acid metabolism in human cancer cells.17 Recent studies demonstrate that LXR activation can regulate tumor cell death,18 and our previous study indicated that LXR ligands, such as TO901317 and GW3965, can reverse acquired gefitinib resistance in NSCLC cells through inhibition of AKT phosphorylation;19 however, the effect of LXR inhibition on EMT has not been previously reported.
The goal of this study was to determine the mechanism by which EMT contributes to acquired gefitinib resistance and to test whether GW3965 can reverse GR cell growth by inhibition of the EMT mesenchymal marker, vimentin. This work could highlight novel potential strategies to reverse acquired gefitinib resistance in NSCLC.
Methods
Reagents
Reagents and suppliers were as follows: gefitinib (AstraZeneca UK Limited, Macclesfield, Cheshire, UK); CCK-8 cell viability kit (Dojindo, Kumamoto, Japan); GW3965 (Sigma-Aldrich Co., St Louis, MO, USA); penicillin–streptomycin solution (Thermo Fisher Scientific, Waltham, MA, USA); antibodies against vimentin, E-cadherin, β-actin, and EGFR (Cell Signaling Technology, Danvers, MA, USA); cell lines, HCC827, H1299, H1975, H1650, and H358 (Shanghai Institutes for Biological Sciences, China Academy of Cell Resource Center).
Cell culture
HCC827 GR cells were generated from HCC827 (gefitinib-sensitive) cells and maintained in the Affiliated Cancer Hospital of Nanjing Medical University. This was achieved by exposure of HCC827 cells to increasing concentrations of gefitinib, as previously described.20 All cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated FBS (Wisent Inc. Montreal, QC, Canada) and 1% penicillin–streptomycin solution.
Drug sensitivity assay
Cells were seeded in 96-well plates in 100 μL media at a density of 5×103 per well. After 24 h of incubation, cells were treated with indicated concentrations of gefitinib (1, 5, 10, 20, and 40 μM for resistant cell lines; 1, 5, 10, 20, and 40 nM for sensitive cell lines). GW3965 (5 μM) and gefitinib (5 μM) were used to treat cells separately or in combination, as previously described.19 After incubation for 72 h, CCK-8 assay reagent (0.5 mg/mL) was mixed with RPMI 1640 medium, added to cells, and the plates then read at 450 nm on an enzyme-linked immunosorbent assay plate reader to determine cell viability. Three individual experiments were performed.
Observation of morphologic changes
Cells were seeded into six-well flat-bottomed plates. Briefly, cells were synchronized by culture in serum-free RPMI 1640 medium for 1 h before the experiment and then washed twice with ice-cold phosphate-buffered saline (PBS). After synchronization, the cell morphology was analyzed under an inverted microscope. Three individual experiments were performed.
RNA isolation and quantitative real-time polymerase chain reaction (RT-PCR) analysis
Total RNA samples were extracted from sorted cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Total RNA was quantified using a spectrophotometer (NanoDrop Technologies). The mRNA levels were determined by quantitative RT-PCR and each sample was analyzed in triplicate. Complementary DNA was synthesized using RT Master Mix (Takara, Dalian, People’s Republic of China) according to the manufacturer’s instructions. Quantitative PCR was performed using SYBR Green PCR Mix (Roche, Mannheim, Germany) on a StepOnePlus RT PCR system (Applied Biosystems, Foster City, CA, USA). Primer sequences are presented in Table 1. Gene expression levels were calculated using the ΔΔCt method. Three individual experiments were performed.
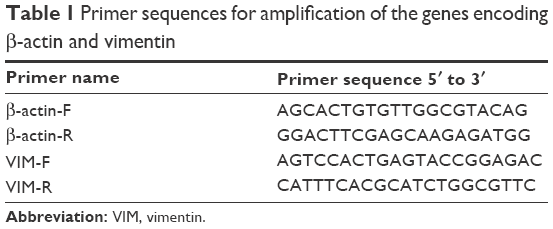 | Table 1 Primer sequences for amplification of the genes encoding β-actin and vimentin |
Transwell cell migration assay
A total of 5×104 cells were seeded into the top chamber of polycarbonate transwell filters. Transwell inserts were incubated at 37°C for at least 12 h before seeding cells. After incubation at 37°C for 24 h, the cells that adhered to the inserts and those inside the upper chamber were carefully wiped with cotton swabs. The cells that had migrated to the lower membrane surface were fixed with 70% methanol, stained with 0.1% crystal violet, and ten random 100× magnification fields per well were counted.
Protein extraction and Western blot analysis
Proteins were extracted using RIPA lysis buffer (Beyotime Biotechnology, Jiangsu, People’s Republic of China), resuspended in SDS-containing buffer (Beyotime Biotechnology, Jiangsu, People’s Republic of China), and denatured at 100°C for 10 min. Protein lysates were separated by 10% SDS-polyacrylamide gel electrophoresis gels for 90 min at 110 V and then transferred to polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). Membranes were blocked with 5% bovine serum albumin and then incubated with primary antibodies (1:1000) at 4°C overnight. Next, the membranes were incubated with secondary antibody for 2 h at room temperature. Bands were detected by chemiluminescence with BeyoECL Plus kits (Beyotime, People’s Republic of China). Three individual experiments were performed.
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde for 30 min and then incubated with primary antibodies (1:1000). Cells were then incubated with horseradish peroxidase-conjugated IgG antibody (Cell Signaling Technology, Danvers, MA, USA) for 1 h at room temperature. Then, the cells were incubated with anti-rabbit biotin-conjugated secondary antibody for 1 h, followed by treatment with Vectastain ABC reagent for 30 min at 4°C, and then stained with DAB and hematoxylin. Images were captured using a light microscope imaging system, and the selected regions of interest were outlined manually. Three individual experiments were performed.
Statistical analysis
The results of all experiments are expressed as mean ± standard deviation of at least three separate tests. Statistical comparisons between groups were performed using Student’s t-tests.
Results
Analysis of gefitinib sensitivity
We classified the cell lines into those that were insensitive (H1299, H1975), sensitive (HCC827), and moderately sensitive (H358) to gefitinib-mediated growth inhibition in vitro. Sensitivity was defined as >50% in vitro growth inhibition at a gefitinib concentration of 5 μmol/L. IC50 values of gefitinib for the H1299, H1975, and H358 cell lines are presented in Table 2. Next, we established cell lines with acquired gefitinib resistance. After a continuous treatment with gefitinib at gradually increasing concentrations, HCC827/GR cell lines grew steadily in culture medium containing gefitinib. Next, we compared the drug sensitivity of the HCC827/GR cell lines to that of the parental (HCC827) cells. IC50 values of HCC827/GR cell lines are presented in Table 2. The results indicate that GR cell lines were successfully established from parental gefitinib-sensitive HCC827 cells.
 | Table 2 Gefitinib IC50 values of H1975, H1299, H358, HCC827, HCC827/GR-8-1, HCC827/GR-8-2, and HCC827/GR-8-12 cell lines |
Acquisition of gefitinib resistance is associated with changes in cell morphology and migration
GR cell lines exhibited significant morphologic differences compared with sensitive cells (Figure 1A). GR cells typically showed varying degrees of shrinkage, blebbing, aggregation, and lack of obvious boundaries, whereas gefitinib-sensitive cells had clear, regular boundaries. Cell lines with both intrinsic and acquired gefitinib resistance exhibited morphologic changes consistent with EMT, consistent with the fact that GR cells acquire migratory properties, contributing to cancer metastasis. The mesenchymal phenotype of these resistant cells was confirmed by their increased capacity for migration. We observed that resistant cells acquired increased migratory ability, as determined by transwell migration assays (Figure 1B). Compared with sensitive HCC827 cells and moderately sensitive H358 cells, the GR H1299 and H1975 NSCLC cell lines with morphology consistent with EMT displayed higher migration capability. Furthermore, our data demonstrate that cells with acquired gefitinib resistance also showed moderately increased migration capacity, despite the poor migration ability of the parental HCC827 cell line (Figure 1B). These results reveal that the phenotype associated with intrinsic or acquired gefitinib resistance involves EMT, resulting in higher migration capacity. Our results demonstrate a potential link between migration capacity and gefitinib resistance.
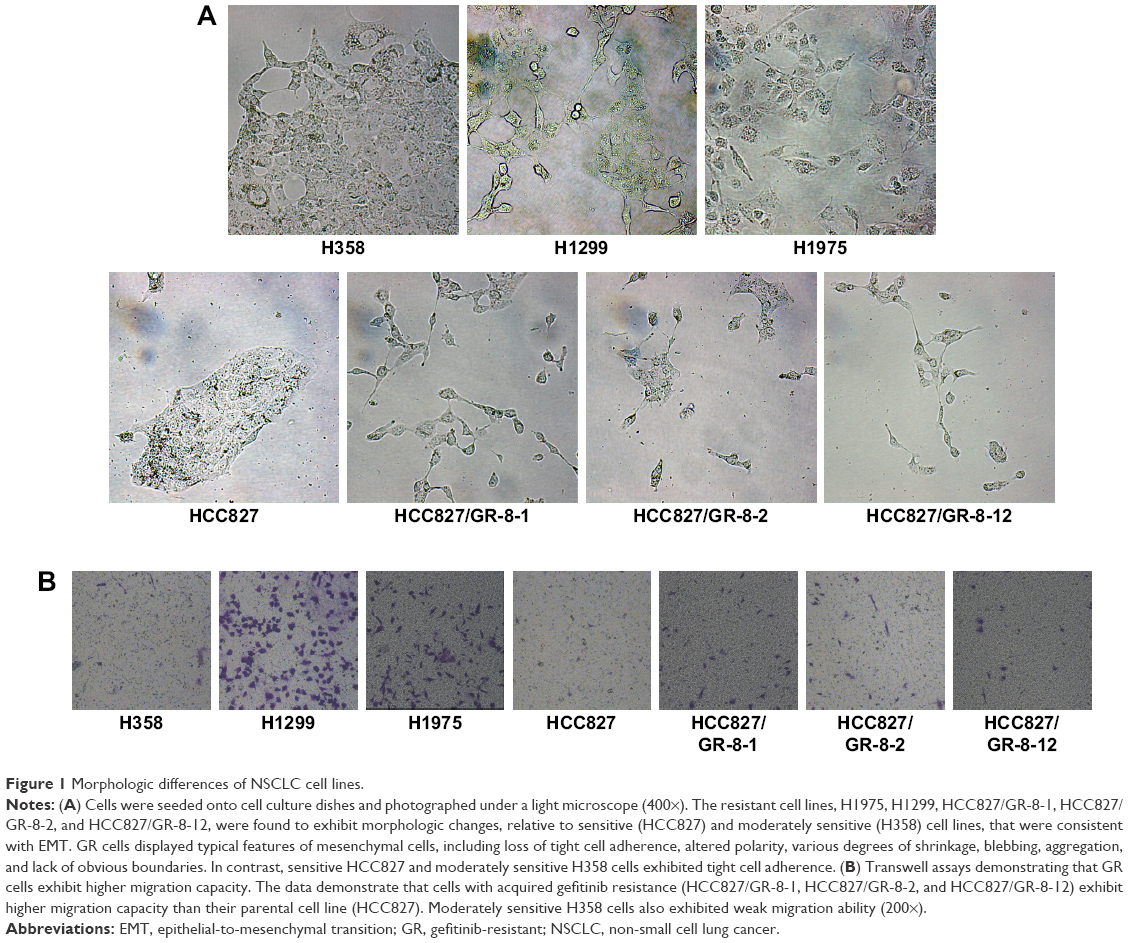 | Figure 1 Morphologic differences of NSCLC cell lines. |
Altered expression of the epithelial and mesenchymal cell marker, vimentin, is correlated with sensitivity to gefitinib
To confirm EMT in GR cells, we performed genome-wide mRNA expression analysis of the parental HCC827 and the GR HCC827/GR-8-1 cells, and EMT markers with altered expression were subject to further analysis. A total of 18 mRNAs, including the EMT-related gene VIM, were upregulated in the GR HCC827/GR-8-1 cell line relative to gefitinib-sensitive HCC827 cells (Figure 2A). In addition, the GR cell lines H1299 and H1975 exhibited substantial upregulation of VIM. Similarly, insensitive cell lines, including HCC827/GR-8-1, HCC827/GR-8-2, and HCC827/GR-8-12, appeared to have undergone EMT, as judged by VIM expression. Hence, consistent with the observed morphologic changes, GR cells showed upregulation of the mesenchymal marker vimentin. As shown in Figure 2B, VIM expression levels were dramatically higher in GR than in parental cells. These results suggest that gefitinib resistance is associated with EMT.
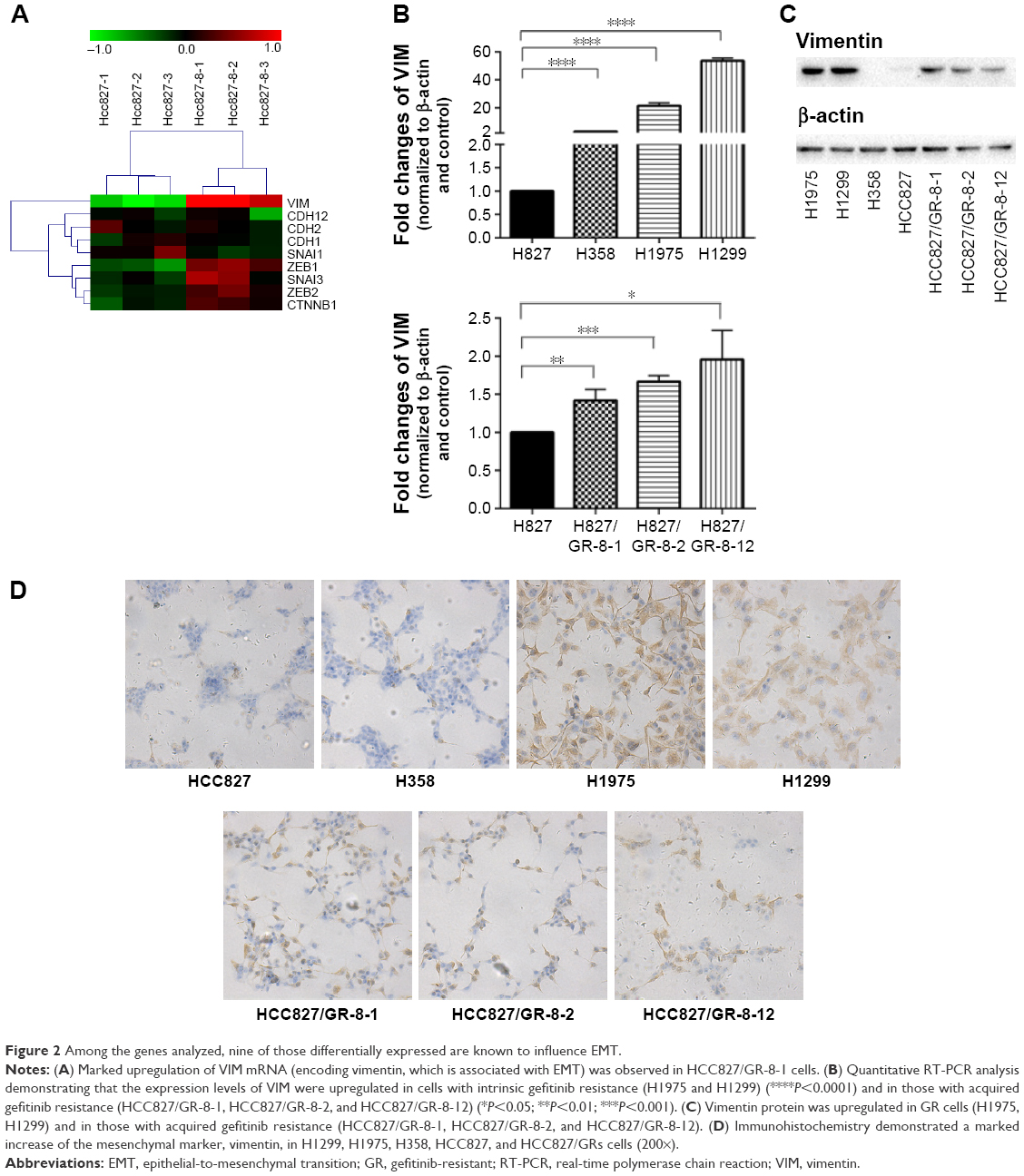 | Figure 2 Among the genes analyzed, nine of those differentially expressed are known to influence EMT. |
To better understand the mechanisms of acquired gefitinib resistance, we examined differences in protein expression of mesenchymal markers between gefitinib-sensitive and -insensitive NSCLC lines. An obvious difference in expression of vimentin was observed between gefitinib-sensitive and -resistant NSCLC cells (Figure 2C). The expression of vimentin was elevated in GR H1299 and H1975 cells, whereas relatively low levels of vimentin expression were observed in gefitinib-sensitive NSCLC lines. The substantial upregulation of vimentin associated with gefitinib resistance was subsequently studied in HCC827 parental/resistant cell line pairs, revealing that vimentin protein was significantly upregulated in HCC827/GR-8-1, HCC827/GR-8-2, and HCC827/GR-8-12 cells compared with the parental gefitinib-sensitive cell line. The majority of cell lines analyzed retained the expression of E-cadherin. Hence, although the insensitive cells maintained expression of some epithelial cell proteins, they also began to express mesenchymal proteins.
We conducted further investigation of the expression levels of vimentin in gefitinib-sensitive and -insensitive NSCLC lines by immunohistochemistry. A marked increase in expression of mesenchymal markers, including vimentin, was observed in the GR lines, H1299, H1975, H358, and HCC827/GRs (Figure 2D). In addition, the ratio of mesenchymal vimentin to epithelial E-cadherin was enhanced in cells with acquired gefitinib resistance, compared with their parental counterparts.
GW3965 treatment alleviates gefitinib sensitivity and is associated with suppression of vimentin expression
In our previous study, we demonstrated that GW3965 can reverse GR cell growth of HCC827/GR cells.19 Treatment of HCC827/GR cells with a combination of gefitinib (5 μM) and GW3965 (5 μM) led to significant suppression of cell viability.19 In lung cancer, EMT is not required for metastasis, but can contribute to chemoresistance.21 A recent study revealed that the EMT phenotype is associated with both intrinsic and acquired resistance to EGFR-specific TKIs in NSCLC cell lines.22 Upon induction of EMT, HCC827 cells become significantly more resistant to EGFR-TKI.7
In this study, EMT-induced cells were found to be insensitive to treatment with gefitinib, while cotreatment with GW3965 resensitized the GR cells. The exposure of HCC827 cells to gefitinib for several days resulted in the expected EMT, as assessed by increased vimentin expression (Figure 3). We next sought to determine the mechanism of drug resistance following EMT, and tested the possibility that recombinant GW3965 could alleviate gefitinib resistance. HCC827/GR-8-1 cells were exposed to gefitinib and GW3965, either in combination or separately, in 72 h viability assays. We found that vimentin was significantly overexpressed in the gefitinib-treated group, compared with the control group. GW3965 alone had no effect on vimentin levels, while treatment of HCC827/GR-8-1 cells with GW3965 (5 μM) and gefitinib (5 μM) indicated that GW3965 decreased vimentin levels in the presence of gefitinib, suggesting that GW3965 inhibits EMT in gefitinib-treated HCC827/GR-8-1 cells. Moreover, GW3965 inhibition of vimentin expression in HCC827/GR-8-1 cells was dose-dependent in the presence of gefitinib. These findings suggest that combined treatment with GW3965 can reverse acquired drug resistance in HCC827/GR-8-1 cancer cells by inhibition of the gefitinib-induced increase in vimentin expression.
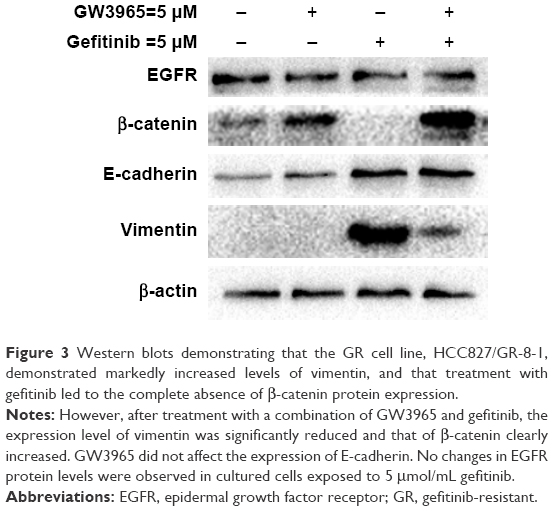 | Figure 3 Western blots demonstrating that the GR cell line, HCC827/GR-8-1, demonstrated markedly increased levels of vimentin, and that treatment with gefitinib led to the complete absence of β-catenin protein expression. |
Discussion
The EGFR-TKI, gefitinib, is used as a second- or third-line treatment for patients with mutant EGFR.23 Despite frequent dramatic responses to gefitinib, nearly all patients develop resistance and undergo disease progression.22 Acquisition of drug resistance is a common phenomenon during tumor progression, and can arise through a wide range of mechanisms, including changes in drug efflux or drug metabolism and specific mutations of drug targets, among others.
Data suggest that EMT may be a general biologic switch rendering NSCLC sensitive or insensitive to EGFR inhibition. Our study revealed that the expression of epithelial- and mesenchymal-associated proteins differs between gefitinib-sensitive and -resistant NSCLC cell lines. Genes associated with mesenchymal proteins, such as vimentin, were highly expressed in NSCLC cell lines resistant to gefitinib, while they exhibited low expression levels in sensitive lines.
The GR cell model established in our laboratory provided us with a unique opportunity to study the contribution of EMT to acquired EGFR-TKI resistance. Our results demonstrate that long-term exposure of NSCLC cells to gefitinib results in the acquisition of phenotypic features associated with tumor EMT. In summary, sensitive cells remaining after long-term exposure to gefitinib exhibited an EMT phenotype accompanied by acquired drug resistance. This was demonstrated by the substantial upregulation of markers, including vimentin, typically associated with the mesenchymal phenotype in tumor cells resistant to gefitinib. Our results show that the expression levels of the vimentin protein and its encoding gene are significantly increased in GR cells. These results indicated that vimentin could be associated with the mechanism underlying acquired gefitinib resistance.
In the context of gefitinib resistance, we demonstrated that GW3965 can reverse mesenchymal tumor features and resensitize tumor cells to the cytotoxic effects of gefitinib. We previously determined that GW3965 can enhance the inhibitory effects of gefitinib on the growth of GR HCC827/GR-8-1, and our results demonstrated that HCC827/GR-8-1 cells treated with gefitinib at different concentrations in the presence of GW3965 at 5 μM had a reduced IC50 for gefitinib. These data suggest that GW3965 has synergistic effects with gefitinib in HCC827-8-1 cells.19 During treatment with gefitinib, the HCC827/GR-8-1 cells exhibit upregulation of E-cadherin. E-cadherin expression is also significantly elevated following treatment with TKIs. A previous study indicated that restoring E-cadherin expression increases sensitivity to EGFR-TKIs.12 However, although the cell lines were gefitinib resistant, treatment with the drug was still able to induce upregulation of E-cadherin expression.
Moreover, gefitinib-treated HCC827/GR-8-1 cells exhibited significantly upregulated expression of vimentin. Together with other recent studies, which suggest an association between EMT and drug resistance,24 our results confirm the association of vimentin expression with gefitinib resistance in NSCLC cell lines. We also assessed the effects of GW3965 combined with gefitinib. Combination treatment led to a reduction of vimentin upregulation stimulated by gefitinib in HCC827/GR-8-1, and restoration of β-catenin expression, which was ablated by gefitinib treatment. The decreased expression of vimentin is likely to contribute to the restoration of drug sensitivity; however, the combination of the two drugs was also associated with maintenance of E-cadherin upregulation, which is also associated with decreased resistance to gefitinib.
A recent report showed that LXR ligands can be used in combined therapeutics with other anticancer drugs.25 However, it remains unclear how GW3965 interferes with EMT. Considering the marked changes in vimentin expression after treatment with gefitinib in resistant cells, there may be a threshold for the function of LXR agonists. LXR ligands trigger cell signaling pathway regulation, which may also participate in this process. Meanwhile, as a drug affecting lipid metabolism, GW3965 may interfere with lipid-dependent processes, such as cholesterol pump effusion, to influence cell signaling. Here, we explored the ability of GW3965 to reverse drug resistance in GR cells. Understanding of the specific role of GW3965 in this context will require further investigation.
In conclusion, GR human NSCLC cell lines were established and found to exhibit dysregulated expression of vimentin, which may have a prominent role in gefitinib resistance of NSCLC cells. This study investigated the resistance mechanisms of NSCLC and provided a theoretical basis for the reversal of gefitinib resistance through inhibition of vimentin using LXR ligands, such as GW3965. Until recently, the majority of data connecting EMT with drug resistance was primarily derived from in vitro studies;26 therefore, additional research will be required in the future to confirm the applicability of these findings in vivo and, ultimately, their clinical utility.
Acknowledgment
This work was supported by grants from the National Nature Science Foundation of China (81372396).
Disclosure
The authors report no conflicts of interest in this work.
References
Pan H, Jiang T, Cheng N, et al. Long non-coding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget.2016;7(31):49948–49960. | ||
Chapman AM, Sun KY, Ruestow P, Cowan DM, Madl AK. Lung cancer mutation profile of EGFR, ALK, and KRAS: Meta-analysis and comparison of never and ever smokers. Lung Cancer.2016;102:122–134. | ||
Shrader M, Pino MS, Brown G, et al. Molecular correlates of gefitinib responsiveness in human bladder cancer cells. Mol Cancer Ther.2007;6(1):277–285. | ||
Liu M, Zhou C, Zheng J. Cigarette smoking impairs the response of EGFR-TKIs therapy in lung adenocarcinoma patients by promoting EGFR signaling and epithelial-mesenchymal transition. Am J Transl Res.2015;7(10):2026–2035. | ||
Li D, Zhang L, Zhou J, Chen H. Cigarette smoke extract exposure induces EGFR-TKI resistance in EGFR-mutated NSCLC via mediating Src activation and EMT. Lung Cancer.2016;93:35–42. | ||
Vu T, Jin L, Datta PK. Effect of cigarette smoking on epithelial to mesenchymal transition (EMT) in lung cancer. J Clin Med.2016;5(4). | ||
Wilson C, Nicholes K, Bustos D, et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget.2014;5(17):7328–7341. | ||
Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules.2016;21(7). | ||
Yin H, Wang Y, Chen W, Zhong S, Liu Z, Zhao J. Drug-resistant CXCR4-positive cells have the molecular characteristics of EMT in NSCLC. Gene.2016;594(1):23–29. | ||
Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene.2010;29(34):4741–4751. | ||
Thomson S, Buck E, Petti F, et al. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res.2005;65(20):9455–9462. | ||
Bai XY, Zhang XC, Yang SQ, et al. Blockade of hedgehog signaling synergistically increases sensitivity to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer cell lines. PLoS One.2016;11(3):e0149370. | ||
Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther.2007;6(6):1683–1691. | ||
Xu J, Yang H, Zhou X, Wang H, Gong L, Tang C. Bisdemethoxycurcumin suppresses migration and invasion of highly metastatic 95D lung cancer cells by regulating E-cadherin and vimentin expression, and inducing autophagy. Mol Med Rep.2015;12(5):7603–7608. | ||
Yan YR, Xie Q, Li F, et al. Epithelial-to-mesenchymal transition is involved in BCNU resistance in human glioma cells. Neuropathology.2014;34(2):128–134. | ||
Liu H, Zhang HW, Sun XF, et al. Tamoxifen-resistant breast cancer cells possess cancer stem-like cell properties. Chin Med J (Engl).2013;126(16):3030–3034. | ||
Shrestha E, Hussein MA, Savas JN, et al. Poly(ADP-ribose) polymerase 1 represses liver X receptor-mediated ABCA1 expression and cholesterol efflux in macrophages. J Biol Chem.2016;291(21):11172–11184. | ||
Zhang R, Liu Z, Li Y, Wu B. LXR agonist regulates the proliferation and apoptosis of human T-Cell acute lymphoblastic leukemia cells via the SOCS3 pathway. Int J Biochem Cell Biol.2016;78:180–185. | ||
Wu Y, Yu DD, Hu Y, et al. LXR ligands sensitize EGFR-TKI-resistant human lung cancer cells in vitro by inhibiting Akt activation. Biochem Biophys Res Commun.2015;467(4):900–905. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science.2007;316(5827):1039–1043. | ||
Fischer KR, Durrans A, Lee S, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature.2015;527(7579):472–476. | ||
Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis.2013;2:e39. | ||
Fernando RI, Hamilton DH, Dominguez C, David JM, McCampbell KK, Palena C. IL-8 signaling is involved in resistance of lung carcinoma cells to erlotinib. Oncotarget.2016;7(27):42031–42044. | ||
Ma JL, Zeng S, Zhang Y, Deng GL, Shen H. Epithelial-mesenchymal transition plays a critical role in drug resistance of hepatocellular carcinoma cells to oxaliplatin. Tumour Biol.2016;37(5):6177–6184. | ||
Wairagu PM, Park KH, Kim J, et al. Combined therapeutic potential of nuclear receptors with receptor tyrosine kinase inhibitors in lung cancer. Biochem Biophys Res Commun.2014;447(3):490–495. | ||
Kim H, Yoo SB, Sun P, et al. Alteration of the E-cadherin/beta-catenin complex is an Independent poor prognostic factor in lung adenocarcinoma. Korean J Pathol.2013;47(1):44–51. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.